
The Glioma Immune Landscape: A Double-Edged Sword for Treatment Regimens



Abstract
:Simple Summary
Abstract
1. Introduction
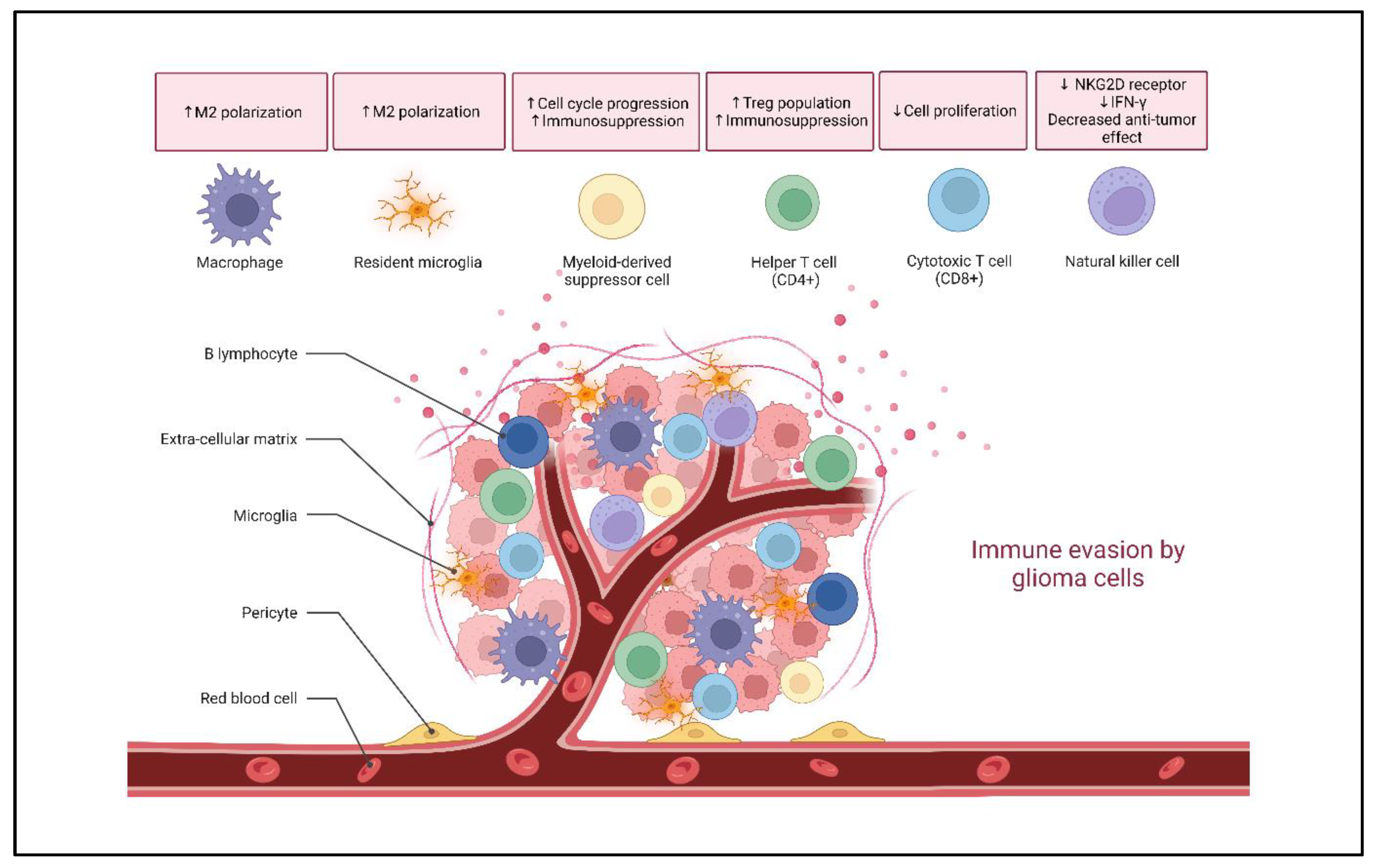
2. Glioma Immune Landscape
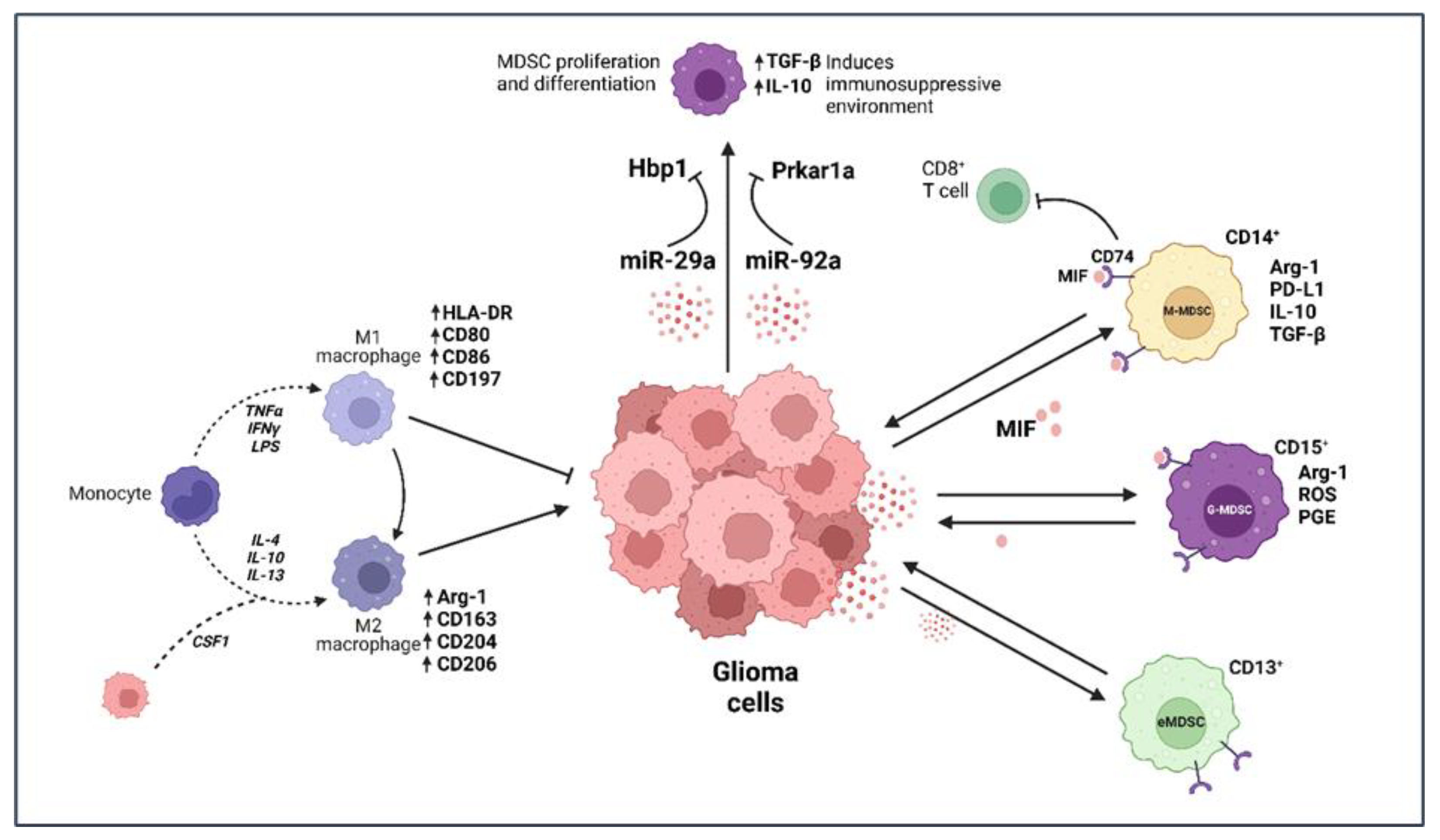
2.1. Microglia and Macrophages
2.2. Myeloid-Derived Suppressor Cells (MDSCs)
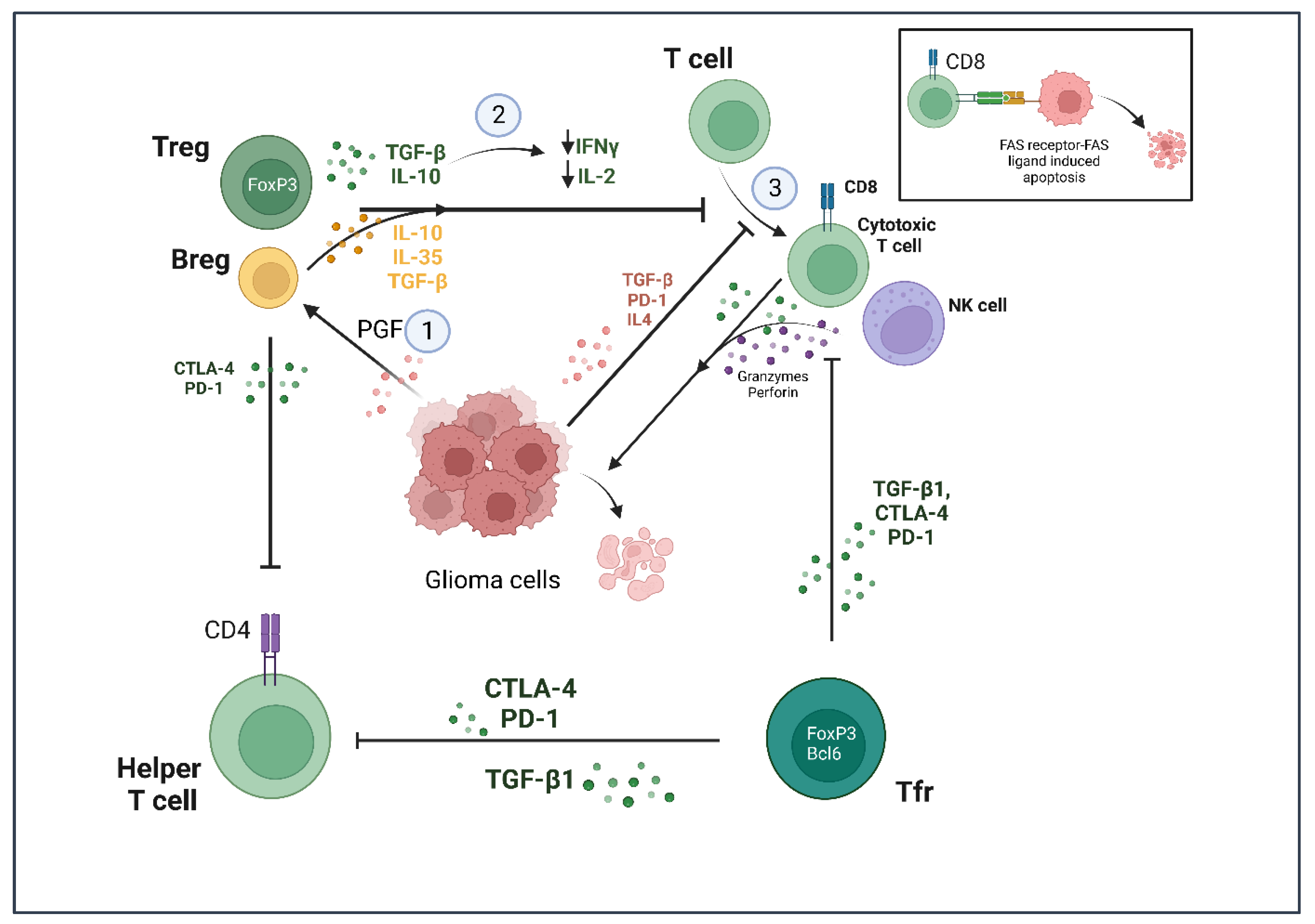
2.3. T Lymphocytes
2.4. B Lymphocytes
2.5. Natural Killer (NK) Cells
3. Currently Known Immunotherapies
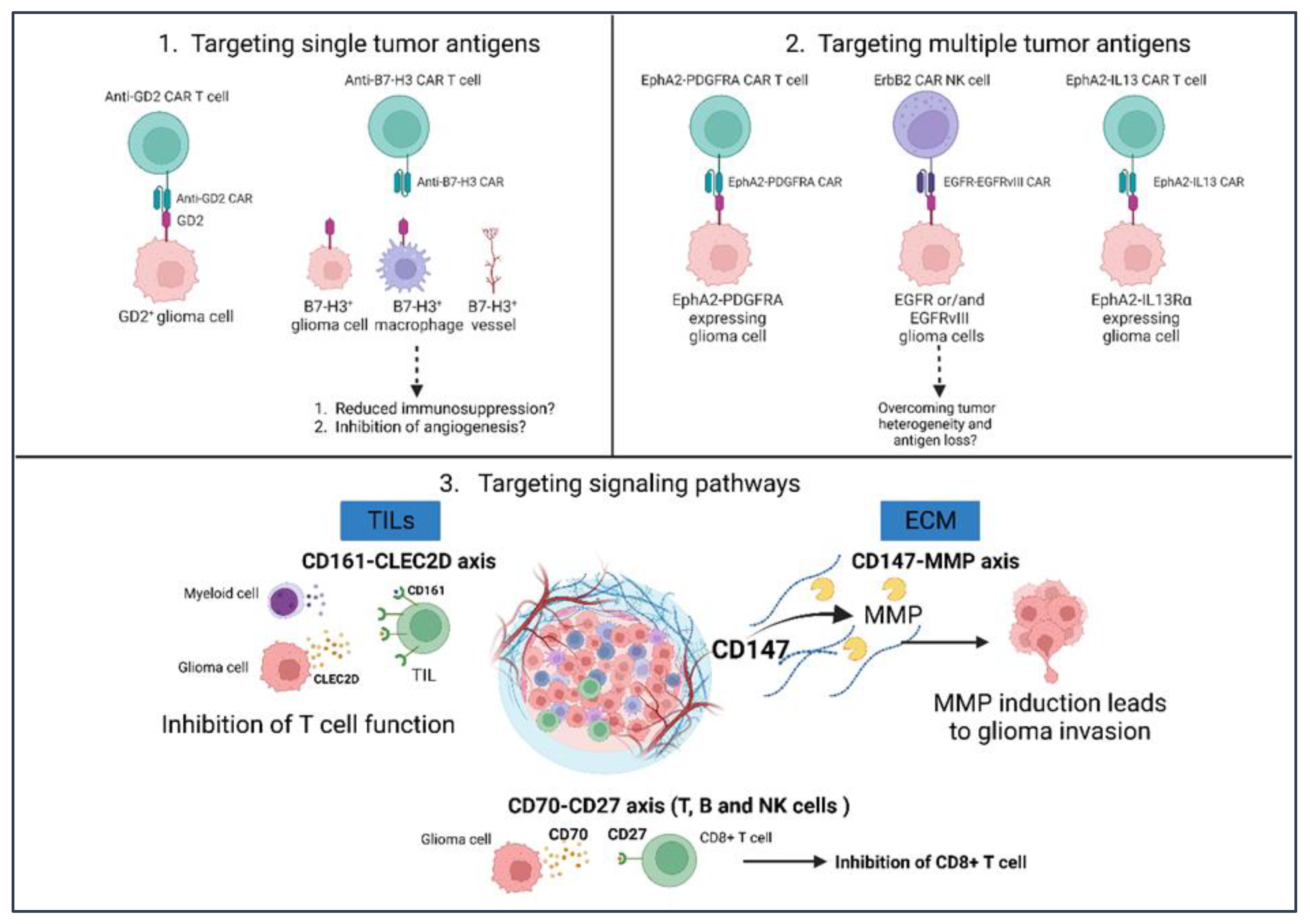
3.1. CAR-T Cells
3.2. Tumor Vaccines
3.3. Immune Checkpoint Inhibitors
3.4. Oncolytic Viruses
4. Potential Glioma Immunotherapies
5. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alexander, B.M.; Cloughesy, T.F. Adult Glioblastoma. J. Clin. Oncol. 2017, 35, 2402–2409. [Google Scholar] [CrossRef] [PubMed]
- Rapôso, C.; Vitorino-Araujo, J.L.; Barreto, N. Molecular Markers of Gliomas to Predict Treatment and Prognosis: Current State and Future Directions. In Gliomas; Debinski, W., Ed.; Exon Publications: Brisbane, Australia, 2021; Chapter 10. Available online: https://www.ncbi.nlm.nih.gov/books/NBK570713/ (accessed on 8 January 2023). [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A Summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- De Vleeschouwer, S. (Ed.) Glioblastoma; Exon Publications: Brisbane, Australia, 2017. Available online: https://www.ncbi.nlm.nih.gov/books/NBK469998/ (accessed on 8 January 2023). [CrossRef]
- Molinaro, A.M.; Taylor, J.W.; Wiencke, J.K.; Wrensch, M.R. Genetic and Molecular Epidemiology of Adult Diffuse Glioma. Nat. Rev. Neurol. 2019, 15, 405–417. [Google Scholar] [CrossRef]
- Fernandes, C.; Costa, A.; Osório, L.; Lago, R.C.; Linhares, P.; Carvalho, B.; Caeiro, C. Current Standards of Care in Glioblastoma Therapy. In Glioblastoma; De Vleeschouwer, S., Ed.; Exon Publications: Brisbane, Australia, 2017; Chapter 11. Available online: https://www.ncbi.nlm.nih.gov/books/NBK469987/ (accessed on 8 January 2023). [CrossRef] [Green Version]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.M.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients with Glioblastoma a Randomized Clinical Trial. JAMA—J. Am. Med. Assoc. 2017, 318, 2306–2316. [Google Scholar] [CrossRef] [Green Version]
- Hotchkiss, K.M.; Sampson, J.H. Temozolomide treatment outcomes and immunotherapy efficacy in brain tumor. J. Neurooncol. 2021, 151, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant Astrocytic Glioma: Genetics, Biology, and Paths to Treatment. Genes Dev. 2007, 21, 2683–2710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenger, C.; Miranda, P.C.; Salvador, R.; Thielscher, A.; Bomzon, Z.; Giladi, M.; Mrugala, M.M.; Korshoej, A.R. A Review on Tumor-Treating Fields (TTFields): Clinical Implications Inferred from Computational Modeling. IEEE Rev. Biomed. Eng. 2018, 11, 195–207. [Google Scholar] [CrossRef] [Green Version]
- Prabhu, S.S.; Broaddus, W.C.; Oveissi, C.; Berr, S.S.; Gillies, G.T. Determination of Intracranial Tumor Volumes in a Rodent Brain Using Magnetic Resonance Imaging, Evans Blue, and Histology: A Comparative Study. IEEE Trans. Biomed. Eng. 2000, 47, 259–265. [Google Scholar] [CrossRef]
- Cao, Y.; Sundgren, P.C.; Tsien, C.I.; Chenevert, T.T.; Junck, L. Physiologic and Metabolic Magnetic Resonance Imaging in Gliomas. J. Clin. Oncol. 2006, 24, 1228–1235. [Google Scholar] [CrossRef]
- Parker, N.R.; Khong, P.; Parkinson, J.F.; Howell, V.M.; Wheeler, H.R. Molecular Heterogeneity in Glioblastoma: Potential Clinical Implications. Front. Oncol. 2015, 5, 55. [Google Scholar] [CrossRef]
- Nicholson, J.G.; Fine, H.A. Diffuse Glioma Heterogeneity and Its Therapeutic Implications. Cancer Discov. 2021, 11, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Vessoni, A.T.; Filippi-Chiela, E.C.; Lenz, G.; Batista, L.F.Z. Tumor Propagating Cells: Drivers of Tumor Plasticity, Heterogeneity, and Recurrence. Oncogene 2020, 39, 2055–2068. [Google Scholar] [CrossRef]
- Wintterle, S.; Schreiner, B.; Mitsdoerffer, M.; Schneider, D.; Chen, L.; Meyermann, R.; Weller, M.; Wiendl, H. Expression of the B7-Related Molecule B7-H1 by Glioma Cells: A Potential Mechanism of Immune Paralysis. Cancer Res. 2003, 63, 7462–7467. [Google Scholar]
- Xue, S.; Hu, M.; Iyer, V.; Yu, J. Blocking the PD-1/PD-L1 Pathway in Glioma: A Potential New Treatment Strategy. J. Hematol. Oncol. 2017, 10, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, R.S.; Anand, A.; Harwood, D.S.L.; Kristensen, B.W. Tumor-Associated Microglia and Macrophages in the Glioblastoma Microenvironment and Their Implications for Therapy. Cancers 2021, 13, 4255. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Schmidt, S.v.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; DeNardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-Based Network Analysis Reveals a Spectrum Model of Human Macrophage Activation. Immunity 2014, 40, 274–288. [Google Scholar] [CrossRef] [Green Version]
- DeNardo, D.G.; Ruffell, B. Macrophages as Regulators of Tumour Immunity and Immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef]
- Bowman, R.L.; Klemm, F.; Akkari, L.; Pyonteck, S.M.; Sevenich, L.; Quail, D.F.; Dhara, S.; Simpson, K.; Gardner, E.E.; Iacobuzio-Donahue, C.A.; et al. Macrophage Ontogeny Underlies Differences in Tumor-Specific Education in Brain Malignancies. Cell Rep. 2016, 17, 2445–2459. [Google Scholar] [CrossRef] [Green Version]
- Vidyarthi, A.; Agnihotri, T.; Khan, N.; Singh, S.; Tewari, M.K.; Radotra, B.D.; Chatterjee, D.; Agrewala, J.N. Predominance of M2 Macrophages in Gliomas Leads to the Suppression of Local and Systemic Immunity. Cancer Immunol. Immunother. 2019, 68, 1995–2004. [Google Scholar] [CrossRef]
- Akkari, L.; Quail, D.F.; Quick, M.L.; Huse, J.T.; Sutton, J.C.; Joyce, J.A. Abstract A33: Combinatorial Targeting of Tumor-Associated Macrophages/ Microglia and Radiotherapy in Gliomas. Cancer Res. 2015, 75, A33. [Google Scholar] [CrossRef]
- Tomaszewski, W.; Sanchez-Perez, L.; Gajewski, T.F.; Sampson, J.H. Brain Tumor Microenvironment and Host State: Implications for Immunotherapy. Clin. Cancer Res. 2019, 25, 4202–4210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabrilovich, D.I. Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mi, Y.; Guo, N.; Luan, J.; Cheng, J.; Hu, Z.; Jiang, P.; Jin, W.; Gao, X. The Emerging Role of Myeloid-Derived Suppressor Cells in the Glioma Immune Suppressive Microenvironment. Front. Immunol. 2020, 11, 737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate Mapping Analysis Reveals That Adult Microglia Derive from Primitive Macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Barres, B.A. Microglia and Macrophages in Brain Homeostasis and Disease. Nat. Rev. Immunol. 2018, 18, 225–242. [Google Scholar] [CrossRef]
- Prinz, M.; Erny, D.; Hagemeyer, N. Ontogeny and Homeostasis of CNS Myeloid Cells. Nat. Immunol. 2017, 18, 385–392. [Google Scholar] [CrossRef]
- Zeiner, P.S.; Preusse, C.; Golebiewska, A.; Zinke, J.; Iriondo, A.; Muller, A.; Kaoma, T.; Filipski, K.; Müller-Eschner, M.; Bernatz, S.; et al. Distribution and Prognostic Impact of Microglia/Macrophage Subpopulations in Gliomas. Brain Pathol. 2019, 29, 513–529. [Google Scholar] [CrossRef]
- Andersen, J.K.; Miletic, H.; Hossain, J.A. Tumor-Associated Macrophages in Gliomas—Basic Insights and Treatment Opportunities. Cancers 2022, 14, 1319. [Google Scholar] [CrossRef]
- Mira, E.; Carmona-Rodríguez, L.; Tardáguila, M.; Azcoitia, I.; González-Martín, A.; Almonacid, L.; Casas, J.; Fabriás, G.; Mañes, S. A Lovastatin-Elicited Genetic Program Inhibits M2 Macrophage Polarization and Enhances T Cell Infiltration into Spontaneous Mouse Mammary Tumors. Oncotarget 2013, 4, 2288–2301. [Google Scholar] [CrossRef] [Green Version]
- Tong, N.; He, Z.; Ma, Y.; Wang, Z.; Huang, Z.; Cao, H.; Xu, L.; Zou, Y.; Wang, W.; Yi, C.; et al. Tumor Associated Macrophages, as the Dominant Immune Cells, Are an Indispensable Target for Immunologically Cold Tumor—Glioma Therapy? Front. Cell Dev. Biol. 2021, 9, 706286. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, M.; Liu, S.; Guo, J.; Lu, Y.; Cheng, J.; Liu, J. Macrophage-Derived Extracellular Vesicles: Diverse Mediators of Pathology and Therapeutics in Multiple Diseases. Cell Death Dis. 2020, 11, 924. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Smith, W.; Hao, D.; He, B.; Kong, L. M1 and M2 Macrophage Polarization and Potentially Therapeutic Naturally Occurring Compounds. Int. Immunopharmacol. 2019, 70, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 Polarization and Metabolic States. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef] [Green Version]
- Ellert-Miklaszewska, A.; Dabrowski, M.; Lipko, M.; Sliwa, M.; Maleszewska, M.; Kaminska, B. Molecular Definition of the Pro-Tumorigenic Phenotype of Glioma-Activated Microglia. Glia 2013, 61, 1178–1190. [Google Scholar] [CrossRef]
- Ellert-Miklaszewska, A.; Wisniewski, P.; Kijewska, M.; Gajdanowicz, P.; Pszczolkowska, D.; Przanowski, P.; Dabrowski, M.; Maleszewska, M.; Kaminska, B. Tumour-Processed Osteopontin and Lactadherin Drive the Protumorigenic Reprogramming of Microglia and Glioma Progression. Oncogene 2016, 35, 6366–6377. [Google Scholar] [CrossRef]
- Wu, S.Y.; Xing, F.; Sharma, S.; Wu, K.; Tyagi, A.; Liu, Y.; Zhao, D.; Deshpande, R.P.; Shiozawa, Y.; Ahmed, T.; et al. Nicotine Promotes Brain Metastasis by Polarizing Microglia and Suppressing Innate Immune Function. J. Exp. Med. 2020, 217, e20191131. [Google Scholar] [CrossRef]
- Walker, D.G.; Lue, L.F. Immune Phenotypes of Microglia in Human Neurodegenerative Disease: Challenges to Detecting Microglial Polarization in Human Brains. Alzheimers Res. Ther. 2015, 7, 56. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Geng, X.; Hou, J.; Wu, G. New Insights into M1/M2 Macrophages: Key Modulators in Cancer Progression. Cancer Cell Int. 2021, 21, 389. [Google Scholar] [CrossRef]
- Smith, T.D.; Nagalla, R.R.; Chen, E.Y.; Liu, W.F. Harnessing Macrophage Plasticity for Tissue Regeneration. Adv. Drug Deliv. Rev. 2017, 114, 193–205. [Google Scholar] [CrossRef]
- Kubota, K.; Moriyama, M.; Furukawa, S.; Rafiul, H.A.S.M.; Maruse, Y.; Jinno, T.; Tanaka, A.; Ohta, M.; Ishiguro, N.; Yamauchi, M.; et al. CD163+CD204+ Tumor-Associated Macrophages Contribute to T Cell Regulation via Interleukin-10 and PD-L1 Production in Oral Squamous Cell Carcinoma. Sci. Rep. 2017, 7, 1755. [Google Scholar] [CrossRef] [Green Version]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R Inhibition Alters Macrophage Polarization and Blocks Glioma Progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Xue, H.; Shao, Q.; Wang, J.; Guo, X.; Chen, X.; Zhang, J.; Xu, S.; Li, T.; Zhang, P.; et al. Hypoxia Promotes Glioma-Associated Macrophage Infiltration via Periostin and Subsequent M2 Polarization by Upregulating TGF-Beta and M-CSFR. Oncotarget 2016, 7, 80521–80542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azambuja, J.H.; Ludwig, N.; Yerneni, S.S.; Braganhol, E.; Whiteside, T.L. Arginase-1+ Exosomes from Reprogrammed Macrophages Promote Glioblastoma Progression. Int. J. Mol. Sci. 2020, 21, 3990. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Sun, Y.; Sun, X.; Zhao, X.; Ma, Y.; Wang, Y.; Zhang, X. AEG-1 Silencing Attenuates M2-Polarization of Glioma-Associated Microglia/Macrophages and Sensitizes Glioma Cells to Temozolomide. Sci. Rep. 2021, 11, 17348. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The Chemokine System in Diverse Forms of Macrophage Activation and Polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Specht, H.; Emmott, E.; Petelski, A.A.; Huffman, R.G.; Perlman, D.H.; Serra, M.; Kharchenko, P.; Koller, A.; Slavov, N. Single-Cell Proteomic and Transcriptomic Analysis of Macrophage Heterogeneity Using SCoPE2. Genome Biol. 2021, 22, 50. [Google Scholar] [CrossRef]
- Hume, D.A.; Freeman, T.C. Transcriptomic Analysis of Mononuclear Phagocyte Differentiation and Activation. Immunol. Rev. 2014, 262, 74–84. [Google Scholar] [CrossRef]
- Derlindati, E.; Cas, A.D.; Montanini, B.; Spigoni, V.; Curella, V.; Aldigeri, R.; Ardigò, D.; Zavaroni, I.; Bonadonna, R.C. Transcriptomic Analysis of Human Polarized Macrophages: More than One Role of Alternative Activation? PLoS ONE 2015, 10, e0119751. [Google Scholar] [CrossRef] [Green Version]
- Ke, X.; Chen, C.; Song, Y.; Cai, Q.; Li, J.; Tang, Y.; Han, X.; Qu, W.; Chen, A.; Wang, H.; et al. Hypoxia Modifies the Polarization of Macrophages and Their Inflammatory Microenvironment, and Inhibits Malignant Behavior in Cancer Cells. Oncol. Lett. 2019, 18, 5871–5878. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Si, Y.; Wang, L.; Ding, M.; Yu, S.; Lu, L.; Guo, Y.; Zong, M.; Fan, L. The Regulation of Macrophage Polarization by Hypoxia-PADI4 Coordination in Rheumatoid Arthritis. Int. Immunopharmacol. 2021, 99, 107988. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Horner, J.W.; Paul, E.; Shang, X.; Troncoso, P.; Deng, P.; Jiang, S.; Chang, Q.; Spring, D.J.; Sharma, P.; et al. Effective Combinatorial Immunotherapy for Castration-Resistant Prostate Cancer. Nature 2017, 543, 728–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farshidpour, M.; Ahmed, M.; Junna, S.; Merchant, J.L. Myeloid-Derived Suppressor Cells in Gastrointestinal Cancers: A Systemic Review. World J. Gastrointest. Oncol. 2021, 13, 1. [Google Scholar] [CrossRef] [PubMed]
- Weber, R.; Riester, Z.; Hüser, L.; Sticht, C.; Siebenmorgen, A.; Groth, C.; Hu, X.; Altevogt, P.; Utikal, J.S.; Umansky, V. IL-6 Regulates CCR5 Expression and Immunosuppressive Capacity of MDSC in Murine Melanoma. J. Immunother. Cancer 2020, 8, e000949. [Google Scholar] [CrossRef]
- Raychaudhuri, B.; Rayman, P.; Huang, P.; Grabowski, M.; Hambardzumyan, D.; Finke, J.H.; Vogelbaum, M.A. Myeloid Derived Suppressor Cell Infiltration of Murine and Human Gliomas Is Associated with Reduction of Tumor Infiltrating Lymphocytes. J. Neurooncol. 2015, 122, 293–301. [Google Scholar] [CrossRef]
- Ugel, S.; de Sanctis, F.; Mandruzzato, S.; Bronte, V. Tumor-Induced Myeloid Deviation: When Myeloid-Derived Suppressor Cells Meet Tumor-Associated Macrophages. J. Clin. Investig. 2015, 125, 3365–3376. [Google Scholar] [CrossRef] [Green Version]
- Groth, C.; Hu, X.; Weber, R.; Fleming, V.; Altevogt, P.; Utikal, J.; Umansky, V. Immunosuppression Mediated by Myeloid-Derived Suppressor Cells (MDSCs) during Tumour Progression. Br. J. Cancer 2019, 120, 16–25. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Zhu, D.; Tian, J.; Tang, X.; Guo, H.; Ma, J.; Xu, H.; Wang, S. Granulocytic Myeloid-Derived Suppressor Cell Exosomal Prostaglandin E2 Ameliorates Collagen-Induced Arthritis by Enhancing IL-10+ B Cells. Front. Immunol. 2020, 11, 588500. [Google Scholar] [CrossRef]
- Rodriguez, P.C.; Ernstoff, M.S.; Hernandez, C.; Atkins, M.; Zabaleta, J.; Sierra, R.; Ochoa, A.C. Arginase I-Producing Myeloid-Derived Suppressor Cells in Renal Cell Carcinoma Are a Subpopulation of Activated Granulocytes. Cancer Res. 2009, 69, 1553–1560. [Google Scholar] [CrossRef] [Green Version]
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-Derived Suppressor Cells in the Era of Increasing Myeloid Cell Diversity. Nat. Rev. Immunol. 2021, 21, 485–498. [Google Scholar] [CrossRef]
- Bayik, D.; Zhou, Y.; Park, C.; Hong, C.; Vail, D.; Silver, D.J.; Lauko, A.; Roversi, G.; Watson, D.C.; Lo, A.; et al. Myeloid-Derived Suppressor Cell Subsets Drive Glioblastoma Growth in a Sex-Specific Manner. Cancer Discov. 2020, 10, 1210–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Li, X.; Ji, C.; Liu, P.; Zhou, L.; Xu, D.; Wang, D.; Li, J.; Yu, J. Early Myeloid-Derived Suppressor Cells Accelerate Epithelial-Mesenchymal Transition by Downregulating ARID1A in Luminal A Breast Cancer. Front. Bioeng. Biotechnol. 2022, 10, 973731. [Google Scholar] [CrossRef] [PubMed]
- Okła, K.; Czerwonka, A.; Wawruszak, A.; Bobiński, M.; Bilska, M.; Tarkowski, R.; Bednarek, W.; Wertel, I.; Kotarski, J. Clinical Relevance and Immunosuppressive Pattern of Circulating and Infiltrating Subsets of Myeloid-Derived Suppressor Cells (MDSCs) in Epithelial Ovarian Cancer. Front. Immunol. 2019, 10, 691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, S.; Bruderek, K.; Kaspar, C.; Höing, B.; Kanaan, O.; Dominas, N.; Hussain, T.; Droege, F.; Eyth, C.; Hadaschik, B.; et al. Clinical Relevance and Suppressive Capacity of Human Myeloid-Derived Suppressor Cell Subsets. Clin. Cancer Res. 2018, 24, 4834–4844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.N.H.; Emmons, T.R.; Wong, J.T.; Alqassim, E.; Singel, K.L.; Mark, J.; Smith, B.E.; Tario, J.D.; Eng, K.H.; Moysich, K.B.; et al. Quantification of Early-Stage Myeloid-Derived Suppressor Cells in Cancer Requires Excluding Basophils. Cancer Immunol. Res. 2020, 8, 819–828. [Google Scholar] [CrossRef] [Green Version]
- Dubinski, D.; Wölfer, J.; Hasselblatt, M.; Schneider-Hohendorf, T.; Bogdahn, U.; Stummer, W.; Wiendl, H.; Grauer, O.M. CD4+ T Effector Memory Cell Dysfunction Is Associated with the Accumulation of Granulocytic Myeloid-Derived Suppressor Cells in Glioblastoma Patients. Neuro Oncol. 2016, 18, 807–818. [Google Scholar] [CrossRef] [Green Version]
- Gielen, P.R.; Schulte, B.M.; Kers-Rebel, E.D.; Verrijp, K.; Petersen-Baltussen, H.M.J.M.; ter Laan, M.; Wesseling, P.; Adema, G.J. Increase in Both CD14-Positive and CD15-Positive Myeloid-Derived Suppressor Cell Subpopulations in the Blood of Patients with Glioma but Predominance of CD15-Positive Myeloid-Derived Suppressor Cells in Glioma Tissue. J. Neuropathol. Exp. Neurol. 2015, 74, 390–400. [Google Scholar] [CrossRef] [Green Version]
- Alban, T.J.; Bayik, D.; Otvos, B.; Rabljenovic, A.; Leng, L.; Jia-Shiun, L.; Roversi, G.; Lauko, A.; Momin, A.A.; Mohammadi, A.M.; et al. Glioblastoma Myeloid-Derived Suppressor Cell Subsets Express Differential Macrophage Migration Inhibitory Factor Receptor Profiles That Can Be Targeted to Reduce Immune Suppression. Front. Immunol. 2020, 11, 1191. [Google Scholar] [CrossRef]
- Yaddanapudi, K.; Rendon, B.E.; Lamont, G.; Kim, E.J.; Al Rayyan, N.; Richie, J.; Albeituni, S.; Waigel, S.; Wise, A.; Mitchell, R.A. MIF Is Necessary for Late-Stage Melanoma Patient MDSC Immune Suppression and Differentiation. Cancer Immunol. Res. 2016, 4, 101–112. [Google Scholar] [CrossRef] [Green Version]
- Mawhinney, L.; Armstrong, M.E.; O’ Reilly, C.; Bucala, R.; Leng, L.; Fingerle-Rowson, G.; Fayne, D.; Keane, M.P.; Tynan, A.; Maher, L.; et al. Macrophage Migration Inhibitory Factor (MIF) Enzymatic Activity and Lung Cancer. Mol. Med. 2014, 20, 729–735. [Google Scholar] [CrossRef] [Green Version]
- Richard, V.; Kindt, N.; Saussez, S. Macrophage Migration Inhibitory Factor Involvement in Breast Cancer (Review). Int. J. Oncol. 2015, 47, 1627–1633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otvos, B.; Silver, D.J.; Mulkearns-Hubert, E.E.; Alvarado, A.G.; Turaga, S.M.; Sorensen, M.D.; Rayman, P.; Flavahan, W.A.; Hale, J.S.; Stoltz, K.; et al. Cancer Stem Cell-Secreted Macrophage Migration Inhibitory Factor Stimulates Myeloid Derived Suppressor Cell Function and Facilitates Glioblastoma Immune Evasion. Stem Cells 2016, 34, 2026–2039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noe, J.T.; Mitchell, R.A. MIF-Dependent Control of Tumor Immunity. Front. Immunol. 2020, 11, 609948. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Qiu, W.; Wang, J.; Liu, Q.; Qian, M.; Wang, S.; Zhang, Z.; Gao, X.; Chen, Z.; Guo, Q.; et al. Glioma Exosomes Mediate the Expansion and Function of Myeloid-Derived Suppressor Cells through MicroRNA-29a/Hbp1 and MicroRNA-92a/Prkar1a Pathways. Int. J. Cancer 2019, 144, 3111–3126. [Google Scholar] [CrossRef]
- Xue, S.; Song, G.; Yu, J. The Prognostic Significance of PD-L1 Expression in Patients with Glioma: A Meta-Analysis. Sci. Rep. 2017, 7, 4231. [Google Scholar] [CrossRef] [Green Version]
- Eschweiler, S.; Clarke, J.; Ramírez-Suástegui, C.; Panwar, B.; Madrigal, A.; Chee, S.J.; Karydis, I.; Woo, E.; Alzetani, A.; Elsheikh, S.; et al. Intratumoral Follicular Regulatory T Cells Curtail Anti-PD-1 Treatment Efficacy. Nat. Immunol. 2021, 22, 1052–1063. [Google Scholar] [CrossRef]
- Lu, L.; Sun, J.; Su, H.; Luo, S.; Chen, J.; Qiu, S.; Chi, Y.; Lin, J.; Xu, X.; Zheng, D. Antitumor CD8 T Cell Responses in Glioma Patients Are Effectively Suppressed by T Follicular Regulatory Cells. Exp. Cell Res. 2021, 407, 112808. [Google Scholar] [CrossRef]
- Han, S.; Feng, S.; Ren, M.; Ma, E.; Wang, X.; Xu, L.; Xu, M. Glioma Cell-Derived Placental Growth Factor Induces Regulatory B Cells. Int. J. Biochem. Cell Biol. 2014, 57, 63–68. [Google Scholar] [CrossRef]
- Crane, C.A.; Han, S.J.; Barry, J.J.; Ahn, B.J.; Lanier, L.L.; Parsa, A.T. TGF-β Downregulates the Activating Receptor NKG2D on NK Cells and CD8+ T Cells in Glioma Patients. Neuro Oncol. 2010, 12, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Fu, W.; Wang, W.; Li, H.; Jiao, Y.; Huo, R.; Yan, Z.; Wang, J.; Wang, S.; Wang, J.; Chen, D.; et al. Single-Cell Atlas Reveals Complexity of the Immunosuppressive Microenvironment of Initial and Recurrent Glioblastoma. Front. Immunol. 2020, 11, 835. [Google Scholar] [CrossRef]
- Kohanbash, G.; McKaveney, K.; Sakaki, M.; Ueda, R.; Mintz, A.H.; Amankulor, N.; Fujita, M.; Ohlfest, J.R.; Okada, H. GM-CSF Promotes the Immunosuppressive Activity of Glioma-Infiltrating Myeloid Cells through Interleukin-4 Receptor-α. Cancer Res. 2013, 73, 6413–6423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alban, T.J.; Alvarado, A.G.; Sorensen, M.D.; Bayik, D.; Volovetz, J.; Serbinowski, E.; Mulkearns-Hubert, E.E.; Sinyuk, M.; Hale, J.S.; Onzi, G.R.; et al. Global Immune Fingerprinting in Glioblastoma Patient Peripheral Blood Reveals Immune-Suppression Signatures Associated with Prognosis. JCI Insight 2018, 3, e122264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gielen, P.R.; Schulte, B.M.; Kers-Rebel, E.D.; Verrijp, K.; Bossman, S.A.J.F.H.; ter Laan, M.; Wesseling, P.; Adema, G.J. Elevated Levels of Polymorphonuclear Myeloid-Derived Suppressor Cells in Patients with Glioblastoma Highly Express S100A8/9 and Arginase and Suppress T Cell Function. Neuro Oncol. 2016, 18, 1253–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegde, S.; Leader, A.M.; Merad, M. MDSC: Markers, Development, States, and Unaddressed Complexity. Immunity 2021, 54, 875–884. [Google Scholar] [CrossRef]
- Yang, W.; Li, Y.; Gao, R.; Xiu, Z.; Sun, T. MHC Class I Dysfunction of Glioma Stem Cells Escapes from CTL-Mediated Immune Response via Activation of Wnt/β-Catenin Signaling Pathway. Oncogene 2020, 39, 1098–1111. [Google Scholar] [CrossRef]
- Grabowski, M.M.; Sankey, E.W.; Ryan, K.J.; Chongsathidkiet, P.; Lorrey, S.J.; Wilkinson, D.S.; Fecci, P.E. Immune Suppression in Gliomas. J. Neurooncol. 2021, 151, 3–12. [Google Scholar] [CrossRef]
- Ravi, V.M.; Neidert, N.; Will, P.; Joseph, K.; Maier, J.P.; Kückelhaus, J.; Vollmer, L.; Goeldner, J.M.; Behringer, S.P.; Scherer, F.; et al. T-Cell Dysfunction in the Glioblastoma Microenvironment Is Mediated by Myeloid Cells Releasing Interleukin-10. Nat. Commun. 2022, 13, 925. [Google Scholar] [CrossRef]
- Chongsathidkiet, P.; Jackson, C.; Koyama, S.; Loebel, F.; Cui, X.; Farber, S.H.; Woroniecka, K.; Elsamadicy, A.A.; Dechant, C.A.; Kemeny, H.R.; et al. Sequestration of T Cells in Bone Marrow in the Setting of Glioblastoma and Other Intracranial Tumors. Nat. Med. 2018, 24, 1459–1468. [Google Scholar] [CrossRef]
- Lu, L.; Barbi, J.; Pan, F. The Regulation of Immune Tolerance by FOXP3. Nat. Rev. Immunol. 2017, 17, 703–717. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, J.F.M.; Idema, A.J.; Bol, K.F.; Nierkens, S.; Grauer, O.M.; Wesseling, P.; Grotenhuis, J.A.; Hoogerbrugge, P.M.; de Vries, I.J.M.; Adema, G.J. Regulatory T Cells and the PD-L1/PD-1 Pathway Mediate Immune Suppression in Malignant Human Brain Tumors. Neuro Oncol. 2009, 11, 394–402. [Google Scholar] [CrossRef] [Green Version]
- Brown, N.F.; Carter, T.J.; Ottaviani, D.; Mulholland, P. Harnessing the Immune System in Glioblastoma. Br. J. Cancer 2018, 119, 1171–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maraskovsky, E.; Chen, W.F.; Shortman, K. IL-2 and IFN-Gamma Are Two Necessary Lymphokines in the Development of Cytolytic T Cells. J. Immunol. 1989, 143, 1210–1214. [Google Scholar] [CrossRef] [PubMed]
- Raskov, H.; Orhan, A.; Christensen, J.P.; Gögenur, I. Cytotoxic CD8+ T Cells in Cancer and Cancer Immunotherapy. Br. J. Cancer 2021, 124, 359–367. [Google Scholar] [CrossRef]
- Tokunaga, R.; Naseem, M.; Lo, J.H.; Battaglin, F.; Soni, S.; Puccini, A.; Berger, M.D.; Zhang, W.; Baba, H.; Lenz, H.J. B Cell and B Cell-Related Pathways for Novel Cancer Treatments. Cancer Treat. Rev. 2019, 73, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Rosser, E.C.; Mauri, C. Regulatory B Cells: Origin, Phenotype, and Function. Immunity 2015, 42, 607–612. [Google Scholar] [CrossRef] [Green Version]
- Diefenbach, A. Chapter 4—Natural Killer Cells; Ackerman, M.E., Nimmerjahn, F., Fc, A., Eds.; Academic Press: Cambridge, MA, USA, 2014; pp. 75–93. ISBN 9780123948021. [Google Scholar] [CrossRef]
- Pegram, H.J.; Andrews, D.M.; Smyth, M.J.; Darcy, P.K.; Kershaw, M.H. Activating and Inhibitory Receptors of Natural Killer Cells. Immunol. Cell Biol. 2011, 89, 216–224. [Google Scholar] [CrossRef]
- Golán, I.; de La Fuente, L.R.; Costoya, J.A. NK Cell-Based Glioblastoma Immunotherapy. Cancers 2018, 10, 522. [Google Scholar] [CrossRef] [Green Version]
- Takeda, K.; Hayakawa, Y.; Smyth, M.J.; Kayagaki, N.; Yamaguchi, N.; Kakuta, S.; Iwakura, Y.; Yagita, H.; Okumura, K. Involvement of Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand in Surveillance of Tumor Metastasis by Liver Natural Killer Cells. Nat. Med. 2001, 7, 94–100. [Google Scholar] [CrossRef]
- Guerra, N.; Tan, Y.X.; Joncker, N.T.; Choy, A.; Gallardo, F.; Xiong, N.; Knoblaugh, S.; Cado, D.; Greenberg, N.R.; Raulet, D.H. NKG2D-Deficient Mice Are Defective in Tumor Surveillance in Models of Spontaneous Malignancy. Immunity 2008, 28, 571–580. [Google Scholar] [CrossRef] [Green Version]
- Takeda, K.; Smyth, M.J.; Cretney, E.; Hayakawa, Y.; Kayagaki, N.; Yagita, H.; Okumura, K. Critical Role for Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand in Immune Surveillance against Tumor Development. J. Exp. Med. 2002, 195, 161–169. [Google Scholar] [CrossRef] [Green Version]
- Smyth, M.J.; Thia, K.Y.T.; Street, S.E.A.; MacGregor, D.; Godfrey, D.I.; Trapani, J.A. Perforin-Mediated Cytotoxicity Is Critical for Surveillance of Spontaneous Lymphoma. J. Exp. Med. 2000, 192, 755–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramanathan, A.; Lorimer, I.A.J. Engineered Cells as Glioblastoma Therapeutics. Cancer Gene Ther. 2022, 29, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Wensveen, F.M.; Jelenčić, V.; Polić, B. NKG2D: A Master Regulator of Immune Cell Responsiveness. Front. Immunol. 2018, 9, 441. [Google Scholar] [CrossRef]
- Weiss, T.; Weller, M.; Guckenberger, M.; Sentman, C.L.; Roth, P. NKG2D-Based CAR T Cells and Radiotherapy Exert Synergistic Efficacy in Glioblastoma. Cancer Res. 2018, 78, 1031–1043. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, Y.; Nakazawa, T.; Nakamura, M.; Nishimura, F.; Matsuda, R.; Omoto, K.; Shida, Y.; Murakami, T.; Nakagawa, I.; Motoyama, Y.; et al. Ex Vivo-Expanded Highly Purified Natural Killer Cells in Combination with Temozolomide Induce Antitumor Effects in Human Glioblastoma Cells in Vitro. PLoS ONE 2019, 14, e0212455. [Google Scholar] [CrossRef] [PubMed]
- Shaim, H.; Shanley, M.; Basar, R.; Daher, M.; Gumin, J.; Zamler, D.B.; Uprety, N.; Wang, F.; Huang, Y.; Gabrusiewicz, K.; et al. Targeting the Av Integrin/TGF-β Axis Improves Natural Killer Cell Function against Glioblastoma Stem Cells. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Reardon, D.A.; Desjardins, A.; Vredenburgh, J.J.; O’Rourke, D.M.; Tran, D.D.; Fink, K.L.; Nabors, L.B.; Li, G.; Bota, D.A.; Lukas, R.V.; et al. Rindopepimut with Bevacizumab for Patients with Relapsed EGFRvIII-Expressing Glioblastoma (REACT): Results of a Double-Blind Randomized Phase II Trial. Clin. Cancer Res. 2020, 26, 1586–1594. [Google Scholar] [CrossRef]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F.; et al. Rindopepimut with Temozolomide for Patients with Newly Diagnosed, EGFRvIII-Expressing Glioblastoma (ACT IV): A Randomised, Double-Blind, International Phase 3 Trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef] [Green Version]
- Schuster, J.; Lai, R.K.; Recht, L.D.; Reardon, D.A.; Paleologos, N.A.; Groves, M.D.; Mrugala, M.M.; Jensen, R.; Baehring, J.M.; Sloan, A.; et al. A Phase II, Multicenter Trial of Rindopepimut (CDX-110) in Newly Diagnosed Glioblastoma: The ACT III Study. Neuro Oncol. 2015, 17, 854–861. [Google Scholar] [CrossRef] [Green Version]
- Migliorini, D.; Dutoit, V.; Allard, M.; Grandjean Hallez, N.; Marinari, E.; Widmer, V.; Philippin, G.; Corlazzoli, F.; Gustave, R.; Kreutzfeldt, M.; et al. Phase I/II Trial Testing Safety and Immunogenicity of the Multipeptide IMA950/Poly-ICLC Vaccine in Newly Diagnosed Adult Malignant Astrocytoma Patients. Neuro Oncol. 2019, 21, 923–933. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.T.; Nie, Y.; Sun, S.N.; Lin, T.; Han, R.J.; Jiang, J.; Li, Z.; Li, J.Q.; Xiao, Y.P.; Fan, Y.Y.; et al. Tumor-Associated Antigen-Based Personalized Dendritic Cell Vaccine in Solid Tumor Patients. Cancer Immunol. Immunother. 2020, 69, 1375–1387. [Google Scholar] [CrossRef] [PubMed]
- Bloch, O.; Lim, M.; Sughrue, M.E.; Komotar, R.J.; Abrahams, J.M.; O’Rourke, D.M.; D’Ambrosio, A.; Bruce, J.N.; Parsa, A.T. Autologous Heat Shock Protein Peptide Vaccination for Newly Diagnosed Glioblastoma: Impact of Peripheral PD-L1 Expression on Response to Therapy. Clin. Cancer Res. 2017, 23, 3575–3584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bota, D.; Taylor, T.; Picconi, D.; Duma, C.; Aiken, R.; LaRocca, R.; Xiao-Tang, K.; Fu, B.; Alsharif, M.; Hsieh, C.; et al. ATIM-28. Phase II trial of AV-GBM-1 (Autologous Dendritic Cells Loaded with Tumor Associated Antigens) as Adjunctive Therapy following Surgery Plus Concurrent Chemoradiation in Newly Diagnosed GBM Patients. Neuro Oncol. 2019, 21, vi7. [Google Scholar] [CrossRef]
- Ranjan, S.; Quezado, M.; Garren, N.; Boris, L.; Siegel, C.; Lopes Abath Neto, O.; Theeler, B.J.; Park, D.M.; Nduom, E.; Zaghloul, K.A.; et al. Clinical Decision Making in the Era of Immunotherapy for High Grade-Glioma: Report of Four Cases. BMC Cancer 2018, 18, 239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desjardins, A.; Gromeier, M.; Herndon, J.E.; Beaubier, N.; Bolognesi, D.P.; Friedman, A.H.; Friedman, H.S.; McSherry, F.; Muscat, A.M.; Nair, S.; et al. Recurrent Glioblastoma Treated with Recombinant Poliovirus. N. Engl. J. Med. 2018, 379, 150–161. [Google Scholar] [CrossRef]
- Markert, J.M.; Medlock, M.D.; Rabkin, S.D.; Gillespie, G.Y.; Todo, T.; Hunter, W.D.; Palmer, C.A.; Feigenbaum, F.; Tornatore, C.; Tufaro, F.; et al. Conditionally Replicating Herpes Simplex Virus Mutant G207 for the Treatment of Malignant Glioma: Results of a Phase I Trial. Gene Ther. 2000, 7, 867–874. [Google Scholar] [CrossRef] [Green Version]
- Markert, J.M.; Razdan, S.N.; Kuo, H.C.; Cantor, A.; Knoll, A.; Karrasch, M.; Nabors, L.B.; Markiewicz, M.; Agee, B.S.; Coleman, J.M.; et al. A Phase 1 Trial of Oncolytic HSV-1, G207, given in Combination with Radiation for Recurrent GBM Demonstrates Safety and Radiographic Responses. Mol. Ther. 2014, 22, 1048–1055. [Google Scholar] [CrossRef] [Green Version]
- Chiocca, E.A.; Gelb, A.B.; Chen, C.C.; Rao, G.; Reardon, D.A.; Wen, P.Y.; Bi, W.L.; Peruzzi, P.; Amidei, C.; Triggs, D.; et al. Combined Immunotherapy with Controlled Interleukin-12 Gene Therapy and Immune Checkpoint Blockade in Recurrent Glioblastoma: An Open-Label, Multi-Institutional Phase I Trial. Neuro Oncol. 2022, 24, 951–963. [Google Scholar] [CrossRef]
- An, Z.; Aksoy, O.; Zheng, T.; Fan, Q.W.; Weiss, W.A. Epidermal Growth Factor Receptor and EGFRvIII in Glioblastoma: Signaling Pathways and Targeted Therapies. Oncogene 2018, 37, 1561–1575. [Google Scholar] [CrossRef]
- Keller, S.; Schmidt, M.H.H. EGFR and EGFRvIII Promote Angiogenesis and Cell Invasion in Glioblastoma: Combination Therapies for an Effective Treatment. Int. J. Mol. Sci. 2017, 18, 1295. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Yu, S.; Xu, H.; Zheng, Y.; Lin, J.; Wu, M.; Wang, J.; Wang, A.; Lan, Q.; Furnari, F.; et al. FHL2 Interacts with EGFR to Promote Glioblastoma Growth. Oncogene 2018, 37, 1386–1398. [Google Scholar] [CrossRef] [PubMed]
- Bonavia, R.; Inda, M.M.; Vandenberg, S.; Cheng, S.Y.; Nagane, M.; Hadwiger, P.; Tan, P.; Sah, D.W.Y.; Cavenee, W.K.; Furnari, F.B. EGFRvIII Promotes Glio.oma Angiogenesis and Growth through the NF-B, Interleukin-8 Pathway. Oncogene 2012, 31, 4054–4066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutkowska, A.; Stoczyńska-Fidelus, E.; Janik, K.; Włodarczyk, A.; Rieske, P. EGFRvIII: An Oncogene with Ambiguous Role. J. Oncol. 2019, 2019, 1092587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Yan, J.; Liu, B. Targeting EGFRvIII for Glioblastoma Multiforme. Cancer Lett. 2017, 403, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Kong, S.; Sengupta, S.; Tyler, B.; Bais, A.J.; Ma, Q.; Doucette, S.; Zhou, J.; Sahin, A.; Carter, B.S.; Brem, H.; et al. Suppression of Human Glioma Xenografts with Second-Generation IL13R-Specific Chimeric Antigen Receptor-Modified T Cells. Clin. Cancer Res. 2012, 18, 5949–5960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, J.; Zhang, J.; Yang, Y.Z.; Wang, F.; Jiang, H.; Chen, H.D.; Wu, H.Y.; Sai, K.; Hu, W.M. IL13RA2 Is Overexpressed in Malignant Gliomas and Related to Clinical Outcome of Patients. Am. J. Transl. Res. 2020, 12, 4702–4714. [Google Scholar]
- Brown, C.E.; Warden, C.D.; Starr, R.; Deng, X.; Badie, B.; Yuan, Y.C.; Forman, S.J.; Barish, M.E. Glioma IL13Rα2 Is Associated with Mesenchymal Signature Gene Expression and Poor Patient Prognosis. PLoS ONE 2013, 8, e77769. [Google Scholar] [CrossRef]
- Tu, M.; Wange, W.; Cai, L.; Zhu, P.; Gao, Z.; Zheng, W. IL-13 Receptor A2 Stimulates Human Glioma Cell Growth and Metastasis through the Src/PI3K/Akt/MTOR Signaling Pathway. Tumor Biol. 2016, 37, 14701–14709. [Google Scholar] [CrossRef]
- Newman, J.P.; Wang, G.Y.; Arima, K.; Guan, S.P.; Waters, M.R.; Cavenee, W.K.; Pan, E.; Aliwarga, E.; Chong, S.T.; Kok, C.Y.L.; et al. Interleukin-13 Receptor Alpha 2 Cooperates with EGFRvIII Signaling to Promote Glioblastoma Multiforme. Nat. Commun. 2017, 8, 1913. [Google Scholar] [CrossRef] [Green Version]
- Mineo, J.F.; Bordron, A.; Baroncini, M.; Maurage, C.A.; Ramirez, C.; Siminski, R.M.; Berthou, C.; Dam Hieu, P. Low HER2-Expressing Glioblastomas Are More Often Secondary to Anaplastic Transformation of Low-Grade Glioma. J. Neurooncol. 2007, 85, 281–287. [Google Scholar] [CrossRef]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor–Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017, 3, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Söderberg-Nauclér, C. New Mechanistic Insights of the Pathogenicity of High-Risk Cytomegalovirus (CMV) Strains Derived from Breast Cancer: Hope for New Cancer Therapy Options Comment. EBioMedicine 2022, 81, 104103. [Google Scholar] [CrossRef] [PubMed]
- Krenzlin, H.; Behera, P.; Lorenz, V.; Passaro, C.; Zdioruk, M.; Nowicki, M.O.; Grauwet, K.; Zhang, H.; Skubal, M.; Ito, H.; et al. Cytomegalovirus Promotes Murine Glioblastoma Growth via Pericyte Recruitment and Angiogenesis. J. Clin. Investig. 2019, 129, 1671–1683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, C.E.; Badie, B.; Barish, M.E.; Weng, L.; Ostberg, J.R.; Chang, W.C.; Naranjo, A.; Starr, R.; Wagner, J.; Wright, C.; et al. Bioactivity and Safety of IL13Rα2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2015, 21, 4062–4072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef]
- Durgin, J.S.; Henderson, F.; Nasrallah, M.P.; Mohan, S.; Wang, S.; Lacey, S.F.; Melenhorst, J.J.; Desai, A.S.; Lee, J.Y.K.; Maus, M.V.; et al. Case Report: Prolonged Survival Following EGFRvIII CAR T Cell Treatment for Recurrent Glioblastoma. Front. Oncol. 2021, 11, 669071. [Google Scholar] [CrossRef]
- Karschnia, P.; Teske, N.; Thon, N.; Subklewe, M.; Tonn, J.C.; Dietrich, J.; von Baumgarten, L. Chimeric Antigen Receptor T Cells for Glioblastoma: Current Concepts, Challenges, and Future Perspectives. Neurology 2021, 97, 218–230. [Google Scholar] [CrossRef]
- Land, C.A.; Musich, P.R.; Haydar, D.; Krenciute, G.; Xie, Q. Chimeric Antigen Receptor T-Cell Therapy in Glioblastoma: Charging the T Cells to Fight. J. Transl. Med. 2020, 18, 428. [Google Scholar] [CrossRef]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A Single Dose of Peripherally Infused EGFRvIII-Directed CAR T Cells Mediates Antigen Loss and Induces Adaptive Resistance in Patients with Recurrent Glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef] [Green Version]
- Pirozzi, C.J.; Carpenter, A.B.; Hennika, T.; Becher, O.J.; Yan, H. Tumor-Specific Mutations in Gliomas and Their Implications for Immunotherapy. In Translational Immunotherapy of Brain Tumors; Samson, J., Ed.; Elsevier Inc.: Frisco, CO, USA, 2017. [Google Scholar]
- Swartz, A.M.; Li, Q.J.; Sampson, J.H. Rindopepimut: A Promising Immunotherapeutic for the Treatment of Glioblastoma Multiforme. Immunotherapy 2014, 6, 679–690. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.W.; Chow, K.K.H.; Lim, M.; Li, G. Current Vaccine Trials in Glioblastoma: A Review. J. Immunol. Res. 2014, 2014, 796856. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Liu, Y.; Cai, S.J.; Qian, M.; Ding, J.; Larion, M.; Gilbert, M.R.; Yang, C. IDH Mutation in Glioma: Molecular Mechanisms and Potential Therapeutic Targets. Br. J. Cancer 2020, 122, 1580–1589. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Weller, M. Vaccination for IDH-Mutant Tumors: A Novel Therapeutic Approach Applied to Glioma. Med 2021, 2, 450–452. [Google Scholar] [CrossRef] [PubMed]
- Platten, M.; Bunse, L.; Wick, A.; Bunse, T.; le Cornet, L.; Harting, I.; Sahm, F.; Sanghvi, K.; Tan, C.L.; Poschke, I.; et al. A Vaccine Targeting Mutant IDH1 in Newly Diagnosed Glioma. Nature 2021, 592, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, A.; Zhai, G.G.; Zhang, M.; Malhotra, R.; Latham, D.E.; Delaney, M.A.; Robe, P.; Nestler, U.; Song, Q.; Loeffler, J. Survivin Enhances Radiation Resistance in Primary Human Glioblastoma Cells via Caspase-Independent Mechanisms. Oncogene 2004, 23, 7494–7506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dohi, T.; Beltrami, E.; Wall, N.R.; Plescia, J.; Altieri, D.C. Mitochondrial Survivin Inhibits Apoptosis and Promotes Tumorigenesis. J. Clin. Investig. 2004, 114, 1117–1127. [Google Scholar] [CrossRef] [Green Version]
- Ahluwalia, M.S.; Reardon, D.A.; Abad, A.P.; Curry, W.T.; Wong, E.T.; Figel, S.A.; Mechtler, L.L.; Peereboom, M.; Hutson, A.D.; Withers, H.G.; et al. Phase IIa Study of SurVaxM Plus Adjuvant Temozolomide for Newly Diagnosed Glioblastoma. J. Clin. Oncol. 2022, 41, 1453–1465. [Google Scholar] [CrossRef]
- Oji, Y.; Suzuki, T.; Nakano, Y.; Maruno, M.; Nakatsuka, S.I.; Jomgeow, T.; Abeno, S.; Tatsumi, N.; Yokota, A.; Aoyagi, S.; et al. Overexpression of the Wilms’ Tumor Gene WT1 in Primary Astrocytic Tumors. Cancer Sci. 2004, 95, 822–827. [Google Scholar] [CrossRef]
- Menssen, H.D.; Bertelmann, E.; Bartelt, S.; Schmidt, R.A.; Pecher, G.; Schramm, K.; Thiel, E. Wilms’ Tumor Gene (WT1) Expression in Lung Cancer, Colon Cancer and Glioblastoma Cell Lines Compared to Freshly Isolated Tumor Specimens. J. Cancer Res. Clin. Oncol. 2000, 126, 226–232. [Google Scholar] [CrossRef]
- Somasundaram, A.; Ardanowski, N.; Opalak, C.F.; Fillmore, H.L.; Chidambaram, A.; Broaddus, W.C. Wilms Tumor 1 Gene, CD97, and the Emerging Biogenetic Profile of Glioblastoma. Neurosurg. Focus 2014, 37, E14. [Google Scholar] [CrossRef] [Green Version]
- de Groot, J.F.; Cloughesy, T.F.; Pitz, M.W.; Narita, Y.; Nonomura, T. A Randomized, Multicenter Phase 2 Study of DSP-7888 Dosing Emulsion in Combination with Bevacizumab (Bev) versus Bev Alone in Patients with Recurrent or Progressive Glioblastoma. J. Clin. Oncol. 2018, 36, TPS2071. [Google Scholar] [CrossRef]
- NCT03149003 A Study of DSP-7888 Dosing Emulsion in Combination With Bevacizumab in Patients With Recurrent or Progressive Glioblastoma Following Initial Therapy. 2017. Available online: https://clinicaltrials.gov/show/NCT03149003 (accessed on 31 January 2023).
- Spira, A.; Hansen, A.R.; Harb, W.A.; Curtis, K.K.; Koga-Yamakawa, E.; Origuchi, M.; Li, Z.; Ertik, B.; Shaib, W.L. Multicenter, Open-Label, Phase I Study of DSP-7888 Dosing Emulsion in Patients with Advanced Malignancies. Target. Oncol. 2021, 16, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Goto, M.; Nakamura, M.; Suginobe, N.; Takasu, H.; Takanashi, Y.; Ban, H.; Li, C.J. DSP-7888, a Novel Cocktail Design of WT1 Peptide Vaccine, and Its Combinational Immunotherapy with Immune Checkpoint-Blocking Antibody Against PD-1. Blood 2016, 128, 4715. [Google Scholar] [CrossRef]
- Hu, T.; Xie, N.; Qin, C.; Wang, J.; You, Y. Glucose-Regulated Protein 94 Is a Novel Glioma Biomarker and Promotes the Aggressiveness of Glioma via Wnt/β-Catenin Signaling Pathway. Tumor Biol. 2015, 36, 9357–9364. [Google Scholar] [CrossRef]
- Memmel, S.; Sisario, D.; Zöller, C.; Fiedler, V.; Katzer, A.; Heiden, R.; Becker, N.; Eing, L.; Ferreira, F.L.R.; Zimmermann, H.; et al. Migration Pattern, Actin Cytoskeleton Organization and Response to PI3K-, MTOR-, and Hsp90-Inhibition of Glioblastoma Cells with Different Invasive Capacities. Oncotarget 2017, 8, 45298–45310. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Mudgal, P.; Wang, L.; Wu, H.; Huang, N.; Alexander, P.B.; Gao, Z.; Ji, N.; Li, Q.J. T Cell Receptor Repertoire as a Prognosis Marker for Heat Shock Protein Peptide Complex-96 Vaccine Trial against Newly Diagnosed Glioblastoma. Oncoimmunology 2020, 9, 1749476. [Google Scholar] [CrossRef] [Green Version]
- Eton, O.; Ross, M.I.; East, M.J.; Mansfield, P.F.; Papadopoulos, N.; Ellerhorst, J.A.; Bedikian, A.Y.; Lee, J.E. Autologous Tumor-Derived Heat-Shock Protein Peptide Complex-96 (HSPPC-96) in Patients with Metastatic Melanoma. J. Transl. Med. 2010, 8, 9. [Google Scholar] [CrossRef] [Green Version]
- Batich, K.A.; Reap, E.A.; Archer, G.E.; Sanchez-Perez, L.; Nair, S.K.; Schmittling, R.J.; Norberg, P.; Xie, W.; Herndon, J.E.; Healy, P.; et al. Long-Term Survival in Glioblastoma with Cytomegalovirus Pp65-Targeted Vaccination. Clin. Cancer Res. 2017, 23, 1898–1909. [Google Scholar] [CrossRef] [Green Version]
- Lawler, S.E. Cytomegalovirus and Glioblastoma; Controversies and Opportunities. J. Neurooncol. 2015, 123, 465–471. [Google Scholar] [CrossRef]
- Datsi, A.; Sorg, R.v. Dendritic Cell Vaccination of Glioblastoma: Road to Success or Dead End. Front. Immunol. 2021, 12, 770390. [Google Scholar] [CrossRef]
- Lee, E.Q. Immune Checkpoint Inhibitors in GBM. J. Neurooncol. 2021, 155, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, A.H.; Pauken, K.E. The Diverse Functions of the PD1 Inhibitory Pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 Blockade with Nivolumab in Relapsed or Refractory Hodgkin’s Lymphoma. N. Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferris, R.L.; Blumenschein, G.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N. Engl. J. Med. 2016, 375, 1856–1867. [Google Scholar] [CrossRef] [Green Version]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bähr, O.; et al. Effect of Nivolumab vs Bevacizumab in Patients with Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.; Weller, M.; Idbaih, A.; Steinbach, J.; Finocchiaro, G.; Raval, R.R.; Ansstas, G.; Baehring, J.; Taylor, J.W.; Honnorat, J.; et al. Phase III Trial of Chemoradiotherapy with Temozolomide plus Nivolumab or Placebo for Newly Diagnosed Glioblastoma with Methylated MGMT Promoter. Neuro Oncol. 2022, 24, 1935–1949. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant Anti-PD-1 Immunotherapy Promotes a Survival Benefit with Intratumoral and Systemic Immune Responses in Recurrent Glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef]
- de Groot, J.; Penas-Prado, M.; Alfaro-Munoz, K.; Hunter, K.; Pei, B.L.; O’Brien, B.; Weathers, S.P.; Loghin, M.; Kamiya Matsouka, C.; Yung, W.K.A.; et al. Window-of-Opportunity Clinical Trial of Pembrolizumab in Patients with Recurrent Glioblastoma Reveals Predominance of Immune-Suppressive Macrophages. Neuro Oncol. 2020, 22, 539–549. [Google Scholar] [CrossRef]
- Nayak, L.; Molinaro, A.M.; Peters, K.; Clarke, J.L.; Jordan, J.T.; de Groot, J.; Nghiemphu, L.; Kaley, T.; Colman, H.; McCluskey, C.; et al. Randomized Phase II and Biomarker Study of Pembrolizumab plus Bevacizumab versus Pembrolizumab Alone for Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2021, 27, 1048–1057. [Google Scholar] [CrossRef]
- Chikuma, S. CTLA-4, an Essential Immune-Checkpoint for T-Cell Activation. In Current Topics in Microbiology and Immunology; Yoshimura, A., Ed.; Springer International Publishing AG: Cham, Switzerland, 2017; Volume 410. [Google Scholar]
- Belcaid, Z.; Phallen, J.A.; Zeng, J.; See, A.P.; Mathios, D.; Gottschalk, C.; Nicholas, S.; Kellett, M.; Ruzevick, J.; Jackson, C.; et al. Focal Radiation Therapy Combined with 4-1BB Activation and CTLA-4 Blockade Yields Long-Term Survival and a Protective Antigen-Specific Memory Response in a Murine Glioma Model. PLoS ONE 2014, 9, e101764. [Google Scholar] [CrossRef] [Green Version]
- Fecci, P.E.; Ochiai, H.; Mitchell, D.A.; Grossi, P.M.; Sweeney, A.E.; Archer, G.E.; Cummings, T.; Allison, J.P.; Bigner, D.D.; Sampson, J.H. Systemic CTLA-4 Blockade Ameliorates Glioma-Induced Changes to the CD4 + T Cell Compartment without Affecting Regulatory T-Cell Function. Clin. Cancer Res. 2007, 13, 2158–2167. [Google Scholar] [CrossRef] [Green Version]
- Brown, N.F.; Ng, S.M.; Brooks, C.; Coutts, T.; Holmes, J.; Roberts, C.; Elhussein, L.; Hoskin, P.; Maughan, T.; Blagden, S.; et al. A Phase II Open Label, Randomised Study of Ipilimumab with Temozolomide versus Temozolomide Alone after Surgery and Chemoradiotherapy in Patients with Recently Diagnosed Glioblastoma: The Ipi-Glio Trial Protocol. BMC Cancer 2020, 20, 198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, T.; Shaw, H.; Cohn-Brown, D.; Chester, K.; Mulholland, P. Ipilimumab and Bevacizumab in Glioblastoma. Clin. Oncol. 2016, 28, 622–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wainwright, D.A.; Balyasnikova, I.v.; Chang, A.L.; Ahmed, A.U.; Moon, K.S.; Auffinger, B.; Tobias, A.L.; Han, Y.; Lesniak, M.S. IDO Expression in Brain Tumors Increases the Recruitment of Regulatory T Cells and Negatively Impacts Survival. Clin. Cancer Res. 2012, 18, 6110–6121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, L.; Ladomersky, E.; Lauing, K.L.; Wu, M.; Genet, M.; Gritsina, G.; Győrffy, B.; Brastianos, P.K.; Binder, D.C.; Sosman, J.A.; et al. Infiltrating T Cells Increase IDO1 Expression in Glioblastoma and Contribute to Decreased Patient Survival. Clin. Cancer Res. 2017, 23, 6650–6660. [Google Scholar] [CrossRef] [Green Version]
- Anderson, A.C.; Anderson, D.E.; Bregoli, L.; Hastings, W.D.; Kassam, N.; Lei, C.; Chandwaskar, R.; Karman, J.; Su, E.W.; Hirashima, M.; et al. Promotion of Tissue Inflammation by the Immune Receptor Tim-3 Expressed on Innate Immune Cells. Science 2007, 318, 1141–1143. [Google Scholar] [CrossRef]
- Ndhlovu, L.C.; Lopez-Vergès, S.; Barbour, J.D.; Brad Jones, R.; Jha, A.R.; Long, B.R.; Schoeffler, E.C.; Fujita, T.; Nixon, D.F.; Lanier, L.L. Tim-3 Marks Human Natural Killer Cell Maturation and Suppresses Cell-Mediated Cytotoxicity. Blood 2012, 119, 3734–3743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Zhu, Y.; Li, G.; Huang, H.; Zhang, G.; Wang, F.; Sun, J.; Yang, Q.; Zhang, X.; Lu, B. TIM-3 Expression Characterizes Regulatory T Cells in Tumor Tissues and Is Associated with Lung Cancer Progression. PLoS ONE 2012, 7, e30676. [Google Scholar] [CrossRef] [Green Version]
- Monney, L.; Sabatos, C.A.; Gaglia, J.L.; Ryu, A.; Waldner, H.; Chernova, T.; Manning, S.; Greenfield, E.A.; Coyle, A.J.; Sobel, R.A.; et al. Th1-Specific Cell Surface Protein Tim-3 Regulates Macrophage Activation and Severity of an Autoimmune Disease. Nature 2002, 415, 536–541. [Google Scholar] [CrossRef]
- Guo, Q.; Shen, S.; Guan, G.; Zhu, C.; Zou, C.; Cao, J.; Cheng, W.; Xu, X.; Yu, J.; Lin, Z.; et al. Cancer Cell Intrinsic TIM-3 Induces Glioblastoma Progression. iScience 2022, 25, 105329. [Google Scholar] [CrossRef]
- He, Y.; Cao, J.; Zhao, C.; Li, X.; Zhou, C.; Hirsch, F.R. TIM-3, a Promising Target for Cancer Immunotherapy. Onco Targets Ther. 2018, 11, 7005–7009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, L.; Ruan, Z. Tim-3 and Tim-4 as the Potential Targets for Antitumor Therapy. Hum. Vaccines Immunother. 2015, 11, 2458–2462. [Google Scholar] [CrossRef] [PubMed]
- Martuza, R.L.; Malick, A.; Markert, J.M.; Ruffner, K.L.; Coen, D.M. Experimental Therapy of Human Glioma by Means of a Genetically Engineered Virus Mutant. Science 1991, 252, 854–856. [Google Scholar] [CrossRef]
- Lang, F.F.; Conrad, C.; Gomez-Manzano, C.; Alfred Yung, W.K.; Sawaya, R.; Weinberg, J.S.; Prabhu, S.S.; Rao, G.; Fuller, G.N.; Aldape, K.D.; et al. Phase I Study of DNX-2401 (Delta-24-RGD) Oncolytic Adenovirus: Replication and Immunotherapeutic Effects in Recurrent Malignant Glioma. J. Clin. Oncol. 2018, 36, 1419–1427. [Google Scholar] [CrossRef]
- van den Bossche, W.B.L.; Kleijn, A.; Teunissen, C.E.; Voerman, J.S.A.; Teodosio, C.; Noske, D.P.; van Dongen, J.J.M.; Dirven, C.M.F.; Lamfers, M.L.M. Oncolytic Virotherapy in Glioblastoma Patients Induces a Tumor Macrophage Phenotypic Shift Leading to an Altered Glioblastoma Microenvironment. Neuro Oncol. 2018, 20, 1494–1504. [Google Scholar] [CrossRef] [PubMed]
- Zadeh, G.; Daras, M.; Cloughesy, T.F.; Colman, H.; Kumthekar, P.U.; Chen, C.C.; Aiken, R.; Groves, M.D.; Ong, S.; Ramakrishna, R.; et al. LTBK-04. Phase 2 multicenter study of the oncolytic adenovirus DNX-2401 (tasadenoturev) in combination with pembrolizumab for recurrent glioblastoma; captive study (KEYNOTE-192). Neuro Oncol. 2020, 22, ii237. [Google Scholar] [CrossRef]
- Ino, Y.; Todo, T. Clinical Development of a Third-Generation Oncolytic HSV-1 (G47Δ) for Malignant Glioma. Gene Ther. Regul. 2010, 5, 101–111. [Google Scholar] [CrossRef]
- Todo, T.; Ito, H.; Ino, Y.; Ohtsu, H.; Ota, Y.; Shibahara, J.; Tanaka, M. Intratumoral Oncolytic Herpes Virus G47∆ for Residual or Recurrent Glioblastoma: A Phase 2 Trial. Nat. Med. 2022, 28, 1630–1639. [Google Scholar] [CrossRef]
- Chiocca, E.A.; Nakashima, H.; Kasai, K.; Fernandez, S.A.; Oglesbee, M. Preclinical Toxicology of RQNestin34.5v.2: An Oncolytic Herpes Virus with Transcriptional Regulation of the ICP34.5 Neurovirulence Gene. Mol. Ther. Methods Clin. Dev. 2020, 17, 871–893. [Google Scholar] [CrossRef]
- Otani, Y.; Yoo, J.Y.; Lewis, C.T.; Chao, S.; Swanner, J.; Shimizu, T.; Kang, J.M.; Murphy, S.A.; Rivera-Caraballo, K.; Hong, B.; et al. NOTCH-Induced MDSC Recruitment after OHSV Virotherapy in CNS Cancer Models Modulates Antitumor Immunotherapy. Clin. Cancer Res. 2022, 28, 1460–1473. [Google Scholar] [CrossRef]
- Geletneky, K.; Hajda, J.; Angelova, A.L.; Leuchs, B.; Capper, D.; Bartsch, A.J.; Neumann, J.O.; Schöning, T.; Hüsing, J.; Beelte, B.; et al. Oncolytic H-1 Parvovirus Shows Safety and Signs of Immunogenic Activity in a First Phase I/IIa Glioblastoma Trial. Mol. Ther. 2017, 25, 2620–2634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shalhout, S.Z.; Miller, D.M.; Emerick, K.S.; Kaufman, H.L. Therapy with Oncolytic Viruses: Progress and Challenges. Nat. Rev. Clin. Oncol. 2023, 20, 160–177. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, A.B.; Carpenter, A.M.; Aiken, R.; Hanft, S. Oncolytic Virus in Gliomas: A Review of Human Clinical Investigations. Ann. Oncol. 2021, 32, 968–982. [Google Scholar] [CrossRef] [PubMed]
- Samson, A.; Scott, K.J.; Taggart, D.; West, E.J.; Wilson, E.; Nuovo, G.J.; Thomson, S.; Corns, R.; Mathew, R.K.; Fuller, M.J.; et al. Intravenous Delivery of Oncolytic Reovirus to Brain Tumor Patients Immunologically Primes for Subsequent Checkpoint Blockade. Sci. Transl. Med. 2018, 10, eaam7577. [Google Scholar] [CrossRef] [PubMed]
- Nazha, B.; Inal, C.; Owonikoko, T.K. Disialoganglioside GD2 Expression in Solid Tumors and Role as a Target for Cancer Therapy. Front. Oncol. 2020, 10, 1000. [Google Scholar] [CrossRef]
- Hamaoka, Y.; Negishi, M.; Katoh, H. EphA2 Is a Key Effector of the MEK/ERK/RSK Pathway Regulating Glioblastoma Cell Proliferation. Cell Signal. 2016, 28, 937–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; Yuan, Y.; Zhang, H.; Yan, M.; Wang, S.; Feng, F.; Ji, P.; Li, Y.; Li, B.; Gao, G.; et al. Prognostic Significance of CD147 in Patients with Glioblastoma. J. Neurooncol. 2013, 115, 19–26. [Google Scholar] [CrossRef]
- Golinelli, G.; Grisendi, G.; Prapa, M.; Bestagno, M.; Spano, C.; Rossignoli, F.; Bambi, F.; Sardi, I.; Cellini, M.; Horwitz, E.M.; et al. Targeting GD2-Positive Glioblastoma by Chimeric Antigen Receptor Empowered Mesenchymal Progenitors. Cancer Gene Ther. 2020, 27, 558–570. [Google Scholar] [CrossRef] [Green Version]
- Marx, S.; Wilken, F.; Wagner, I.; Marx, M.; Troschke-Meurer, S.; Zumpe, M.; Bien-Moeller, S.; Weidemeier, M.; Baldauf, J.; Fleck, S.K.; et al. GD2 Targeting by Dinutuximab Beta Is a Promising Immunotherapeutic Approach against Malignant Glioma. J. Neurooncol. 2020, 147, 577–585. [Google Scholar] [CrossRef]
- Tang, X.; Zhao, S.; Zhang, Y.; Wang, Y.; Zhang, Z.; Yang, M.; Zhu, Y.; Zhang, G.; Guo, G.; Tong, A.; et al. B7-H3 as a Novel CAR-T Therapeutic Target for Glioblastoma. Mol. Ther. Oncolytics 2019, 14, 279–287. [Google Scholar] [CrossRef] [Green Version]
- Paulsson, J.; Lindh, M.B.; Jarvius, M.; Puputti, M.; Nistér, M.; Nupponen, N.N.; Paulus, W.; Söderberg, O.; Dresemann, G.; von Deimling, A.; et al. Prognostic but Not Predictive Role of Platelet-Derived Growth Factor Receptors in Patients with Recurrent Glioblastoma. Int. J. Cancer 2011, 128, 1981–1988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Witt Hamer, P.C. Small Molecule Kinase Inhibitors in Glioblastoma: A Systematic Review of Clinical Studies. Neuro Oncol. 2010, 12, 304–316. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Tan, X.; Dong, Y.; Giese, A.; Chou, T.C.; Rainov, N.; Yang, B. Differential Effect of Imatinib and Synergism of Combination Treatment with Chemotherapeutic Agents in Malignant Glioma Cells. Basic Clin. Pharmacol. Toxicol. 2009, 104, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Ranza, E.; Mazzini, G.; Facoetti, A.; Nano, R. In-Vitro Effects of the Tyrosine Kinase Inhibitor Imatinib on Glioblastoma Cell Proliferation. J. Neurooncol. 2010, 96, 349–357. [Google Scholar] [CrossRef]
- Gai, Q.J.; Fu, Z.; He, J.; Mao, M.; Yao, X.X.; Qin, Y.; Lan, X.; Zhang, L.; Miao, J.Y.; Wang, Y.X.; et al. EPHA2 Mediates PDGFA Activity and Functions Together with PDGFRA as Prognostic Marker and Therapeutic Target in Glioblastoma. Signal. Transduct. Target. Ther. 2022, 7, 33. [Google Scholar] [CrossRef]
- Li, H.; Xi, Z.; Dai, X.; Wu, W.; Li, Y.; Liu, Y.; Zhang, H. CD147 and Glioma: A Meta-Analysis. J. Neurooncol. 2017, 134, 145–156. [Google Scholar] [CrossRef]
- Nabeshima, K.; Iwasaki, H.; Koga, K.; Hojo, H.; Suzumiya, J.; Kikuchi, M. Emmprin (Basigin/CD147): Matrix Metalloproteinase Modulator and Multifunctional Cell Recognition Molecule That Plays a Critical Role in Cancer Progression. Pathol. Int. 2006, 56, 359–367. [Google Scholar] [CrossRef]
- Mathewson, N.D.; Ashenberg, O.; Tirosh, I.; Gritsch, S.; Perez, E.M.; Marx, S.; Jerby-Arnon, L.; Chanoch-Myers, R.; Hara, T.; Richman, A.R.; et al. Inhibitory CD161 Receptor Identified in Glioma-Infiltrating T Cells by Single-Cell Analysis. Cell 2021, 184, 1281–1298.e26. [Google Scholar] [CrossRef]
- Flieswasser, T.; van den Eynde, A.; van Audenaerde, J.; de Waele, J.; Lardon, F.; Riether, C.; de Haard, H.; Smits, E.; Pauwels, P.; Jacobs, J. The CD70-CD27 Axis in Oncology: The New Kids on the Block. J. Exp. Clin. Cancer Res. 2022, 41, 12. [Google Scholar] [CrossRef]
- Jin, L.; Ge, H.; Long, Y.; Yang, C.; Chang, Y.E.; Mu, L.; Sayour, E.J.; de Leon, G.; Wang, Q.J.; Yang, J.C.; et al. CD70, a Novel Target of CAR T-Cell Therapy for Gliomas. Neuro Oncol. 2018, 20, 55–65. [Google Scholar] [CrossRef] [Green Version]
- Muhammad, N.; Wang, R.; Li, W.; Zhang, Z.; Chang, Y.; Hu, Y.; Zhao, J.; Zheng, X.; Mao, Q.; Xia, H. A Novel TanCAR Targeting IL13Rα2 and EphA2 for Enhanced Glioblastoma Therapy. Mol. Ther. Oncolytics 2022, 24, 729–741. [Google Scholar] [CrossRef] [PubMed]
- Genßler, S.; Burger, M.C.; Zhang, C.; Oelsner, S.; Mildenberger, I.; Wagner, M.; Steinbach, J.P.; Wels, W.S. Dual Targeting of Glioblastoma with Chimeric Antigen Receptor-Engineered Natural Killer Cells Overcomes Heterogeneity of Target Antigen Expression and Enhances Antitumor Activity and Survival. Oncoimmunology 2016, 5, e1119354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Burger, M.C.; Jennewein, L.; Genßler, S.; Schönfeld, K.; Zeiner, P.; Hattingen, E.; Harter, P.N.; Mittelbronn, M.; Tonn, T.; et al. ErbB2/HER2-Specific NK Cells for Targeted Therapy of Glioblastoma. J. Natl. Cancer Inst. 2016, 108, djv375. [Google Scholar] [CrossRef] [Green Version]
- Genoud, V.; Marinari, E.; Nikolaev, S.I.; Castle, J.C.; Bukur, V.; Dietrich, P.Y.; Okada, H.; Walker, P.R. Responsiveness to Anti-PD-1 and Anti-CTLA-4 Immune Checkpoint Blockade in SB28 and GL261 Mouse Glioma Models. Oncoimmunology 2018, 7, e1501137. [Google Scholar] [CrossRef] [Green Version]
- Haddad, A.F.; Young, J.S.; Amara, D.; Berger, M.S.; Raleigh, D.R.; Aghi, M.K.; Butowski, N.A. Mouse Models of Glioblastoma for the Evaluation of Novel Therapeutic Strategies. Neurooncol. Adv. 2021, 3, vdab100. [Google Scholar] [CrossRef] [PubMed]
- Karimi, E.; Yu, M.W.; Maritan, S.M.; Perus, L.J.M.; Rezanejad, M.; Sorin, M.; Dankner, M.; Fallah, P.; Doré, S.; Zuo, D.; et al. Single-Cell Spatial Immune Landscapes of Primary and Metastatic Brain Tumours. Nature 2023, 614, 555–563. [Google Scholar] [CrossRef]
- Abdelfattah, N.; Kumar, P.; Wang, C.; Leu, J.S.; Flynn, W.F.; Gao, R.; Baskin, D.S.; Pichumani, K.; Ijare, O.B.; Wood, S.L.; et al. Single-Cell Analysis of Human Glioma and Immune Cells Identifies S100A4 as an Immunotherapy Target. Nat. Commun. 2022, 13, 767. [Google Scholar] [CrossRef]
- Chen, A.X.; Gartrell, R.D.; Zhao, J.; Upadhyayula, P.S.; Zhao, W.; Yuan, J.; Minns, H.E.; Dovas, A.; Bruce, J.N.; Lasorella, A.; et al. Single-Cell Characterization of Macrophages in Glioblastoma Reveals MARCO as a Mesenchymal pro-Tumor Marker. Genome Med. 2021, 13, 88. [Google Scholar] [CrossRef]
- Kaminska, B.; Ochocka, N.; Segit, P. Single-Cell Omics in Dissecting Immune Microenvironment of Malignant Gliomas—Challenges and Perspectives. Cells 2021, 10, 2264. [Google Scholar] [CrossRef]
- Koh, L.; Novera, W.; Lim, S.W.; Chong, Y.K.; Pang, Q.Y.; Low, D.; Ang, B.T.; Tang, C. Integrative Multi-Omics Approach to Targeted Therapy for Glioblastoma. Pharmacol. Res. 2022, 182, 106308. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Immune Cells Involved | Molecule(s) Significant for Glioma Research | Model Systems Used | References |
|---|---|---|---|
| Microglia and macrophages | Glioma derived CSF-1 | Mouse models, human GBM tumor spheres, cell lines | [45,46] |
| Arg1+ exosomes | Cell culture | [47] | |
| AEG1 | Bioinformatic analysis (TCGA, GTex, CGGA) of human samples, cell lines, co-culture analysis | [48] | |
| MDSCs | NA | GBM patient blood samples + tumor tissue, mouse models | [58] |
| MIF | Co-culture assays, GBM patient samples, syngeneic mouse models | [71] | |
| Cytotoxic T cells | PD-1 | Metadata analysis of glioma samples from published studies | [78] |
| Tregs + Tfr cells | PD-1, CTLA-4 | Human tumor samples, syngeneic mouse models, tumor cell lines | [79] |
| Tfr cells | NA | Resected glioma samples from patients | [80] |
| B lymphocytes | Glioma derived PFG | Primary cell culture | [81] |
| NK cells | TGF-β and NKG2D | Blood samples from glioma patients | [82] |
| IFN-γ | Human GBM tissue samples | [83] |
| Immunotherapy | Clinical Trial | Immune Response | Reference |
|---|---|---|---|
| CAR-T therapies | |||
| IL13Rα2-CAR-T cells | Phase I | Naïve and memory T cells | NCT02208362 NCT04003649 (with immune checkpoint inhibitors) |
| CMV-specific T cells | Phase I/II | Cytotoxic T cells | NCT02661282 |
| HER2-CAR-CMV-T cells | Phase I | T cells | NCT01109095 |
| Vaccines | |||
| Rindopepimut | Phase II Phase III Phase II | EGFRvIII-specific humoral immune response | NCT01498328 [111] NCT01480479 [112] NCT00458601 [113] |
| IMA950 | Phase I/II | CD8+ response and sustained T helper 1 CD4+ T cell response | NCT01920191 [114] NCT01222221 |
| DCs vaccine (PERCELLVAC) | Phase I | Tumor-associated antigen specific CD4+ and CD8+ T cell response | NCT02709616 [115] |
| CMV pp65 DC vaccine | Phase I Phase II Phase I | Expected activation of CD4+ and CD8+ cells | NCT03615404 NCT02366728 NCT02529072 |
| SurVaxM peptide vaccine | Phase II | Preliminary results do not discuss immune response | NCT02455557 |
| HSPCC-96 vaccine | Phase II | Low PD-L1 expression on myeloid immune cells showed better survival | NCT00905060 [116] |
| DSP-7888 |
Phase III
Phase I | 1. WT-1 specific CTL induction activity not observed in a dose-dependent manner (NCT02498665). 2. Higher WT1-specific CTL induction was observed intradermally than subcutaneously (NCT02498665). | NCT03149003 NCT02498665 |
| AV-GBM1 | Phase II | Pro-inflammatory response: increase of Th1, Th2 and Th17 pathway markers as well as B-cells, NK cells and cytotoxic T-lymphocytes | NCT03400917 [117] |
| Immune checkpoint inhibitors | |||
| Nivolumab | Phase II | NA | NCT02550249 |
| Pembrolizumab | Phase II | Infiltration of T cells but CD68+ macrophages predominate (NCT02337686) | NCT02337491 NCT02337686 |
| Ipilimumab | Phase I | NA | NCT02311920 [118] (with Nivolumab) |
| Oncolytic viruses | |||
| PVSRIPO | Phase I | Reduction of Tregs and onset of homeostatic reconstitution of effector T cells | NCT01491893 [119] |
| DNX-2401 (Tasadenoturev) |
Phase I
Phase II | CD8+ and T-bet+ cell infiltration (NCT00805376) | NCT01956734 NCT02798406 NCT00805376 |
| G207 |
Phase I/II
Phase I | Short term CD4+ and CD8+ T cell response and a low humoral response (NCT00157703) | NCT00028158 [120] NCT00157703 [121] |
| Ad-RTS-hIL-12 + Veledimex | Phase I | 1. Sustained increase of IFN-γ. 2. Increase in percentage of CD3+CD8+ T cells in peripheral blood. 3. No change in CD3+CD4+ T cells or NK cells (NCT03636477). | NCT02026271 NCT03636477 [122] |
| NCT Number | Phase | Type of Immunotherapy | Status | Glioma Targeted |
|---|---|---|---|---|
| NCT05474378 | I | B7-H3 CAR-T | Recruiting | Recurrent IDH wildtype GBM |
| NCT04099797 | I | C7R-GD2 CAR-T | Recruiting | High grade glioma Diffuse intrinsic pontine glioma |
| NCT04077866 | I II | B7-H3 CAR-T + Temozolomide B7-H3 CAR-T | Recruiting | Recurrent GBM Refractory GBM |
| NCT02575261 | I II | EphA2 CAR-T | Withdrawn | Recurrent GBM Metastatic GBM |
| NCT04045847 | I | CD147 CAR-T | Recruiting | Recurrent GBM |
| NCT05465954 | II | Immune checkpoint inhibitor + IL-7 | Recruiting | Recurrent GBM |
| NCT03360708 | I | Tumor lysate-pulsed autologous dendritic cell vaccine | Active, not recruiting | Recurrent GBM |
| NCT01567202 | II | DC vaccine with antigens from glioma stem-like cells (A2B5+) | Recruiting | Newly diagnosed GBM Secondary GBM |
| NCT05100641 | III | DC vaccine (AV-GBM1) | Not yet recruiting | Primary GBM |
| NCT03661723 | II | Immune checkpoint inhibitor + bevacizumab + radiation | Active, not recruiting | Bevacizumab resistant recurrent GBM |
| NCT04214392 | I | Chlorotoxin (EQ)-CD28-CD3zeta-CD19t-expressing CAR T-lymphocytes | Recruiting | MMP2+ recurrent and progressive GBM |
| NCT03423992 | I | CAR-T based on EGFRVIII, IL13Rα2, Her-2, EphA2, CD133, GD2 expression | Recruiting | Malignant glioma |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahajan, S.; Schmidt, M.H.H.; Schumann, U. The Glioma Immune Landscape: A Double-Edged Sword for Treatment Regimens. Cancers 2023, 15, 2024. https://doi.org/10.3390/cancers15072024
Mahajan S, Schmidt MHH, Schumann U. The Glioma Immune Landscape: A Double-Edged Sword for Treatment Regimens. Cancers. 2023; 15(7):2024. https://doi.org/10.3390/cancers15072024
Chicago/Turabian StyleMahajan, Sukrit, Mirko H. H. Schmidt, and Ulrike Schumann. 2023. "The Glioma Immune Landscape: A Double-Edged Sword for Treatment Regimens" Cancers 15, no. 7: 2024. https://doi.org/10.3390/cancers15072024
APA StyleMahajan, S., Schmidt, M. H. H., & Schumann, U. (2023). The Glioma Immune Landscape: A Double-Edged Sword for Treatment Regimens. Cancers, 15(7), 2024. https://doi.org/10.3390/cancers15072024