
Biosynthesis and Significance of Fatty Acids, Glycerophospholipids, and Triacylglycerol in the Processes of Glioblastoma Tumorigenesis
 , and
, and
Abstract
:Simple Summary
Abstract
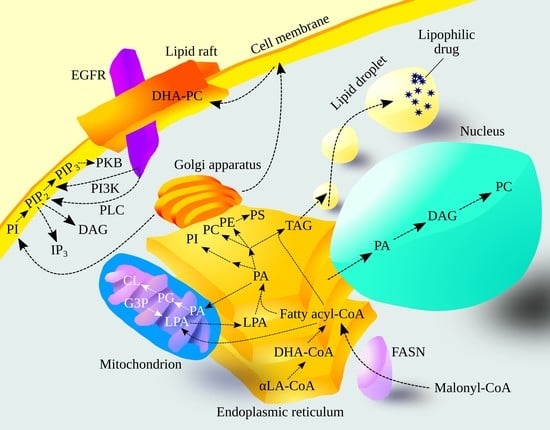
1. Introduction
2. Synthesis of Fatty Acids and Glioblastoma
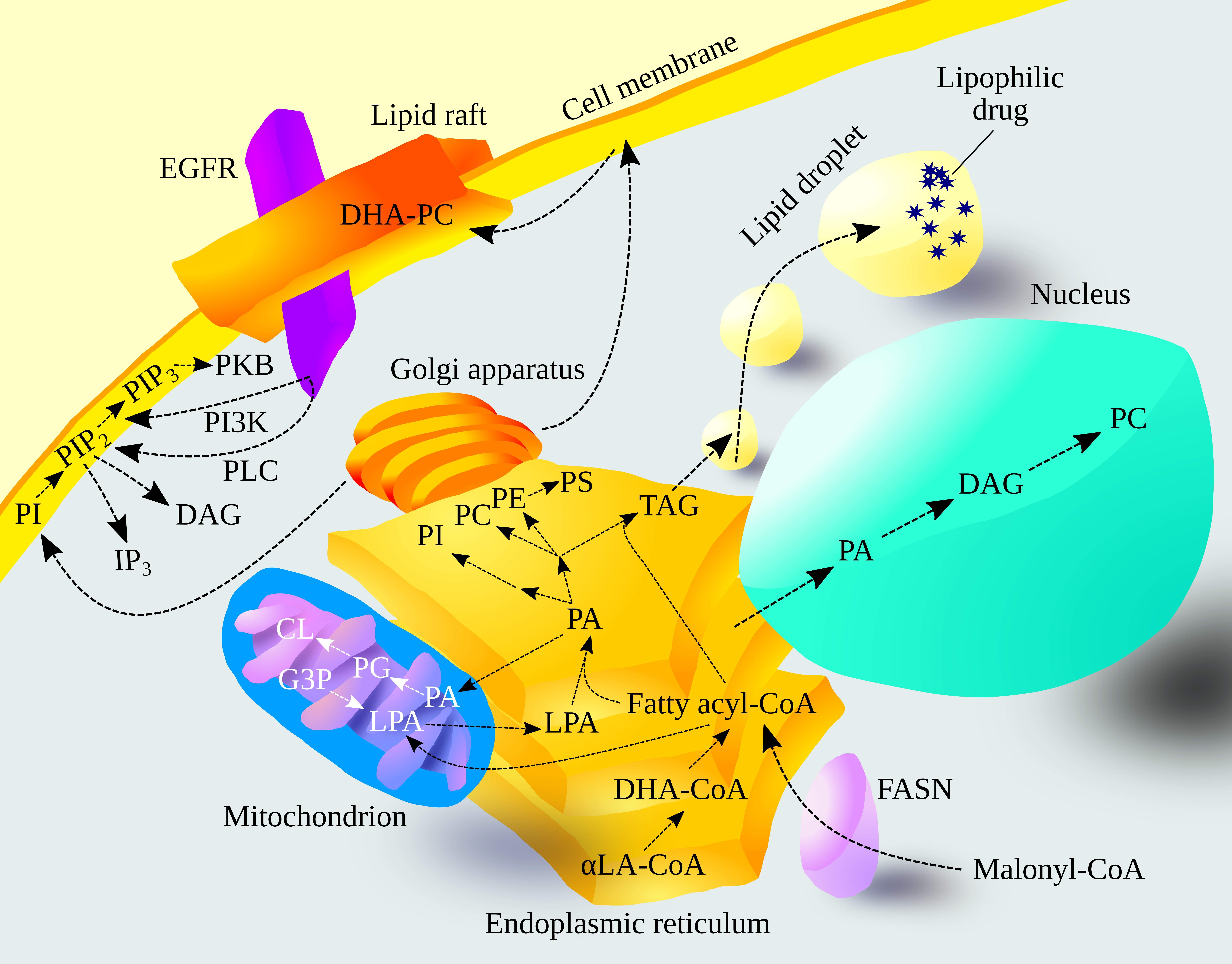
2.1. Synthesis of Fatty Acids
2.2. Significance of the Fatty Acid Synthesis Enzymes in Glioblastoma
2.2.1. Fatty Acid Synthase and Acetyl-CoA Carboxylase in Glioblastoma
2.2.2. Elongases in Glioblastoma
2.2.3. Desaturases in Glioblastoma
2.2.4. DHA, EGFR, and Glioblastoma
2.2.5. The Effect of Hypoxia on Fatty Acid Synthesis in Glioblastoma
2.2.6. The Impact of IDH1 Mutation on Fatty Acid Synthesis
3. Synthesis of Glycerophospholipids and Glioblastoma Tumorigenesis
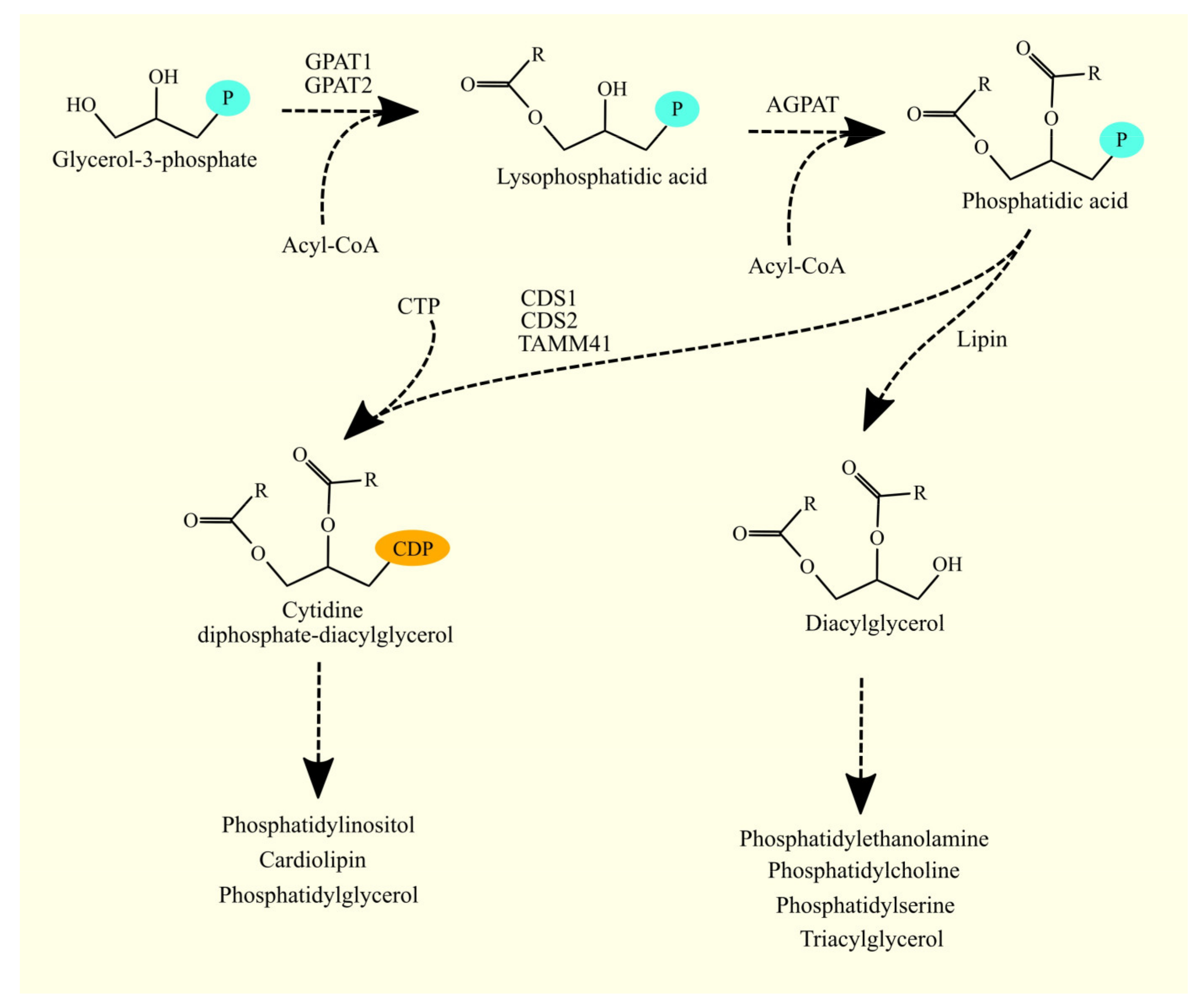
3.1. Glycerol-3-Phosphate Acyltransferases and Glioblastoma Tumorigenesis
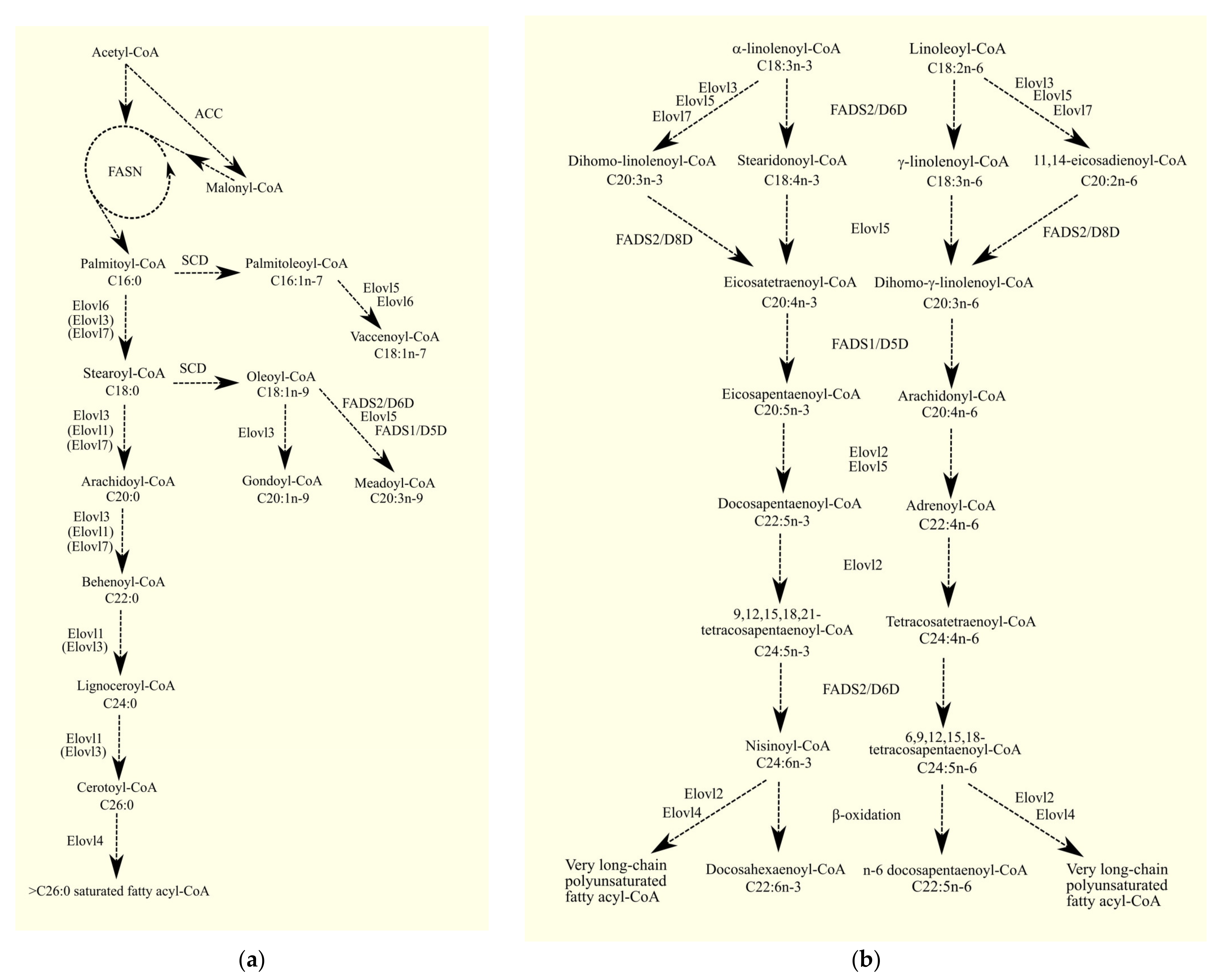
3.1.1. Glycerol-3-Phosphate Acyltransferases
3.1.2. Glycerol-3-Phosphate Acyltransferases in Glioblastoma
3.2. Dihydroxyacetone Phosphate Pathway and Glioblastoma Tumorigenesis
3.2.1. Dihydroxyacetone Phosphate Pathway
3.2.2. Dihydroxyacetone Phosphate Pathway in Glioblastoma
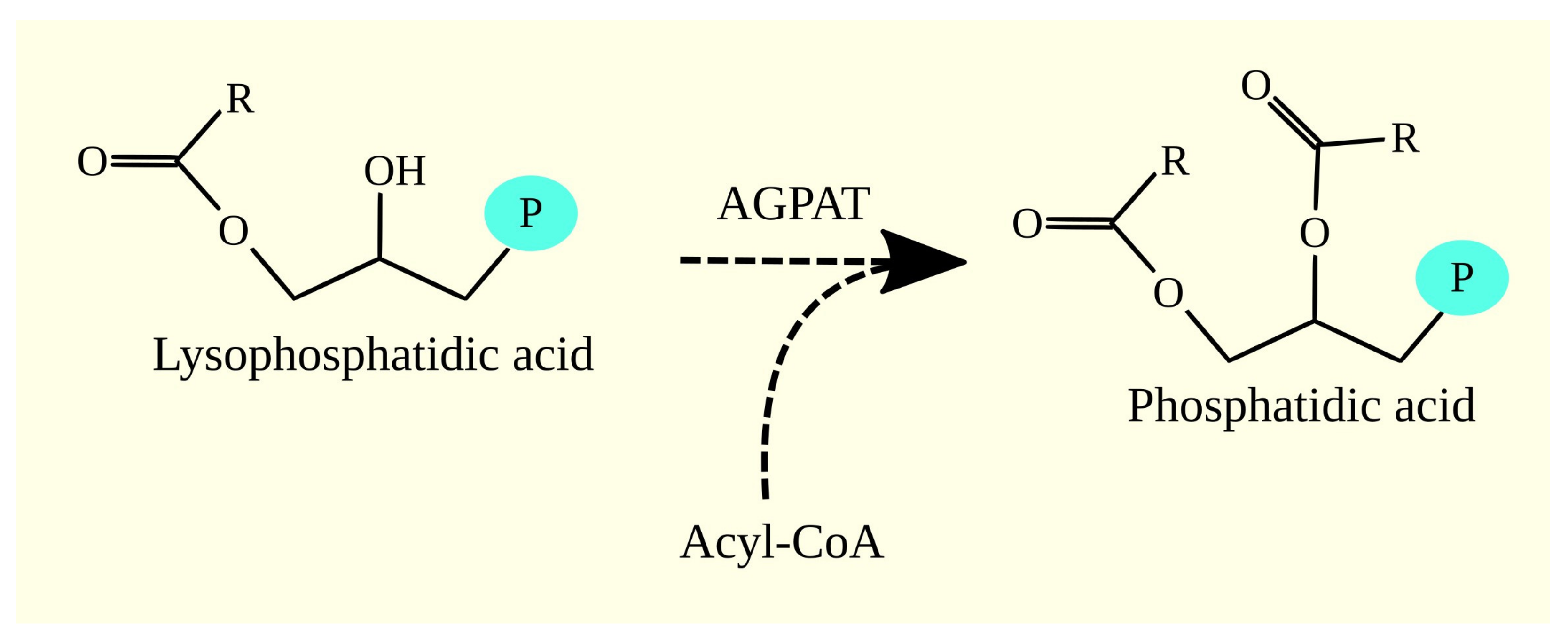
3.3. 1-Acylglycerol-3-phosphate O-acyltransferases and Glioblastoma Tumorigenesis
3.3.1. 1-Acylglycerol-3-phosphate O-acyltransferases
3.3.2. 1-Acylglycerol-3-phosphate O-acyltransferases in Glioblastoma
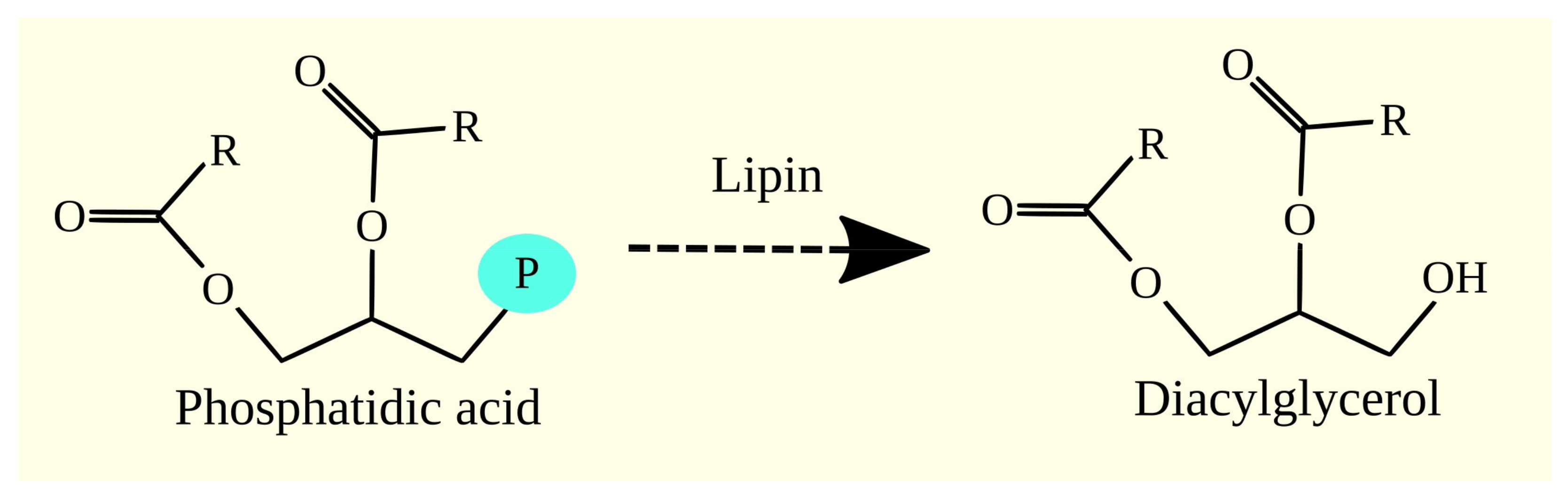
3.4. Synthesis of Glycerophospholipids and Triacylglycerol from Phosphatidic Acid
3.5. Lipins and Glioblastoma Tumorigenesis
3.5.1. Lipins
3.5.2. Lipins in Glioblastoma
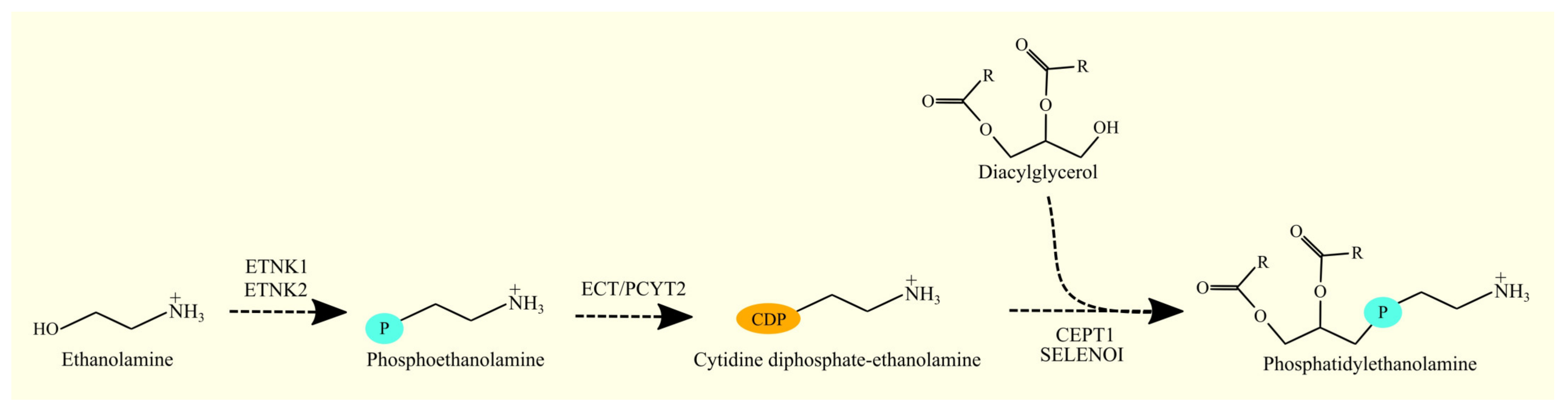
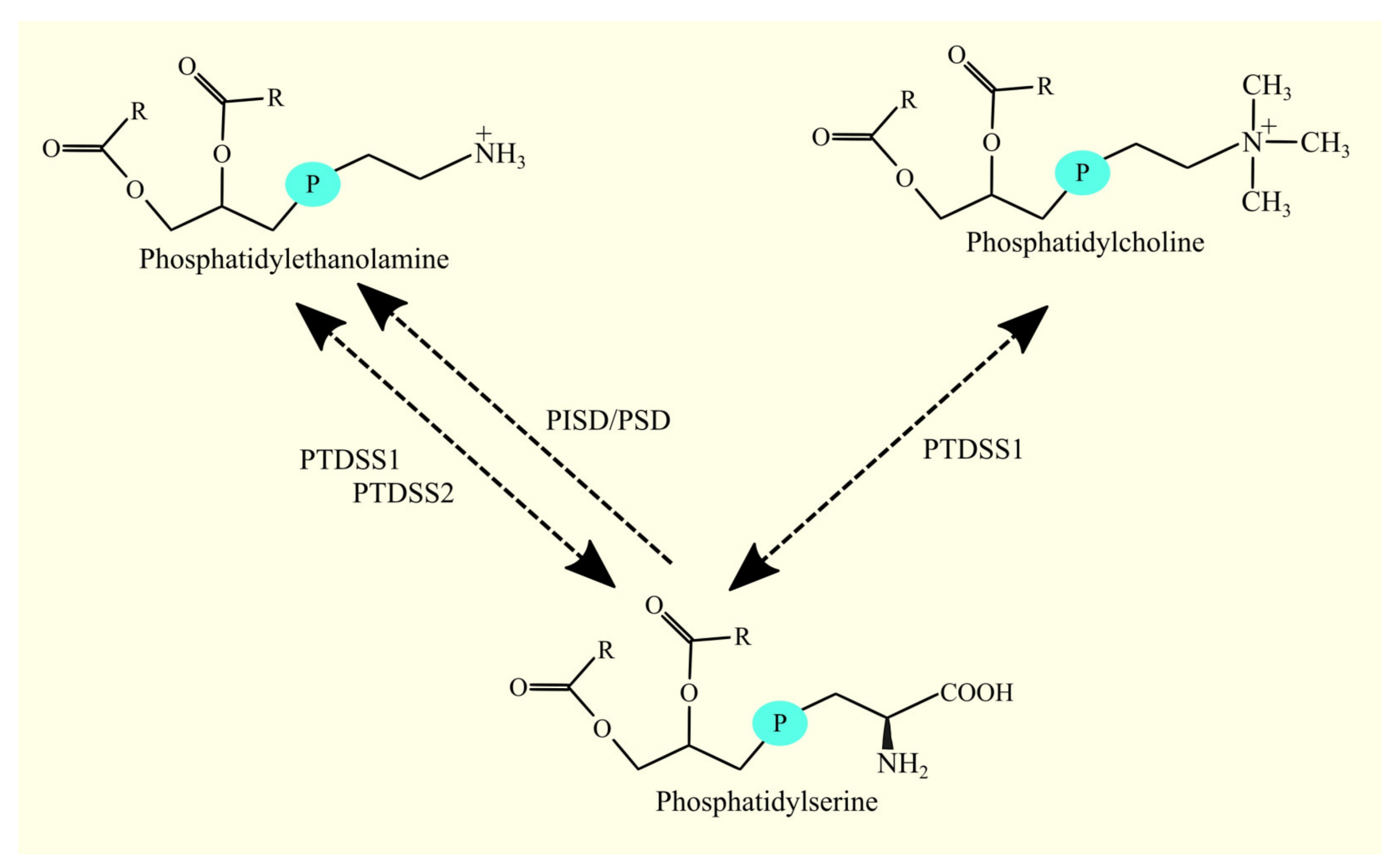
3.6. Biosynthesis of Phosphatidylethanolamine and Glioblastoma Tumorigenesis
3.6.1. Biosynthesis of Phosphatidylethanolamine
3.6.2. Biosynthesis of Phosphatidylethanolamine in Glioblastoma
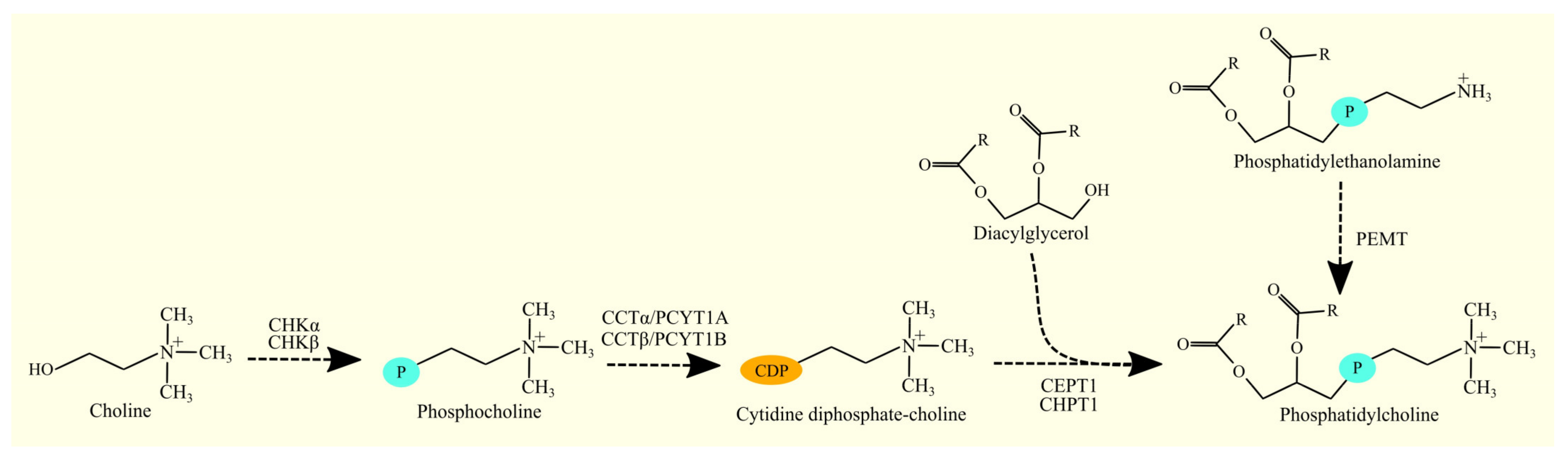
3.7. Biosynthesis of Phosphatidylcholine and Glioblastoma Tumorigenesis
3.7.1. Biosynthesis of Phosphatidylcholine
3.7.2. Biosynthesis of Phosphatidylcholine in Glioblastoma
3.8. Biosynthesis of Phosphatidylserine and Glioblastoma Tumorigenesis
3.8.1. Biosynthesis of Phosphatidylserine
3.8.2. Biosynthesis of Phosphatidylserine in Glioblastoma
3.9. CDP-DAG Synthases and Glioblastoma Tumorigenesis
3.9.1. CDP-DAG Synthases
3.9.2. CDP-DAG Synthases in Glioblastoma
3.10. Biosynthesis of Phosphatidylinositol and Phosphatidylinositol Phosphate and Glioblastoma Tumorigenesis
3.10.1. Biosynthesis of Phosphatidylinositol and Phosphatidylinositol Phosphate
3.10.2. Biosynthesis of Phosphatidylinositol and Phosphatidylinositol Phosphate in Glioblastoma
- Phosphatidylinositol 4-kinase type III, α (PI4KIIIα)/PI4KA;
- Phosphatidylinositol-4-phosphate 5-kinase, type I, β (PIP5KIβ)/PIP5K1B;
- PIP5KIγ/PIP5K1C.
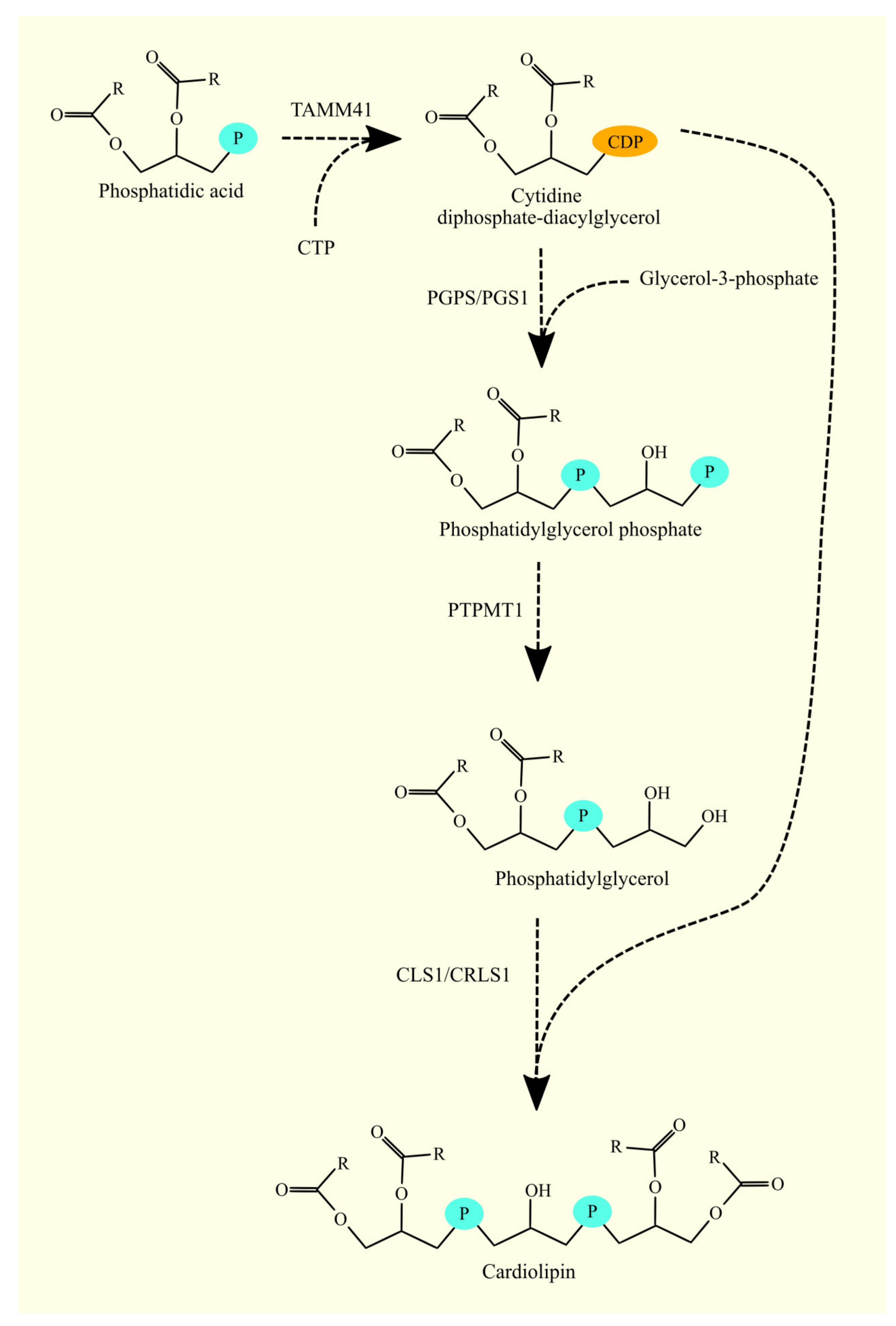
3.11. Biosynthesis of Phosphatidylglycerol and Cardiolipin and Glioblastoma Tumorigenesis
3.11.1. Biosynthesis of Phosphatidylglycerol and Cardiolipin
3.11.2. Biosynthesis of Phosphatidylglycerol and Cardiolipin in Glioblastoma
4. Synthesis of Triacylglycerol and Glioblastoma Tumorigenesis
4.1. Diacylglycerol O-acyltransferases and Glioblastoma Tumorigenesis
4.1.1. Diacylglycerol O-acyltransferases
4.1.2. Diacylglycerol O-acyltransferases in Glioblastoma
4.2. Monoacylglycerol Acyltransferases and Glioblastoma Tumorigenesis
4.2.1. Monoacylglycerol Acyltransferases
4.2.2. Monoacylglycerol Acyltransferases in Glioblastoma
4.3. Lipid Droplets and Triacylglycerol in Glioblastoma
5. Conclusions and Perspective for Future Research
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Brodbelt, A.; Greenberg, D.; Winters, T.; Williams, M.; Vernon, S.; Collins, V.P. (UK) National Cancer Information Network Brain Tumour Group. Glioblastoma in England: 2007–2011. Eur. J. Cancer 2015, 51, 533–542. [Google Scholar] [CrossRef] [Green Version]
- Ostrom, Q.T.; Gittleman, H.; Liao, P.; Vecchione-Koval, T.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary brain and other central nervous system tumors diagnosed in the United States in 2010–2014. Neuro-Oncol. 2017, 19, v1–v88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grech, N.; Dalli, T.; Mizzi, S.; Meilak, L.; Calleja, N.; Zrinzo, A. Rising Incidence of Glioblastoma Multiforme in a Well-Defined Population. Cureus 2020, 12, e8195. [Google Scholar] [CrossRef]
- Johnson, D.R.; Omuro, A.M.P.; Ravelo, A.; Sommer, N.; Guerin, A.; Ionescu-Ittu, R.; Shi, S.; Macalalad, A.; Uhm, J.H. Overall survival in patients with glioblastoma before and after bevacizumab approval. Curr. Med. Res. Opin. 2018, 34, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Bilgin, E.; Duman, B.B.; Altintas, S.; Cil, T.; Gezercan, Y.; Okten, A.I. Predictors of Survival in Turkish Patients with Primary Glioblastoma. Turk. Neurosurg. 2021, 31, 641–653. [Google Scholar] [CrossRef]
- Mirimanoff, R.O.; Gorlia, T.; Mason, W.; Van den Bent, M.J.; Kortmann, R.D.; Fisher, B.; Reni, M.; Brandes, A.A.; Curschmann, J.; Villa, S.; et al. Radiotherapy and temozolomide for newly diagnosed glioblastoma: Recursive partitioning analysis of the EORTC 26981/22981-NCIC CE3 phase III randomized trial. J. Clin. Oncol. 2006, 24, 2563–2569. [Google Scholar] [CrossRef] [Green Version]
- Fisher, J.P.; Adamson, D.C. Current FDA-Approved Therapies for High-Grade Malignant Gliomas. Biomedicines 2021, 9, 324. [Google Scholar] [CrossRef]
- Cohen, M.H.; Shen, Y.L.; Keegan, P.; Pazdur, R. FDA drug approval summary: Bevacizumab (Avastin) as treatment of recurrent glioblastoma multiforme. Oncologist 2009, 14, 1131–1138. [Google Scholar] [CrossRef]
- Desjardins, A.; Herndon, J.E., 2nd; McSherry, F.; Ravelo, A.; Lipp, E.S.; Healy, P.; Peters, K.B.; Sampson, J.H.; Randazzo, D.; Sommer, N.; et al. Single-institution retrospective review of patients with recurrent glioblastoma treated with bevacizumab in clinical practice. Health Sci. Rep. 2019, 2, e114. [Google Scholar] [CrossRef] [Green Version]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grube, S.; Dünisch, P.; Freitag, D.; Klausnitzer, M.; Sakr, Y.; Walter, J.; Kalff, R.; Ewald, C. Overexpression of fatty acid synthase in human gliomas correlates with the WHO tumor grade and inhibition with Orlistat reduces cell viability and triggers apoptosis. J. Neurooncol. 2014, 118, 277–287. [Google Scholar] [CrossRef]
- Zhou, Y.; Jin, G.; Mi, R.; Zhang, J.; Zhang, J.; Xu, H.; Cheng, S.; Zhang, Y.; Song, W.; Liu, F. Inhibition of fatty acid synthase suppresses neovascularization via regulating the expression of VEGF-A in glioma. J. Cancer Res. Clin. Oncol. 2016, 142, 2447–2459. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Wang, Y.; Hao, W.; Zhao, M.; Peng, S. In vitro inhibition of fatty acid synthase by 1,2,3,4,6-penta-O-galloyl-β-D-glucose plays a vital role in anti-tumour activity. Biochem. Biophys. Res. Commun. 2014, 445, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Libertini, L.J.; Smith, S. Synthesis of long chain acyl-enzyme thioesters by modified fatty acid synthetases and their hydrolysis by a mammary gland thioesterase. Arch. Biochem. Biophys. 1979, 192, 47–60. [Google Scholar] [CrossRef]
- Beaty, N.B.; Lane, M.D. Acetyl coenzyme A carboxylase. Rapid purification of the chick liver enzyme and steady state kinetic analysis of the carboxylase-catalyzed reaction. J. Biol. Chem. 1982, 257, 924–929. [Google Scholar] [CrossRef]
- Guillou, H.; Zadravec, D.; Martin, P.G.; Jacobsson, A. The key roles of elongases and desaturases in mammalian fatty acid metabolism: Insights from transgenic mice. Prog. Lipid Res. 2010, 49, 186–199. [Google Scholar] [CrossRef]
- Moon, Y.A.; Horton, J.D. Identification of two mammalian reductases involved in the two-carbon fatty acyl elongation cascade. J. Biol. Chem. 2003, 278, 7335–7343. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, M.; Kanao, Y.; Yamanaka, M.; Sakuraba, H.; Mizutani, Y.; Igarashi, Y.; Kihara, A. Characterization of four mammalian 3-hydroxyacyl-CoA dehydratases involved in very long-chain fatty acid synthesis. FEBS Lett. 2008, 582, 2435–2440. [Google Scholar] [CrossRef] [Green Version]
- Kitazawa, H.; Miyamoto, Y.; Shimamura, K.; Nagumo, A.; Tokita, S. Development of a high-density assay for long-chain fatty acyl-CoA elongases. Lipids 2009, 44, 765–773. [Google Scholar] [CrossRef]
- Jump, D.B.; Torres-Gonzalez, M.; Olson, L.K. Soraphen A, an inhibitor of acetyl CoA carboxylase activity, interferes with fatty acid elongation. Biochem. Pharmacol. 2011, 81, 649–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohno, Y.; Suto, S.; Yamanaka, M.; Mizutani, Y.; Mitsutake, S.; Igarashi, Y.; Sassa, T.; Kihara, A. ELOVL1 production of C24 acyl-CoAs is linked to C24 sphingolipid synthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 18439–18444. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Yu, L.; Schmidt, R.E.; Su, C.; Huang, X.; Gould, K.; Cao, G. Characterization of HSCD5, a novel human stearoyl-CoA desaturase unique to primates. Biochem. Biophys. Res. Commun. 2005, 332, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Man, W.C.; Miyazaki, M.; Chu, K.; Ntambi, J. Colocalization of SCD1 and DGAT2: Implying preference for endogenous monounsaturated fatty acids in triglyceride synthesis. J. Lipid Res. 2006, 47, 1928–1939. [Google Scholar] [CrossRef] [Green Version]
- Enoch, H.G.; Catalá, A.; Strittmatter, P. Mechanism of rat liver microsomal stearyl-CoA desaturase. Studies of the substrate specificity, enzyme-substrate interactions, and the function of lipid. J. Biol. Chem. 1976, 251, 5095–5103. [Google Scholar] [CrossRef]
- Sinner, D.I.; Kim, G.J.; Henderson, G.C.; Igal, R.A. StearoylCoA Desaturase-5: A Novel Regulator of Neuronal Cell Proliferation and Differentiation. PLoS ONE 2012, 7, e39787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellenghi, M.; Puglisi, R.; Pedini, F.; De Feo, A.; Felicetti, F.; Bottero, L.; Sangaletti, S.; Errico, M.C.; Petrini, M.; Gesumundo, C.; et al. SCD5-induced oleic acid production reduces melanoma malignancy by intracellular retention of SPARC and cathepsin B. J. Pathol. 2015, 236, 315–325. [Google Scholar] [CrossRef]
- Bellenghi, M.; Talarico, G.; Botti, L.; Puglisi, R.; Tabolacci, C.; Portararo, P.; Piva, A.; Pontecorvi, G.; Carè, A.; Colombo, M.P.; et al. SCD5-dependent inhibition of SPARC secretion hampers metastatic spreading and favors host immunity in a TNBC murine model. Oncogene 2022, 41, 4055–4065. [Google Scholar] [CrossRef] [PubMed]
- Ves Losada, A.; Brenner, R.R. Incorporation of delta 5 desaturase substrate (dihomogammalinolenic acid, 20:3 n-6) and product (arachidonic acid 20:4 n-6) into rat liver cell nuclei. Prostaglandins Leukot. Essent. Fatty Acids 1998, 59, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.P.; Nakamura, M.; Clarke, S.D. Cloning, expression, and fatty acid regulation of the human delta-5 desaturase. J. Biol. Chem. 1999, 274, 37335–37339. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.P.; Nakamura, M.T.; Clarke, S.D. Cloning, expression, and nutritional regulation of the mammalian Delta-6 desaturase. J. Biol. Chem. 1999, 274, 471–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Antueno, R.J.; Knickle, L.C.; Smith, H.; Elliot, M.L.; Allen, S.J.; Nwaka, S.; Winther, M.D. Activity of human Delta5 and Delta6 desaturases on multiple n-3 and n-6 polyunsaturated fatty acids. FEBS Lett. 2001, 509, 77–80. [Google Scholar] [CrossRef] [PubMed]
- D’andrea, S.; Guillou, H.; Jan, S.; Catheline, D.; Thibault, J.N.; Bouriel, M.; Rioux, V.; Legrand, P. The same rat Delta6-desaturase not only acts on 18- but also on 24-carbon fatty acids in very-long-chain polyunsaturated fatty acid biosynthesis. Biochem. J. 2002, 364, 49–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nitta, S.; Kandori, S.; Tanaka, K.; Sakka, S.; Siga, M.; Nagumo, Y.; Negoro, H.; Kojima, T.; Mathis, B.J.; Shimazui, T.; et al. ELOVL5-mediated fatty acid elongation promotes cellular proliferation and invasion in renal cell carcinoma. Cancer Sci. 2022, 113, 2738–2752. [Google Scholar] [CrossRef] [PubMed]
- Rioux, V.; Pédrono, F.; Blanchard, H.; Duby, C.; Boulier-Monthéan, N.; Bernard, L.; Beauchamp, E.; Catheline, D.; Legrand, P. Trans-vaccenate is Δ13-desaturated by FADS3 in rodents. J. Lipid Res. 2013, 54, 3438–3452. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.Y.; Qin, X.; Liang, A.; Kim, E.; Lawrence, P.; Park, W.J.; Kothapalli, K.S.D.; Brenna, J.T. Fads3 modulates docosahexaenoic acid in liver and brain. Prostaglandins Leukot. Essent. Fatty Acids 2017, 123, 25–32. [Google Scholar] [CrossRef]
- Garcia, C.; Guillocheau, E.; Richard, L.; Drouin, G.; Catheline, D.; Legrand, P.; Rioux, V. Conversion of dietary trans-vaccenic acid to trans11,cis13-conjugated linoleic acid in the rat lactating mammary gland by Fatty Acid Desaturase 3-catalyzed methyl-end Δ13-desaturation. Biochem. Biophys. Res. Commun. 2018, 505, 385–391. [Google Scholar] [CrossRef]
- Karsai, G.; Lone, M.; Kutalik, Z.; Brenna, J.T.; Li, H.; Pan, D.; von Eckardstein, A.; Hornemann, T. FADS3 is a Δ14Z sphingoid base desaturase that contributes to gender differences in the human plasma sphingolipidome. J. Biol. Chem. 2020, 295, 1889–1897. [Google Scholar] [CrossRef] [Green Version]
- Maher, E.A.; Marin-Valencia, I.; Bachoo, R.M.; Mashimo, T.; Raisanen, J.; Hatanpaa, K.J.; Jindal, A.; Jeffrey, F.M.; Choi, C.; Madden, C.; et al. Metabolism of [U-13 C]glucose in human brain tumors in vivo. NMR Biomed. 2012, 25, 1234–1244. [Google Scholar] [CrossRef] [Green Version]
- Ta, N.L.; Seyfried, T.N. Influence of Serum and Hypoxia on Incorporation of [(14)C]-D-Glucose or [(14)C]-L-Glutamine into Lipids and Lactate in Murine Glioblastoma Cells. Lipids 2015, 50, 1167–1184. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell. Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, B.B.; He, H.; Shi, X.H.; Wang, C.L.; Li, W.Q.; Li, B.; Dong, Y.; Hu, G.H.; Hou, L.J.; Luo, C.; et al. Up-regulation of USP2a and FASN in gliomas correlates strongly with glioma grade. J. Clin. Neurosci. 2013, 20, 717–720. [Google Scholar] [CrossRef] [PubMed]
- Ricklefs, F.L.; Maire, C.L.; Matschke, J.; Dührsen, L.; Sauvigny, T.; Holz, M.; Kolbe, K.; Peine, S.; Herold-Mende, C.; Carter, B.; et al. FASN Is a Biomarker Enriched in Malignant Glioma-Derived Extracellular Vesicles. Int. J. Mol. Sci. 2020, 21, 1931. [Google Scholar] [CrossRef] [Green Version]
- Lita, A.; Pliss, A.; Kuzmin, A.; Yamasaki, T.; Zhang, L.; Dowdy, T.; Burks, C.; de Val, N.; Celiku, O.; Ruiz-Rodado, V.; et al. IDH1 mutations induce organelle defects via dysregulated phospholipids. Nat. Commun. 2021, 12, 614. [Google Scholar] [CrossRef]
- Yasumoto, Y.; Miyazaki, H.; Vaidyan, L.K.; Kagawa, Y.; Ebrahimi, M.; Yamamoto, Y.; Ogata, M.; Katsuyama, Y.; Sadahiro, H.; Suzuki, M.; et al. Inhibition of Fatty Acid Synthase Decreases Expression of Stemness Markers in Glioma Stem Cells. PLoS ONE 2016, 11, e0147717. [Google Scholar] [CrossRef]
- Ricklefs, F.; Mineo, M.; Rooj, A.K.; Nakano, I.; Charest, A.; Weissleder, R.; Breakefield, X.O.; Chiocca, E.A.; Godlewski, J.; Bronisz, A. Extracellular Vesicles from High-Grade Glioma Exchange Diverse Pro-oncogenic Signals That Maintain Intratumoral Heterogeneity. Cancer Res. 2016, 76, 2876–2881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.; Kridel, S.; Thorburn, A.; Kooshki, M.; Little, J.; Hebbar, S.; Robbins, M. Fatty acid synthase: A novel target for antiglioma therapy. Br. J. Cancer 2006, 95, 869–878. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [Green Version]
- Seifert, M.; Garbe, M.; Friedrich, B.; Mittelbronn, M.; Klink, B. Comparative transcriptomics reveals similarities and differences between astrocytoma grades. BMC Cancer 2015, 15, 952. [Google Scholar] [CrossRef]
- Jones, J.E.; Esler, W.P.; Patel, R.; Lanba, A.; Vera, N.B.; Pfefferkorn, J.A.; Vernochet, C. Inhibition of Acetyl-CoA Carboxylase 1 (ACC1) and 2 (ACC2) Reduces Proliferation and De Novo Lipogenesis of EGFRvIII Human Glioblastoma Cells. PLoS ONE 2017, 12, e0169566. [Google Scholar] [CrossRef] [Green Version]
- Scott, K.E.; Wheeler, F.B.; Davis, A.L.; Thomas, M.J.; Ntambi, J.M.; Seals, D.F.; Kridel, S.J. Metabolic regulation of invadopodia and invasion by acetyl-CoA carboxylase 1 and de novo lipogenesis. PLoS ONE 2012, 7, e29761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korbecki, J.; Simińska, D.; Jeżewski, D.; Kojder, K.; Tomasiak, P.; Tarnowski, M.; Chlubek, D.; Baranowska-Bosiacka, I. Glioblastoma Multiforme Tumors in Women Have a Lower Expression of Fatty Acid Elongases ELOVL2, ELOVL5, ELOVL6, and ELOVL7 than in Men. Brain Sci. 2022, 12, 1356. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, M.; Koparan, M.; Sari, A.; Ozturk, S.; Kaplan, S.K.; Erol, F.S. Can Behenic Acid (C22:0) Levels be a Prognostic Factor in Glial Tumors? Can. J. Neurol. Sci. 2013, 40, 854–856. [Google Scholar] [CrossRef] [Green Version]
- Saurty-Seerunghen, M.S.; Bellenger, L.; El-Habr, E.A.; Delaunay, V.; Garnier, D.; Chneiweiss, H.; Antoniewski, C.; Morvan-Dubois, G.; Junier, M.P. Capture at the single cell level of metabolic modules distinguishing aggressive and indolent glioblastoma cells. Acta Neuropathol. Commun. 2019, 7, 155. [Google Scholar] [CrossRef] [Green Version]
- Gimple, R.C.; Kidwell, R.L.; Kim, L.J.Y.; Sun, T.; Gromovsky, A.D.; Wu, Q.; Wolf, M.; Lv, D.; Bhargava, S.; Jiang, L.; et al. Glioma Stem Cell-Specific Superenhancer Promotes Polyunsaturated Fatty-Acid Synthesis to Support EGFR Signaling. Cancer Discov. 2019, 9, 1248–1267. [Google Scholar] [CrossRef]
- Zhang, Y.; Pang, S.; Sun, B.; Zhang, M.; Jiao, X.; Lai, L.; Qian, Y.; Yang, N.; Yang, W. ELOVLs Predict Distinct Prognosis Value and Immunotherapy Efficacy In Patients With Hepatocellular Carcinoma. Front. Oncol. 2022, 12, 884066. [Google Scholar] [CrossRef] [PubMed]
- Furse, K.E.; Pratt, D.A.; Porter, N.A.; Lybrand, T.P. Molecular dynamics simulations of arachidonic acid complexes with COX-1 and COX-2: Insights into equilibrium behavior. Biochemistry 2006, 45, 3189–3205. [Google Scholar] [CrossRef] [Green Version]
- Joki, T.; Heese, O.; Nikas, D.C.; Bello, L.; Zhang, J.; Kraeft, S.K.; Seyfried, N.T.; Abe, T.; Chen, L.B.; Carroll, R.S.; et al. Expression of cyclooxygenase 2 (COX-2) in human glioma and in vitro inhibition by a specific COX-2 inhibitor, NS-398. Cancer Res. 2000, 60, 4926–4931. [Google Scholar]
- Nakatsugi, S.; Sugimoto, N.; Furukawa, M. Effects of non-steroidal anti-inflammatory drugs on prostaglandin E2 production by cyclooxygenase-2 from endogenous and exogenous arachidonic acid in rat peritoneal macrophages stimulated with lipopolysaccharide. Prostaglandins Leukot. Essent. Fatty Acids 1996, 55, 451–457. [Google Scholar] [CrossRef]
- Hawcroft, G.; Loadman, P.M.; Belluzzi, A.; Hull, M.A. Effect of eicosapentaenoic acid on E-type prostaglandin synthesis and EP4 receptor signaling in human colorectal cancer cells. Neoplasia 2010, 12, 618–627. [Google Scholar] [CrossRef] [Green Version]
- Kardosh, A.; Blumenthal, M.; Wang, W.J.; Chen, T.C.; Schönthal, A.H. Differential effects of selective COX-2 inhibitors on cell cycle regulation and proliferation of glioblastoma cell lines. Cancer Biol. Ther. 2004, 3, 55–62. [Google Scholar] [CrossRef] [Green Version]
- Brocard, E.; Oizel, K.; Lalier, L.; Pecqueur, C.; Paris, F.; Vallette, F.M.; Oliver, L. Radiation-induced PGE2 sustains human glioma cells growth and survival through EGF signaling. Oncotarget 2015, 6, 6840–6849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, M.T.; Miyake, J.A.; Gomes, R.N.; Feitoza, F.; Stevannato, P.B.; da Cunha, A.S.; Serachi, F.O.; Panagopoulos, A.T.; Colquhoun, A. Cyclooxygenase Inhibition Alters Proliferative, Migratory, and Invasive Properties of Human Glioblastoma Cells In Vitro. Int. J. Mol. Sci. 2021, 22, 4297. [Google Scholar] [CrossRef] [PubMed]
- Cook, P.J.; Thomas, R.; Kingsley, P.J.; Shimizu, F.; Montrose, D.C.; Marnett, L.J.; Tabar, V.S.; Dannenberg, A.J.; Benezra, R. Cox-2-derived PGE2 induces Id1-dependent radiation resistance and self-renewal in experimental glioblastoma. Neuro-Oncol. 2016, 18, 1379–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, Y.T.; Lo, W.L.; Chen, P.Y.; Ko, C.Y.; Chuang, J.Y.; Kao, T.J.; Yang, W.B.; Chang, K.Y.; Hung, C.Y.; Kikkawa, U.; et al. Reprogramming of arachidonate metabolism confers temozolomide resistance to glioblastoma through enhancing mitochondrial activity in fatty acid oxidation. J. Biomed. Sci. 2022, 29, 21. [Google Scholar] [CrossRef]
- Vyazunova, I.; Maklakova, V.I.; Berman, S.; De, I.; Steffen, M.D.; Hong, W.; Lincoln, H.; Morrissy, A.S.; Taylor, M.D.; Akagi, K.; et al. Sleeping Beauty mouse models identify candidate genes involved in gliomagenesis. PLoS ONE 2014, 9, e113489. [Google Scholar] [CrossRef] [Green Version]
- Leonardi, F.; Attorri, L.; Di Benedetto, R.; Di Biase, A.; Sanchez, M.; Nardini, M.; Salvati, S. Effect of arachidonic, eicosapentaenoic and docosahexaenoic acids on the oxidative status of C6 glioma cells. Free. Radic. Res. 2005, 39, 865–874. [Google Scholar] [CrossRef]
- Korbecki, J.; Kojder, K.; Jeżewski, D.; Simińska, D.; Tarnowski, M.; Kopytko, P.; Safranow, K.; Gutowska, I.; Goschorska, M.; Kolasa-Wołosiuk, A.; et al. Expression of SCD and FADS2 Is Lower in the Necrotic Core and Growing Tumor Area than in the Peritumoral Area of Glioblastoma Multiforme. Biomolecules 2020, 10, 727. [Google Scholar] [CrossRef]
- Oatman, N.; Dasgupta, N.; Arora, P.; Choi, K.; Gawali, M.V.; Gupta, N.; Parameswaran, S.; Salomone, J.; Reisz, J.A.; Lawler, S.; et al. Mechanisms of stearoyl CoA desaturase inhibitor sensitivity and acquired resistance in cancer. Sci. Adv. 2021, 7, eabd7459. [Google Scholar] [CrossRef]
- Marszałek, R.; Pisklak, M.; Jankowski, W.; Łukaszkiewicz, J.; Horsztyński, D.; Wawer, I. NMR and gas chromatography studies of lyophilized human brain the tumors. Acta Pol. Pharm. 2010, 67, 129–136. [Google Scholar]
- Guo, D.; Prins, R.M.; Dang, J.; Kuga, D.; Iwanami, A.; Soto, H.; Lin, K.Y.; Huang, T.T.; Akhavan, D.; Hock, M.B.; et al. EGFR signaling through an Akt-SREBP-1-dependent, rapamycin-resistant pathway sensitizes glioblastomas to antilipogenic therapy. Sci. Signal. 2009, 2, ra82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, D.; Nutt, C.L.; Shanehsaz, P.; Peng, X.; Louis, D.N.; Kaetzel, D.M. Autocrine platelet-derived growth factor-dependent gene expression in glioblastoma cells is mediated largely by activation of the transcription factor sterol regulatory element binding protein and is associated with altered genotype and patient survival in human brain the tumors. Cancer Res. 2005, 65, 5523–5534. [Google Scholar] [PubMed] [Green Version]
- Pinkham, K.; Park, D.J.; Hashemiaghdam, A.; Kirov, A.B.; Adam, I.; Rosiak, K.; da Hora, C.C.; Teng, J.; Cheah, P.S.; Carvalho, L.; et al. Stearoyl CoA Desaturase Is Essential for Regulation of Endoplasmic Reticulum Homeostasis and Tumor Growth in Glioblastoma Cancer Stem Cells. Stem Cell. Rep. 2019, 12, 712–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, C.A.; Brault, C.; Peck, B.; Bensaad, K.; Griffiths, B.; Mitter, R.; Chakravarty, P.; East, P.; Dankworth, B.; Alibhai, D.; et al. SREBP maintains lipid biosynthesis and viability of cancer cells under lipid- and oxygen-deprived conditions and defines a gene signature associated with poor survival in glioblastoma multiforme. Oncogene 2015, 34, 5128–5140. [Google Scholar] [CrossRef]
- Mellai, M.; Piazzi, A.; Caldera, V.; Monzeglio, O.; Cassoni, P.; Valente, G.; Schiffer, D. IDH1 and IDH2 mutations, immunohistochemistry and associations in a series of brain the tumors. J. Neurooncol 2011, 105, 345–357. [Google Scholar] [CrossRef]
- Deng, L.; Xiong, P.; Luo, Y.; Bu, X.; Qian, S.; Zhong, W.; Lv, S. Association between IDH1/2 mutations and brain glioma grade. Oncol. Lett. 2018, 16, 5405–5409. [Google Scholar] [CrossRef] [Green Version]
- Dai, S.; Yan, Y.; Xu, Z.; Zeng, S.; Qian, L.; Huo, L.; Li, X.; Sun, L.; Gong, Z. SCD1 Confers Temozolomide Resistance to Human Glioma Cells via the Akt/GSK3β/β-Catenin Signaling Axis. Front. Pharmacol. 2018, 8, 960. [Google Scholar] [CrossRef] [Green Version]
- Shakya, S.; Gromovsky, A.D.; Hale, J.S.; Knudsen, A.M.; Prager, B.; Wallace, L.C.; Penalva, L.O.F.; Brown, H.A.; Kristensen, B.W.; Rich, J.N.; et al. Altered lipid metabolism marks glioblastoma stem and non-stem cells in separate tumor niches. Acta Neuropathol. Commun. 2021, 9, 101. [Google Scholar] [CrossRef]
- Wang, J.; Liang, H.; Sun, M.; Zhang, L.; Xu, H.; Liu, W.; Li, Y.; Zhou, Y.; Li, Y.; Li, M. Delta-6-desaturase inhibitor enhances radiation therapy in glioblastoma in vitro and in vivo. Cancer Manag. Res. 2018, 10, 6779–6790. [Google Scholar] [CrossRef] [Green Version]
- Ariotti, N.; Liang, H.; Xu, Y.; Zhang, Y.; Yonekubo, Y.; Inder, K.; Du, G.; Parton, R.G.; Hancock, J.F.; Plowman, S.J. Epidermal growth factor receptor activation remodels the plasma membrane lipid environment to induce nanocluster formation. Mol. Cell. Biol. 2010, 30, 3795–3804. [Google Scholar] [CrossRef] [Green Version]
- Langelier, B.; Linard, A.; Bordat, C.; Lavialle, M.; Heberden, C. Long chain-polyunsaturated fatty acids modulate membrane phospholipid composition and protein localization in lipid rafts of neural stem cell cultures. J. Cell. Biochem. 2010, 110, 1356–1364. [Google Scholar] [CrossRef] [PubMed]
- Demar, J.C., Jr.; Ma, K.; Chang, L.; Bell, J.M.; Rapoport, S.I. alpha-Linolenic acid does not contribute appreciably to docosahexaenoic acid within brain phospholipids of adult rats fed a diet enriched in docosahexaenoic acid. J. Neurochem. 2005, 94, 1063–1076. [Google Scholar] [CrossRef] [PubMed]
- Schley, P.D.; Brindley, D.N.; Field, C.J. (n-3) PUFA alter raft lipid composition and decrease epidermal growth factor receptor levels in lipid rafts of human breast cancer cells. J. Nutr. 2007, 137, 548–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuentes, N.R.; Mlih, M.; Wang, X.; Webster, G.; Cortes-Acosta, S.; Salinas, M.L.; Corbin, I.R.; Karpac, J.; Chapkin, R.S. Membrane therapy using DHA suppresses epidermal growth factor receptor signaling by disrupting nanocluster formation. J. Lipid Res. 2021, 62, 100026. [Google Scholar] [CrossRef]
- Turk, H.F.; Barhoumi, R.; Chapkin, R.S. Alteration of EGFR spatiotemporal dynamics suppresses signal transduction. PLoS ONE 2012, 7, e39682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.J.; Yun, U.J.; Koo, K.H.; Sung, J.Y.; Shim, J.; Ye, S.K.; Hong, K.M.; Kim, Y.N. Down-regulation of lipid raft-associated onco-proteins via cholesterol-dependent lipid raft internalization in docosahexaenoic acid-induced apoptosis. Biochim. Biophys. Acta 2014, 1841, 190–203. [Google Scholar] [CrossRef] [PubMed]
- Corsetto, P.A.; Montorfano, G.; Zava, S.; Jovenitti, I.E.; Cremona, A.; Berra, B.; Rizzo, A.M. Effects of n-3 PUFAs on breast cancer cells through their incorporation in plasma membrane. Lipids Health Dis. 2011, 10, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turk, H.F.; Monk, J.M.; Fan, Y.Y.; Callaway, E.S.; Weeks, B.; Chapkin, R.S. Inhibitory effects of omega-3 fatty acids on injury-induced epidermal growth factor receptor transactivation contribute to delayed wound healing. Am. J. Physiol. Cell. Physiol. 2013, 304, C905–C917. [Google Scholar] [CrossRef] [Green Version]
- Park, M.; Lim, J.W.; Kim, H. Docoxahexaenoic Acid Induces Apoptosis of Pancreatic Cancer Cells by Suppressing Activation of STAT3 and NF-κB. Nutrients 2018, 10, 1621. [Google Scholar] [CrossRef] [Green Version]
- Patel, K.P.; Ravandi, F.; Ma, D.; Paladugu, A.; Barkoh, B.A.; Medeiros, L.J.; Luthra, R. Acute myeloid leukemia with IDH1 or IDH2 mutation: Frequency and clinicopathologic features. Am. J. Clin. Pathol. 2011, 135, 35–45. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Chen, M.; Hua, H.; Qin, W.; Zhang, R.; Lu, X.; Chao, H. Additional mutations in IDH1/2-mutated patients with acute myeloid leukemia. Int. J. Lab. Hematol. 2021, 43, 1483–1490. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wei, H.; Tang, K.; Lin, D.; Zhang, C.; Mi, Y.; Wang, L.; Wang, C.; Wang, M.; Wang, J. Mutation analysis of isocitrate dehydrogenase in acute lymphoblastic leukemia. Genet. Test. Mol. Biomark. 2012, 16, 991–995. [Google Scholar] [CrossRef] [Green Version]
- Ježek, P. 2-Hydroxyglutarate in Cancer Cells. Antioxid. Redox Signal. 2020, 33, 903–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wendel, A.A.; Lewin, T.M.; Coleman, R.A. Glycerol-3-phosphate acyltransferases: Rate limiting enzymes of triacylglycerol biosynthesis. Biochim. Biophys. Acta 2009, 1791, 501–506. [Google Scholar] [CrossRef] [Green Version]
- Sukumaran, S.; Barnes, R.I.; Garg, A.; Agarwal, A.K. Functional characterization of the human 1-acylglycerol-3-phosphate-O-acyltransferase isoform 10/glycerol-3-phosphate acyltransferase isoform 3. J. Mol. Endocrinol. 2009, 42, 469–478. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.Q.; Kuo, M.S.; Li, S.; Bui, H.H.; Peake, D.A.; Sanders, P.E.; Thibodeaux, S.J.; Chu, S.; Qian, Y.W.; Zhao, Y.; et al. AGPAT6 is a novel microsomal glycerol-3-phosphate acyltransferase. J. Biol. Chem. 2008, 283, 10048–10057. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, A.K. Lysophospholipid acyltransferases: 1-acylglycerol-3-phosphate O-acyltransferases. From discovery to disease. Curr. Opin. Lipidol. 2012, 23, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Fisher, A.B.; Huber, G.A.; Furia, L.; Bassett, D.; Rabinowitz, J.L. Evidence for lipid synthesis by the dihydroxyacetone phosphate pathway in rabbit lung subcellular fractions. J. Lab. Clin. Med. 1976, 87, 1033–1040. [Google Scholar]
- Athenstaedt, K.; Weys, S.; Paltauf, F.; Daum, G. Redundant systems of phosphatidic acid biosynthesis via acylation of glycerol-3-phosphate or dihydroxyacetone phosphate in the yeast Saccharomyces cerevisiae. J. Bacteriol. 1999, 181, 1458–1463. [Google Scholar] [CrossRef] [Green Version]
- Vance, J.E. Phospholipid synthesis and transport in mammalian cells. Traffic 2015, 16, 1–18. [Google Scholar] [CrossRef]
- Nagan, N.; Zoeller, R.A. Plasmalogens: Biosynthesis and functions. Prog. Lipid Res. 2001, 40, 199–229. [Google Scholar] [CrossRef]
- Webber, K.O.; Hajra, A.K. Purification of dihydroxyacetone phosphate acyltransferase from guinea pig liver peroxisomes. Arch. Biochem. Biophys. 1993, 300, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.C.; Ghosh, M.K.; Hajra, A.K. Purification and properties of acyl/alkyl dihydroxyacetone-phosphate reductase from guinea pig liver peroxisomes. J. Biol. Chem. 1990, 265, 8268–8274. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.K.; Sukumaran, S.; Bartz, R.; Barnes, R.I.; Garg, A. Functional characterization of human 1-acylglycerol-3-phosphate-O-acyltransferase isoform 9: Cloning, tissue distribution, gene structure, and enzymatic activity. J. Endocrinol. 2007, 193, 445–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, A.K.; Garg, A. Enzymatic activity of the human 1-acylglycerol-3-phosphate-O-acyltransferase isoform 11: Upregulated in breast and cervical cancers. J. Lipid Res. 2010, 51, 2143–2152. [Google Scholar] [CrossRef] [Green Version]
- Prasad, S.S.; Garg, A.; Agarwal, A.K. Enzymatic activities of the human AGPAT isoform 3 and isoform 5: Localization of AGPAT5 to mitochondria. J. Lipid Res. 2011, 52, 451–462. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, A.K.; Barnes, R.I.; Garg, A. Functional characterization of human 1-acylglycerol-3-phosphate acyltransferase isoform 8: Cloning, tissue distribution, gene structure, and enzymatic activity. Arch. Biochem. Biophys. 2006, 449, 64–76. [Google Scholar] [CrossRef]
- Harayama, T.; Shindou, H.; Shimizu, T. Biosynthesis of phosphatidylcholine by human lysophosphatidylcholine acyltransferase 1. J. Lipid Res. 2009, 50, 1824–1831. [Google Scholar] [CrossRef] [Green Version]
- Eto, M.; Shindou, H.; Yamamoto, S.; Tamura-Nakano, M.; Shimizu, T. Lysophosphatidylethanolamine acyltransferase 2 (LPEAT2) incorporates DHA into phospholipids and has possible functions for fatty acid-induced cell death. Biochem. Biophys. Res. Commun. 2020, 526, 246–252. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, H. Expression of Cytosolic Phospholipase A2 Alpha in Glioblastoma Is Associated with Resistance to Chemotherapy. Am. J. Med. Sci. 2018, 356, 391–398. [Google Scholar] [CrossRef]
- Wu, C.; Su, J.; Wang, X.; Wang, J.; Xiao, K.; Li, Y.; Xiao, Q.; Ling, M.; Xiao, Y.; Qin, C.; et al. Overexpression of the phospholipase A2 group V gene in glioma tumors is associated with poor patient prognosis. Cancer Manag. Res. 2019, 11, 3139–3152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blunsom, N.J.; Cockcroft, S. CDP-Diacylglycerol Synthases (CDS): Gateway to Phosphatidylinositol and Cardiolipin Synthesis. Front. Cell. Dev. Biol. 2020, 8, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csaki, L.S.; Dwyer, J.R.; Fong, L.G.; Tontonoz, P.; Young, G.S.; Reue, K. Lipins, lipinopathies, and the modulation of cellular lipid storage and signaling. Prog. Lipid Res. 2013, 52, 305–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donkor, J.; Sariahmetoglu, M.; Dewald, J.; Brindley, D.N.; Reue, K. Three mammalian lipins act as phosphatidate phosphatases with distinct tissue expression patterns. J. Biol. Chem. 2007, 282, 3450–3457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mylonis, I.; Sembongi, H.; Befani, C.; Liakos, P.; Siniossoglou, S.; Simos, G. Hypoxia causes triglyceride accumulation by HIF-1-mediated stimulation of lipin 1 expression. J. Cell. Sci. 2012, 125, 3485–3493. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wang, S.; Yan, C.; Deng, Y.; Huang, Z.; Shi, P. Mass spectrometry-based proteomic analysis reveals the interacting partners of lipin1. IUBMB Life 2018, 70, 753–762. [Google Scholar] [CrossRef] [Green Version]
- Lordén, G.; Sanjuán-García, I.; de Pablo, N.; Meana, C.; Alvarez-Miguel, I.; Pérez-García, M.T.; Pelegrín, P.; Balsinde, J.; Balboa, M.A. Lipin-2 regulates NLRP3 inflammasome by affecting P2X7 receptor activation. J. Exp. Med. 2017, 214, 511–528. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, E.P.; Weiss, S.B. The function of cytidine coenzymes in the biosynthesis of phospholipides. J. Biol. Chem. 1956, 222, 193–214. [Google Scholar] [CrossRef]
- Gibellini, F.; Smith, T.K. The Kennedy pathway--De novo synthesis of phosphatidylethanolamine and phosphatidylcholine. IUBMB Life 2010, 62, 414–428. [Google Scholar] [CrossRef]
- Lykidis, A.; Wang, J.; Karim, M.A.; Jackowski, S. Overexpression of a mammalian ethanolamine-specific kinase accelerates the CDP-ethanolamine pathway. J. Biol. Chem. 2001, 276, 2174–2179. [Google Scholar] [CrossRef] [Green Version]
- Draus, E.; Niefind, J.; Vietor, K.; Havsteen, B. Isolation and characterization of the human liver ethanolamine kinase. Biochim. Biophys. Acta 1990, 1045, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, A.; Hosaka, K.; Nikawa, J. Cloning of a human cDNA for CTP-phosphoethanolamine cytidylyltransferase by complementation in vivo of a yeast mutant. J. Biol. Chem. 1997, 272, 9567–9572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, S.; Ohtsuka, J.; Wang, S.; Nagata, K.; Tanokura, M.; Ohta, A.; Horiuchi, H.; Fukuda, R. Human CTP:phosphoethanolamine cytidylyltransferase: Enzymatic properties and unequal catalytic roles of CTP-binding motifs in two cytidylyltransferase domains. Biochem. Biophys. Res. Commun. 2014, 449, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Henneberry, A.L.; McMaster, C.R. Cloning and expression of a human choline/ethanolaminephosphotransferase: Synthesis of phosphatidylcholine and phosphatidylethanolamine. Biochem. J. 1999, 339, 291–298. [Google Scholar] [CrossRef]
- Wright, M.M.; McMaster, C.R. PC and PE synthesis: Mixed micellar analysis of the cholinephosphotransferase and ethanolaminephosphotransferase activities of human choline/ethanolamine phosphotransferase 1 (CEPT1). Lipids 2002, 37, 663–672. [Google Scholar] [CrossRef]
- Horibata, Y.; Hirabayashi, Y. Identification and characterization of human ethanolaminephosphotransferase1. J. Lipid Res. 2007, 48, 503–508. [Google Scholar] [CrossRef] [Green Version]
- Kuge, O.; Nishijima, M.; Akamatsu, Y. A cloned gene encoding phosphatidylserine decarboxylase complements the phosphatidylserine biosynthetic defect of a Chinese hamster ovary cell mutant. J. Biol. Chem. 1991, 266, 6370–6376. [Google Scholar] [CrossRef]
- Hovius, R.; Faber, B.; Brigot, B.; Nicolay, K.; de Kruijff, B. On the mechanism of the mitochondrial decarboxylation of phosphatidylserine. J. Biol. Chem. 1992, 267, 16790–16795. [Google Scholar] [CrossRef]
- Steenbergen, R.; Nanowski, T.S.; Beigneux, A.; Kulinski, A.; Young, S.G.; Vance, J.E. Disruption of the phosphatidylserine decarboxylase gene in mice causes embryonic lethality and mitochondrial defects. J. Biol. Chem. 2005, 280, 40032–40040. [Google Scholar] [CrossRef] [Green Version]
- Tasseva, G.; Bai, H.D.; Davidescu, M.; Haromy, A.; Michelakis, E.; Vance, J.E. Phosphatidylethanolamine deficiency in Mammalian mitochondria impairs oxidative phosphorylation and alters mitochondrial morphology. J. Biol. Chem. 2013, 288, 4158–4173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viswanath, P.; Radoul, M.; Izquierdo-Garcia, J.L.; Luchman, H.A.; Gregory Cairncross, J.; Pieper, R.O.; Phillips, J.J.; Ronen, S.M. Mutant IDH1 gliomas downregulate phosphocholine and phosphoethanolamine synthesis in a 2-hydroxyglutarate-dependent manner. Cancer Metab. 2018, 6, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Mou, Y.P.; Wang, Y.Y.; Wang, H.J.; Mou, X.Z. miR-199a-3p targets ETNK1 to promote invasion and migration in gastric cancer cells and is associated with poor prognosis. Pathol. Res. Pract. 2019, 215, 152511. [Google Scholar] [CrossRef] [PubMed]
- Miwa, T.; Kanda, M.; Shimizu, D.; Umeda, S.; Sawaki, K.; Tanaka, H.; Tanaka, C.; Hattori, N.; Hayashi, M.; Yamada, S.; et al. Hepatic metastasis of gastric cancer is associated with enhanced expression of ethanolamine kinase 2 via the p53-Bcl-2 intrinsic apoptosis pathway. Br. J. Cancer 2021, 124, 1449–1460. [Google Scholar] [CrossRef]
- Geng, F.; Cheng, X.; Wu, X.; Yoo, J.Y.; Cheng, C.; Guo, J.Y.; Mo, X.; Ru, P.; Hurwitz, B.; Kim, S.H.; et al. Inhibition of SOAT1 Suppresses Glioblastoma Growth via Blocking SREBP-1-Mediated Lipogenesis. Clin. Cancer Res. 2016, 22, 5337–5348. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Geng, F.; Pan, M.; Wu, X.; Zhong, Y.; Wang, C.; Tian, Z.; Cheng, C.; Zhang, R.; Puduvalli, V.; et al. Targeting DGAT1 Ameliorates Glioblastoma by Increasing Fat Catabolism and Oxidative Stress. Cell. Metab. 2020, 32, 229–242.e8. [Google Scholar] [CrossRef] [PubMed]
- Hosaka, K.; Tanaka, S.; Nikawa, J.; Yamashita, S. Cloning of a human choline kinase cDNA by complementation of the yeast cki mutation. FEBS Lett. 1992, 304, 229–232. [Google Scholar] [CrossRef] [Green Version]
- Uchida, T.; Yamashita, S. Molecular cloning, characterization, and expression in Escherichia coli of a cDNA encoding mammalian choline kinase. J. Biol. Chem. 1992, 267, 10156–10162. [Google Scholar] [CrossRef]
- Aoyama, C.; Yamazaki, N.; Terada, H.; Ishidate, K. Structure and characterization of the genes for murine choline/ethanolamine kinase isozymes alpha and beta. J. Lipid Res. 2000, 41, 452–464. [Google Scholar] [CrossRef]
- Kalmar, G.B.; Kay, R.J.; LaChance, A.C.; Cornell, R.B. Primary structure and expression of a human CTP:phosphocholine cytidylyltransferase. Biochim. Biophys. Acta 1994, 1219, 328–334. [Google Scholar] [CrossRef]
- Lykidis, A.; Murti, K.G.; Jackowski, S. Cloning and characterization of a second human CTP:phosphocholine cytidylyltransferase. J. Biol. Chem. 1998, 273, 14022–14029. [Google Scholar] [CrossRef] [Green Version]
- Henneberry, A.L.; Wistow, G.; McMaster, C.R. Cloning, genomic organization, and characterization of a human cholinephosphotransferase. J. Biol. Chem. 2000, 275, 29808–29815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henneberry, A.L.; Wright, M.M.; McMaster, C.R. The major sites of cellular phospholipid synthesis and molecular determinants of Fatty Acid and lipid head group specificity. Mol. Biol. Cell 2002, 13, 3148–3161. [Google Scholar] [CrossRef] [PubMed]
- Ridgway, N.D.; Vance, D.E. Purification of phosphatidylethanolamine N-methyltransferase from rat liver. J. Biol. Chem. 1987, 262, 17231–17239. [Google Scholar] [CrossRef]
- Ridgway, N.D.; Vance, D.E. Kinetic mechanism of phosphatidylethanolamine N-methyltransferase. J. Biol. Chem. 1988, 263, 16864–16871. [Google Scholar] [CrossRef]
- Vance, D.E. Physiological roles of phosphatidylethanolamine N-methyltransferase. Biochim. Biophys. Acta 2013, 1831, 626–632. [Google Scholar] [CrossRef] [Green Version]
- DeLong, C.J.; Shen, Y.J.; Thomas, M.J.; Cui, Z. Molecular distinction of phosphatidylcholine synthesis between the CDP-choline pathway and phosphatidylethanolamine methylation pathway. J. Biol. Chem. 1999, 274, 29683–29688. [Google Scholar] [CrossRef] [Green Version]
- Ridgway, N.D.; Vance, D.E. Specificity of rat hepatic phosphatidylethanolamine N-methyltransferase for molecular species of diacyl phosphatidylethanolamine. J. Biol. Chem. 1988, 263, 16856–16863. [Google Scholar] [CrossRef]
- Righi, V.; Roda, J.M.; Paz, J.; Mucci, A.; Tugnoli, V.; Rodriguez-Tarduchy, G.; Barrios, L.; Schenetti, L.; Cerdán, S.; García-Martín, M.L. 1H HR-MAS and genomic analysis of human tumor biopsies discriminate between high and low grade astrocytomas. NMR Biomed. 2009, 22, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Asim, M.; Massie, C.E.; Orafidiya, F.; Pértega-Gomes, N.; Warren, A.Y.; Esmaeili, M.; Selth, L.A.; Zecchini, H.I.; Luko, K.; Qureshi, A.; et al. Choline Kinase Alpha as an Androgen Receptor Chaperone and Prostate Cancer Therapeutic Target. J. Natl. Cancer Inst. 2015, 108, djv371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, N.K.; Pradhan, S.; Gowda, G.A.; Kumar, R. In vitro, high-resolution 1H and 31P NMR based analysis of the lipid components in the tissue, serum, and CSF of the patients with primary brain the tumors: One possible diagnostic view. NMR Biomed. 2010, 23, 113–122. [Google Scholar]
- Koch, K.; Hartmann, R.; Schröter, F.; Suwala, A.K.; Maciaczyk, D.; Krüger, A.C.; Willbold, D.; Kahlert, U.D.; Maciaczyk, J. Reciprocal regulation of the cholinic phenotype and epithelial-mesenchymal transition in glioblastoma cells. Oncotarget 2016, 7, 73414–73431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, F.; Zou, Y.; Sun, S.; Wang, Z.; Huang, L.; Ma, H. Knockdown of choline kinase α (CHKA) inhibits the proliferation, invasion and migration of human U87MG glioma cells. Xi Bao Yu Fen. Zi Mian Yi Xue Za Zhi 2020, 36, 724–728. [Google Scholar] [PubMed]
- Zou, Y.; Huang, L.; Sun, S.; Yue, F.; Li, Z.; Ma, Y.; Ma, H. Choline Kinase Alpha Promoted Glioma Development by Activating PI3K/AKT Signaling Pathway. Cancer Biother. Radiopharm. 2021; ahead of print. [Google Scholar] [CrossRef]
- Liu, R.; Lee, J.H.; Li, J.; Yu, R.; Tan, L.; Xia, Y.; Zheng, Y.; Bian, X.L.; Lorenzi, P.L.; Chen, Q.; et al. Choline kinase alpha 2 acts as a protein kinase to promote lipolysis of lipid droplets. Mol. Cell 2021, 81, 2722–2735.e9. [Google Scholar] [CrossRef] [PubMed]
- Miyake, T.; Parsons, S.J. Functional interactions between Choline kinase α, epidermal growth factor receptor and c-Src in breast cancer cell proliferation. Oncogene 2012, 31, 1431–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faulkner, C.; Palmer, A.; Williams, H.; Wragg, C.; Haynes, H.R.; White, P.; DeSouza, R.M.; Williams, M.; Hopkins, K.; Kurian, K.M. EGFR and EGFRvIII analysis in glioblastoma as therapeutic biomarkers. Br. J. Neurosurg. 2015, 29, 23–29. [Google Scholar] [CrossRef]
- Tomohiro, S.; Kawaguti, A.; Kawabe, Y.; Kitada, S.; Kuge, O. Purification and characterization of human phosphatidylserine synthases 1 and 2. Biochem. J. 2009, 418, 421–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuge, O.; Saito, K.; Nishijima, M. Cloning of a Chinese hamster ovary (CHO) cDNA encoding phosphatidylserine synthase (PSS) II, overexpression of which suppresses the phosphatidylserine biosynthetic defect of a PSS I-lacking mutant of CHO-K1 cells. J. Biol. Chem. 1997, 272, 19133–19139. [Google Scholar] [CrossRef] [Green Version]
- Maimó-Barceló, A.; Martín-Saiz, L.; Fernández, J.A.; Pérez-Romero, K.; Garfias-Arjona, S.; Lara-Almúnia, M.; Piérola-Lopetegui, J.; Bestard-Escalas, J.; Barceló-Coblijn, G. Polyunsaturated Fatty Acid-Enriched Lipid Fingerprint of Glioblastoma Proliferative Regions Is Differentially Regulated According to Glioblastoma Molecular Subtype. Int. J. Mol. Sci. 2022, 23, 2949. [Google Scholar] [CrossRef]
- Segawa, K.; Kurata, S.; Yanagihashi, Y.; Brummelkamp, T.R.; Matsuda, F.; Nagata, S. Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science 2014, 344, 1164–1168. [Google Scholar] [CrossRef] [Green Version]
- Graeber, T.G.; Osmanian, C.; Jacks, T.; Housman, D.E.; Koch, C.J.; Lowe, S.W.; Giaccia, A.J. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature 1996, 379, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Castillo Ferrer, C.; Berthenet, K.; Ichim, G. Apoptosis—Fueling the oncogenic fire. FEBS J. 2021, 288, 4445–4463. [Google Scholar] [CrossRef] [PubMed]
- Sekar, D.; Dillmann, C.; Sirait-Fischer, E.; Fink, A.F.; Zivkovic, A.; Baum, N.; Strack, E.; Klatt, S.; Zukunft, S.; Wallner, S.; et al. Phosphatidylserine Synthase PTDSS1 Shapes the Tumor Lipidome to Maintain the tumor-Promoting Inflammation. Cancer Res. 2022, 82, 1617–1632. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Luo, M.; Shao, B.; Yang, J.Y.; Tong, A.; Wang, R.B.; Liu, Y.T.; Jun, R.; Liu, T.; Yi, T.; et al. Phosphatidylserine released from apoptotic cells in the tumor induces M2-like macrophage polarization through the PSR-STAT3-JMJD3 axis. Cancer Commun. 2022, 42, 205–222. [Google Scholar] [CrossRef] [PubMed]
- Weeks, R.; Dowhan, W.; Shen, H.; Balantac, N.; Meengs, B.; Nudelman, E.; Leung, D.W. Isolation and expression of an isoform of human CDP-diacylglycerol synthase cDNA. DNA Cell Biol. 1997, 16, 281–289. [Google Scholar] [CrossRef]
- Volta, M.; Bulfone, A.; Gattuso, C.; Rossi, E.; Mariani, M.; Consalez, G.G.; Zuffardi, O.; Ballabio, A.; Banfi, S.; Franco, B. Identification and characterization of CDS2, a mammalian homolog of the Drosophila CDP-diacylglycerol synthase gene. Genomics 1999, 55, 68–77. [Google Scholar] [CrossRef]
- Tamura, Y.; Harada, Y.; Nishikawa, S.; Yamano, K.; Kamiya, M.; Shiota, T.; Kuroda, T.; Kuge, O.; Sesaki, H.; Imai, K.; et al. Tam41 is a CDP-diacylglycerol synthase required for cardiolipin biosynthesis in mitochondria. Cell Metab. 2013, 17, 709–718. [Google Scholar] [CrossRef] [Green Version]
- Blunsom, N.J.; Gomez-Espinosa, E.; Ashlin, T.G.; Cockcroft, S. Mitochondrial CDP-diacylglycerol synthase activity is due to the peripheral protein, TAMM41 and not due to the integral membrane protein, CDP-diacylglycerol synthase 1. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 284–298. [Google Scholar] [CrossRef]
- Inglis-Broadgate, S.L.; Ocaka, L.; Banerjee, R.; Gaasenbeek, M.; Chapple, J.P.; Cheetham, M.E.; Clark, B.J.; Hunt, D.M.; Halford, S. Isolation and characterization of murine Cds (CDP-diacylglycerol synthase) 1 and 2. Gene 2005, 356, 19–31. [Google Scholar] [CrossRef]
- Mak, H.Y.; Ouyang, Q.; Tumanov, S.; Xu, J.; Rong, P.; Dong, F.; Lam, S.M.; Wang, X.; Lukmantara, I.; Du, X.; et al. AGPAT2 interaction with CDP-diacylglycerol synthases promotes the flux of fatty acids through the CDP-diacylglycerol pathway. Nat. Commun. 2021, 12, 6877. [Google Scholar] [CrossRef]
- Antonsson, B.E. Purification and characterization of phosphatidylinositol synthase from human placenta. Biochem. J. 1994, 297, 517–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, S.; Nikawa, J.; Imai, H.; Yamashita, S.; Hosaka, K. Molecular cloning of rat phosphatidylinositol synthase cDNA by functional complementation of the yeast Saccharomyces cerevisiae pis mutation. FEBS Lett. 1996, 393, 89–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blunsom, N.J.; Cockcroft, S. Phosphatidylinositol synthesis at the endoplasmic reticulum. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158471. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, K.; Epand, R.M. The phosphatidylinositol synthase-catalyzed formation of phosphatidylinositol does not exhibit acyl chain specificity. Biochemistry 2015, 54, 1151–1153. [Google Scholar] [CrossRef] [PubMed]
- Van den Bout, I.; Divecha, N. PIP5K-driven PtdIns(4,5)P2 synthesis: Regulation and cellular functions. J. Cell Sci. 2009, 122, 3837–3850. [Google Scholar] [CrossRef]
- Minogue, S.; Waugh, M.G. The phosphatidylinositol 4-kinases: Don’t call it a comeback. Subcell. Biochem. 2012, 58, 1–24. [Google Scholar]
- Waugh, M.G. Phosphatidylinositol 4-kinases, phosphatidylinositol 4-phosphate and cancer. Cancer Lett. 2012, 325, 125–131. [Google Scholar] [CrossRef]
- Kawasaki, K.; Kuge, O.; Chang, S.C.; Heacock, P.N.; Rho, M.; Suzuki, K.; Nishijima, M.; Dowhan, W. Isolation of a chinese hamster ovary (CHO) cDNA encoding phosphatidylglycerophosphate (PGP) synthase, expression of which corrects the mitochondrial abnormalities of a PGP synthase-defective mutant of CHO-K1 cells. J. Biol. Chem. 1999, 274, 1828–1834. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, K.; Kuge, O.; Yamakawa, Y.; Nishijima, M. Purification of phosphatidylglycerophosphate synthase from Chinese hamster ovary cells. Biochem. J. 2001, 354, 9–15. [Google Scholar] [CrossRef]
- Zhang, J.; Guan, Z.; Murphy, A.N.; Wiley, S.E.; Perkins, G.A.; Worby, C.A.; Engel, J.L.; Heacock, P.; Nguyen, O.K.; Wang, J.H.; et al. Mitochondrial phosphatase PTPMT1 is essential for cardiolipin biosynthesis. Cell Metab. 2011, 13, 690–700. [Google Scholar] [CrossRef] [Green Version]
- Schlame, M.; Hostetler, K.Y. Solubilization, purification, and characterization of cardiolipin synthase from rat liver mitochondria. Demonstration of its phospholipid requirement. J. Biol. Chem. 1991, 266, 22398–22403. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Zhang, X.Y.; Shi, Y. Identification and functional characterization of hCLS1, a human cardiolipin synthase localized in mitochondria. Biochem. J. 2006, 398, 169–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, B.; Xu, F.Y.; Jiang, Y.J.; Choy, P.C.; Hatch, G.M.; Grunfeld, C.; Feingold, K.R. Cloning and characterization of a cDNA encoding human cardiolipin synthase (hCLS1). J. Lipid Res. 2006, 47, 1140–1145. [Google Scholar] [CrossRef] [Green Version]
- Nie, J.; Hao, X.; Chen, D.; Han, X.; Chang, Z.; Shi, Y. A novel function of the human CLS1 in phosphatidylglycerol synthesis and remodeling. Biochim. Biophys. Acta 2010, 1801, 438–445. [Google Scholar] [CrossRef] [Green Version]
- Ohlig, T.; Le, D.V.; Gardemann, A.; Wolke, C.; Gürtler, S.; Peter, D.; Schild, L.; Lendeckel, U. Effects of siRNA-dependent knock-down of cardiolipin synthase and tafazzin on mitochondria and proliferation of glioma cells. Biochim. Biophys. Acta Mol. Cell. Biol. Lipids 2018, 1863, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Gürtler, S.; Wolke, C.; Otto, O.; Heise, N.; Scholz, F.; Laporte, A.; Elsner, M.; Jörns, A.; Weinert, S.; Döring, M.; et al. Tafazzin-dependent cardiolipin composition in C6 glioma cells correlates with changes in mitochondrial and cellular functions, and cellular proliferation. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 452–465. [Google Scholar] [CrossRef]
- Schild, L.; Döring, M.; Jansing, S.; Peter, D.; Jagirdar, G.; Wolke, C.; Gardemann, A.; Lendeckel, U. Proliferation of C6 glioma cells requires the phospholipid remodeling enzyme tafazzin independent of cardiolipin composition. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158596. [Google Scholar] [CrossRef]
- Cases, S.; Smith, S.J.; Zheng, Y.W.; Myers, H.M.; Lear, S.R.; Sande, E.; Novak, S.; Collins, C.; Welch, C.B.; Lusis, A.J.; et al. Identification of a gene encoding an acyl CoA:diacylglycerol acyltransferase, a key enzyme in triacylglycerol synthesis. Proc. Natl. Acad. Sci. USA 1998, 95, 13018–13023. [Google Scholar] [CrossRef] [Green Version]
- Cases, S.; Stone, S.J.; Zhou, P.; Yen, E.; Tow, B.; Lardizabal, K.D.; Voelker, T.; Farese, R.V., Jr. Cloning of DGAT2, a second mammalian diacylglycerol acyltransferase, and related family members. J. Biol. Chem. 2001, 276, 38870–38876. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Onorato, J.M.; Chen, L.; Nelson, D.W.; Yen, C.E.; Cheng, D. Synthesis of neutral ether lipid monoalkyl-diacylglycerol by lipid acyltransferases. J. Lipid Res. 2017, 58, 1091–1099. [Google Scholar] [CrossRef] [Green Version]
- Chitraju, C.; Walther, T.C.; Farese, R.V., Jr. The triglyceride synthesis enzymes DGAT1 and DGAT2 have distinct and overlapping functions in adipocytes. J. Lipid Res. 2019, 60, 1112–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turchetto-Zolet, A.C.; Maraschin, F.S.; de Morais, G.L.; Cagliari, A.; Andrade, C.M.; Margis-Pinheiro, M.; Margis, R. Evolutionary view of acyl-CoA diacylglycerol acyltransferase (DGAT), a key enzyme in neutral lipid biosynthesis. BMC Evol. Biol. 2011, 11, 263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, I.; Cui, Y.; Amiri, A.; Ding, Y.; Campbell, R.E.; Maysinger, D. Pharmacological inhibition of lipid droplet formation enhances the effectiveness of curcumin in glioblastoma. Eur. J. Pharm. Biopharm. 2016, 100, 66–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, J.; Cheng, L.; Shi, Y. Catalytic properties of MGAT3, a putative triacylgycerol synthase. J. Lipid Res. 2007, 48, 583–591. [Google Scholar] [CrossRef] [Green Version]
- Yen, C.L.; Stone, S.J.; Cases, S.; Zhou, P.; Farese, R.V., Jr. Identification of a gene encoding MGAT1, a monoacylglycerol acyltransferase. Proc. Natl. Acad. Sci. USA 2002, 99, 8512–8517. [Google Scholar] [CrossRef] [Green Version]
- Yen, C.L.; Farese, R.V., Jr. MGAT2, a monoacylglycerol acyltransferase expressed in the small intestine. J. Biol. Chem. 2003, 278, 18532–18537. [Google Scholar] [CrossRef] [Green Version]
- Cheng, D.; Nelson, T.C.; Chen, J.; Walker, S.G.; Wardwell-Swanson, J.; Meegalla, R.; Taub, R.; Billheimer, J.T.; Ramaker, M.; Feder, J.N. Identification of acyl coenzyme A:monoacylglycerol acyltransferase 3, an intestinal specific enzyme implicated in dietary fat absorption. J. Biol. Chem. 2003, 278, 13611–13614. [Google Scholar] [CrossRef] [Green Version]
- Walther, T.C.; Chung, J.; Farese, R.V., Jr. Lipid Droplet Biogenesis. Annu. Rev. Cell Dev. Biol. 2017, 33, 491–510. [Google Scholar] [CrossRef] [Green Version]
- Laurenti, G.; Benedetti, E.; D’Angelo, B.; Cristiano, L.; Cinque, B.; Raysi, S.; Alecci, M.; Cerù, M.P.; Cifone, M.G.; Galzio, R.; et al. Hypoxia induces peroxisome proliferator-activated receptor α (PPARα) and lipid metabolism peroxisomal enzymes in human glioblastoma cells. J. Cell Biochem. 2011, 112, 3891–3901. [Google Scholar] [CrossRef]
- Lahrech, H.; Zoula, S.; Farion, R.; Rémy, C.; Décorps, M. In vivo measurement of the size of lipid droplets in an intracerebral glioma in the rat. Magn. Reson. Med. 2001, 45, 409–414. [Google Scholar] [CrossRef]
- Zoula, S.; Hérigault, G.; Ziegler, A.; Farion, R.; Décorps, M.; Rémy, C. Correlation between the occurrence of 1H-MRS lipid signal, necrosis and lipid droplets during C6 rat glioma development. NMR Biomed. 2003, 16, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Cabodevilla, A.G.; Sánchez-Caballero, L.; Nintou, E.; Boiadjieva, V.G.; Picatoste, F.; Gubern, A.; Claro, E. Cell survival during complete nutrient deprivation depends on lipid droplet-fueled β-oxidation of fatty acids. J. Biol. Chem. 2013, 288, 27777–27788. [Google Scholar] [CrossRef] [Green Version]
- Ogasawara, Y.; Cheng, J.; Tatematsu, T.; Uchida, M.; Murase, O.; Yoshikawa, S.; Ohsaki, Y.; Fujimoto, T. Long-term autophagy is sustained by activation of CCTβ3 on lipid droplets. Nat. Commun. 2020, 11, 4480. [Google Scholar] [CrossRef] [PubMed]
- Cerezo-Magaña, M.; Christianson, H.C.; van Kuppevelt, T.H.; Forsberg-Nilsson, K.; Belting, M. Hypoxic Induction of Exosome Uptake through Proteoglycan-Dependent Endocytosis Fuels the Lipid Droplet Phenotype in Glioma. Mol. Cancer Res. 2021, 19, 528–540. [Google Scholar] [CrossRef]
- Wu, X.; Geng, F.; Cheng, X.; Guo, Q.; Zhong, Y.; Cloughesy, T.F.; Yong, W.H.; Chakravarti, A.; Guo, D. Lipid Droplets Maintain Energy Homeostasis and Glioblastoma Growth via Autophagic Release of Stored Fatty Acids. iScience 2020, 23, 101569. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Shah, N.; Almohaisin, M.I.; Saha, S.; Lu, F. Assessing fatty acid-induced lipotoxicity and its therapeutic potential in glioblastoma using stimulated Raman microscopy. Sci. Rep. 2021, 11, 7422. [Google Scholar] [CrossRef]
- Taïb, B.; Aboussalah, A.M.; Moniruzzaman, M.; Chen, S.; Haughey, N.J.; Kim, S.F.; Ahima, R.S. Lipid accumulation and oxidation in glioblastoma multiforme. Sci. Rep. 2019, 9, 19593. [Google Scholar] [CrossRef] [Green Version]
- Nau, R.; Sörgel, F.; Eiffert, H. Penetration of drugs through the blood-cerebrospinal fluid/blood-brain barrier for treatment of central nervous system infections. Clin. Microbiol. Rev. 2010, 23, 858–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Properties | Expression Level in Glioblastoma Tumor Relative to Healthy Brain Tissue | Impact on Survival Rate | Comments | ||
|---|---|---|---|---|---|---|
| Source | GEPIA [48] | Seifert et al. [49] | Other Data Source | GEPIA [48] | 1. | |
| FASN | Synthesis of acyl-CoA to a length of 16 carbons | Expression does not change | Expression does not change | Higher expression in the tumor [12,13,42,43] | No significant impact on prognosis | Expression higher by IDH1 mutation, hypoxia reduces expression, and higher expression in glioblastoma cancer stem cells |
| ACC | Production of malonyl-CoA, a substrate for FASN and elongase | Expression does not change | Lower expression in the tumor | No significant impact on prognosis | Hypoxia reduces expression | |
| Enzyme | Properties | Expression Level in Glioblastoma Tumor Relative to Healthy Brain Tissue | Impact on Survival Rate | Comments | ||
|---|---|---|---|---|---|---|
| Source | GEPIA [48] | Seifert et al. [49] | Other Data Source | GEPIA [48] | 2. | |
| Elovl1 | Elongation of saturated acyl-CoA | Higher expression in the tumor | Expression does not change | Lower expression in the tumor [52] | Worse prognosis | Hypoxia reduces expression |
| Elovl2 | Elongation of 20- and 22-carbon polyunsaturated acyl-CoA | Higher expression in the tumor | Expression does not change | Higher expression in the tumor [54]; | No significant impact on prognosis | Higher expression in glioblastoma cancer stem cells |
| Expression does not change [52] | ||||||
| Elovl3 | Elongation of saturated acyl-CoA | Expression does not change | Expression does not change | Expression does not change [52] | Worse prognosis | Hypoxia reduces expression |
| Elovl4 | Elongation of very long-chain fatty acyl-CoA | Expression does not change | Expression does not change | Expression does not change [52] | No significant impact on prognosis | |
| Elovl5 | Elongation of 18- and 2-carbon polyunsaturated acyl-CoA | Higher expression in the tumor | Higher expression in the tumor | Expression does not change [52] | No significant impact on prognosis | |
| Elovl6 | Elongation of palmitoyl-CoA C16:0 | Expression does not change | Expression does not change | Expression does not change [52] | No significant impact on prognosis | |
| Elovl7 | Elongation of saturated acyl-CoA | Lower expression in the tumor | Lower expression in the tumor | Lower expression in the tumor [52] | No significant impact on prognosis | Hypoxia reduces expression |
| Enzyme | Properties | Expression Level in the Glioblastoma Tumor Relative to Healthy Brain Tissue | Impact on Survival Rate | Comments | ||
|---|---|---|---|---|---|---|
| Source | GEPIA [48] | Seifert et al. [49] | Other Data Source | GEPIA [48] | 3. | |
| SCD | Desaturation of saturated acyl-CoA, MUFA formation | Expression does not change | Lower expression in the tumor | Lower expression in the tumor [68,69] | No significant impact on prognosis | Hypoxia increases expression; Higher expression in IDH1 mutation |
| SCD5 | Desaturation of saturated acyl-CoA, formation of MUFAs | Higher expression in the tumor | Expression does not change | Expression does not change [68] | No significant impact on prognosis | |
| FADS1 | Insertion of a double bond into polyunsaturated acyl-CoA | Expression does not change | Expression does not change | Expression does not change [68] | No significant impact on prognosis | Higher expression in glioblastoma cancer stem cells |
| FADS2 | Insertion of a double bond into polyunsaturated acyl-CoA | Higher expression in the tumor | Higher expression in the tumor | Lower expression in the tumor [68] | No significant impact on prognosis | Higher expression in glioblastoma cancer stem cells |
| FADS3 | Little known | Expression does not change | Expression does not change | Expression does not change [68] | Worse prognosis | |
| Enzyme | Properties | Expression Level in the Glioblastoma Tumor Relative to Healthy Brain Tissue | Impact on Survival Rate | |
|---|---|---|---|---|
| Source | GEPIA [48] | Seifert et al. [49] | GEPIA [48] | |
| GPAT1 | Mitochondrial enzyme | Expression does not change | Expression does not change | No significant impact on prognosis |
| GPAT2 | Mitochondrial enzyme | Expression does not change | No significant impact on prognosis | |
| GPAT3 | Enzyme in endoplasmic reticulum, also 1-acylglycerol-3-phosphate O-acyltransferase activity; Questionable GPAT activity; Other name AGPAT10 and AGPAT9 | Expression does not change | Worse prognosis | |
| GPAT4 | An enzyme in the endoplasmic reticulum, also 1-acylglycerol-3-phosphate O-acyltransferase activity; Other name AGPAT6 | Higher expression in the tumor | Expression does not change | No significant impact on prognosis |
| Enzyme | Properties | Expression Level in the Glioblastoma Tumor Relative to Healthy Brain Tissue | Impact on Survival Rate | |
|---|---|---|---|---|
| Source | GEPIA [48] | Seifert et al. [49] | GEPIA [48] | |
| AGPAT1 | Localization in the endoplasmic reticulum | Expression does not change | Expression does not change | No significant impact on prognosis |
| AGPAT2 | Localization in the endoplasmic reticulum | Expression does not change | Expression does not change | No significant impact on prognosis |
| AGPAT3 | Localization in the endoplasmic reticulum, lysophospholipids acyltransferase activity | Expression does not change | Lower expression in the tumor | No significant impact on prognosis |
| AGPAT4 | Localization in the endoplasmic reticulum | Expression does not change | Lower expression in the tumor | No significant impact on prognosis |
| AGPAT5 | Localization in the endoplasmic reticulum and mitochondria, lysophospholipids acyltransferase activity | Higher expression in the tumor | Higher expression in the tumor | No significant impact on prognosis |
| AGPAT6 | Localization in the endoplasmic reticulum and on lipid droplets; GPAT activity, also called GPAT4 | Higher expression in the tumor | Expression does not change | No significant impact on prognosis |
| AGPAT7 | Localization in the endoplasmic reticulum, Introduces DHA C22:6n-3 into lysophospholipids; Other name LPEAT2 and LPCAT4 | Lower expression in the tumor | Lower expression in the tumor | No significant impact on prognosis |
| AGPAT8 | Localization in the endoplasmic reticulum, acyl-CoA:lysocardiolipin acyltransferase activity; Other name ALCAT1 and LCLAT1 | Higher expression in the tumor | Expression does not change | No significant impact on prognosis |
| AGPAT9 | Localization in the endoplasmic reticulum and on lipid droplets, lysophospholipids acyltransferase activity, production of dipalmitoylphosphatidylcholine; Other name LPCAT1 | Higher expression in the tumor | Higher expression in the tumor | No significant impact on prognosis |
| AGPAT10 | Localization in the endoplasmic reticulum, GPAT activity, also called GPAT3, AGPAT9 | Expression does not change | Worse prognosis | |
| AGPAT11 | Localization in the endoplasmic reticulum and on lipid droplets, lysophospholipids acyltransferase activity another name for LPCAT2 | Higher expression in the tumor | Expression does not change | No significant impact on prognosis |
| Enzyme | Properties | Expression Level in the Glioblastoma Tumor Relative to Healthy Brain Tissue | Impact on Survival Rate | |
|---|---|---|---|---|
| Source | GEPIA [48] | Seifert et al. [49] | GEPIA [48] | |
| Lipin 1 | Expression increased by hypoxia [115] It interacts with about 30 proteins, including PPARα and PPARγ [116] | Expression does not change | Lower expression in the tumor | No significant impact on prognosis |
| Lipin 2 | Reduction in NLRP3 activation, decrease in P2X7 activation [117] | Expression does not change | Expression does not change | No significant impact on prognosis |
| Lipin 3 | Expression does not change | Expression does not change | No significant impact on prognosis | |
| Enzyme | Properties | Expression Level in the Glioblastoma Tumor Relative to Healthy Brain Tissue | Impact on Survival Rate | |
|---|---|---|---|---|
| Source | GEPIA [48] | Seifert et al. [49] | GEPIA [48] | |
| ETNK1 | Production of phosphoethanolamine | Expression does not change | Expression does not change | Better prognosis |
| ETNK2 | Generation of phosphoethanolamine, slight choline kinase activity, expression reduced by IDH1 mutation | Expression does not change | Expression does not change | Worse prognosis |
| ECT/PCYT2 | Production of CDP-ethanolamine | Expression does not change | Lower expression in the tumor | No significant impact on prognosis |
| CEPT1 | Production of PE and PC in Kennedy pathway | Higher expression in the tumor | Expression does not change | Better prognosis (p = 0.062) |
| SELENOI | Production of PE and plasmanyl-PE in the Kennedy pathway | Expression does not change | Lower expression in the tumor | No significant impact on prognosis |
| PISD/PSD | Production of PE from PS | Expression does not change | Expression does not change | No significant impact on prognosis |
| Enzyme | Properties | Expression Level in the Glioblastoma Tumor Relative to Healthy Brain Tissue | Impact on Survival Rate | |
|---|---|---|---|---|
| Source | GEPIA [48] | Seifert et al. [49] | GEPIA [48] | |
| CHKα | Production of phosphocholine, pro-oncogenic properties; Androgen receptor chaperone [149]; Protein kinase activity | Lower expression in the tumor | Expression does not change | No significant impact on prognosis |
| CHKβ | Production of phosphocholine | Expression does not change | Expression does not change | No significant impact on prognosis |
| CCTα/PCYT1A | Generation of CDP-choline, localization in the endoplasmic reticulum and in the cell nucleus | Expression does not change | Expression does not change | No significant impact on prognosis |
| CCTβ/PCYT1B | Generation of CDP-choline, localization in the endoplasmic reticulum | Expression does not change | Expression does not change | No significant impact on prognosis |
| CEPT1 | Production of PC and PE in Kennedy pathway | Higher expression in the tumor | Expression does not change | Better prognosis (p = 0.062) |
| CHPT1 | Production of PC in Kennedy pathway | Expression does not change | Higher expression in the tumor | No significant impact on prognosis |
| PEMT | Production PC from PE | Higher expression in the tumor | Higher expression in the tumor | No significant impact on prognosis |
| Enzyme | Properties | Expression Level in the Glioblastoma Tumor Relative to Healthy Brain Tissue | Impact on Survival Rate | |
|---|---|---|---|---|
| Source | GEPIA [48] | Seifert et al. [49] | GEPIA [48] | |
| PTDSS1 | Replacing PC choline with serine, activity is not reduced by PS | Higher expression in the tumor | Lower expression in the tumor | No significant impact on prognosis |
| PTDSS2 | Replacing choline in PC and ethanolamine in PE with serine, activity is reduced by PS | Expression does not change | Higher expression in the tumor | Worse prognosis |
| Enzyme | Properties | Expression Level in the Glioblastoma Tumor Relative to Healthy Brain Tissue | Impact on Survival Rate | |
|---|---|---|---|---|
| Source | GEPIA [48] | Seifert et al. [49] | GEPIA [48] | |
| CDS1 | An enzyme in the endoplasmic reticulum, PI biosynthesis pathway | Lower expression in the tumor | Lower expression in the tumor | No significant impact on prognosis |
| CDS2 | An enzyme in the endoplasmic reticulum, PI biosynthesis pathway | Expression does not change | Expression does not change | Better prognosis |
| TAMM41 | Enzyme in the mitochondrion, CL and PG biosynthesis pathway | Expression does not change | Expression does not change | No significant impact on prognosis |
| Enzyme | Properties | Expression Level in the Glioblastoma Tumor Relative to Healthy Brain Tissue | Impact on Survival Rate | |
|---|---|---|---|---|
| Source | GEPIA [48] | Seifert et al. [49] | GEPIA [48] | |
| PGPS/PGS1 | Biosynthesis of phosphatidylglycerol phosphate from CDP-DAG and glycerol-3-phosphate | Expression does not change | Expression does not change | No significant impact on prognosis |
| PTPMT1 | Generation of phosphatidylglycerol from phosphatidylglycerol phosphate | Higher expression in the tumor | Higher expression in the tumor | No significant impact on prognosis |
| CLS1/CRLS1 | Biosynthesis of CL from phosphatidylglycerol and CDP-DAG lysophosphatidylglycerol acyltransferase activity | Higher expression in the tumor | Higher expression in the tumor | No significant impact on prognosis |
| Enzyme | Properties | Expression Level in the Glioblastoma Tumor Relative to Healthy Brain Tissue | Impact on Survival Rate | ||
|---|---|---|---|---|---|
| Source | GEPIA [48] | Seifert et al. [49] | Cheng et al. [135] | ||
| DGAT1 | TAG biosynthesis from DAG starvation-induced lipid droplets formation | Expression does not change | Expression does not change | Higher expression in the tumor | Worse prognosis [135] |
| No significant impact on prognosis [48] | |||||
| DGAT2 | TAG biosynthesis from DAG lipid droplets formation | Expression does not change | Lower expression in the tumor | No significant impact on prognosis [48] | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korbecki, J.; Bosiacki, M.; Gutowska, I.; Chlubek, D.; Baranowska-Bosiacka, I. Biosynthesis and Significance of Fatty Acids, Glycerophospholipids, and Triacylglycerol in the Processes of Glioblastoma Tumorigenesis. Cancers 2023, 15, 2183. https://doi.org/10.3390/cancers15072183
Korbecki J, Bosiacki M, Gutowska I, Chlubek D, Baranowska-Bosiacka I. Biosynthesis and Significance of Fatty Acids, Glycerophospholipids, and Triacylglycerol in the Processes of Glioblastoma Tumorigenesis. Cancers. 2023; 15(7):2183. https://doi.org/10.3390/cancers15072183
Chicago/Turabian StyleKorbecki, Jan, Mateusz Bosiacki, Izabela Gutowska, Dariusz Chlubek, and Irena Baranowska-Bosiacka. 2023. "Biosynthesis and Significance of Fatty Acids, Glycerophospholipids, and Triacylglycerol in the Processes of Glioblastoma Tumorigenesis" Cancers 15, no. 7: 2183. https://doi.org/10.3390/cancers15072183
APA StyleKorbecki, J., Bosiacki, M., Gutowska, I., Chlubek, D., & Baranowska-Bosiacka, I. (2023). Biosynthesis and Significance of Fatty Acids, Glycerophospholipids, and Triacylglycerol in the Processes of Glioblastoma Tumorigenesis. Cancers, 15(7), 2183. https://doi.org/10.3390/cancers15072183