



Regulation of Autophagy via Carbohydrate and Lipid Metabolism in Cancer
 , , , , and
, , , , and 
Abstract
:Simple Summary
Abstract
1. Introduction
General Metabolism
2. Glycolysis and Autophagy Regulation in Cancer
2.1. Hexokinase 2 (HK2)
2.2. Phosphofructo-Kinase/Fructose Biphosphatases (PFKFBs)
2.3. Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH)
2.4. Phosphoglycerate Kinase (PGK)
2.5. Pyruvate Kinase (PK)
2.6. Lactate Dehydrogenase (LDH)
2.7. Lactate
2.8. Hypoxia-Inducible Factor-1 Alpha (HIF-1α) and Glycolysis
3. Lipid Metabolism and Regulation of Autophagy in Cancer
3.1. Enzymes in Lipid Metabolism Can Regulate Autophagy in Different Cancers
3.1.1. Fatty Acid Synthase (FASN)
3.1.2. 3-Hydroxy-3-MethylGlutaryl-CoA Reductase (HMGCR) Inhibitors
3.1.3. Sphingosine-1-Kinase (SPHK1)
3.1.4. Fatty Acid Translocase/Cluster of Differentiation 36 (FAT/CD36)
3.2. Lipid Metabolites Can Regulate Autophagy in Different Cancers
3.2.1. Phosphatidic Acid (PA)
3.2.2. Sphingolipids
3.2.3. Peroxisome Proliferator-Activating Receptors (PPARs)
3.2.4. Free Fatty Acids (FFAs)
3.2.5. Omega-3 Polyunsaturated Fatty Acids (n-3 PUFAs)
3.2.6. Cholesterol
4. Lung Cancer, Metabolism and Autophagy
4.1. Etiology, Pathogenesis, and Natural History of Lung Cancer
4.2. Modulation of Autophagy through Genetic Changes in NSCLC
4.3. Autophagy, Glucose Metabolism, and Lung Cancer
4.4. Lipid Metabolism Regulation of Autophagy in Lung Cancer
4.4.1. HMGCR Inhibition
4.4.2. SPHK1
4.4.3. PA
4.4.4. Peroxisome Proliferator-Activating Receptors (PPARs)
4.4.5. Omega-3 Polyunsaturated Fatty Acids (n-3 PUFAs) (Docosahexaenoic Acid (DHA), Docosahexaenoyl Ethanolamine (DHEA), EPA)
5. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Esmaeili, Y.; Yarjanli, Z.; Pakniya, F.; Bidram, E.; Los, M.J.; Eshraghi, M.; Klionsky, D.J.; Ghavami, S.; Zarrabi, A. Targeting autophagy, oxidative stress, and ER stress for neurodegenerative disease treatment. J. Control. Release 2022, 345, 147–175. [Google Scholar] [CrossRef]
- Shojaei, S.; Suresh, M.; Klionsky, D.J.; Labouta, H.I.; Ghavami, S. Autophagy and SARS-CoV-2 infection: Apossible smart targeting of the autophagy pathway. Virulence 2020, 11, 805–810. [Google Scholar] [CrossRef]
- Dalvand, A.; da Silva Rosa, S.C.; Ghavami, S.; Marzban, H. Potential role of TGFBeta and autophagy in early crebellum development. Biochem. Biophys. Rep. 2022, 32, 101358. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [Green Version]
- Ghavami, S.; Gupta, S.; Ambrose, E.; Hnatowich, M.; Freed, D.H.; Dixon, I.M. Autophagy and heart disease: Implications for cardiac ischemia-reperfusion damage. Curr. Mol. Med. 2014, 14, 616–629. [Google Scholar] [CrossRef] [PubMed]
- Ghavami, S.; Zamani, M.; Ahmadi, M.; Erfani, M.; Dastghaib, S.; Darbandi, M.; Darbandi, S.; Vakili, O.; Siri, M.; Grabarek, B.O.; et al. Epigenetic regulation of autophagy in gastrointestinal cancers. Biochim. Biophys. Acta Mol. Basis Dis. 2022, 1868, 166512. [Google Scholar] [CrossRef]
- Li, W.W.; Li, J.; Bao, J.K. Microautophagy: Lesser-known self-eating. Cell. Mol. Life Sci. CMLS 2012, 69, 1125–1136. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Wong, E. Chaperone-mediated autophagy: Roles in disease and aging. Cell Res. 2014, 24, 92–104. [Google Scholar] [CrossRef] [Green Version]
- Mokarram, P.; Albokashy, M.; Zarghooni, M.; Moosavi, M.A.; Sepehri, Z.; Chen, Q.M.; Hudecki, A.; Sargazi, A.; Alizadeh, J.; Moghadam, A.R.; et al. New frontiers in the treatment of colorectal cancer: Autophagy and the unfolded protein response as promising targets. Autophagy 2017, 13, 781–819. [Google Scholar] [CrossRef] [PubMed]
- Seo, A.Y.; Lau, P.W.; Feliciano, D.; Sengupta, P.; Gros, M.A.L.; Cinquin, B.; Larabell, C.A.; Lippincott-Schwartz, J. AMPK and vacuole-associated Atg14p orchestrate mu-lipophagy for energy production and long-term survival under glucose starvation. eLife 2017, 6, e21690. [Google Scholar] [CrossRef]
- Martelli, A.; Omrani, M.; Zarghooni, M.; Citi, V.; Brogi, S.; Calderone, V.; Sureda, A.; Lorzadeh, S.; da Silva Rosa, S.C.; Grabarek, B.O.; et al. New Visions on Natural Products and Cancer Therapy: Autophagy and Related Regulatory Pathways. Cancers 2022, 14, 5839. [Google Scholar] [CrossRef]
- Jung, S.; Choe, S.; Woo, H.; Jeong, H.; An, H.K.; Moon, H.; Ryu, H.Y.; Yeo, B.K.; Lee, Y.W.; Choi, H.; et al. Autophagic death of neural stem cells mediates chronic stress-induced decline of adult hippocampal neurogenesis and cognitive deficits. Autophagy 2020, 16, 512–530. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; He, J.; Ye, X.; Zhu, J.; Hu, X.; Shen, M.; Ma, Y.; Mao, Z.; Song, H.; Chen, F. β-Thujaplicin induces autophagic cell death, apoptosis, and cell cycle arrest through ROS-mediated Akt and p38/ERK MAPK signaling in human hepatocellular carcinoma. Cell Death Dis. 2019, 10, 255. [Google Scholar] [CrossRef] [Green Version]
- Ruolan, W.; Liangjiao, C.; Longquan, S. The mTOR/ULK1 signaling pathway mediates the autophagy-promoting and osteogenic effects of dicalcium silicate nanoparticles. J. Nanobiotechnol. 2020, 18, 119. [Google Scholar] [CrossRef]
- Guevara-Cruz, M.; Godinez-Salas, E.T.; Sanchez-Tapia, M.; Torres-Villalobos, G.; Pichardo-Ontiveros, E.; Guizar-Heredia, R.; Arteaga-Sanchez, L.; Gamba, G.; Mojica-Espinosa, R.; Schcolnik-Cabrera, A.; et al. Genistein stimulates insulin sensitivity through gut microbiota reshaping and skeletal muscle AMPK activation in obese subjects. BMJ Open Diabetes Res. Care 2020, 8, e000948. [Google Scholar] [CrossRef] [Green Version]
- Rothschild, J.A.; Islam, H.; Bishop, D.J.; Kilding, A.E.; Stewart, T.; Plews, D.J. Factors Influencing AMPK Activation During Cycling Exercise: A Pooled Analysis and Meta-Regression. Sport. Med. 2022, 52, 1273–1294. [Google Scholar] [CrossRef]
- Goldsmith, J.; Levine, B.; Debnath, J. Autophagy and cancer metabolism. Methods Enzymol. 2014, 542, 25–57. [Google Scholar]
- Liu, Y.; Shoji-Kawata, S.; Sumpter, R., Jr.; Wei, Y.; Ginet, V.; Zhang, L.; Posner, B.; Tran, K.A.; Green, D.R.; Xavier, R.J.; et al. Autosis is a Na+, K+-ATPase-regulated form of cell death triggered by autophagy-962 inducing peptides, starvation, and hypoxia-ischemia. Proc. Natl. Acad. Sci. USA 2013, 110, 964. [Google Scholar] [CrossRef] [Green Version]
- Baghaei, K.; Mazhari, S.; Tokhanbigli, S.; Parsamanesh, G.; Alavifard, H.; Schaafsma, D.; Ghavami, S. Therapeutic potential of targeting regulatory mechanisms of hepatic stellate cell activation in liver fibrosis. Drug Discov. Today 2022, 27, 1044–1061. [Google Scholar] [CrossRef]
- Eshraghi, M.; Ahmadi, M.; Afshar, S.; Lorzadeh, S.; Adlimoghaddam, A.; Rezvani Jalal, N.; West, R.; Dastghaib, S.; Igder, S.; Torshizi, S.R.N.; et al. Enhancing autophagy in Alzheimer’s disease through drug repositioning. Pharmacol. Ther. 2022, 237, 108171. [Google Scholar] [CrossRef]
- Habibzadeh, P.; Dastsooz, H.; Eshraghi, M.; Los, M.J.; Klionsky, D.J.; Ghavami, S. Autophagy: The Potential Link between SARS-CoV-2 and Cancer. Cancers 2021, 13, 5721. [Google Scholar] [CrossRef] [PubMed]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2019, 38, e101812. [Google Scholar] [CrossRef] [PubMed]
- Barzegar Behrooz, A.; Talaie, Z.; Jusheghani, F.; Los, M.J.; Klonisch, T.; Ghavami, S. Wnt and PI3K/Akt/mTOR Survival Pathways as Therapeutic Targets in Glioblastoma. Int. J. Mol. Sci. 2022, 23, 1353. [Google Scholar] [CrossRef]
- Sharifzad, F.; Ghavami, S.; Verdi, J.; Mardpour, S.; Mollapour Sisakht, M.; Azizi, Z.; Taghikhani, A.; Los, M.J.; Fakharian, E.; Ebrahimi, M.; et al. Glioblastoma cancer stem cell biology: Potential theranostic targets. Drug Resist. Updates 2019, 42, 35–45. [Google Scholar] [CrossRef]
- Alizadeh, J.; Lorzadeh, S.; Ghavami, S. Autophagy and cancer metastasis: A Trojan horse. J. Investig. Med. 2021, 69, 1145–1147. [Google Scholar] [CrossRef] [PubMed]
- Brun, P.; Tarricone, E.; Di Stefano, A.; Mattiuzzo, E.; Mehrbod, P.; Ghavami, S.; Leonardi, A. The regulatory activity of autophagy in conjunctival fibroblasts and its possible role in vernal keratoconjunctivitis. J. Allergy Clin. Immunol. 2020, 146, 1210–1213.e1219. [Google Scholar] [CrossRef]
- Samiei, E.; Seyfoori, A.; Toyota, B.; Ghavami, S.; Akbari, M. Investigating Programmed Cell Death and Tumor Invasion in a Three-Dimensional (3D) Microfluidic Model of Glioblastoma. Int. J. Mol. Sci. 2020, 21, 3162. [Google Scholar] [CrossRef]
- Shojaei, S.; Alizadeh, J.; Thliveris, J.; Koleini, N.; Kardami, E.; Hatch, G.M.; Xu, F.; Hombach-Klonisch, S.; Klonisch, T.; Ghavami, S. Statins: A new approach to combat temozolomide chemoresistance in glioblastoma. J. Investig. Med. 2018, 66, 1083. [Google Scholar] [CrossRef]
- Shojaei, S.; Koleini, N.; Samiei, E.; Aghaei, M.; Cole, L.K.; Alizadeh, J.; Islam, M.I.; Vosoughi, A.R.; Albokashy, M.; Butterfield, Y.; et al. Simvastatin increases temozolomide-induced cell death by targeting the fusion of autophagosomes and lysosomes. FEBS J. 2020, 287, 1005–1034. [Google Scholar] [CrossRef]
- Kawalec, P.; Martens, M.D.; Field, J.T.; Mughal, W.; Caymo, A.M.; Chapman, D.; Xiang, B.; Ghavami, S.; Dolinsky, V.W.; Gordon, J.W. Differential impact of doxorubicin dose on cell death and autophagy pathways during acute cardiotoxicity. Toxicol. Appl. Pharmacol. 2022, 453, 116210. [Google Scholar] [CrossRef]
- Dastghaib, S.; Shojaei, S.; Mostafavi-Pour, Z.; Sharma, P.; Patterson, J.B.; Samali, A.; Mokarram, P.; Ghavami, S. Simvastatin Induces Unfolded Protein Response and Enhances Temozolomide-Induced Cell Death in Glioblastoma Cells. Cells 2020, 9, 2339. [Google Scholar] [CrossRef]
- Stefanek, E.; Samiei, E.; Kavoosi, M.; Esmaeillou, M.; Roustai Geraylow, K.; Emami, A.; Ashrafizadeh, M.; Perrin, D.; Gordon, J.W.; Akbari, M.; et al. A bioengineering method for modeling alveolar Rhabdomyosarcoma and assessing chemotherapy responses. MethodsX 2021, 8, 101473. [Google Scholar] [CrossRef] [PubMed]
- Moghadam, A.R.; da Silva Rosa, S.C.; Samiei, E.; Alizadeh, J.; Field, J.; Kawalec, P.; Thliveris, J.; Akbari, M.; Ghavami, S.; Gordon, J.W. Autophagy modulates temozolomide-induced cell death in alveolar Rhabdomyosarcoma cells. Cell Death Discov. 2018, 4, 52. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhang, Y.; Wang, L.; Li, M.; Yang, J.; Chen, P.; Zhu, J.; Li, X.; Zeng, Z.; Li, G.; et al. FOXA1 prevents nutrients deprivation induced autophagic cell death through inducing loss of imprinting of IGF2 in lung adenocarcinoma. Cell Death Dis. 2022, 13, 711. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Li, X.; Wang, K.; Zhu, D.; Meng, B.; Liu, J.; Liang, X.; Jin, Y.; Liu, X.; Wen, Q.; et al. PURPL represses autophagic cell death to promote cutaneous melanoma by modulating ULK1 phosphorylation. Cell Death Dis. 2021, 12, 1070. [Google Scholar] [CrossRef] [PubMed]
- Long, J.S.; Kania, E.; McEwan, D.G.; Barthet, V.J.A.; Brucoli, M.; Ladds, M.; Nossing, C.; Ryan, K.M. ATG7 is a haploinsufficient repressor of tumor progression and promoter of metastasis. Proc. Natl. Acad. Sci. USA 2022, 119, e2113465119. [Google Scholar] [CrossRef] [PubMed]
- Bermudez, O.; Marchetti, S.; Pages, G.; Gimond, C. Post-translational regulation of the ERK phosphatase DUSP6/MKP3 by the mTOR pathway. Oncogene 2008, 27, 3685–3691. [Google Scholar] [CrossRef] [Green Version]
- Alizadeh, J.; Glogowska, A.; Thliveris, J.; Kalantari, F.; Shojaei, S.; Hombach-Klonisch, S.; Klonisch, T.; Ghavami, S. Autophagy modulates transforming growth factor beta 1 induced epithelial to mesenchymal transition in non-small cell lung cancer cells. Biochim. Et Biophys. Acta (BBA) Mol. Cell Res. 2018, 1865, 749–768. [Google Scholar] [CrossRef]
- Alizadeh, J.; Shojaei, S.; Sepanjnia, A.; Hashemi, M.; Eftekharpour, E.; Ghavami, S. Simultaneous Detection of Autophagy and Epithelial to Mesenchymal Transition in the Non-small Cell Lung Cancer Cells. Methods Mol. Biol. 2019, 1854, 87–103. [Google Scholar] [CrossRef]
- Gundamaraju, R.; Lu, W.; Paul, M.K.; Jha, N.K.; Gupta, P.K.; Ojha, S.; Chattopadhyay, I.; Rao, P.V.; Ghavami, S. Autophagy and EMT in cancer and metastasis: Who controls whom? Biochim. Biophys. Acta Mol. Basis Dis. 2022, 1868, 166431. [Google Scholar] [CrossRef]
- Fell, D.; Cornish-Bowden, A. Understanding the Control of Metabolism; Portland Press: London, UK, 1997. [Google Scholar]
- Newsholme, E.A.; Start, C. Regulation in Metabolism; John Wiley and Sons: Hoboken, NJ, USA, 1973. [Google Scholar]
- Morrison, A.J. Cancer cell metabolism connects epigenetic modifications to transcriptional regulation. FEBS J. 2022, 289, 1302–1314. [Google Scholar] [CrossRef]
- Wang, X.; An, P.; Gu, Z.; Luo, Y.; Luo, J. Mitochondrial Metal Ion Transport in Cell Metabolism and Disease. Int. J. Mol. Sci. 2021, 22, 7525. [Google Scholar] [CrossRef] [PubMed]
- Sekar, M.; Thirumurugan, K. Autophagy: A molecular switch to regulate adipogenesis and lipolysis. Mol. Cell. Biochem. 2022, 477, 727–742. [Google Scholar] [CrossRef]
- Watanabe, S.; Tsujino, S. Applications of Medium-Chain Triglycerides in Foods. Front. Nutr. 2022, 9, 802805. [Google Scholar] [CrossRef] [PubMed]
- Vance, J.E.; Vance, D.E. Biochemistry of Lipids, Lipoproteins and Membranes; Elsevier: Amsterdam, The Netherlands, 2008. [Google Scholar]
- Solinas, G.; Borén, J.; Dulloo, A.G. De novo lipogenesis in metabolic homeostasis: More friend than foe? Mol. Metab. 2015, 4, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Ameer, F.; Scandiuzzi, L.; Hasnain, S.; Kalbacher, H.; Zaidi, N. De novo lipogenesis in health and disease. Metabolism 2014, 63, 895–902. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, W.; Li, S.; Guo, D.; He, J.; Wang, Y. Acetyl-CoA Carboxylases and Diseases. Front. Oncol. 2022, 12, 836058. [Google Scholar] [CrossRef]
- Berg, J.; Tymoczko, J.; Stryer, L.; Gatto, G. Chapter 17: The citric acid cycle. In Biochemistry; WH Freeman and Company: New York, NY, USA, 2002. [Google Scholar]
- Cases, S.; Smith, S.J.; Zheng, Y.-W.; Myers, H.M.; Lear, S.R.; Sande, E.; Novak, S.; Collins, C.; Welch, C.B.; Lusis, A.J. Identification of a gene encoding an acyl CoA: Diacylglycerol acyltransferase, a key enzyme in triacylglycerol synthesis. Proc. Natl. Acad. Sci. USA 1998, 95, 13018–13023. [Google Scholar] [CrossRef] [Green Version]
- Yen, C.-L.E.; Stone, S.J.; Koliwad, S.; Harris, C.; Farese, R.V., Jr. Thematic review series: Glycerolipids. DGAT enzymes and triacylglycerol biosynthesis. J. Lipid Res. 2008, 49, 2283–2301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fillmore, N.; Alrob, O.A.; Lopaschuk, G.D. Fatty Acid beta-Oxidation; AOCS Lipid library: Urbana, IL, USA, 2011. [Google Scholar]
- Calder, P.C. Omega-3 fatty acids and metabolic partitioning of fatty acids within the liver in the context of nonalcoholic fatty liver disease. Curr. Opin. Clin. Nutr. Metab. Care 2022, 25, 248–255. [Google Scholar] [CrossRef]
- Gao, Z.; Chen, X. Fatty Acid beta-Oxidation in Kidney Diseases: Perspectives on Pathophysiological Mechanisms and Therapeutic Opportunities. Front. Pharmacol. 2022, 13, 805281. [Google Scholar] [CrossRef]
- Coleman, R.A.; Lee, D.P. Enzymes of triacylglycerol synthesis and their regulation. Prog. Lipid Res. 2004, 43, 134–176. [Google Scholar] [CrossRef]
- Aguado, B.; Campbell, R.D. Characterization of a human lysophosphatidic acid acyltransferase that is encoded by a gene located in the class III region of the human major histocompatibility complex. J. Biol. Chem. 1998, 273, 4096–4105. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Cheng, D. Beyond triglyceride synthesis: The dynamic functional roles of MGAT and DGAT enzymes in energy metabolism. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E10–E18. [Google Scholar] [CrossRef] [Green Version]
- Anne, L. The Heterogeneity of Cancer Metabolism; Springer: Berlin/Heidelberg, Germany, 2021. [Google Scholar]
- El Sayed, S.M. Biochemical Origin of the Warburg Effect in Light of 15 Years of Research Experience: A Novel Evidence-Based View (An Expert Opinion Article). OncoTargets Ther. 2023, 16, 143–155. [Google Scholar] [CrossRef]
- Vaupel, P.; Schmidberger, H.; Mayer, A. The Warburg effect: Essential part of metabolic reprogramming and central contributor to cancer progression. Int. J. Radiat. Biol. 2019, 95, 912–919. [Google Scholar] [CrossRef] [PubMed]
- Gambardella, J.; Fiordelisi, A.; Cerasuolo, F.A.; Buonaiuto, A.; Avvisato, R.; Viti, A.; Sommella, E.; Merciai, F.; Salviati, E.; Campiglia, P.; et al. Experimental evidence and clinical implications of Warburg effect in the skeletal muscle of Fabry disease. iScience 2023, 26, 106074. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- San-Millán, I.; Brooks, G.A. Reexamining cancer metabolism: Lactate production for carcinogenesis could be the purpose and explanation of the Warburg Effect. Carcinogenesis 2017, 38, 119–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, R.; Zong, X. Aberrant cancer metabolism in epithelial-mesenchymal transition and cancer metastasis: Mechanisms in cancer progression. Crit. Rev. Oncol./Hematol. 2017, 115, 13–22. [Google Scholar] [CrossRef]
- Sanders, F.W.; Griffin, J.L. De novo lipogenesis in the liver in health and disease: More than just a shunting yard for glucose. Biol. Rev. Camb. Philos. Soc. 2016, 91, 452–468. [Google Scholar] [CrossRef] [Green Version]
- Han, H.-S.; Kang, G.; Kim, J.S.; Choi, B.H.; Koo, S.-H. Regulation of glucose metabolism from a liver-centric perspective. Exp. Mol. Med. 2016, 48, e218. [Google Scholar] [CrossRef] [Green Version]
- Xia, C.; Li, M.; Ran, G.; Wang, X.; Lu, Z.; Li, T.; Tang, X.; Zhang, Z.; He, Q. Redox-responsive nanoassembly restrained myeloid-derived suppressor cells recruitment through autophagy-involved lactate dehydrogenase A silencing for enhanced cancer immunochemotherapy. J. Control. Release 2021, 335, 557–574. [Google Scholar] [CrossRef] [PubMed]
- Gwangwa, M.V.; Joubert, A.M.; Visagie, M.H. Crosstalk between the Warburg effect, redox regulation and autophagy induction in tumourigenesis. Cell. Mol. Biol. Lett. 2018, 23, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, T.; Song, X.; Yang, Y.; Wan, X.; Alvarez, A.A.; Sastry, N.; Feng, H.; Hu, B.; Cheng, S.Y. Autophagy and Hallmarks of Cancer. Crit. Rev. Oncog. 2018, 23, 247–267. [Google Scholar] [CrossRef]
- Peng, X.; Gong, F.; Chen, Y.; Jiang, Y.; Liu, J.; Yu, M.; Zhang, S.; Wang, M.; Xiao, G.; Liao, H. Autophagy promotes paclitaxel resistance of cervical cancer cells: Involvement of Warburg effect activated hypoxia-induced factor 1-α-mediated signaling. Cell Death Dis. 2014, 5, e1367. [Google Scholar] [CrossRef] [Green Version]
- Guido, C.; Whitaker-Menezes, D.; Capparelli, C.; Balliet, R.; Lin, Z.; Pestell, R.G.; Howell, A.; Aquila, S.; Andò, S.; Martinez-Outschoorn, U. Metabolic reprogramming of cancer-associated fibroblasts by TGF-β drives tumor growth: Connecting TGF-β signaling with “Warburg-like” cancer metabolism and L-lactate production. Cell Cycle 2012, 11, 3019–3035. [Google Scholar] [CrossRef] [Green Version]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [Green Version]
- Luo, X.; Cheng, C.; Tan, Z.; Li, N.; Tang, M.; Yang, L.; Cao, Y. Emerging roles of lipid metabolism in cancer metastasis. Mol. Cancer 2017, 16, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; Zhang, Y.; Ren, J. Autophagic Regulation of Lipid Homeostasis in Cardiometabolic Syndrome. Front. Cardiovasc. Med. 2018, 5, 38. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Li, J.; Kang, R.; Tang, D. Interplay Between Lipid Metabolism and Autophagy. Front. Cell Dev. Biol. 2020, 8, 431. [Google Scholar] [CrossRef] [PubMed]
- Jaishy, B.; Abel, E.D. Lipids, lysosomes, and autophagy. J. Lipid Res. 2016, 57, 1619–1635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dall’Armi, C.; Devereaux, K.A.; Di Paolo, G. The Role of Lipids in the Control of Autophagy. Curr. Biol. 2013, 23, R33–R45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, T.; Li, X.; Yang, M.; Shao, Q.; Zhao, Y.; Ma, C.; Wang, P. Autophagy: An Intracellular Degradation Pathway Regulating Plant Survival and Stress Response. Front. Plant Sci. 2020, 11, 164. [Google Scholar] [CrossRef] [Green Version]
- Soto-Avellaneda, A.; Morrison, B.E. Signaling and other functions of lipids in autophagy: A review. Lipids Health Dis. 2020, 19, 214. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Peng, X.; Yang, S.; Li, X.; Huang, M.; Wei, S.; Liu, J.; He, G.; Zheng, H.; Yang, L.; et al. The regulation, function, and role of lipophagy, a form of selective autophagy, in metabolic disorders. Cell Death Dis. 2022, 13, 132. [Google Scholar] [CrossRef]
- Paik, S.; Jo, E.-K. An Interplay Between Autophagy and Immunometabolism for Host Defense Against Mycobacterial Infection. Front. Immunol. 2020, 11, 603951. [Google Scholar] [CrossRef]
- Lahiri, V.; Hawkins, W.D.; Klionsky, D.J. Watch What You (Self-) Eat: Autophagic Mechanisms that Modulate Metabolism. Cell Metab. 2019, 29, 803–826. [Google Scholar] [CrossRef] [Green Version]
- Zong, W.X.; Rabinowitz, J.D.; White, E. Mitochondria and Cancer. Mol. Cell 2016, 61, 667–676. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [Green Version]
- Missiroli, S.; Perrone, M.; Genovese, I.; Pinton, P.; Giorgi, C. Cancer metabolism and mitochondria: Finding novel mechanisms to fight tumours. EBioMedicine 2020, 59, 102943. [Google Scholar] [CrossRef]
- Shan, W.; Zhou, Y.; Tam, K.Y. The development of small-molecule inhibitors targeting hexokinase 2. Drug Discov. Today 2022, 27, 2574–2585. [Google Scholar] [CrossRef] [PubMed]
- Du, D.; Liu, C.; Qin, M.; Zhang, X.; Xi, T.; Yuan, S.; Hao, H.; Xiong, J. Metabolic dysregulation and emerging therapeutical targets for hepatocellular carcinoma. Acta Pharm. Sin. B 2022, 12, 558–580. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P.; Pereira, G.C.; Pasdois, P. The role of hexokinase in cardioprotection—Mechanism and potential for translation. Br. J. Pharmacol. 2015, 172, 2085–2100. [Google Scholar] [CrossRef] [Green Version]
- DeWaal, D.; Nogueira, V.; Terry, A.R.; Patra, K.C.; Jeon, S.-M.; Guzman, G.; Au, J.; Long, C.P.; Antoniewicz, M.R.; Hay, N. Hexokinase-2 depletion inhibits glycolysis and induces oxidative phosphorylation in hepatocellular carcinoma and sensitizes to metformin. Nat. Commun. 2018, 9, 1–14. [Google Scholar]
- Palmieri, D.; Fitzgerald, D.; Shreeve, S.M.; Hua, E.; Bronder, J.L.; Weil, R.J.; Davis, S.; Stark, A.M.; Merino, M.J.; Kurek, R. Analyses of resected human brain metastases of breast cancer reveal the association between up-regulation of hexokinase 2 and poor prognosis. Mol. Cancer Res. 2009, 7, 1438–1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Zhang, X.; Wang, Y.; Sun, Q.; Chen, M.; Liu, S.; Zou, X. Licochalcone A suppresses hexokinase 2-mediated tumor glycolysis in gastric cancer via downregulation of the Akt signaling pathway. Oncol. Rep. 2018, 39, 1181–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.; Jin, J.; Yu, H.; Zhao, Z.; Ma, D.; Zhang, C.; Jiang, H. Chrysin inhibited tumor glycolysis and induced apoptosis in hepatocellular carcinoma by targeting hexokinase-2. J. Exp. Clin. Cancer Res. 2017, 36, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Zhang, S.; Li, Y.; Tang, Z.; Kong, W. Hexokinase 2 overexpression promotes the proliferation and survival of laryngeal squamous cell carcinoma. Tumor Biol. 2014, 35, 3743–3753. [Google Scholar] [CrossRef]
- Yoshino, H.; Enokida, H.; Itesako, T.; Kojima, S.; Kinoshita, T.; Tatarano, S.; Chiyomaru, T.; Nakagawa, M.; Seki, N. Tumor-suppressive micro RNA-143/145 cluster targets hexokinase-2 in renal cell carcinoma. Cancer Sci. 2013, 104, 1567–1574. [Google Scholar] [CrossRef]
- Arundhathi, J.R.D.; Mathur, S.R.; Gogia, A.; Deo, S.V.S.; Mohapatra, P.; Prasad, C.P. Metabolic changes in triple negative breast cancer-focus on aerobic glycolysis. Mol. Biol. Rep. 2021, 48, 4733–4745. [Google Scholar] [CrossRef] [PubMed]
- Mathupala, S.; Ko, Y.A.; Pedersen, P. Hexokinase II: Cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene 2006, 25, 4777–4786. [Google Scholar] [CrossRef] [Green Version]
- Fan, K.; Fan, Z.; Cheng, H.; Huang, Q.; Yang, C.; Jin, K.; Luo, G.; Yu, X.; Liu, C. Hexokinase 2 dimerization and interaction with voltage-dependent anion channel promoted resistance to cell apoptosis induced by gemcitabine in pancreatic cancer. Cancer Med. 2019, 8, 5903–5915. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.J.; Singh, A.; Xue, K.; Mavis, C.; Barth, M.; Yanamadala, V.; Lenz, P.; Grau, M.; Lenz, G.; Czuczman, M.S. Up-regulation of hexokinase II contributes to rituximab-chemotherapy resistance and is a clinically relevant target for therapeutic development. Oncotarget 2018, 9, 4020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suh, D.H.; Kim, M.A.; Kim, H.; Kim, M.-K.; Kim, H.S.; Chung, H.H.; Kim, Y.-B.; Song, Y.S. Association of overexpression of hexokinase II with chemoresistance in epithelial ovarian cancer. Clin. Exp. Med. 2014, 14, 345–353. [Google Scholar] [CrossRef]
- Varghese, E.; Samuel, S.M.; Liskova, A.; Samec, M.; Kubatka, P.; Busselberg, D. Targeting Glucose Metabolism to Overcome Resistance to Anticancer Chemotherapy in Breast Cancer. Cancers 2020, 12, 2252. [Google Scholar] [CrossRef]
- Patra, K.C.; Wang, Q.; Bhaskar, P.T.; Miller, L.; Wang, Z.; Wheaton, W.; Chandel, N.; Laakso, M.; Muller, W.J.; Allen, E.L. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell 2013, 24, 213–228. [Google Scholar] [CrossRef] [Green Version]
- Gregersen, L.H.; Jacobsen, A.; Frankel, L.B.; Wen, J.; Krogh, A.; Lund, A.H. MicroRNA-143 down-regulates Hexokinase 2 in colon cancer cells. BMC Cancer 2012, 12, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Zhang, L.F.; Zhang, H.W.; Hu, S.; Lu, M.H.; Liang, S.; Li, B.; Li, Y.; Li, D.; Wang, E.D. A novel miR-155/miR-143 cascade controls glycolysis by regulating hexokinase 2 in breast cancer cells. EMBO J. 2012, 31, 1985–1998. [Google Scholar] [CrossRef] [Green Version]
- Kojima, S.; Enokida, H.; Yoshino, H.; Itesako, T.; Chiyomaru, T.; Kinoshita, T.; Fuse, M.; Nishikawa, R.; Goto, Y.; Naya, Y. The tumor-suppressive microRNA-143/145 cluster inhibits cell migration and invasion by targeting GOLM1 in prostate cancer. J. Hum. Genet. 2014, 59, 78–87. [Google Scholar] [CrossRef]
- Tan, V.P.; Miyamoto, S. HK2/hexokinase-II integrates glycolysis and autophagy to confer cellular protection. Autophagy 2015, 11, 963–964. [Google Scholar] [CrossRef] [Green Version]
- Pilkis, S.J.; Claus, T.H.; Kurland, I.J.; Lange, A.J. 6-Phosphofructo-2-kinase/fructose-2,6-bisphosphatase: A metabolic signaling enzyme. Annu. Rev. Biochem. 1995, 64, 799–835. [Google Scholar] [CrossRef]
- El-Maghrabi, M.R.; Noto, F.; Wu, N.; Manes, N. 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase: Suiting structure to need, in a family of tissue-specific enzymes. Curr. Opin. Clin. Nutr. Metab. Care 2001, 4, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Okar, D.A.; Manzano, A.; Navarro-Sabate, A.; Riera, L.; Bartrons, R.; Lange, A.J. PFK-2/FBPase-2: Maker and breaker of the essential biofactor fructose-2,6-bisphosphate. Trends Biochem. Sci. 2001, 26, 30–35. [Google Scholar] [CrossRef]
- O’Neal, J.; Clem, A.; Reynolds, L.; Dougherty, S.; Imbert-Fernandez, Y.; Telang, S.; Chesney, J.; Clem, B.F. Inhibition of 6-phosphofructo-2-kinase (PFKFB3) suppresses glucose metabolism and the growth of HER2+ breast cancer. Breast Cancer Res. Treat. 2016, 160, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Imbert-Fernandez, Y.; Clem, B.F.; O’Neal, J.; Kerr, D.A.; Spaulding, R.; Lanceta, L.; Clem, A.L.; Telang, S.; Chesney, J. Estradiol stimulates glucose metabolism via 6-phosphofructo-2-kinase (PFKFB3). J. Biol. Chem. 2014, 289, 9440–9448. [Google Scholar] [CrossRef] [Green Version]
- Novellasdemunt, L.; Obach, M.; Millán-Ariño, L.; Manzano, A.; Ventura, F.; Rosa, J.L.; Jordan, A.; Navarro-Sabate, À.; Bartrons, R. Progestins activate 6-phosphofructo-2-kinase/fructose-2, 6-bisphosphatase 3 (PFKFB3) in breast cancer cells. Biochem. J. 2012, 442, 345–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasgupta, S.; Rajapakshe, K.; Zhu, B.; Nikolai, B.C.; Yi, P.; Putluri, N.; Choi, J.M.; Jung, S.Y.; Coarfa, C.; Westbrook, T.F. Metabolic enzyme PFKFB4 activates transcriptional coactivator SRC-3 to drive breast cancer. Nature 2018, 556, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Liu, Y.; Li, D.; Xun, J.; Zhou, W.; Wang, P.; Liu, C.; Li, X.; Shen, W.; Su, W. PFKFB4 promotes breast cancer metastasis via induction of hyaluronan production in a p38-dependent manner. Cell. Physiol. Biochem. 2018, 50, 2108–2123. [Google Scholar] [CrossRef]
- Yao, L.; Wang, L.; Cao, Z.-G.; Hu, X.; Shao, Z.-M. High expression of metabolic enzyme PFKFB4 is associated with poor prognosis of operable breast cancer. Cancer Cell Int. 2019, 19, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Li, D.; Xun, J.; Zhou, W.; Li, J.; Wang, J.; Liu, C.; Li, X.; Shen, W.; Qiao, H. CD44ICD promotes breast cancer stemness via PFKFB4-mediated glucose metabolism. Theranostics 2018, 8, 6248. [Google Scholar] [CrossRef]
- Hu, K.Y.; Wang, D.G.; Liu, P.F.; Cao, Y.W.; Wang, Y.H.; Yang, X.C.; Hu, C.X.; Sun, L.J.; Niu, H.T. Targeting of MCT1 and PFKFB3 influences cell proliferation and apoptosis in bladder cancer by altering the tumor microenvironment. Oncol. Rep. 2016, 36, 945–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, S.J.; Jo, S.-W.; Ha, Y.-S.; Lee, O.-J.; Kim, W.T.; Kim, Y.-J.; Lee, S.-C.; Kim, W.-J. (Eds.) PFKFB4 as a prognostic marker in non-muscle-invasive bladder cancer. In Urologic Oncology: Seminars and Original Investigations; Elsevier: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Bobarykina, A.Y.; Minchenko, D.O.; Opentanova, I.L.; Moenner, M.; Caro, J.; Esumi, H.; Minchenko, O.H. Hypoxic regulation of PFKFB-3 and PFKFB-4 gene expression in gastric and pancreatic cancer cell lines and expression of PFKFB genes in gastric cancers. Acta Biochim. Pol. 2006, 53, 789–799. [Google Scholar] [CrossRef] [Green Version]
- Minchenko, O.H.; Tsuchihara, K.; Minchenko, D.O.; Bikfalvi, A.; Esumi, H. Mechanisms of regulation of PFKFB expression in pancreatic and gastric cancer cells. World J. Gastroenterol. 2014, 20, 13705. [Google Scholar] [CrossRef] [PubMed]
- Bolaños, J.P. Adapting glycolysis to cancer cell proliferation: The MAPK pathway focuses on PFKFB3. Biochem. J. 2013, 452, e7–e9. [Google Scholar] [CrossRef] [Green Version]
- Klarer, A.C.; O’Neal, J.; Imbert-Fernandez, Y.; Clem, A.; Ellis, S.R.; Clark, J.; Clem, B.; Chesney, J.; Telang, S. Inhibition of 6-phosphofructo-2-kinase (PFKFB3) induces autophagy as a survival mechanism. Cancer Metab. 2014, 2, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Strohecker, A.M.; Joshi, S.; Possemato, R.; Abraham, R.T.; Sabatini, D.M.; White, E. Identification of 6-phosphofructo-2-kinase/fructose-2, 6-bisphosphatase as a novel autophagy regulator by high content shRNA screening. Oncogene 2015, 34, 5662–5676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Goronzy, J.J.; Weyand, C.M. The glycolytic enzyme PFKFB3/phosphofructokinase regulates autophagy. Autophagy 2014, 10, 382–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ripple, M.O.; Wilding, G. Alteration of glyceraldehyde-3-phosphate dehydrogenase activity and messenger RNA content by androgen in human prostate carcinoma cells. Cancer Res. 1995, 55, 4234–4236. [Google Scholar]
- Marcucci, F.; Rumio, C. Tumor Cell Glycolysis-At the Crossroad of Epithelial-Mesenchymal Transition and Autophagy. Cells 2022, 11, 1041. [Google Scholar] [CrossRef]
- Butera, G.; Mullappilly, N.; Masetto, F.; Palmieri, M.; Scupoli, M.T.; Pacchiana, R.; Donadelli, M. Regulation of Autophagy by Nuclear GAPDH and Its Aggregates in Cancer and Neurodegenerative Disorders. Int. J. Mol. Sci. 2019, 20, 2062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.; Su, H.; Zhang, D.; Wang, Y.; Shen, Q.; Liu, B.; Huang, R.; Zhou, T.; Peng, C.; Wong, C.C. AMPK-dependent phosphorylation of GAPDH triggers Sirt1 activation and is necessary for autophagy upon glucose starvation. Mol. Cell 2015, 60, 930–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokunaga, K.; Nakamura, Y.; Sakata, K.; Fujimori, K.; Ohkubo, M.; Sawada, K.; Sakiyama, S. Enhanced expression of a glyceraldehyde-3-phosphate dehydrogenase gene in human lung cancers. Cancer Res. 1987, 47, 5616–5619. [Google Scholar] [PubMed]
- Phadke, M.; Krynetskaia, N.; Mishra, A.; Krynetskiy, E. Accelerated cellular senescence phenotype of GAPDH-depleted human lung carcinoma cells. Biochem. Biophys. Res. Commun. 2011, 411, 409–415. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.; Lan, F.; Zheng, Z.; Xie, F.; Han, J.; Dong, L.; Xie, Y.; Zheng, F. Akt2 kinase suppresses glyceraldehyde-3-phosphate dehydrogenase (GAPDH)-mediated apoptosis in ovarian cancer cells via phosphorylating GAPDH at threonine 237 and decreasing its nuclear translocation. J. Biol. Chem. 2011, 286, 42211–42220. [Google Scholar] [CrossRef] [Green Version]
- Harada, N.; Yasunaga, R.; Higashimura, Y.; Yamaji, R.; Fujimoto, K.; Moss, J.; Inui, H.; Nakano, Y. Glyceraldehyde-3-phosphate dehydrogenase enhances transcriptional activity of androgen receptor in prostate cancer cells. J. Biol. Chem. 2007, 282, 22651–22661. [Google Scholar] [CrossRef] [Green Version]
- Correa, C.; Bertollo, C.; Zouain, C.; Goes, A. Glyceraldehyde-3-phosphate dehydrogenase as a surface associated antigen on human breast cancer cell lines MACL-1 and MGSO-3. Oncol. Rep. 2010, 24, 677–685. [Google Scholar]
- Pacchiana, R.; Mullappilly, N.; Pinto, A.; Bova, S.; Forciniti, S.; Cullia, G.; Dalla Pozza, E.; Bottani, E.; Decimo, I.; Dando, I.; et al. 3-Bromo-Isoxazoline Derivatives Inhibit GAPDH Enzyme in PDAC Cells Triggering Autophagy and Apoptotic Cell Death. Cancers 2022, 14, 3153. [Google Scholar] [CrossRef]
- Dando, I.; Pacchiana, R.; Dalla Pozza, E.; Cataldo, I.; Bruno, S.; Conti, P.; Cordani, M.; Grimaldi, A.; Butera, G.; Caraglia, M. UCP2 inhibition induces ROS/Akt/mTOR axis: Role of GAPDH nuclear translocation in genipin/everolimus anticancer synergism. Free Radic. Biol. Med. 2017, 113, 176–189. [Google Scholar] [CrossRef]
- Wang, S.; Jiang, B.; Zhang, T.; Liu, L.; Wang, Y.; Wang, Y.; Chen, X.; Lin, H.; Zhou, L.; Xia, Y. Insulin and mTOR pathway regulate HDAC3-mediated deacetylation and activation of PGK1. PLoS Biol. 2015, 13, e1002243. [Google Scholar]
- Rojas-Pirela, M.; Andrade-Alviarez, D.; Rojas, V.; Kemmerling, U.; Caceres, A.J.; Michels, P.A.; Concepcion, J.L.; Quinones, W. Phosphoglycerate kinase: Structural aspects and functions, with special emphasis on the enzyme from Kinetoplastea. Open Biol. 2020, 10, 200302. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Li, X.; Cai, Q.; Zhang, C.; Yu, Q.; Jiang, Y.; Lee, J.-H.; Hawke, D.; Wang, Y.; Xia, Y. Phosphoglycerate kinase 1 phosphorylates BECLIN1 to induce autophagy. Mol. Cell 2017, 65, 917–931.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Wang, Y.; Yu, H.; Zhang, T.; Guo, L.; Xu, J.; Wei, X.; Wang, N.; Wu, Y.; Wang, X.; et al. PGK1 represses autophagy-mediated cell death to promote the proliferation of liver cancer cells by phosphorylating PRAS40. Cell Death Dis. 2022, 13, 68. [Google Scholar] [CrossRef] [PubMed]
- Altenberg, B.A.; Greulich, K. Genes of glycolysis are ubiquitously overexpressed in 24 cancer classes. Genomics 2004, 84, 1014–1020. [Google Scholar] [CrossRef] [PubMed]
- Hardt, P.; Mazurek, S.; Toepler, M.; Schlierbach, P.; Bretzel, R.; Eigenbrodt, E.; Kloer, H. Faecal tumour M2 pyruvate kinase: A new, sensitive screening tool for colorectal cancer. Br. J. Cancer 2004, 91, 980–984. [Google Scholar] [CrossRef] [Green Version]
- Verma, H.; Cholia, R.P.; Kaur, S.; Dhiman, M.; Mantha, A.K. A short review on cross-link between pyruvate kinase (PKM2) and Glioblastoma Multiforme. Metab. Brain Dis. 2021, 36, 751–765. [Google Scholar] [CrossRef]
- Dey, P.; Kundu, A.; Sachan, R.; Park, J.; Ahn, M.Y.; Yoon, K.; Lee, J.; Kim, N.D.; Kim, I.S.; Lee, B.M. PKM2 Knockdown Induces Autophagic Cell Death via the AKT/mTOR Pathway in Human Prostate Cancer Cells. Preprints 2019. [Google Scholar] [CrossRef] [Green Version]
- Lin, M. Pyruvate Kinase Isoform M2 Influences Autophagy and Related Processes in Hepatocellular Carcinoma Cells. 2018. Available online: https://opencommons.uconn.edu/usp_projects/42/ (accessed on 1 December 2022).
- Tamada, M.; Suematsu, M.; Saya, H. Pyruvate kinase M2: Multiple faces for conferring benefits on cancer cells. Clin. Cancer Res. 2012, 18, 5554–5561. [Google Scholar] [CrossRef] [Green Version]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef]
- Kumar, Y.; Tapuria, N.; Kirmani, N.; Davidson, B.R. Tumour M2-pyruvate kinase: A gastrointestinal cancer marker. Eur. J. Gastroenterol. Hepatol. 2007, 19, 265–276. [Google Scholar] [CrossRef]
- Lv, L.; Li, D.; Zhao, D.; Lin, R.; Chu, Y.; Zhang, H.; Zha, Z.; Liu, Y.; Li, Z.; Xu, Y. Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol. Cell 2011, 42, 719–730. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Yang, L.; Yang, Z.; Tang, Y.; Tao, Y.; Zhan, Q.; Lei, L.; Jing, Y.; Jiang, X.; Jin, H. Glycolytic enzyme PKM2 mediates autophagic activation to promote cell survival in NPM1-mutated leukemia. Int. J. Biol. Sci. 2019, 15, 882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Z.; Wang, D.; Tang, Y. PKM2 promotes cell metastasis and inhibits autophagy via the JAK/STAT3 pathway in hepatocellular carcinoma. Mol. Cell. Biochem. 2021, 476, 2001–2010. [Google Scholar] [CrossRef] [PubMed]
- Dawson, D.M.; Goodfriend, T.L.; Kaplan, N.O.; Kaplan, N.O. Lactic Dehydrogenases: Functions of the Two Types Rates of Synthesis of the Two Major Forms Can Be Correlated with Metabolic Differentiation. Science 1964, 143, 929–933. [Google Scholar] [CrossRef]
- Valvona, C.J.; Fillmore, H.L.; Nunn, P.B.; Pilkington, G.J. The regulation and function of lactate dehydrogenase A: Therapeutic potential in brain tumor. Brain Pathol. 2016, 26, 3–17. [Google Scholar] [CrossRef] [Green Version]
- Gordon, J.S.; Wood, C.T.; Luc, J.G.Y.; Watson, R.A.; Maynes, E.J.; Choi, J.H.; Morris, R.J.; Massey, H.T.; Throckmorton, A.L.; Tchantchaleishvili, V. Clinical implications of LDH isoenzymes in hemolysis and continuous-flow left ventricular assist device-induced thrombosis. Artif. Organs 2020, 44, 231–238. [Google Scholar] [CrossRef]
- Sheng, S.L.; Liu, J.J.; Dai, Y.H.; Sun, X.G.; Xiong, X.P.; Huang, G. Knockdown of lactate dehydrogenase A suppresses tumor growth and metastasis of human hepatocellular carcinoma. FEBS J. 2012, 279, 3898–3910. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Min, Z.; Sun, K.; Qu, S.; Zhou, J.; Duan, H.; Liu, H.; Liu, X.; Gong, Z.; Li, D. miR-199a-3p/Sp1/LDHA axis controls aerobic glycolysis in testicular tumor cells. Int. J. Mol. Med. 2018, 42, 2163–2174. [Google Scholar] [CrossRef] [Green Version]
- Guddeti, R.K.; Bali, P.; Karyala, P.; Pakala, S.B. MTA1 coregulator regulates LDHA expression and function in breast cancer. Biochem. Biophys. Res. Commun. 2019, 520, 54–59. [Google Scholar] [CrossRef]
- Huang, X.; Li, X.; Xie, X.; Ye, F.; Chen, B.; Song, C.; Tang, H.; Xie, X. High expressions of LDHA and AMPK as prognostic biomarkers for breast cancer. Breast 2016, 30, 39–46. [Google Scholar] [CrossRef]
- Yang, Y.; Su, D.; Zhao, L.; Zhang, D.; Xu, J.; Wan, J.; Fan, S.; Chen, M. Different effects of LDH-A inhibition by oxamate in non-small cell lung cancer cells. Oncotarget 2014, 5, 11886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, C.K.; Parekh, A.; Parida, P.K.; Bhutia, S.K.; Mandal, M. Lactate dehydrogenase A regulates autophagy and tamoxifen resistance in breast cancer. Biochim. Et Biophys. Acta (BBA) Mol. Cell Res. 2019, 1866, 1004–1018. [Google Scholar] [CrossRef] [PubMed]
- Brisson, L.; Bański, P.; Sboarina, M.; Dethier, C.; Danhier, P.; Fontenille, M.-J.; Van Hée, V.F.; Vazeille, T.; Tardy, M.; Falces, J. Lactate dehydrogenase B controls lysosome activity and autophagy in cancer. Cancer Cell 2016, 30, 418–431. [Google Scholar] [CrossRef] [Green Version]
- Pilon-Thomas, S.; Kodumudi, K.N.; El-Kenawi, A.E.; Russell, S.; Weber, A.M.; Luddy, K.; Damaghi, M.; Wojtkowiak, J.W.; Mulé, J.J.; Ibrahim-Hashim, A. Neutralization of tumor acidity improves antitumor responses to immunotherapy. Cancer Res. 2016, 76, 1381–1390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de la Cruz-López, K.G.; Castro-Muñoz, L.J.; Reyes-Hernández, D.O.; García-Carrancá, A.; Manzo-Merino, J. Lactate in the regulation of tumor microenvironment and therapeutic approaches. Front. Oncol. 2019, 9, 1143. [Google Scholar] [CrossRef] [Green Version]
- Rizwan, A.; Serganova, I.; Khanin, R.; Karabeber, H.; Ni, X.; Thakur, S.; Zakian, K.L.; Blasberg, R.; Koutcher, J.A. Relationships between LDH-A, lactate, and metastases in 4T1 breast tumors. Clin. Cancer Res. 2013, 19, 5158–5169. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.; Zuo, H.; Xiong, H.; Kolar, M.J.; Chu, Q.; Saghatelian, A.; Siegwart, D.J.; Wan, Y. Gpr132 sensing of lactate mediates tumor–macrophage interplay to promote breast cancer metastasis. Proc. Natl. Acad. Sci. USA 2017, 114, 580–585. [Google Scholar] [CrossRef] [Green Version]
- Végran, F.; Boidot, R.; Michiels, C.; Sonveaux, P.; Feron, O. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-κB/IL-8 pathway that drives tumor angiogenesis. Cancer Res. 2011, 71, 2550–2560. [Google Scholar] [CrossRef] [Green Version]
- Sonveaux, P.; Copetti, T.; De Saedeleer, C.J.; Végran, F.; Verrax, J.; Kennedy, K.M.; Moon, E.J.; Dhup, S.; Danhier, P.; Frérart, F. Targeting the lactate transporter MCT1 in endothelial cells inhibits lactate-induced HIF-1 activation and tumor angiogenesis. PLoS ONE 2012, 7, e33418. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Dalgard, C.L.; Mohyeldin, A.; McFate, T.; Tait, A.S.; Verma, A. Reversible inactivation of HIF-1 prolyl hydroxylases allows cell metabolism to control basal HIF-1. J. Biol. Chem. 2005, 280, 41928–41939. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Zaguilán, R.; Seftor, E.A.; Seftor, R.E.; Chu, Y.-W.; Gillies, R.J.; Hendrix, M.J. Acidic pH enhances the invasive behavior of human melanoma cells. Clin. Exp. Metastasis 1996, 14, 176–186. [Google Scholar] [CrossRef]
- Calcinotto, A.; Filipazzi, P.; Grioni, M.; Iero, M.; De Milito, A.; Ricupito, A.; Cova, A.; Canese, R.; Jachetti, E.; Rossetti, M. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res. 2012, 72, 2746–2756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, R.; Morris, M. Monocarboxylate transporters: Therapeutic targets and prognostic factors in disease. Clin. Pharmacol. Ther. 2016, 100, 454–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, C.K.; Goldstein, J.L.; Pathak, R.K.; Anderson, R.G.; Brown, M.S. Molecular characterization of a membrane transporter for lactate, pyruvate, and other monocarboxylates: Implications for the Cori cycle. Cell 1994, 76, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, C.; Albergaria, A.; Paredes, J.; Sousa, B.; Dufloth, R.; Vieira, D.; Schmitt, F.; Baltazar, F. Monocarboxylate transporter 1 is up-regulated in basal-like breast carcinoma. Histopathology 2010, 56, 860–867. [Google Scholar] [CrossRef]
- Pinheiro, C.; Longatto-Filho, A.; Ferreira, L.; Pereira, S.M.M.; Etlinger, D.; Moreira, M.A.; Jube, L.F.; Queiroz, G.S.; Schmitt, F.; Baltazar, F. Increasing expression of monocarboxylate transporters 1 and 4 along progression to invasive cervical carcinoma. Int. J. Gynecol. Pathol. 2008, 27, 568–574. [Google Scholar] [CrossRef]
- Pinheiro, C.; Penna, V.; Morais-Santos, F.; Abrahão-Machado, L.F.; Ribeiro, G.; Curcelli, E.C.; Olivieri, M.V.; Morini, S.; Valença, I.; Ribeiro, D. Characterization of monocarboxylate transporters (MCTs) expression in soft tissue sarcomas: Distinct prognostic impact of MCT1 sub-cellular localization. J. Transl. Med. 2014, 12, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Wu, M.-S.; Zou, C.; Tang, Q.; Lu, J.; Liu, D.; Wu, Y.; Yin, J.; Xie, X.; Shen, J. Downregulation of MCT1 inhibits tumor growth, metastasis and enhances chemotherapeutic efficacy in osteosarcoma through regulation of the NF-κB pathway. Cancer Lett. 2014, 342, 150–158. [Google Scholar] [CrossRef]
- Pinheiro, C.; Longatto-Filho, A.; Pereira, S.M.M.; Etlinger, D.; Moreira, M.A.; Jubé, L.F.; Queiroz, G.S.; Schmitt, F.; Baltazar, F. Monocarboxylate transporters 1 and 4 are associated with CD147 in cervical carcinoma. Dis. Mrk. 2009, 26, 97–103. [Google Scholar] [CrossRef]
- Jin, P.; Jiang, J.; Xie, N.; Zhou, L.; Huang, Z.; Zhang, L.; Qin, S.; Fu, S.; Peng, L.; Gao, W. MCT1 relieves osimertinib-induced CRC suppression by promoting autophagy through the LKB1/AMPK signaling. Cell Death Dis. 2019, 10, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Long, Y.; Gao, Z.; Hu, X.; Xiang, F.; Wu, Z.; Zhang, J.; Han, X.; Yin, L.; Qin, J.; Lan, L. Downregulation of MCT 4 for lactate exchange promotes the cytotoxicity of NK cells in breast carcinoma. Cancer Med. 2018, 7, 4690–4700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kierans, S.J.; Taylor, C.T. Regulation of glycolysis by the hypoxia-inducible factor (HIF): Implications for cellular physiology. J. Physiol. 2021, 599, 23–37. [Google Scholar] [CrossRef]
- Mazure, N.M.; Pouysségur, J. Hypoxia-induced autophagy: Cell death or cell survival? Curr. Opin. Cell Biol. 2010, 22, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Janji, B.; Berchem, G.; Mami-Chouaib, F.; Chouaib, S. Hypoxia-induced autophagy: A new player in cancer immunotherapy? Autophagy 2012, 8, 704–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papandreou, I.; Lim, A.; Laderoute, K.; Denko, N. Hypoxia signals autophagy in tumor cells via AMPK activity, independent of HIF-1, BNIP3, and BNIP3L. Cell Death Differ. 2008, 15, 1572–1581. [Google Scholar] [CrossRef]
- de Mello, N.P.; Orellana, A.M.; Mazucanti, C.H.; de Morais Lima, G.; Scavone, C.; Kawamoto, E.M. Insulin and autophagy in neurodegeneration. Front. Neurosci. 2019, 13, 491. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Liu, Y.; Sun, T.; Yang, W. LincRNA-p21 knockdown enhances radiosensitivity of hypoxic tumor cells by reducing autophagy through HIF-1/Akt/mTOR/P70S6K pathway. Exp. Cell Res. 2017, 358, 188–198. [Google Scholar] [CrossRef]
- Talks, K.L.; Turley, H.; Gatter, K.C.; Maxwell, P.H.; Pugh, C.W.; Ratcliffe, P.J.; Harris, A.L. The expression and distribution of the hypoxia-inducible factors HIF-1α and HIF-2α in normal human tissues, cancers, and tumor-associated macrophages. Am. J. Pathol. 2000, 157, 411–421. [Google Scholar] [CrossRef]
- Fu, D.; He, C.; Wei, J.; Zhang, Z.; Luo, Y.; Tan, H.; Ren, C. PGK1 is a Potential Survival Biomarker and Invasion Promoter by Regulating the HIF-1α–Mediated Epithelial-Mesenchymal Transition Process in Breast Cancer. Cell. Physiol. Biochem. 2018, 51, 2434–2444. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, T.; Taniguchi, K.; Matsuhashi, N.; Tajirika, T.; Futamura, M.; Takai, T.; Akao, Y.; Yoshida, K. MiR-133b inhibits growth of human gastric cancer cells by silencing pyruvate kinase muscle-splicer polypyrimidine tract-binding protein 1. Cancer Sci. 2016, 107, 1767–1775. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Yan, H.; An, S.; Shen, M.; Jia, W.; Zhang, R.; Zhao, L.; Huang, G.; Liu, J. SIRT 5-mediated deacetylation of LDHB promotes autophagy and tumorigenesis in colorectal cancer. Mol. Oncol. 2019, 13, 358–375. [Google Scholar] [CrossRef]
- Wang, C.W. Lipid droplets, lipophagy, and beyond. Biochim. Et Biophys. Acta 2016, 1861 (8 Pt B), 793–805. [Google Scholar] [CrossRef]
- Efeyan, A.; Comb, W.C.; Sabatini, D.M. Nutrient-sensing mechanisms and pathways. Nature 2015, 517, 302–310. [Google Scholar] [CrossRef] [Green Version]
- Ward, C.; Martinez-Lopez, N.; Otten, E.G.; Carroll, B.; Maetzel, D.; Singh, R.; Sarkar, S.; Korolchuk, V.I. Autophagy, lipophagy and lysosomal lipid storage disorders. Biochim. Et Biophys. Acta 2016, 1861, 269–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulze, R.J.; Sathyanarayan, A.; Mashek, D.G. Breaking fat: The regulation and mechanisms of lipophagy. Biochim. Biophys. Acta. Mol. Cell Biol. Lipids 2017, 1862 (10 Pt B), 1178–1187. [Google Scholar] [CrossRef]
- Martinez-Lopez, N.; Singh, R. Autophagy and Lipid Droplets in the Liver. Annu. Rev. Nutr. 2015, 35, 215–237. [Google Scholar] [CrossRef] [PubMed]
- Fafián-Labora, J.; Carpintero-Fernández, P.; Jordan, S.J.D.; Shikh-Bahaei, T.; Abdullah, S.M.; Mahenthiran, M.; Rodríguez-Navarro, J.A.; Niklison-Chirou, M.V.; O’Loghlen, A. FASN activity is important for the initial stages of the induction of senescence. Cell Death Dis. 2019, 10, 318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humbert, M.; Seiler, K.; Mosimann, S.; Rentsch, V.; McKenna, S.L.; Tschan, M.P. Autophagy-mediated degradation of Fatty Acid Synthase (FASN) facilitates ATRA-induced granulocytic differentiation of acute myeloid leukemia (AML) cells. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, M.; Impey, S.; Kang, H.; di Ronza, A.; Pelz, C.; Sardiello, M.; Ballabio, A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet. 2011, 20, 3852–3866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Luo, Q.F.; Peng, A.F.; Long, X.H.; Wang, T.F.; Liu, Z.L.; Zhang, G.M.; Zhou, R.P.; Gao, S.; Zhou, Y.; et al. Positive feedback regulation between Akt phosphorylation and fatty acid synthase expression in osteosarcoma. Int. J. Mol. Med. 2014, 33, 633–639. [Google Scholar] [CrossRef] [Green Version]
- Zaytseva, Y.Y.; Harris, J.W.; Mitov, M.I.; Kim, J.T.; Butterfield, D.A.; Lee, E.Y.; Weiss, H.L.; Gao, T.; Evers, B.M. Increased expression of fatty acid synthase provides a survival advantage to colorectal cancer cells via upregulation of cellular respiration. Oncotarget 2015, 6, 18891–18904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alizadeh, J.; Zeki, A.A.; Mirzaei, N.; Tewary, S.; Rezaei Moghadam, A.; Glogowska, A.; Nagakannan, P.; Eftekharpour, E.; Wiechec, E.; Gordon, J.W.; et al. Mevalonate Cascade Inhibition by Simvastatin Induces the Intrinsic Apoptosis Pathway via Depletion of Isoprenoids in Tumor Cells. Sci. Rep. 2017, 7, 44841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmadi, M.; Amiri, S.; Pecic, S.; Machaj, F.; Rosik, J.; Łos, M.J.; Alizadeh, J.; Mahdian, R.; da Silva Rosa, S.C.; Schaafsma, D.; et al. Pleiotropic effects of statins: A focus on cancer. Biochim. Et Biophys. Acta (BBA) Mol. Basis Dis. 2020, 1866, 165968. [Google Scholar] [CrossRef]
- Peymani, P.; Dehesh, T.; Aligolighasemabadi, F.; Sadeghdoust, M.; Kotfis, K.; Ahmadi, M.; Mehrbod, P.; Iranpour, P.; Dastghaib, S.; Nasimian, A.; et al. Statins in patients with COVID-19: A retrospective cohort study in Iranian COVID-19 patients. Transl. Med. Commun. 2021, 6, 3. [Google Scholar] [CrossRef]
- Ghavami, S.; Sharma, P.; Yeganeh, B.; Ojo, O.O.; Jha, A.; Mutawe, M.M.; Kashani, H.H.; Los, M.J.; Klonisch, T.; Unruh, H.; et al. Airway mesenchymal cell death by mevalonate cascade inhibition: Integration of autophagy, unfolded protein response and apoptosis focusing on Bcl2 family proteins. Biochim. Et Biophys. Acta 2014, 1843, 1259–1271. [Google Scholar] [CrossRef] [Green Version]
- Vilimanovich, U.; Bosnjak, M.; Bogdanovic, A.; Markovic, I.; Isakovic, A.; Kravic-Stevovic, T.; Mircic, A.; Trajkovic, V.; Bumbasirevic, V. Statin-mediated inhibition of cholesterol synthesis induces cytoprotective autophagy in human leukemic cells. Eur. J. Pharmacol. 2015, 765, 415–428. [Google Scholar] [CrossRef]
- Guo, J.Y.; Karsli-Uzunbas, G.; Mathew, R.; Aisner, S.C.; Kamphorst, J.J.; Strohecker, A.M.; Chen, G.; Price, S.; Lu, W.; Teng, X.; et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 2013, 27, 1447–1461. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.J.; Park, J.Y.; Kwon, O.; Choe, E.Y.; Kim, C.H.; Hur, K.Y.; Lee, M.S.; Yun, M.; Cha, B.S.; Kim, Y.B.; et al. Chronic HMGCR/HMG-CoA reductase inhibitor treatment contributes to dysglycemia by upregulating hepatic gluconeogenesis through autophagy induction. Autophagy 2015, 11, 2089–2101. [Google Scholar] [CrossRef] [Green Version]
- Parikh, A.; Childress, C.; Deitrick, K.; Lin, Q.; Rukstalis, D.; Yang, W. Statin-induced autophagy by inhibition of geranylgeranyl biosynthesis in prostate cancer PC3 cells. Prostate 2010, 70, 971–981. [Google Scholar] [CrossRef]
- Araki, M.; Motojima, K. Hydrophobic statins induce autophagy in cultured human rhabdomyosarcoma cells. Biochem. Biophys. Res. Commun. 2008, 367, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Araki, M.; Maeda, M.; Motojima, K. Hydrophobic statins induce autophagy and cell death in human rhabdomyosarcoma cells by depleting geranylgeranyl diphosphate. Eur. J. Pharmacol. 2012, 674, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Emami, A.; Shojaei, S.; da Silva Rosa, S.C.; Aghaei, M.; Samiei, E.; Vosoughi, A.R.; Kalantari, F.; Kawalec, P.; Thliveris, J.; Sharma, P.; et al. Mechanisms of simvastatin myotoxicity: The role of autophagy flux inhibition. Eur. J. Pharmacol. 2019, 862, 172616. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.; Zhang, Y.; Huang, Y.; Yi, Q.; Lv, L.; Zhang, T.; Chen, D.; Hao, Q.; Shi, Q. Inhibitors of phosphatidylinositol 3′-kinases promote mitotic cell death in HeLa cells. PLoS ONE 2012, 7, e35665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanzada, U.K.; Pardo, O.E.; Meier, C.; Downward, J.; Seckl, M.J.; Arcaro, A. Potent inhibition of small-cell lung cancer cell growth by simvastatin reveals selective functions of Ras isoforms in growth factor signalling. Oncogene 2006, 25, 877–887. [Google Scholar] [CrossRef] [Green Version]
- Calabro, A.; Tai, J.; Allen, S.L.; Budman, D.R. In-vitro synergism of m-TOR inhibitors, statins, and classical chemotherapy: Potential implications in acute leukemia. Anti-Cancer Drugs 2008, 19, 705–712. [Google Scholar] [CrossRef]
- Cemeus, C.; Zhao, T.T.; Barrett, G.M.; Lorimer, I.A.; Dimitroulakos, J. Lovastatin enhances gefitinib activity in glioblastoma cells irrespective of EGFRvIII and PTEN status. J. Neuro-Oncol. 2008, 90, 9–17. [Google Scholar] [CrossRef]
- Martirosyan, A.; Clendening, J.W.; Goard, C.A.; Penn, L.Z. Lovastatin induces apoptosis of ovarian cancer cells and synergizes with doxorubicin: Potential therapeutic relevance. BMC Cancer 2010, 10, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannavo, A.; Liccardo, D.; Komici, K.; Corbi, G.; de Lucia, C.; Femminella, G.D.; Elia, A.; Bencivenga, L.; Ferrara, N.; Koch, W.J.; et al. Sphingosine Kinases and Sphingosine 1-Phosphate Receptors: Signaling and Actions in the Cardiovascular System. Front. Pharmacol. 2017, 8, 556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, J.; Xie, Y.; Yin, J.; Lu, W.; Fang, S. SphK1 promotes tumor cell migration and invasion in colorectal cancer. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2016, 37, 6831–6836. [Google Scholar] [CrossRef] [PubMed]
- Almejún, M.B.; Borge, M.; Colado, A.; Elías, E.E.; Podaza, E.; Risnik, D.; De Brasi, C.D.; Stanganelli, C.; Slavutsky, I.; Cabrejo, M.; et al. Sphingosine kinase 1 participates in the activation, proliferation and survival of chronic lymphocytic leukemia cells. Haematologica 2017, 102, e257–e260. [Google Scholar] [CrossRef] [Green Version]
- Pchejetski, D.; Doumerc, N.; Golzio, M.; Naymark, M.; Teissié, J.; Kohama, T.; Waxman, J.; Malavaud, B.; Cuvillier, O. Chemosensitizing effects of sphingosine kinase-1 inhibition in prostate cancer cell and animal models. Mol. Cancer Ther. 2008, 7, 1836. [Google Scholar] [CrossRef] [Green Version]
- Kawamori, T.; Kaneshiro, T.; Okumura, M.; Maalouf, S.; Uflacker, A.; Bielawski, J.; Hannun, Y.A.; Obeid, L.M. Role for sphingosine kinase 1 in colon carcinogenesis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2009, 23, 405–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akao, Y.; Banno, Y.; Nakagawa, Y.; Hasegawa, N.; Kim, T.J.; Murate, T.; Igarashi, Y.; Nozawa, Y. High expression of sphingosine kinase 1 and S1P receptors in chemotherapy-resistant prostate cancer PC3 cells and their camptothecin-induced up-regulation. Biochem. Biophys. Res. Commun. 2006, 342, 1284–1290. [Google Scholar] [CrossRef] [PubMed]
- Bao, M.; Chen, Z.; Xu, Y.; Zhao, Y.; Zha, R.; Huang, S.; Liu, L.; Chen, T.; Li, J.; Tu, H.; et al. Sphingosine kinase 1 promotes tumour cell migration and invasion via the S1P/EDG1 axis in hepatocellular carcinoma. Liver Int. Off. J. Int. Assoc. Study Liver 2012, 32, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Moruno Manchon, J.F.; Uzor, N.E.; Finkbeiner, S.; Tsvetkov, A.S. SPHK1/sphingosine kinase 1-mediated autophagy differs between neurons and SH-SY5Y neuroblastoma cells. Autophagy 2016, 12, 1418–1424. [Google Scholar] [CrossRef] [Green Version]
- Lavieu, G.; Scarlatti, F.; Sala, G.; Carpentier, S.; Levade, T.; Ghidoni, R.; Botti, J.; Codogno, P. Regulation of autophagy by sphingosine kinase 1 and its role in cell survival during nutrient starvation. J. Biol. Chem. 2006, 281, 8518–8527. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Zhang, W.; Liu, S.; Wu, W.; Qin, M.; Huang, J. Activation of the SphK1/ERK/p-ERK pathway promotes autophagy in colon cancer cells. Oncol. Lett. 2018, 15, 9719–9724. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Ma, Y.; He, H.-W.; Zhao, W.-L.; Shao, R.-G. SPHK1 (sphingosine kinase 1) induces epithelial-mesenchymal transition by promoting the autophagy-linked lysosomal degradation of CDH1/E-cadherin in hepatoma cells. Autophagy 2017, 13, 900–913. [Google Scholar] [CrossRef]
- Yin, S.; Miao, Z.; Tan, Y.; Wang, P.; Xu, X.; Zhang, C.; Hou, W.; Huang, J.; Xu, H. SPHK1-induced autophagy in peritoneal mesothelial cell enhances gastric cancer peritoneal dissemination. Cancer Med. 2019, 8, 1731–1743. [Google Scholar] [CrossRef]
- Jeppesen, J.; Mogensen, M.; Prats, C.; Sahlin, K.; Madsen, K.; Kiens, B. FAT/CD36 is localized in sarcolemma and in vesicle-like structures in subsarcolemma regions but not in mitochondria. J. Lipid Res. 2010, 51, 1504–1512. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Yang, P.; Zhao, L.; Chen, Y.; Zhang, X.; Zeng, S.; Wei, L.; Varghese, Z.; Moorhead, J.F.; Chen, Y.; et al. CD36 plays a negative role in the regulation of lipophagy in hepatocytes through an AMPK-dependent pathway. J. Lipid Res. 2019, 60, 844–855. [Google Scholar] [CrossRef] [Green Version]
- Su, W.; Frohman, M.A. Chapter 144—Phospholipase D. In Handbook of Cell Signaling, 2nd ed.; Bradshaw, R.A., Dennis, E.A., Eds.; Academic Press: San Diego, CA, USA, 2010; pp. 1167–1176. [Google Scholar] [CrossRef]
- Foster, D.A. Phosphatidic acid signaling to mTOR: Signals for the survival of human cancer cells. Biochim. Et Biophys. Acta 2009, 1791, 949–955. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Vilella-Bach, M.; Bachmann, R.; Flanigan, A.; Chen, J. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science 2001, 294, 1942–1945. [Google Scholar] [CrossRef]
- Shahnazari, S.; Yen, W.-L.; Birmingham, C.L.; Shiu, J.; Namolovan, A.; Zheng, Y.T.; Nakayama, K.; Klionsky, D.J.; Brumell, J.H. A Diacylglycerol-Dependent Signaling Pathway Contributes to Regulation of Antibacterial Autophagy. Cell Host Microbe 2010, 8, 137–146. [Google Scholar] [CrossRef] [Green Version]
- Genc, G.E.; Hipolito, V.E.B.; Botelho, R.J.; Gumuslu, S. Lysophosphatidic acid represses autophagy in prostate carcinoma cells. Biochem. Cell Biol. 2019, 97, 387–396. [Google Scholar] [CrossRef]
- Holland, P.; Knaevelsrud, H.; Soreng, K.; Mathai, B.J.; Lystad, A.H.; Pankiv, S.; Bjorndal, G.T.; Schultz, S.W.; Lobert, V.H.; Chan, R.B.; et al. HS1BP3 negatively regulates autophagy by modulation of phosphatidic acid levels. Nat. Commun. 2016, 7, 13889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmen, B.; Thierry, L.; Patrice, C. Regulation of Autophagy by Sphingolipids. Anti-Cancer Agents Med. Chem. 2011, 11, 844–853. [Google Scholar] [CrossRef]
- Spassieva, S.D.; Mullen, T.D.; Townsend, D.M.; Obeid, L.M. Disruption of ceramide synthesis by CerS2 down-regulation leads to autophagy and the unfolded protein response. Biochem. J. 2009, 424, 273–283. [Google Scholar] [CrossRef] [Green Version]
- Pattingre, S.; Bauvy, C.; Carpentier, S.; Levade, T.; Levine, B.; Codogno, P. Role of JNK1-dependent Bcl-2 phosphorylation in ceramide-induced macroautophagy. J. Biol. Chem. 2009, 284, 2719–2728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarlatti, F.; Bauvy, C.; Ventruti, A.; Sala, G.; Cluzeaud, F.; Vandewalle, A.; Ghidoni, R.; Codogno, P. Ceramide-mediated macroautophagy involves inhibition of protein kinase B and up-regulation of BECLIN 1. J. Biol. Chem. 2004, 279, 18384–18391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guenther, G.G.; Peralta, E.R.; Rosales, K.R.; Wong, S.Y.; Siskind, L.J.; Edinger, A.L. Ceramide starves cells to death by downregulating nutrient transporter proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 17402–17407. [Google Scholar] [CrossRef] [Green Version]
- Lépine, S.; Allegood, J.C.; Park, M.; Dent, P.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate phosphohydrolase-1 regulates ER stress-induced autophagy. Cell Death Differ. 2011, 18, 350–361. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.L.; Ho, M.C.; Lee, P.H.; Hsu, C.Y.; Huang, W.P.; Lee, H. S1P(5) is required for sphingosine 1-phosphate-induced autophagy in human prostate cancer PC-3 cells. Am. J. Physiol. Cell Physiol. 2009, 297, C451–C458. [Google Scholar] [CrossRef]
- Berger, J.; Moller, D.E. The Mechanisms of Action of PPARs. Annu. Rev. Med. 2002, 53, 409–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyata, K.S.; McCaw, S.E.; Marcus, S.L.; Rachubinski, R.A.; Capone, J.P. The peroxisome proliferator-activated receptor interacts with the retinoid X receptor in vivo. Gene 1994, 148, 327–330. [Google Scholar] [CrossRef] [PubMed]
- Bull, A.W.; Steffensen, K.R.; Leers, J.; Rafter, J.J. Activation of PPAR γ in colon tumor cell lines by oxidized metabolites of linoleic acid, endogenous ligands for PPAR γ. Carcinogenesis 2003, 24, 1717–1722. [Google Scholar] [CrossRef] [PubMed]
- Velkov, T. Interactions between Human Liver Fatty Acid Binding Protein and Peroxisome Proliferator Activated Receptor Selective Drugs. PPAR Res. 2013, 2013, 938401. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.S.; Lee, H.-M.; Kim, J.K.; Yang, C.-S.; Kim, T.S.; Jung, M.; Jin, H.S.; Kim, S.; Jang, J.; Oh, G.T.; et al. PPAR-α Activation Mediates Innate Host Defense through Induction of TFEB and Lipid Catabolism. J. Immunol. 2017, 198, 3283–3295. [Google Scholar] [CrossRef] [Green Version]
- Palomer, X.; Capdevila-Busquets, E.; Botteri, G.; Salvadó, L.; Barroso, E.; Davidson, M.M.; Michalik, L.; Wahli, W.; Vázquez-Carrera, M. PPARβ/δ; attenuates palmitate-induced endoplasmic reticulum stress and induces autophagic markers in human cardiac cells. Int. J. Cardiol. 2014, 174, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Assumpção, J.A.F.; Magalhães, K.G.; Corrêa, J.R. The role of pparγ and autophagy in ros production, lipid droplets biogenesis and its involvement with colorectal cancer cells modulation. Cancer Cell Int. 2017, 17, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Zhang, W.; Liang, B.; Casimiro, M.C.; Whitaker-Menezes, D.; Wang, M.; Lisanti, M.P.; Lanza-Jacoby, S.; Pestell, R.G.; Wang, C. PPARγ activation induces autophagy in breast cancer cells. Int. J. Biochem. Cell Biol. 2009, 41, 2334–2342. [Google Scholar] [CrossRef] [Green Version]
- Bajaj, M.; Suraamornkul, S.; Kashyap, S.; Cusi, K.; Mandarino, L.; DeFronzo, R.A. Sustained Reduction in Plasma Free Fatty Acid Concentration Improves Insulin Action without Altering Plasma Adipocytokine Levels in Subjects with Strong Family History of Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2004, 89, 4649–4655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.-W. Lipid droplet dynamics in budding yeast. Cell. Mol. Life Sci. 2015, 72, 2677–2695. [Google Scholar] [CrossRef]
- Carta, G.; Murru, E.; Banni, S.; Manca, C. Palmitic Acid: Physiological Role, Metabolism and Nutritional Implications. Front. Physiol. 2017, 8, 902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, S.H.; Shui, G.; Zhou, J.; Li, J.J.E.; Bay, B.-H.; Wenk, M.R.; Shen, H.-M. Induction of Autophagy by Palmitic Acid via Protein Kinase C-mediated Signaling Pathway Independent of mTOR (Mammalian Target of Rapamycin). J. Biol. Chem. 2012, 287, 14364–14376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukui, M.; Kang, K.S.; Okada, K.; Zhu, B.T. EPA, an omega-3 fatty acid, induces apoptosis in human pancreatic cancer cells: Role of ROS accumulation, caspase-8 activation, and autophagy induction. J. Cell. Biochem. 2013, 114, 192–203. [Google Scholar] [CrossRef]
- Swanson, D.; Block, R.; Mousa, S.A. Omega-3 fatty acids EPA and DHA: Health benefits throughout life. Adv. Nutr. 2012, 3, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krauss-Etschmann, S.; Shadid, R.; Campoy, C.; Hoster, E.; Demmelmair, H.; Jiménez, M.; Gil, A.; Rivero, M.; Veszprémi, B.; Decsi, T.; et al. Effects of fish-oil and folate supplementation of pregnant women on maternal and fetal plasma concentrations of docosahexaenoic acid and eicosapentaenoic acid: A European randomized multicenter trial. Am. J. Clin. Nutr. 2007, 85, 1392–1400. [Google Scholar] [CrossRef] [Green Version]
- Neff, L.M.; Culiner, J.; Cunningham-Rundles, S.; Seidman, C.; Meehan, D.; Maturi, J.; Wittkowski, K.M.; Levine, B.; Breslow, J.L. Algal docosahexaenoic acid affects plasma lipoprotein particle size distribution in overweight and obese adults. J. Nutr. 2011, 141, 207–213. [Google Scholar] [CrossRef] [Green Version]
- Chiu, C.C.; Su, K.P.; Cheng, T.C.; Liu, H.C.; Chang, C.J.; Dewey, M.E.; Stewart, R.; Huang, S.Y. The effects of omega-3 fatty acids monotherapy in Alzheimer’s disease and mild cognitive impairment: A preliminary randomized double-blind placebo-controlled study. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2008, 32, 1538–1544. [Google Scholar] [CrossRef]
- Varela-López, A.; Vera-Ramírez, L.; Giampieri, F.; Navarro-Hortal, M.D.; Forbes-Hernández, T.Y.; Battino, M.; Quiles, J.L. The central role of mitochondria in the relationship between dietary lipids and cancer progression. Semin. Cancer Biol. 2021, 73, 86–100. [Google Scholar] [CrossRef]
- Shin, S.; Jing, K.; Jeong, S.; Kim, N.; Song, K.-S.; Heo, J.-Y.; Park, J.-H.; Seo, K.-S.; Han, J.; Park, J.-I.; et al. The Omega-3 Polyunsaturated Fatty Acid DHA Induces Simultaneous Apoptosis and Autophagy via Mitochondrial ROS-Mediated Akt-mTOR Signaling in Prostate Cancer Cells Expressing Mutant p53. BioMed Res. Int. 2013, 2013, 568671. [Google Scholar] [CrossRef] [Green Version]
- Yao, Q.-H.; Zhang, X.-C.; Fu, T.; Gu, J.-Z.; Wang, L.; Wang, Y.; Lai, Y.-B.; Wang, Y.-Q.; Guo, Y. ω-3 polyunsaturated fatty acids inhibit the proliferation of the lung adenocarcinoma cell line A549 in vitro. Mol. Med. Rep. 2014, 9, 401–406. [Google Scholar] [CrossRef] [Green Version]
- Hsu, H.-C.; Chen, C.-Y.; Chiang, C.-H.; Chen, M.-F. Eicosapentaenoic acid attenuated oxidative stress-induced cardiomyoblast apoptosis by activating adaptive autophagy. Eur. J. Nutr. 2014, 53, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Jing, K.; Song, K.-S.; Shin, S.; Kim, N.; Jeong, S.; Oh, H.-R.; Park, J.-H.; Seo, K.-S.; Heo, J.-Y.; Han, J.; et al. Docosahexaenoic acid induces autophagy through p53/AMPK/mTOR signaling and promotes apoptosis in human cancer cells harboring wild-type p53. Autophagy 2011, 7, 1348–1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Vitale, I.; Djavaheri-Mergny, M.; D’Amelio, M.; Criollo, A.; Morselli, E.; Zhu, C.; Harper, F.; et al. Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol. 2008, 10, 676–687. [Google Scholar] [CrossRef] [Green Version]
- D’Eliseo, D.; Di Renzo, L.; Santoni, A.; Velotti, F. Docosahexaenoic acid (DHA) promotes immunogenic apoptosis in human multiple myeloma cells, induces autophagy and inhibits STAT3 in both tumor and dendritic cells. Genes Cancer 2017, 8, 426–437. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, K.; Monsen, V.T.; Hakvåg Pettersen, C.H.; Overland, H.B.; Pettersen, G.; Samdal, H.; Tesfahun, A.N.; Lundemo, A.G.; Bjørkøy, G.; Schønberg, S.A. DHA-induced stress response in human colon cancer cells—Focus on oxidative stress and autophagy. Free Radic. Biol. Med. 2016, 90, 158–172. [Google Scholar] [CrossRef] [Green Version]
- Kanno, S.-I.; Kurauchi, K.; Tomizawa, A.; Yomogida, S.; Ishikawa, M. Pifithrin-alpha has a p53-independent cytoprotective effect on docosahexaenoic acid-induced cytotoxicity in human hepatocellular carcinoma HepG2 cells. Toxicol. Lett. 2015, 232, 393–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omura, Y. Beneficial effects and side effects of DHEA: True anti-aging and age-promoting effects, as well as anti-cancer and cancer-promoting effects of DHEA evaluated from the effects on the normal and cancer cell telomeres and other parameters. Acupunct. Electro-Ther. Res. 2005, 30, 219–261. [Google Scholar] [CrossRef]
- Rovito, D.; Giordano, C.; Vizza, D.; Plastina, P.; Barone, I.; Casaburi, I.; Lanzino, M.; De Amicis, F.; Sisci, D.; Mauro, L.; et al. Omega-3 PUFA ethanolamides DHEA and EPEA induce autophagy through PPARγ activation in MCF-7 breast cancer cells. J. Cell. Physiol. 2013, 228, 1314–1322. [Google Scholar] [CrossRef] [PubMed]
- Alphonse, P.A.; Jones, P.J. Revisiting Human Cholesterol Synthesis and Absorption: The Reciprocity Paradigm and its Key Regulators. Lipids 2016, 51, 519–536. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.-T.; Kreutzberger, A.J.B.; Lee, J.; Kiessling, V.; Tamm, L.K. The role of cholesterol in membrane fusion. Chem. Phys. Lipids 2016, 199, 136–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerqueira, N.M.; Oliveira, E.F.; Gesto, D.S.; Santos-Martins, D.; Moreira, C.; Moorthy, H.N.; Ramos, M.J.; Fernandes, P.A. Cholesterol Biosynthesis: A Mechanistic Overview. Biochemistry 2016, 55, 5483–5506. [Google Scholar] [CrossRef]
- Liao, P.; Hemmerlin, A.; Bach, T.J.; Chye, M.L. The potential of the mevalonate pathway for enhanced isoprenoid production. Biotechnol. Adv. 2016, 34, 697–713. [Google Scholar] [CrossRef]
- Jiang, S.-Y.; Li, H.; Tang, J.-J.; Wang, J.; Luo, J.; Liu, B.; Wang, J.-K.; Shi, X.-J.; Cui, H.-W.; Tang, J.; et al. Discovery of a potent HMG-CoA reductase degrader that eliminates statin-induced reductase accumulation and lowers cholesterol. Nat. Commun. 2018, 9, 5138. [Google Scholar] [CrossRef] [Green Version]
- Shao, W.Q.; Zhu, W.W.; Luo, M.J.; Fan, M.H.; Li, Q.; Wang, S.H.; Lin, Z.F.; Zhao, J.; Zheng, Y.; Dong, Q.Z.; et al. Cholesterol suppresses GOLM1-dependent selective autophagy of RTKs in hepatocellular carcinoma. Cell Rep. 2022, 39, 110712. [Google Scholar] [CrossRef]
- Kumar, M.; Irungbam, K.; Kataria, M. Depletion of membrane cholesterol compromised caspase-8 imparts in autophagy induction and inhibition of cell migration in cancer cells. Cancer Cell Int. 2018, 18, 23. [Google Scholar] [CrossRef] [Green Version]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- World Health Organization. Cancer; World Health Organization: Geneva, Switzerland, 2022. [Google Scholar]
- American Lung Association. Lung Cancer Fact Sheet; American Lung Association: Chicago, IL, USA, 2020; Available online: https://www.lung.org/lung-health-diseases/lung-disease-lookup/lung-cancer/resource-library/lung-cancer-fact-sheet (accessed on 1 December 2022).
- Canadian Cancer Statistics Advisory Committee. Canadian Cancer Statistics 2018; Canadian Cancer Statistics Advisory Committee: Toronto, ON, USA, 2018. [Google Scholar]
- WHO Classification of Tumours of the Lung, Pleura, Thymus, and Heart. In WHO Classification of Tumours of the Lung, Pleura, Thymus, and Heart; Travis, W.D.; Brambilla, E.; Burke, A.P.; Marx, A.; Nicholson, A.G. (Eds.) WHO: Lyon, France, 2015. [Google Scholar]
- Sakamoto, T.; Furukawa, T.; Pham, H.H.N.; Kuroda, K.; Tabata, K.; Kashima, Y.; Okoshi, E.N.; Morimoto, S.; Bychkov, A.; Fukuoka, J. A collaborative workflow between pathologists and deep learning for the evaluation of tumour cellularity in lung adenocarcinoma. Histopathology 2022, 81, 758–769. [Google Scholar] [CrossRef]
- Oktay, E.; Oflazoglu, U.; Varol, Y.; Tanriverdi, O.; Mermur, N.; Arda, H.U.; Demir, L.; Keskin, O.; Ahmadli, T.; Somali, I.; et al. The prognostic role of thyroid transcription factor-1 in lung adenocarcinoma. J. Cancer Res. Ther. 2020, 16, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Girard, N. New driver mutations in non-small-cell lung cancer. Lancet Oncol. 2011, 12, 175–180. [Google Scholar] [CrossRef]
- Gennen, K.; Kasmann, L.; Taugner, J.; Eze, C.; Karin, M.; Roengvoraphoj, O.; Neumann, J.; Tufman, A.; Orth, M.; Reu, S.; et al. Prognostic value of PD-L1 expression on tumor cells combined with CD8+ TIL density in patients with locally advanced non-small cell lung cancer treated with concurrent chemoradiotherapy. Radiat. Oncol. 2020, 15, 5. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Shi, J.; Lin, D.; Li, X.; Zhao, C.; Wang, Q.; Zhang, L.; Jiang, T.; Zhao, S.; Liu, X.; et al. Prognostic value of PD-L1 expression in combination with CD8(+) TILs density in patients with surgically resected non-small cell lung cancer. Cancer Med. 2018, 7, 32–45. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.; Wang, H.Y.; Liu, Y.; Zhao, M.C.; Zhang, H.J.; Lu, Z.Y.; Fang, Y.C.; Chen, X.F.; Liu, G.T. The prognostic value of PD-L1 expression for non-small cell lung cancer patients: A meta-analysis. Eur. J. Surg. Oncol. 2015, 41, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Daaboul, N.; Nicholas, G.; Laurie, S.A. Algorithm for the treatment of advanced or metastatic squamous non-small-cell lung cancer: An evidence-based overview. Curr. Oncol. 2018, 25 (Suppl. 1), S77–S85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umakanthan, S.; Bukelo, M.M. Concise genetic profile of lung carcinoma. Postgrad. Med. J. 2021. Online ahead of print. [Google Scholar] [CrossRef]
- Alberg, A.J.; Samet, J.M. Epidemiology of lung cancer. Chest 2003, 123 (Suppl. 1), 21S–49S. [Google Scholar] [CrossRef] [Green Version]
- Taylor, R.; Najafi, F.; Dobson, A. Meta-analysis of studies of passive smoking and lung cancer: Effects of study type and continent. Int. J. Epidemiol. 2007, 36, 1048–1059. [Google Scholar] [CrossRef]
- Kim, C.H.; Lee, Y.C.; Hung, R.J.; McNallan, S.R.; Cote, M.L.; Lim, W.Y.; Chang, S.C.; Kim, J.H.; Ugolini, D.; Chen, Y.; et al. Exposure to secondhand tobacco smoke and lung cancer by histological type: A pooled analysis of the International Lung Cancer Consortium (ILCCO). Int. J. Cancer 2014, 135, 1918–1930. [Google Scholar] [CrossRef] [Green Version]
- Darby, S.; Hill, D.; Auvinen, A.; Barros-Dios, J.M.; Baysson, H.; Bochicchio, F.; Deo, H.; Falk, R.; Forastiere, F.; Hakama, M.; et al. Radon in homes and risk of lung cancer: Collaborative analysis of individual data from 13 European case-control studies. BMJ 2005, 330, 223. [Google Scholar] [CrossRef] [Green Version]
- Krewski, D.; Lubin, J.H.; Zielinski, J.M.; Alavanja, M.; Catalan, V.S.; Field, R.W.; Klotz, J.B.; Letourneau, E.G.; Lynch, C.F.; Lyon, J.L.; et al. A combined analysis of North American case-control studies of residential radon and lung cancer. J. Toxicol. Environ. Health A 2006, 69, 533–597. [Google Scholar] [CrossRef] [Green Version]
- Neuberger, J.S.; Field, R.W. Occupation and lung cancer in nonsmokers. Rev. Environ. Health 2003, 18, 251–267. [Google Scholar] [CrossRef]
- van Loon, A.J.; Kant, I.J.; Swaen, G.M.; Goldbohm, R.A.; Kremer, A.M.; van den Brandt, P.A. Occupational exposure to carcinogens and risk of lung cancer: Results from The Netherlands cohort study. Occup. Environ. Med. 1997, 54, 817–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottschall, E.B. Occupational and environmental thoracic malignancies. J. Thorac. Imaging 2002, 17, 189–197. [Google Scholar] [CrossRef]
- Hubbard, R.; Venn, A.; Lewis, S.; Britton, J. Lung cancer and cryptogenic fibrosing alveolitis. A population-based cohort study. Am. J. Respir. Crit. Care Med. 2000, 161, 5–8. [Google Scholar] [CrossRef]
- Chen, Y.C.; Chen, J.H.; Richard, K.; Chen, P.Y.; Christiani, D.C. Lung adenocarcinoma and human papillomavirus infection. Cancer 2004, 101, 1428–1436. [Google Scholar] [CrossRef]
- Kirk, G.D.; Merlo, C.; O’Driscoll, P.; Mehta, S.H.; Galai, N.; Vlahov, D.; Samet, J.; Engels, E.A. HIV infection is associated with an increased risk for lung cancer, independent of smoking. Clin. Infect. Dis. 2007, 45, 103–110. [Google Scholar] [CrossRef] [Green Version]
- Chaturvedi, A.K.; Pfeiffer, R.M.; Chang, L.; Goedert, J.J.; Biggar, R.J.; Engels, E.A. Elevated risk of lung cancer among people with AIDS. AIDS 2007, 21, 207–213. [Google Scholar] [CrossRef] [PubMed]
- D’Jaen, G.A.; Pantanowitz, L.; Bower, M.; Buskin, S.; Neil, N.; Greco, E.M.; Cooley, T.P.; Henry, D.; Stem, J.; Dezube, B.J.; et al. Human immunodeficiency virus-associated primary lung cancer in the era of highly active antiretroviral therapy: A multi-institutional collaboration. Clin. Lung Cancer 2010, 11, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Dziadziuszko, R.; Peters, S.; Ruf, T.; Cardona, A.; Guerini, E.; Kurtsikidze, N.; Smoljanovic, V.; Planchard, D. Clinical experience and management of adverse events in patients with advanced ALK-positive non-small-cell lung cancer receiving alectinib. ESMO Open 2022, 7, 100612. [Google Scholar] [CrossRef]
- Sun, R.; Hou, Z.; Zhang, Y.; Jiang, B. Drug resistance mechanisms and progress in the treatment of EGFR-mutated lung adenocarcinoma. Oncol. Lett. 2022, 24, 408. [Google Scholar] [CrossRef]
- Lindeman, N.I.; Cagle, P.T.; Beasley, M.B.; Chitale, D.A.; Dacic, S.; Giaccone, G.; Jenkins, R.B.; Kwiatkowski, D.J.; Saldivar, J.S.; Squire, J.; et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: Guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. J. Thorac. Oncol. 2013, 8, 823–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dower, C.M.; Wills, C.A.; Frisch, S.M.; Wang, H.G. Mechanisms and context underlying the role of autophagy in cancer metastasis. Autophagy 2018, 14, 1110–1128. [Google Scholar] [CrossRef] [Green Version]
- Chan, E.Y.; Kir, S.; Tooze, S.A. siRNA screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. J. Biol. Chem. 2007, 282, 25464–25474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mleczak, A.; Millar, S.; Tooze, S.A.; Olson, M.F.; Chan, E.Y. Regulation of autophagosome formation by Rho kinase. Cell Signal 2013, 25, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gurkar, A.U.; Chu, K.; Raj, L.; Bouley, R.; Lee, S.-H.; Kim, Y.-B.; Dunn, S.E.; Mandinova, A.; Lee, S.W. Identification of ROCK1 kinase as a critical regulator of BECLIN1-mediated autophagy during metabolic stress. Nat. Commun. 2013, 4, 2189. [Google Scholar] [CrossRef] [Green Version]
- Belaid, A.; Cerezo, M.; Chargui, A.; Corcelle-Termeau, E.; Pedeutour, F.; Giuliano, S.; Ilie, M.; Rubera, I.; Tauc, M.; Barale, S.; et al. Autophagy plays a critical role in the degradation of active RHOA, the control of cell cytokinesis, and genomic stability. Cancer Res. 2013, 73, 4311–4322. [Google Scholar] [CrossRef] [Green Version]
- Belaid, A.; Ndiaye, P.D.; Cerezo, M.; Cailleteau, L.; Brest, P.; Klionsky, D.J.; Carle, G.F.; Hofman, P.; Mograbi, B. Autophagy and SQSTM1 on the RHOA(d) again: Emerging roles of autophagy in the degradation of signaling proteins. Autophagy 2014, 10, 201–208. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, T.; Tsujioka, M.; Honda, S.; Tanaka, M.; Shimizu, S. Autophagy suppresses cell migration by degrading GEF-H1, a RhoA GEF. Oncotarget 2016, 7, 34420–34429. [Google Scholar] [CrossRef]
- Sanz-Moreno, V.; Gadea, G.; Ahn, J.; Paterson, H.; Marra, P.; Pinner, S.; Sahai, E.; Marshall, C.J. Rac activation and inactivation control plasticity of tumor cell movement. Cell 2008, 135, 510–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, K.; Chen, X.; Qiu, F.; Xu, J.; Xiong, H.; Zhang, Z. Serum-Derived Exosomes-Mediated Circular RNA ARHGAP10 Modulates the Progression of Non-Small Cell Lung Cancer Through the miR-638/FAM83F Axis. Cancer Biother. Radiopharm. 2022, 37, 96–110. [Google Scholar] [CrossRef]
- Li, P.; Lv, H.; Xu, M.; Zang, B.; Ma, Y. ARHGAP6 Promotes Apoptosis and Inhibits Glycolysis in Lung Adenocarcinoma Through STAT3 Signaling Pathway. Cancer Manag. Res. 2020, 12, 9665–9678. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Le Toumelin, G.; Criollo, A.; Rain, J.C.; Gautier, F.; Juin, P.; Tasdemir, E.; Pierron, G.; Troulinaki, K.; Tavernarakis, N.; et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in BECLIN1. EMBO J. 2007, 26, 2527–2539. [Google Scholar] [CrossRef]
- Erlich, S.; Mizrachy, L.; Segev, O.; Lindenboim, L.; Zmira, O.; Adi-Harel, S.; Hirsch, J.A.; Stein, R.; Pinkas-Kramarski, R. Differential interactions between BECLIN 1 and Bcl-2 family members. Autophagy 2007, 3, 561–568. [Google Scholar] [CrossRef] [Green Version]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit BECLIN 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [Green Version]
- Adams, J.M.; Cory, S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene 2007, 26, 1324–1337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amundson, S.A.; Myers, T.G.; Scudiero, D.; Kitada, S.; Reed, J.C.; Fornace, A.J., Jr. An informatics approach identifying markers of chemosensitivity in human cancer cell lines. Cancer Res. 2000, 60, 6101–6110. [Google Scholar]
- Lindqvist, L.M.; Simon, A.K.; Baehrecke, E.H. Current questions and possible controversies in autophagy. Cell Death Discov. 2015, 1, 15036. [Google Scholar] [CrossRef] [Green Version]
- Lindqvist, L.M.; Heinlein, M.; Huang, D.C.; Vaux, D.L. Prosurvival Bcl-2 family members affect autophagy only indirectly, by inhibiting Bax and Bak. Proc. Natl. Acad. Sci. USA 2014, 111, 8512–8517. [Google Scholar] [CrossRef] [Green Version]
- Lindqvist, L.M.; Vaux, D.L. BCL2 and related prosurvival proteins require BAK1 and BAX to affect autophagy. Autophagy 2014, 10, 1474–1475. [Google Scholar] [CrossRef] [Green Version]
- Pedro, J.M.B.-S.; Wei, Y.; Sica, V.; Maiuri, M.C.; Zou, Z.; Kroemer, G.; Levine, B. BAX and BAK1 are dispensable for ABT-737-induced dissociation of the BCL2-BECN1 complex and autophagy. Autophagy 2015, 11, 452–459. [Google Scholar] [CrossRef] [Green Version]
- Reljic, B.; Conos, S.; Lee, E.F.; Garnier, J.M.; Dong, L.; Lessene, G.; Fairlie, W.D.; Vaux, D.L.; Lindqvist, L.M. BAX-BAK1-independent LC3B lipidation by BH3 mimetics is unrelated to BH3 mimetic activity and has only minimal effects on autophagic flux. Autophagy 2016, 12, 1083–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Campbell, J.D.; Alexandrov, A.; Kim, J.; Wala, J.; Berger, A.H.; Pedamallu, C.S.; Shukla, S.A.; Guo, G.; Brooks, A.N.; Murray, B.A.; et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 2016, 48, 607–616. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hombach-Klonisch, S.; Mehrpour, M.; Shojaei, S.; Harlos, C.; Pitz, M.; Hamai, A.; Siemianowicz, K.; Likus, W.; Wiechec, E.; Toyota, B.D.; et al. Glioblastoma and chemoresistance to alkylating agents: Involvement of apoptosis, autophagy, and unfolded protein response. Pharmacol. Ther. 2018, 184, 13–41. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [Green Version]
- Rao, S.; Tortola, L.; Perlot, T.; Wirnsberger, G.; Novatchkova, M.; Nitsch, R.; Sykacek, P.; Frank, L.; Schramek, D.; Komnenovic, V.; et al. A dual role for autophagy in a murine model of lung cancer. Nat. Commun. 2014, 5, 3056. [Google Scholar] [CrossRef] [Green Version]
- Feng, Z.; Zhang, H.; Levine, A.J.; Jin, S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc. Natl. Acad. Sci. USA 2005, 102, 8204–8209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, R.J.; Bardeesy, N.; Manning, B.D.; Lopez, L.; Kosmatka, M.; DePinho, R.A.; Cantley, L.C. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell 2004, 6, 91–99. [Google Scholar] [CrossRef] [Green Version]
- Shackelford, D.B.; Vasquez, D.S.; Corbeil, J.; Wu, S.; Leblanc, M.; Wu, C.-L.; Vera, D.R.; Shaw, R.J. mTOR and HIF-1α-mediated tumor metabolism in an LKB1 mouse model of Peutz-Jeghers syndrome. Proc. Natl. Acad. Sci. USA 2009, 106, 11137–11142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faubert, B.; Vincent, E.E.; Griss, T.; Samborska, B.; Izreig, S.; Svensson, R.U.; Mamer, O.A.; Avizonis, D.; Shackelford, D.B.; Shaw, R.J. Loss of the tumor suppressor LKB1 promotes metabolic reprogramming of cancer cells via HIF-1α. Proc. Natl. Acad. Sci. USA 2014, 111, 2554–2559. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Cespedes, M.; Parrella, P.; Esteller, M.; Nomoto, S.; Trink, B.; Engles, J.M.; Westra, W.H.; Herman, J.G.; Sidransky, D. Inactivation of LKB1/STK11 is a common event in adenocarcinomas of the lung. Cancer Res. 2002, 62, 3659–3662. [Google Scholar]
- Ding, L.; Getz, G.; Wheeler, D.A.; Mardis, E.R.; McLellan, M.D.; Cibulskis, K.; Sougnez, C.; Greulich, H.; Muzny, D.M.; Morgan, M.B. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008, 455, 1069–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Momcilovic, M.; Bailey, S.T.; Lee, J.T.; Zamilpa, C.; Jones, A.; Abdelhady, G.; Mansfield, J.; Francis, K.P.; Shackelford, D.B. Utilizing 18F-FDG PET/CT imaging and quantitative histology to measure dynamic changes in the glucose metabolism in mouse models of lung cancer. J. Vis. Exp. JoVE 2018, 137, e57167. [Google Scholar]
- Luo, L.-X.; Li, Y.; Liu, Z.-Q.; Fan, X.-X.; Duan, F.-G.; Li, R.-Z.; Yao, X.-J.; Leung, E.L.-H.; Liu, L. Honokiol induces apoptosis, G1 arrest, and autophagy in KRAS mutant lung cancer cells. Front. Pharmacol. 2017, 8, 199. [Google Scholar] [CrossRef] [Green Version]
- Kapuy, O.; Makk-Merczel, K.; Szarka, A. Therapeutic Approach of KRAS Mutant Tumours by the Combination of Pharmacologic Ascorbate and Chloroquine. Biomolecules 2021, 11, 652. [Google Scholar] [CrossRef]
- Morita, M.; Sato, T.; Nomura, M.; Sakamoto, Y.; Inoue, Y.; Tanaka, R.; Ito, S.; Kurosawa, K.; Yamaguchi, K.; Sugiura, Y.; et al. PKM1 Confers Metabolic Advantages and Promotes Cell-Autonomous Tumor Cell Growth. Cancer Cell 2018, 33, 355–367.e357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fantin, V.R.; Abraham, R.T. Self-eating limits EGFR-dependent tumor growth. Cell 2013, 154, 1184–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, M.; Wang, S.; Wan, T.; Jiang, R.; Qiu, Y.; Pei, L.; Pang, N.; Huang, Y.; Huang, Y.; Zhang, Z. Combined inhibitions of glycolysis and AKT/autophagy can overcome resistance to EGFR-targeted therapy of lung cancer. J. Cancer 2017, 8, 3774. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Yang, D.; Jiang, X.; Tian, L.; Sun, J.; Tian, D.; Bai, W. Final-2 targeted glycolysis mediated apoptosis and autophagy in human lung adenocarcinoma cells but failed to inhibit xenograft in nude mice. Food Chem. Toxicol. 2019, 130, 1–11. [Google Scholar] [CrossRef]
- Xia, S.; Duan, W.; Liu, W.; Zhang, X.; Wang, Q. GRP78 in lung cancer. J. Transl. Med. 2021, 19, 1–14. [Google Scholar] [CrossRef]
- Bello-Perez, M.; Sola, I.; Novoa, B.; Klionsky, D.J.; Falco, A. Canonical and noncanonical autophagy as potential targets for COVID-19. Cells 2020, 9, 1619. [Google Scholar] [CrossRef]
- Wang, M.; Wey, S.; Zhang, Y.; Ye, R.; Lee, A.S. Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxid. Redox Signal. 2009, 11, 2307–2316. [Google Scholar] [CrossRef] [Green Version]
- Kania, E.; Pająk, B.; Orzechowski, A. Calcium homeostasis and ER stress in control of autophagy in cancer cells. BioMed Res. Int. 2015, 2015, 352794. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Guo, W.; Yang, S.; Zhu, X.; Xiang, J.; Li, H. Serum GRP78 as a tumor marker and its prognostic significance in non-small cell lung cancers: A retrospective study. Dis. Mrk. 2015, 2015, 814670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Wu, Y.; Lian, X. Targeted inhibition of GRP78 by HA15 promotes apoptosis of lung cancer cells accompanied by ER stress and autophagy. Biol. Open 2020, 9, bio053298. [Google Scholar] [CrossRef]
- Parker, A.L.; Kavallaris, M.; McCarroll, J.A. Microtubules and their role in cellular stress in cancer. Front. Oncol. 2014, 4, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, A.L.; Turner, N.; McCarroll, J.A.; Kavallaris, M. βIII-Tubulin alters glucose metabolism and stress response signaling to promote cell survival and proliferation in glucose-starved non-small cell lung cancer cells. Carcinogenesis 2016, 37, 787–798. [Google Scholar] [CrossRef] [Green Version]
- Rim, H.-K.; Cho, S.; Shin, D.-H.; Chung, K.-S.; Cho, Y.-W.; Choi, J.-H.; Lee, J.Y.; Lee, K.-T. T-type Ca2+ channel blocker, KYS05090 induces autophagy and apoptosis in A549 cells through inhibiting glucose uptake. Molecules 2014, 19, 9864–9875. [Google Scholar] [CrossRef]
- Bootman, M.D.; Chehab, T.; Bultynck, G.; Parys, J.B.; Rietdorf, K. The regulation of autophagy by calcium signals: Do we have a consensus? Cell Calcium 2018, 70, 32–46. [Google Scholar] [CrossRef]
- Sallán, M.C.; Visa, A.; Shaikh, S.; Nàger, M.; Herreros, J.; Cantí, C. T-type Ca2+ Channels: T for Targetable. Cancer Res. 2018, 78, 603–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villar, V.H.; Durán, R.V. Glutamoptosis: A new cell death mechanism inhibited by autophagy during nutritional imbalance. Autophagy 2017, 13, 1078–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruntz, R.C.; Belshoff, A.C.; Zhang, Y.; Macedo, J.K.; Higashi, R.M.; Lane, A.N.; Fan, T.W.M. Inhibition of anaplerotic glutaminolysis underlies selenite toxicity in human lung cancer. Proteomics 2019, 19, 1800486. [Google Scholar] [CrossRef]
- Pérez-Escuredo, J.; Dadhich, R.K.; Dhup, S.; Cacace, A.; Van Hée, V.F.; De Saedeleer, C.J.; Sboarina, M.; Rodriguez, F.; Fontenille, M.-J.; Brisson, L. Lactate promotes glutamine uptake and metabolism in oxidative cancer cells. Cell Cycle 2016, 15, 72–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, L.; Alesi, G.; Kang, S. Glutaminolysis as a target for cancer therapy. Oncogene 2016, 35, 3619–3625. [Google Scholar] [CrossRef] [Green Version]
- Sonveaux, P.; Végran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 2008, 118, 3930–3942. [Google Scholar] [CrossRef] [Green Version]
- Xie, W.-Y.; Zhou, X.-D.; Li, Q.; Chen, L.-X.; Ran, D.-H. Acid-induced autophagy protects human lung cancer cells from apoptosis by activating ER stress. Exp. Cell Res. 2015, 339, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.-C.; Kim, S.M.; Choi, J.E.; Kim, C.H.; Duong, H.-Q.; Han, S.I.; Kang, H.S. Sodium salicylate switches glucose depletion-induced necrosis to autophagy and inhibits high mobility group box protein 1 release in A549 lung adenocarcinoma cells. Oncol. Rep. 2008, 19, 1165–1171. [Google Scholar] [CrossRef]
- Jiang, M.-J.; Dai, J.-J.; Gu, D.-N.; Huang, Q.; Tian, L. Aspirin in pancreatic cancer: Chemopreventive effects and therapeutic potentials. Biochim. Et Biophys. Acta (BBA) Rev. Cancer 2016, 1866, 163–176. [Google Scholar] [CrossRef]
- Din, F.V.; Valanciute, A.; Houde, V.P.; Zibrova, D.; Green, K.A.; Sakamoto, K.; Alessi, D.R.; Dunlop, M.G. Aspirin inhibits mTOR signaling, activates AMP-activated protein kinase, and induces autophagy in colorectal cancer cells. Gastroenterology 2012, 142, 1504–1515.e1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perera, R.; Son, J.; Lyssiotis, C.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.; Ferrone, C.; Mullarky, E. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar]
- Zhou, X.; Curbo, S.; Li, F.; Krishnan, S.; Karlsson, A. Inhibition of glutamate oxaloacetate transaminase 1 in cancer cell lines results in altered metabolism with increased dependency of glucose. BMC Cancer 2018, 18, 1–14. [Google Scholar] [CrossRef]
- Kang, Y.-T.; Hsu, W.-C.; Wu, C.-H.; Hsin, I.-L.; Wu, P.-R.; Yeh, K.-T.; Ko, J.-L. Metformin alleviates nickel-induced autophagy and apoptosis via inhibition of hexokinase-2, activating lipocalin-2, in human bronchial epithelial cells. Oncotarget 2017, 8, 105536. [Google Scholar] [CrossRef] [Green Version]
- Chaudhri, V.K.; Salzler, G.G.; Dick, S.A.; Buckman, M.S.; Sordella, R.; Karoly, E.D.; Mohney, R.; Stiles, B.M.; Elemento, O.; Altorki, N.K. Metabolic alterations in lung cancer–associated fibroblasts correlated with increased glycolytic metabolism of the tumor. Mol. Cancer Res. 2013, 11, 579–592. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Wang, L.; Zhang, Y.; Wang, J.; Deng, Y.; Lin, D. Inhibition of glycolytic enzyme hexokinase II (HK2) suppresses lung tumor growth. Cancer Cell Int. 2016, 16, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Xi, F.; Ye, J. Inhibition of lung carcinoma A549 cell growth by knockdown of hexokinase 2 in situ and in vivo. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2016, 23, 53–59. [Google Scholar] [CrossRef]
- Wei, J.; Ma, Z.; Li, Y.; Zhao, B.; Wang, D.; Jin, Y.; Jin, Y. miR-143 inhibits cell proliferation by targeting autophagy-related 2B in non-small cell lung cancer H1299 cells. Mol. Med. Rep. 2015, 11, 571–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lypova, N.; Telang, S.; Chesney, J.; Imbert-Fernandez, Y. Increased 6-phosphofructo-2-kinase/fructose-2, 6-bisphosphatase-3 activity in response to EGFR signaling contributes to non–small cell lung cancer cell survival. J. Biol. Chem. 2019, 294, 10530–10543. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zeng, F.; Sun, Y.; Qiu, Q.; Zhang, J.; Huang, W.; Huang, J.; Huang, X.; Guo, L. Etk interaction with PFKFB4 modulates chemoresistance of small-cell lung cancer by regulating autophagy. Clin. Cancer Res. 2018, 24, 950–962. [Google Scholar] [CrossRef] [Green Version]
- Meng, M.-B.; Wang, H.-H.; Guo, W.-H.; Wu, Z.-Q.; Zeng, X.-L.; Zaorsky, N.G.; Shi, H.-S.; Qian, D.; Niu, Z.-M.; Jiang, B. Targeting pyruvate kinase M2 contributes to radiosensitivity of non-small cell lung cancer cells in vitro and in vivo. Cancer Lett. 2015, 356, 985–993. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.-M.; Hu, G.-Y.; Zhao, X.-Q.; Lu, T.; Zhu, F.; Yu, S.-Y.; Xiong, H. Hypoxia-induced autophagy contributes to radioresistance via c-Jun-mediated BECLIN1 expression in lung cancer cells. J. Huazhong Univ. Sci. Technol. [Med. Sci.] 2014, 34, 761–767. [Google Scholar] [CrossRef]
- Hsu, H.-L.; Liao, P.-L.; Cheng, Y.-W.; Huang, S.-H.; Wu, C.-H.; Li, C.-H.; Kang, J.-J. Chloramphenicol induces autophagy and inhibits the hypoxia inducible factor-1 alpha pathway in non-small cell lung cancer cells. Int. J. Mol. Sci. 2019, 20, 157. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, S.F.; Nordestgaard, B.G.; Bojesen, S.E. Statin use and reduced cancer-related mortality. N. Engl. J. Med. 2012, 367, 1792–1802. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Su, Z.; DeWitt, J.P.; Xie, L.; Chen, Y.; Li, X.; Han, L.; Li, D.; Xia, J.; Zhang, Y.; et al. Fluvastatin Prevents Lung Adenocarcinoma Bone Metastasis by Triggering Autophagy. EBioMedicine 2017, 19, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Sanli, T.; Liu, C.; Rashid, A.; Hopmans, S.N.; Tsiani, E.; Schultz, C.; Farrell, T.; Singh, G.; Wright, J.; Tsakiridis, T. Lovastatin sensitizes lung cancer cells to ionizing radiation: Modulation of molecular pathways of radioresistance and tumor suppression. J. Thorac. Oncol. 2011, 6, 439–450. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Wang, Z.; Lin, Y.; Chen, Z.; Liu, H.; Chen, Y.; Wang, N.; Song, X. Sphingosine kinase 1 enhances the invasion and migration of non-small cell lung cancer cells via the AKT pathway. Oncol. Rep. 2015, 33, 1257–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gachechiladze, M.; Tichý, T.; Kolek, V.; Grygárková, I.; Klein, J.; Mgebrishvili, G.; Kharaishvili, G.; Janíková, M.; Smičková, P.; Cierna, L.; et al. Sphingosine kinase-1 predicts overall survival outcomes in non-small cell lung cancer patients treated with carboplatin and navelbine. Oncol. Lett. 2019, 18, 1259–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, L.; Xiong, H.; Li, J.; Liao, W.; Wang, L.; Wu, J.; Li, M. Sphingosine kinase-1 enhances resistance to apoptosis through activation of PI3K/Akt/NF-κB pathway in human non-small cell lung cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 1839–1849. [Google Scholar] [CrossRef] [Green Version]
- Nagahashi, M.; Ramachandran, S.; Kim, E.Y.; Allegood, J.C.; Rashid, O.M.; Yamada, A.; Zhao, R.; Milstien, S.; Zhou, H.; Spiegel, S.; et al. Sphingosine-1-phosphate produced by sphingosine kinase 1 promotes breast cancer progression by stimulating angiogenesis and lymphangiogenesis. Cancer Res. 2012, 72, 726–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, J.H.; Hong, S.K.; Kim, E.Y.; Park, S.Y.; Park, C.H.; Kim, J.M.; Kwon, O.J.; Kwon, S.J.; Lee, K.S.; Han, J.S. Overexpression of phospholipase D suppresses taxotere-induced cell death in stomach cancer cells. Biochim. Et Biophys. Acta 2008, 1783, 912–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, Y.L.; Hung, J.Y.; Ko, Y.C.; Hung, C.H.; Huang, M.S.; Kuo, P.L. Phospholipase D signaling pathway is involved in lung cancer-derived IL-8 increased osteoclastogenesis. Carcinogenesis 2010, 31, 587–596. [Google Scholar] [CrossRef]
- Ahn, M.-J.; Park, S.-Y.; Kim, W.K.; Cho, J.H.; Chang, B.J.; Kim, D.J.; Ahn, J.S.; Park, K.; Han, J.-S. A Single Nucleotide Polymorphism in the Phospholipase D1 Gene is Associated with Risk of Non-Small Cell Lung Cancer. Int. J. Biomed. Sci. 2012, 8, 121–128. [Google Scholar]
- Shatz, O.; Holland, P.; Elazar, Z.; Simonsen, A. Complex Relations Between Phospholipids, Autophagy, and Neutral Lipids. Trends Biochem. Sci. 2016, 41, 907–923. [Google Scholar] [CrossRef]
- Meacci, E.; Vasta, V.; Moorman, J.P.; Bobak, D.A.; Bruni, P.; Moss, J.; Vaughan, M. Effect of Rho and ADP-ribosylation Factor GTPases on Phospholipase D Activity in Intact Human Adenocarcinoma A549 Cells. J. Biol. Chem. 1999, 274, 18605–18612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meacci, E.; Nuti, F.; Catarzi, S.; Vasta, V.; Donati, C.; Bourgoin, S.; Bruni, P.; Moss, J.; Vaughan, M. Activation of Phospholipase D by Bradykinin and Sphingosine 1-Phosphate in A549 Human Lung Adenocarcinoma Cells via Different GTP-Binding Proteins and Protein Kinase C Delta Signaling Pathways. Biochemistry 2003, 42, 284–292. [Google Scholar] [CrossRef]
- Granade, M.E.; Harris, T.E. Purification of Lipin and Measurement of Phosphatidic Acid Phosphatase Activity from Liposomes. Methods Enzymol. 2018, 607, 373–388. [Google Scholar] [CrossRef]
- Fan, X.; Weng, Y.; Bai, Y.; Wang, Z.; Wang, S.; Zhu, J.; Zhang, F. Lipin-1 determines lung cancer cell survival and chemotherapy sensitivity by regulation of endoplasmic reticulum homeostasis and autophagy. Cancer Med. 2018, 7, 2541–2554. [Google Scholar] [CrossRef]
- Nazim, U.M.; Moon, J.-H.; Lee, Y.-J.; Seol, J.-W.; Park, S.-Y. PPARγ activation by troglitazone enhances human lung cancer cells to TRAIL-induced apoptosis via autophagy flux. Oncotarget 2017, 8, 26819–26831. [Google Scholar] [CrossRef] [Green Version]
- To, K.K.W.; Wu, W.K.K.; Loong, H.H.F. PPARgamma agonists sensitize PTEN-deficient resistant lung cancer cells to EGFR tyrosine kinase inhibitors by inducing autophagy. Eur. J. Pharmacol. 2018, 823, 19–26. [Google Scholar] [CrossRef]
- Gkountakos, A.; Sartori, G.; Falcone, I.; Piro, G.; Ciuffreda, L.; Carbone, C.; Tortora, G.; Scarpa, A.; Bria, E.; Milella, M.; et al. PTEN in Lung Cancer: Dealing with the Problem, Building on New Knowledge and Turning the Game Around. Cancers 2019, 11, 1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.Y.; Yuan, H.; Ma, L.T.; Kong, A.W.; Hsin, M.K.; Yip, J.H.; Underwood, M.J.; Chen, G.G. Roles of peroxisome proliferator-activated receptor-alpha and -gamma in the development of non-small cell lung cancer. Am. J. Respir. Cell Mol. Biol. 2010, 43, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Freitas, R.D.S.; Campos, M.M. Protective Effects of Omega-3 Fatty Acids in Cancer-Related Complications. Nutrients 2019, 11, 945. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.; Cartwright, C.; Chan, D.; Ding, J.; Felix, E.; Pan, Y.; Pang, J.; Rhea, P.; Block, K.; Fischer, S.M.; et al. Anticancer activity of fish oils against human lung cancer is associated with changes in formation of PGE2 and PGE3 and alteration of Akt phosphorylation. Mol. Carcinog 2014, 53, 566–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zajdel, A.; Wilczok, A.; Tarkowski, M. Toxic effects of n-3 polyunsaturated fatty acids in human lung A549 cells. Toxicol. Vitr. Int. J. Publ. Assoc. BIBRA 2015, 30 Pt B, 486–491. [Google Scholar] [CrossRef]
- Skácel, J.; Slusher, B.S.; Tsukamoto, T. Small Molecule Inhibitors Targeting Biosynthesis of Ceramide, the Central Hub of the Sphingolipid Network. J. Med. Chem. 2021, 64, 279–297. [Google Scholar] [CrossRef] [PubMed]
- Peterschmitt, M.J.; Crawford, N.P.S.; Gaemers, S.J.M.; Ji, A.J.; Sharma, J.; Pham, T.T. Pharmacokinetics, Pharmacodynamics, Safety, and Tolerability of Oral Venglustat in Healthy Volunteers. Clin. Pharmacol. Drug Dev. 2021, 10, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Loh, K.C.; Leong, W.-I.; Carlson, M.E.; Oskouian, B.; Kumar, A.; Fyrst, H.; Zhang, M.; Proia, R.L.; Hoffman, E.P.; Saba, J.D. Sphingosine-1-Phosphate Enhances Satellite Cell Activation in Dystrophic Muscles through a S1PR2/STAT3 Signaling Pathway. PLoS ONE 2012, 7, e37218. [Google Scholar] [CrossRef]
- Hernández-Tiedra, S.; Fabriàs, G.; Dávila, D.; Salanueva, Í.J.; Casas, J.; Montes, L.R.; Antón, Z.; García-Taboada, E.; Salazar-Roa, M.; Lorente, M.; et al. Dihydroceramide accumulation mediates cytotoxic autophagy of cancer cells via autolysosome destabilization. Autophagy 2016, 12, 2213–2229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, M.; Amato, R.; La Rocca, V.; Bilgin, M.; Freer, G.; Spezia, P.; Quaranta, P.; Piomelli, D.; Pistello, M. Acid ceramidase controls apoptosis and increases autophagy in human melanoma cells treated with doxorubicin. Sci. Rep. 2021, 11, 11221. [Google Scholar] [CrossRef]
- Realini, N.; Palese, F.; Pizzirani, D.; Pontis, S.; Basit, A.; Bach, A.; Ganesan, A.; Piomelli, D. Acid Ceramidase in Melanoma: EXPRESSION, LOCALIZATION, AND EFFECTS OF PHARMACOLOGICAL INHIBITION. J. Biol. Chem. 2016, 291, 2422–2434. [Google Scholar] [CrossRef] [Green Version]
- Lai, M.; La Rocca, V.; Amato, R.; Freer, G.; Pistello, M. Sphingolipid/Ceramide Pathways and Autophagy in the Onset and Progression of Melanoma: Novel Therapeutic Targets and Opportunities. Int. J. Mol. Sci. 2019, 20, 3436. [Google Scholar] [CrossRef] [Green Version]
- Costabile, G.; Della Pepa, G.; Salamone, D.; Luongo, D.; Naviglio, D.; Brancato, V.; Cavaliere, C.; Salvatore, M.; Cipriano, P.; Vitale, M.; et al. Reduction of De Novo Lipogenesis Mediates Beneficial Effects of Isoenergetic Diets on Fatty Liver: Mechanistic Insights from the MEDEA Randomized Clinical Trial. Nutrients 2022, 14, 2178. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.A.; Singh, B.K.; Zhou, J.; Xie, S.; Farah, B.L.; Lesmana, R.; Ohba, K.; Tripathi, M.; Ghosh, S.; Hollenberg, A.N.; et al. Loss of ULK1 increases RPS6KB1-NCOR1 repression of NR1H/LXR-mediated Scd1 transcription and augments lipotoxicity in hepatic cells. Autophagy 2017, 13, 169–186. [Google Scholar] [CrossRef] [Green Version]
- Loucif, H.; Dagenais-Lussier, X.; Beji, C.; Cassin, L.; Jrade, H.; Tellitchenko, R.; Routy, J.P.; Olagnier, D.; van Grevenynghe, J. Lipophagy confers a key metabolic advantage that ensures protective CD8A T-cell responses against HIV-1. Autophagy 2021, 17, 3408–3423. [Google Scholar] [CrossRef]
- Li, W.; Bai, H.; Liu, S.; Cao, D.; Wu, H.; Shen, K.; Tai, Y.; Yang, J. Targeting stearoyl-CoA desaturase 1 to repress endometrial cancer progression. Oncotarget 2018, 9, 12064–12078. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Ren, J.; Yang, L.; Li, Y.; Fu, J.; Li, Y.; Tian, Y.; Qiu, F.; Liu, Z.; Qiu, Y. Stearoyl-CoA desaturase-1 mediated cell apoptosis in colorectal cancer by promoting ceramide synthesis. Sci. Rep. 2016, 6, 19665. [Google Scholar] [CrossRef] [Green Version]
- Piao, C.; Cui, X.; Zhan, B.; Li, J.; Li, Z.; Li, Z.; Liu, X.; Bi, J.; Zhang, Z.; Kong, C. Inhibition of stearoyl CoA desaturase-1 activity suppresses tumour progression and improves prognosis in human bladder cancer. J. Cell. Mol. Med. 2019, 23, 2064–2076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, A.; Sano, O.; Kazetani, K.I.; Muraki, T.; Imamura, K.; Sumi, H.; Matsui, J.; Iwata, H. Feedback activation of AMPK-mediated autophagy acceleration is a key resistance mechanism against SCD1 inhibitor-induced cell growth inhibition. PLoS ONE 2017, 12, e0181243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, G.M.; Jiang, Q.H.; Cai, C.; Qu, M.; Shen, W. SCD1 negatively regulates autophagy-induced cell death in human hepatocellular carcinoma through inactivation of the AMPK signaling pathway. Cancer Lett. 2015, 358, 180–190. [Google Scholar] [CrossRef]
- Drake, L.E.; Springer, M.Z.; Poole, L.P.; Kim, C.J.; Macleod, K.F. Expanding perspectives on the significance of mitophagy in cancer. Semin. Cancer Biol. 2017, 47, 110–124. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ney, P.A. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009, 16, 939–946. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.L.; He, X.S.; Yu, Y.H.; Chen, Z.C. Expression and structure of BNIP3L in lung cancer. Ai Zheng = Aizheng = Chin. J. Cancer 2004, 23, 8–14. [Google Scholar]
- Chourasia, A.H.; Tracy, K.; Frankenberger, C.; Boland, M.L.; Sharifi, M.N.; Drake, L.E.; Sachleben, J.R.; Asara, J.M.; Locasale, J.W.; Karczmar, G.S.; et al. Mitophagy defects arising from BNip3 loss promote mammary tumor progression to metastasis. EMBO Rep. 2015, 16, 1145–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chourasia, A.H.; Macleod, K.F. Tumor suppressor functions of BNIP3 and mitophagy. Autophagy 2015, 11, 1937–1938. [Google Scholar] [CrossRef] [Green Version]
- Picchio, M.C.; Martin, E.S.; Cesari, R.; Calin, G.A.; Yendamuri, S.; Kuroki, T.; Pentimalli, F.; Sarti, M.; Yoder, K.; Kaiser, L.R.; et al. Alterations of the tumor suppressor gene Parkin in non-small cell lung cancer. Clin. Cancer Res. 2004, 10, 2720–2724. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.L.; Wang, N.N.; Ma, Q.L.; Chen, Y.; Yao, L.; Zhang, L.; Li, Q.S.; Shi, M.H.; Wang, H.F.; Ying, Z. Somatic and germline mutations in the tumor suppressor gene PARK2 impair PINK1/Parkin-mediated mitophagy in lung cancer cells. Acta Pharmacol. Sin. 2020, 41, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Bernardini, J.P.; Lazarou, M.; Dewson, G. Parkin and mitophagy in cancer. Oncogene 2016, 36, 1315. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, C.; Zhao, Y.; Yue, X.; Wu, H.; Huang, S.; Chen, J.; Tomsky, K.; Xie, H.; Khella, C.A.; et al. Parkin targets HIF-1α for ubiquitination and degradation to inhibit breast tumor progression. Nat. Commun. 2017, 8, 1823. [Google Scholar] [CrossRef] [PubMed] [Green Version]










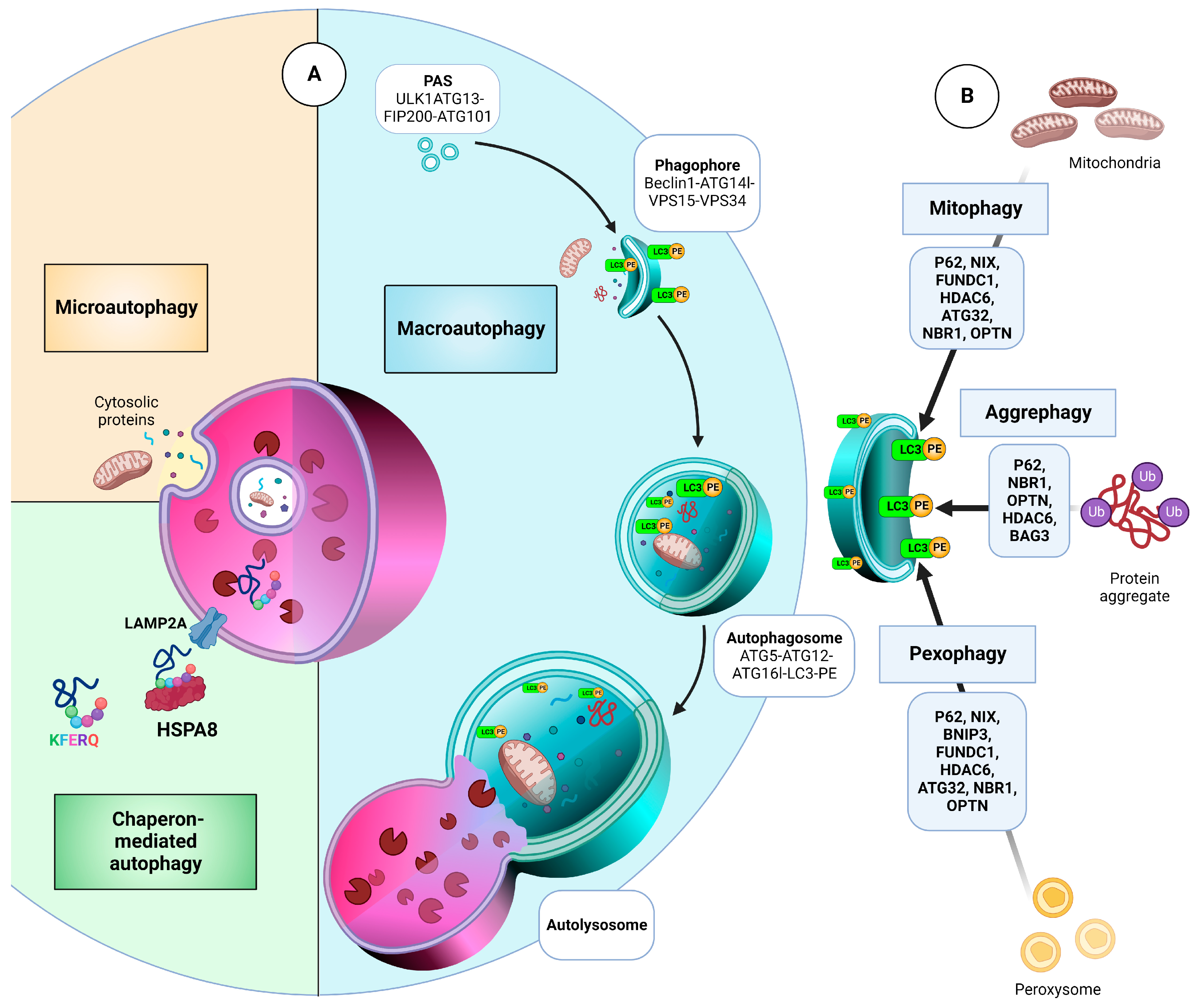{kind=link}
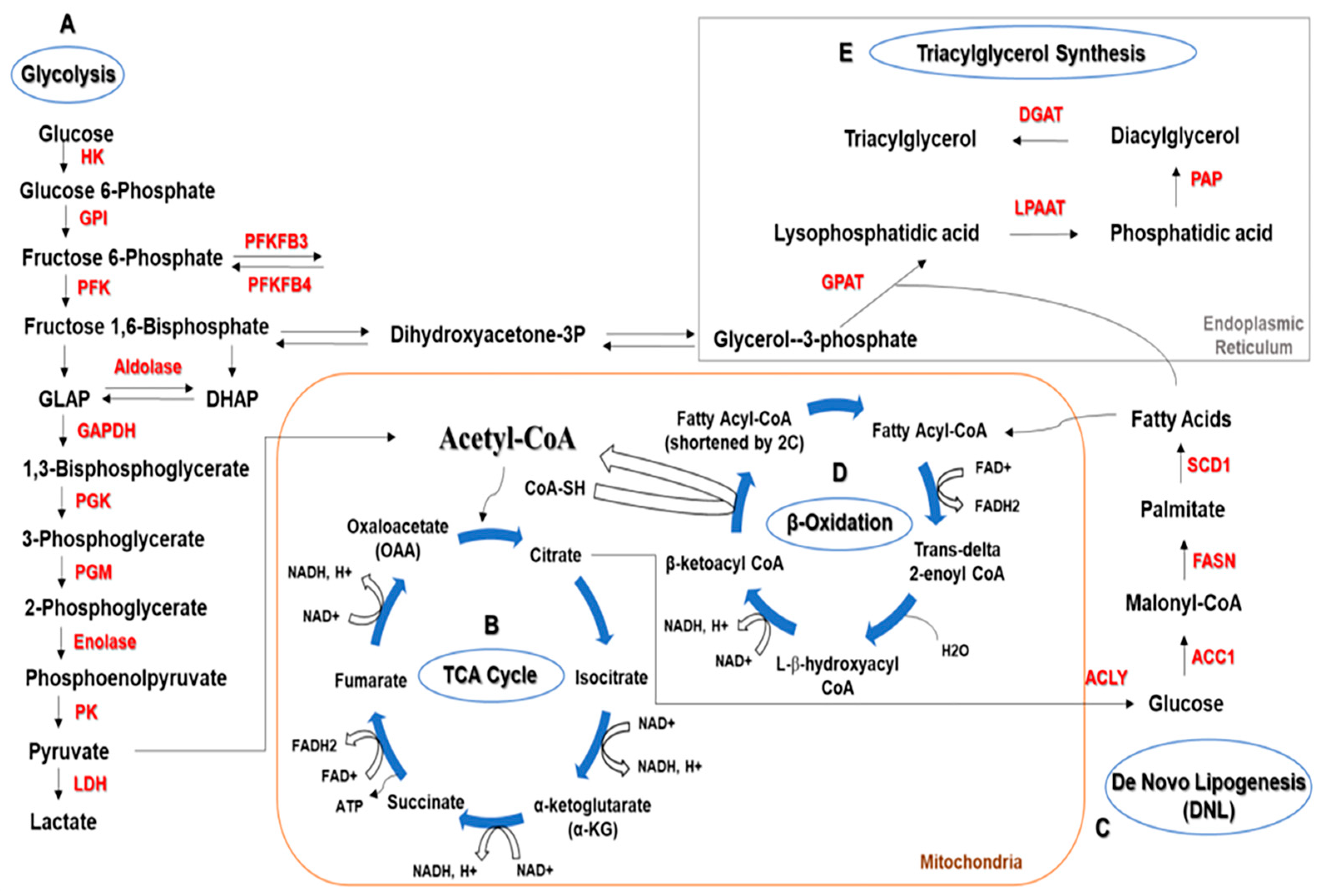{kind=link}
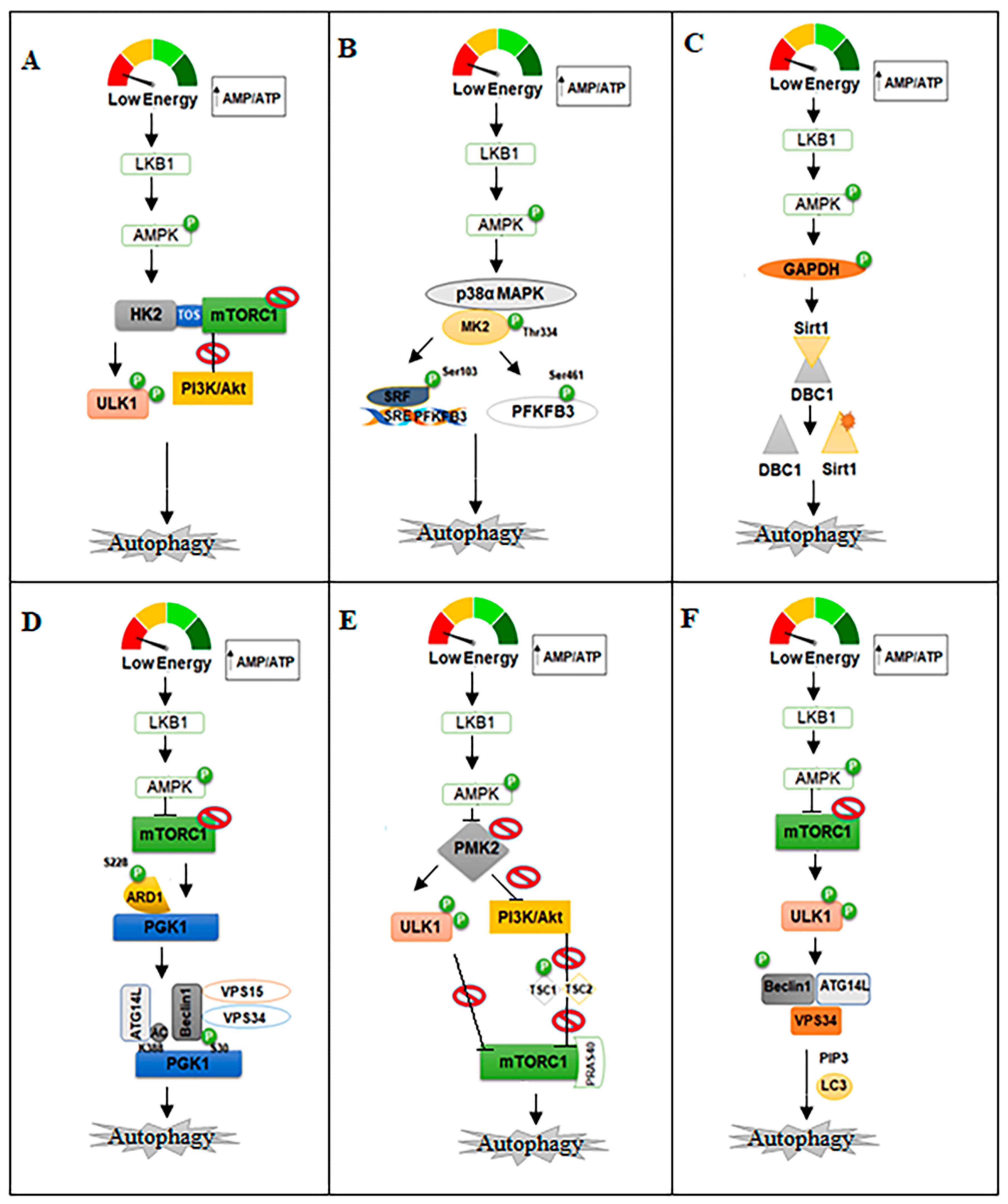{kind=link}
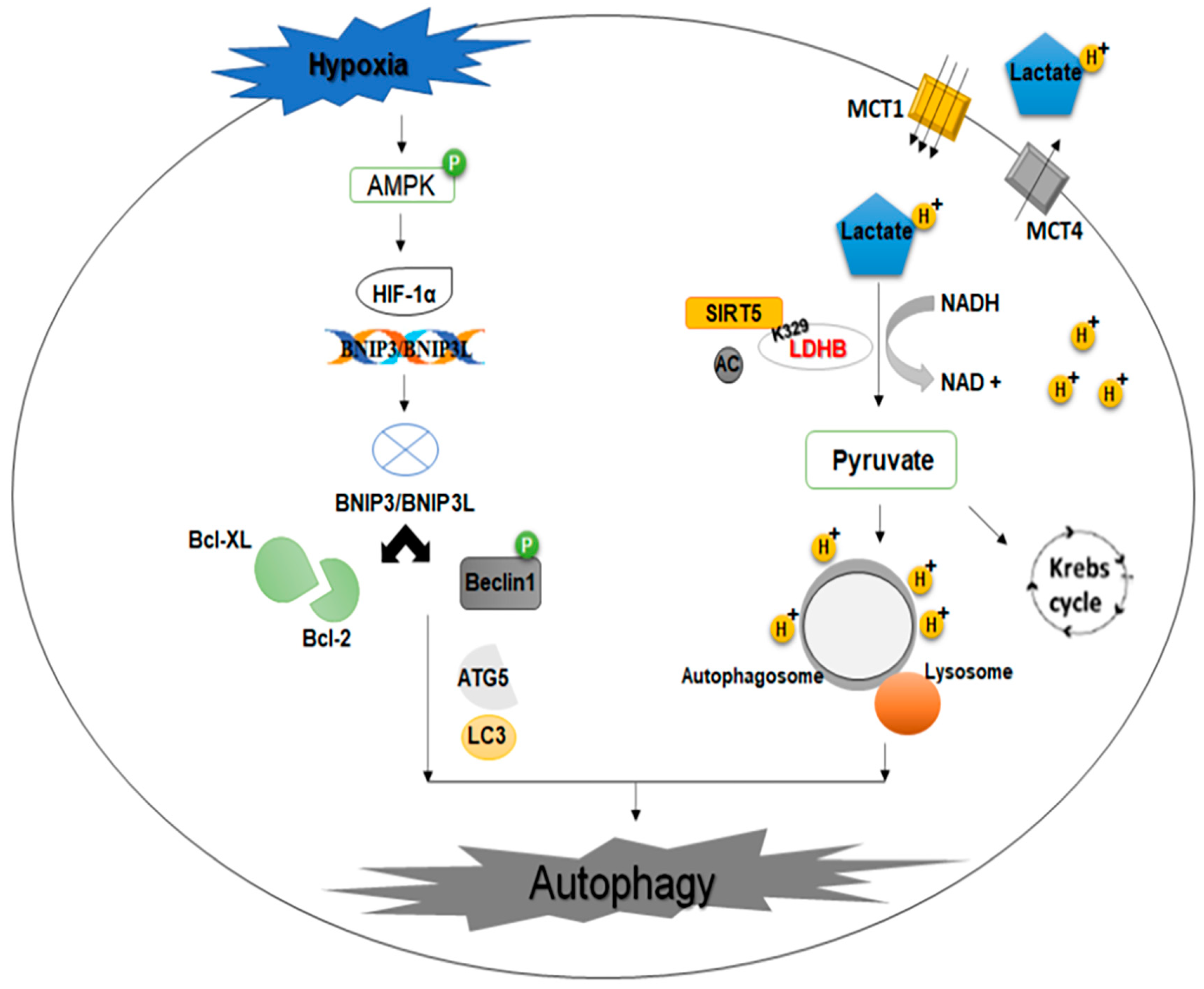{kind=link}
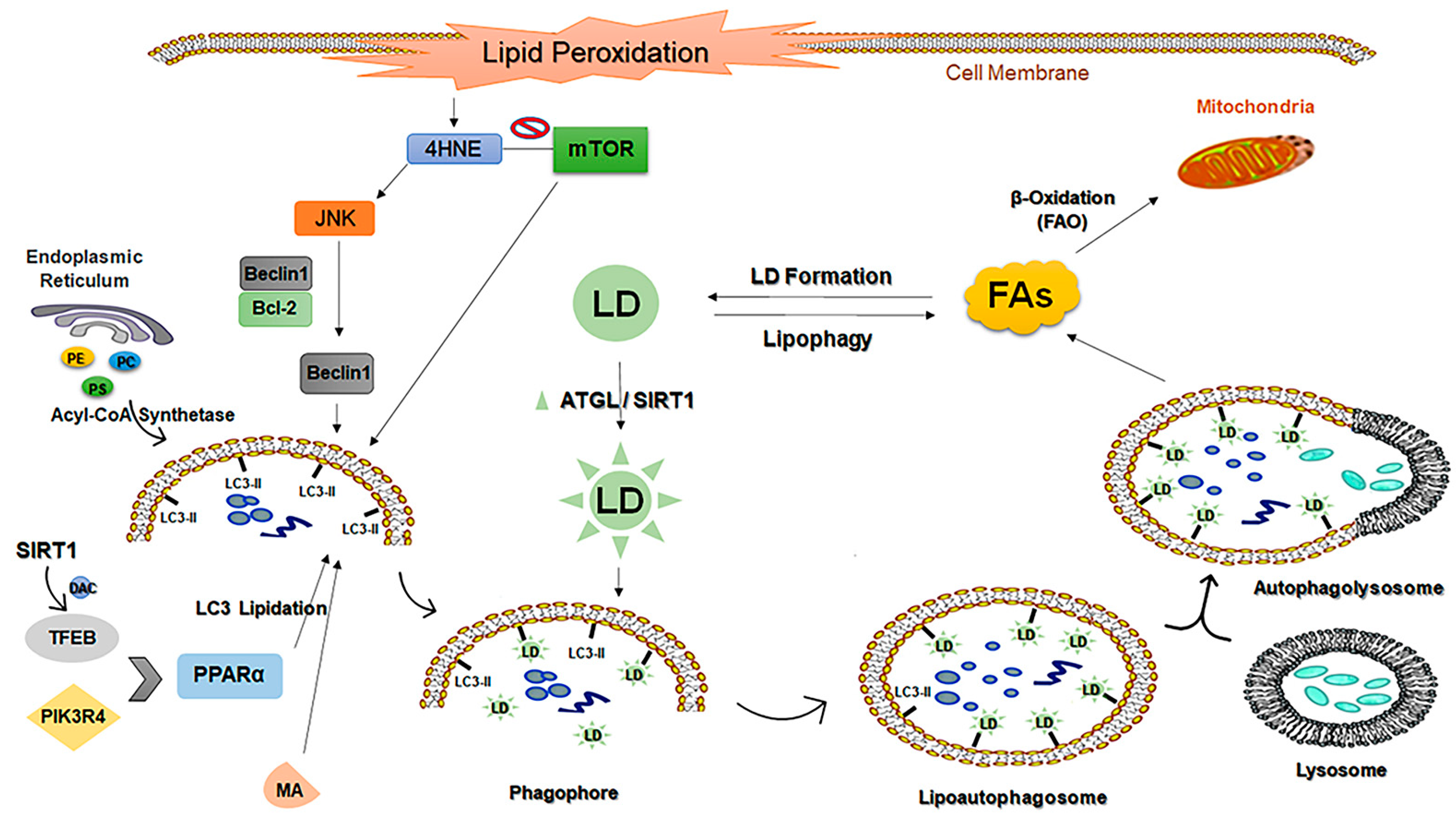{kind=link}
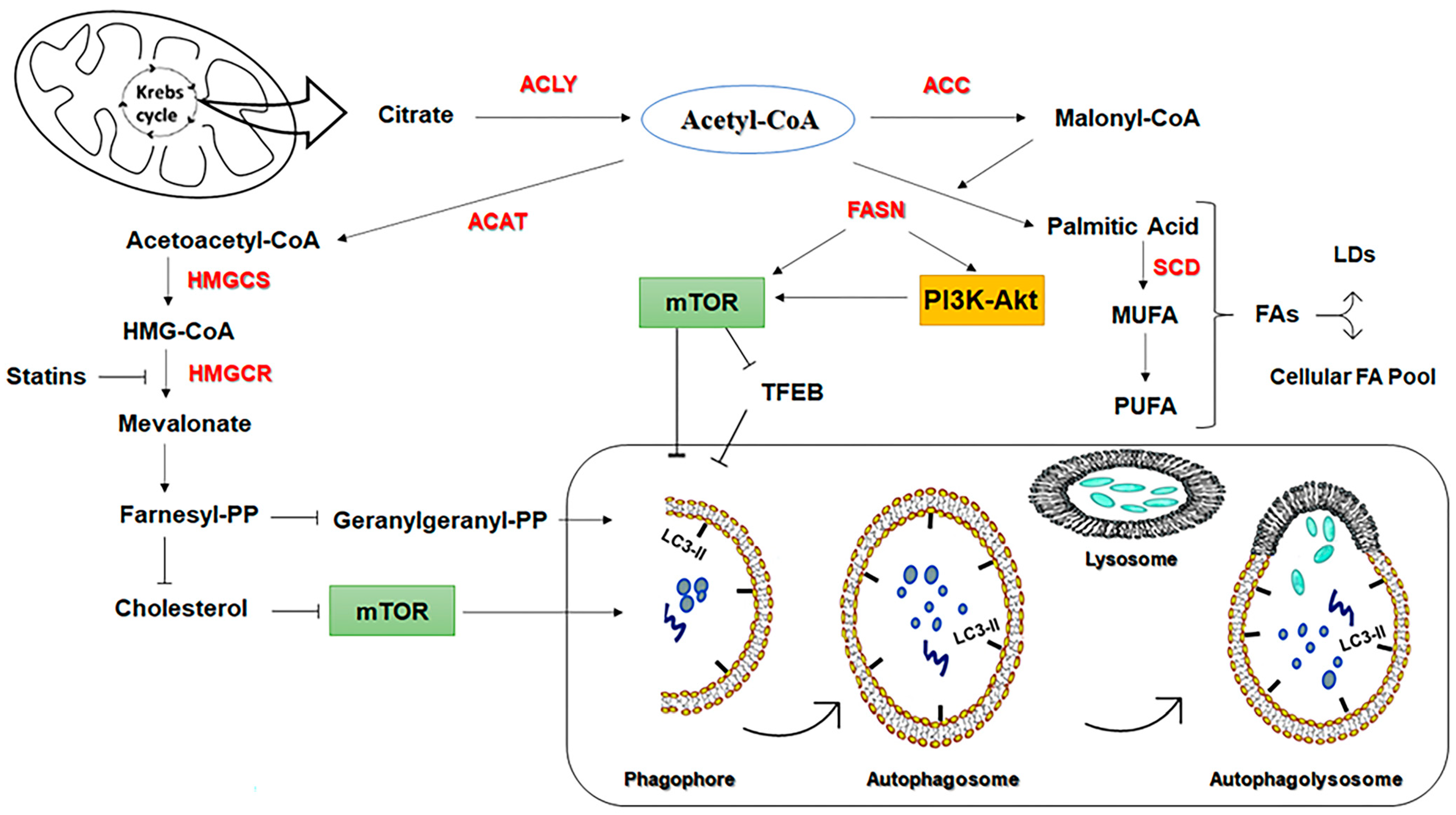{kind=link}
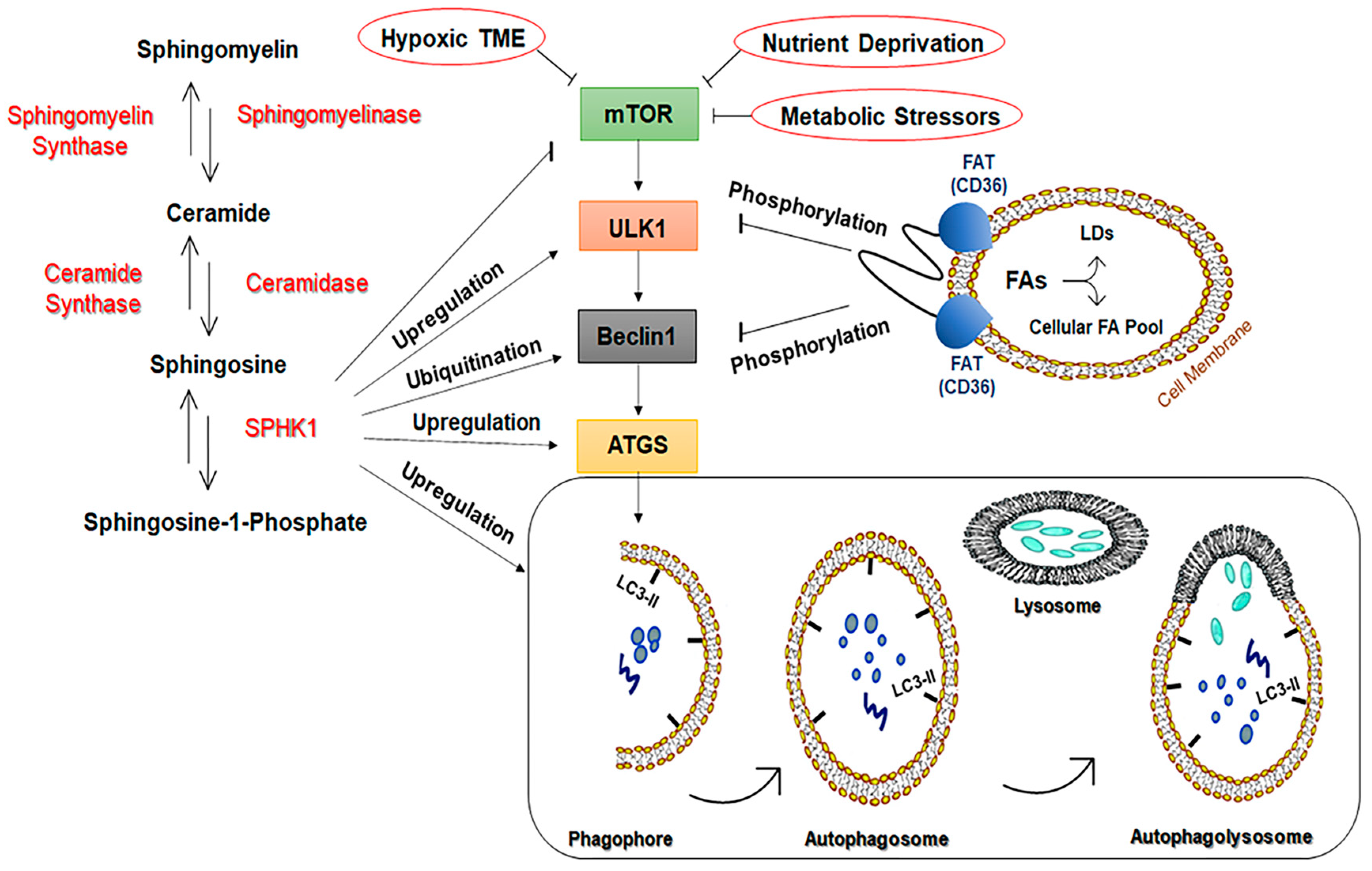{kind=link}
{kind=link}
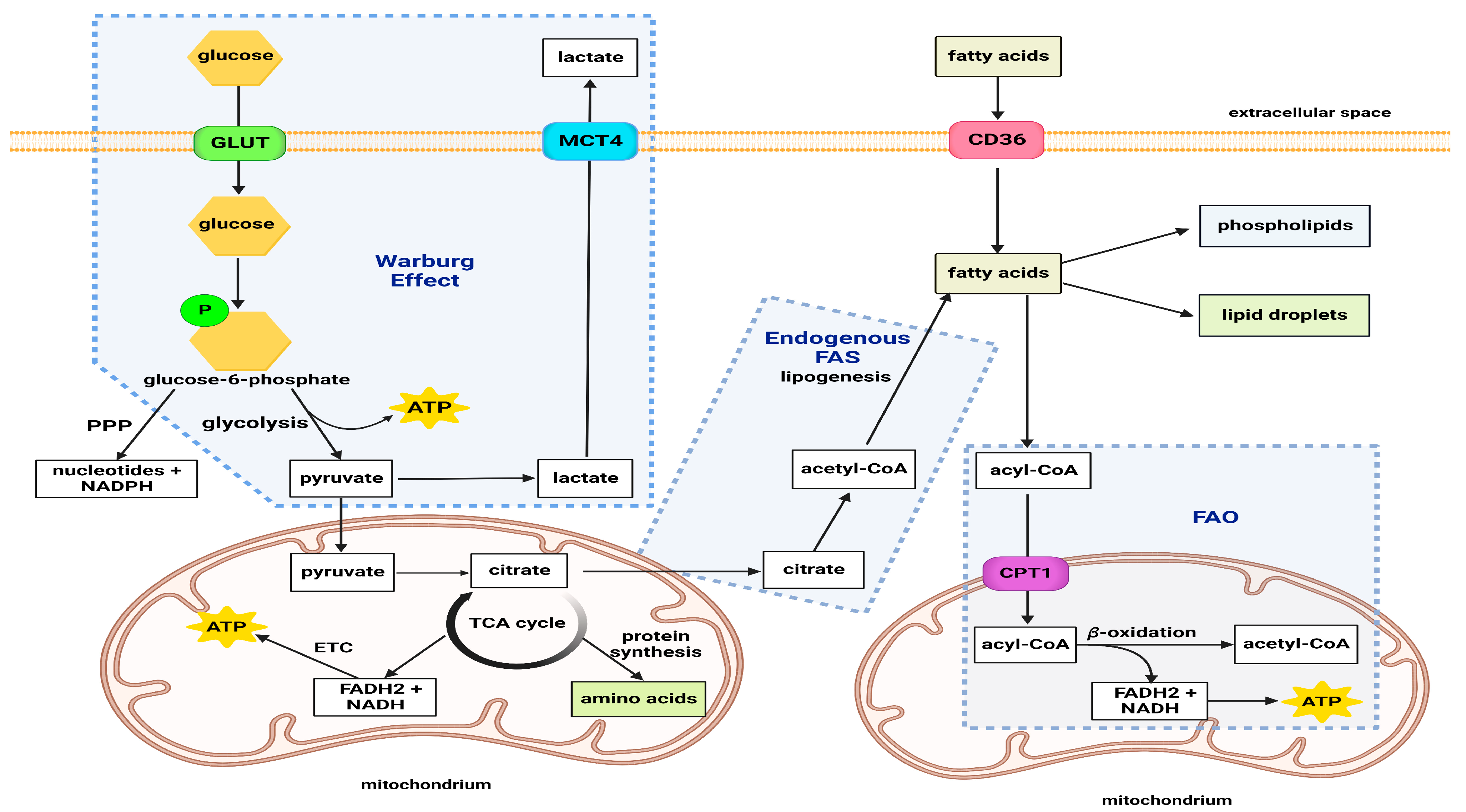{kind=link}
| Enzyme Name | Enzyme Isoform | Type of Cancer | Molecules | Roles in Glycolysis and Autophagy | Ref. |
|---|---|---|---|---|---|
| PFKFBs | PFKFB3 | Colon Adenocarcinoma | SiRNA and 3PO | Knockdown of PFKFB3 induces autophagy. | [123] |
| GAPDH | - | Pancreatic Adenocarcinoma | Genipin and Everolimus/3-MA | Induces Caspase-mediated apoptosis and inhibits BECLIN1-mediated autophagy. | [136] |
| PGK | PGK1 | Breast Cancer | shRNA | Reduces the Warburg effect and reduces invasion. | [187] |
| PK | PKM2 | AML | SiRNA | Reduces BECLIN1-mediated autophagy. | [150] |
| GAPDH | - | Gastric Cancer | miR-133b | Reduces glycolysis and induces autophagy by switching the PKM isoform expression from PKM2 to PKM1. | [188] |
| LDH | LDHA | Breast Cancer | SiRNA | Regulates BECLIN1-mediated autophagy. | [160] |
| PK | LDHB | Colorectal carcinoma | GW5074 | Reduces autophagy by SIRT5 knockout or the inhibition of LDHB acetylation at Lys329. | [189] |
| HIF | HIF-1α | Human Liver and Glioma Cancer | shRNA- lincRNA-p21 | Reduces glycolysis and inhibits autophagy by downregulating the HIF-1 protein. | [185] |
| LDH | LDHA | Cervical Cancer | SiRNA and 2-deoxyD-glucose or koningic acid | Inhibits glycolysis and reduces autophagy. | [72] |
| Lactate | - | Colorectal Carcinoma | Osimertinib | Induces autophagy by regulating the expression of MCT1 and activating LKB1/AMPK signaling. | [178] |
| HIF | HIF-1-α | Breast Cancer | 7acc1 | Induces autophagy via the inhibition of MCT4. | [179] |
| HK | HK2 | Colon, breast, prostate | miR-143 | Induces autophagy and induces proliferation. | [91,92,93,94,95,96,97] |
| Enzyme | Function | Effect on Autophagy | Cancer Type | Ref. | |
|---|---|---|---|---|---|
| Fatty Acid Synthase (FASN) | Synthesizes palmitate from acetyl-CoA and malonyl-CoA |
| Inhibition | Acute Myeloid Leukemia | [196] |
| Inhibition | Osteosarcoma | [198] | ||
| Inhibition | Colorectal Cancer | [199] | ||
| 3-Hydroxy-3-MethylGlutaryl-CoA Reductase (HMGCR) | Synthesizes cholesterol, geranylgeranyl pyrophosphate and farnesyl pyrophosphate, etc. |
| Induction | Acute Lymphocytic Leukemia | [204] |
| Induction | Prostate Cancer, Rhabdomyosarcoma | [207] | ||
| Sphingosine-1-Kinase (SPHK1) | Synthesizes sphingosine 1 phosphate (S1P) from sphingosine |
| Induction | Neuroblastoma | [223] |
| Induction | Breast Cancer | [224] | ||
| Induction | Colon Cancer | [225] | ||
| Induction | Hepatocellular Carcinoma | [226] | ||
| Induction | Gastric Cancer | [227] | ||
| Fatty Acid Translocase (FAT/CD36) | Long-chain fatty acid uptake |
| Inhibition | Hepatocellular Carcinoma | [229] |
| Lipid Metabolite | Effect on Autophagy | Cancer Type | Ref. | |
|---|---|---|---|---|
| Phosphatidic Acid | Induction of autophagosome curvature, inhibiting mTOR | Induction | Breast Cancer | [232] |
| Ceramide | Inhibition of Akt/mTOR; Upregulation of BECLIN1 expression | Induction | Breast Cancer, Neuroblastoma | [237,239] |
| Upregulation of BECLIN1 expression | Induction | Colon Cancer | [239] | |
| Dissociation of BCL2 from BECLIN1 | Induction | Cervical Cancer | [238] | |
| Sphingosine 1-phosphate (S1P) | mTOR inhibition | Induction | Prostate Cancer | [242] |
| mTOR inhibition | Induction | Breast Cancer | [224,241] | |
| Peroxisome Proliferator-Activating Receptors (PPARs) | PTEN activation; mTOR inhibition | Induction | Colorectal Cancer | [249] |
| Increase in lysosomal acidification | Induction | Breast Cancer | [250] | |
| Palmitic Acid | Activation of PKC, increasing the diacylglycerol levels | Induction | Hepatocarcinoma | [254] |
| Docosahexaenoic acid (DHA) | AMPK activation resulting in the inhibition of mTOR | Induction | p53-wild type prostate cancer | [264] |
| Producing mitochondrial reactive oxygen species (ROS) resulting in Akt and mTOR inhibition | Induction | p53-mutant prostate cancer | [264] | |
| Upregulation of LC3 and ATG14 expression | Induction | Colon Cancer | [267] | |
| mTOR inhibition | Induction | Glioblastoma | [264] | |
| mTOR inhibition | Induction | Cervix Carcinoma | [264] | |
| Docosahexaenoyl ethanolamine (DHEA) | Upregulation of PPARγ, which upregulates PTEN and, consequently, inhibits Akt-mTOR; promotes BCL2 phosphorylation and, therefore, its dissociation from BECLIN1 | Induction | Breast Cancer | [270] |
| Cholesterol | mTOR inhibition | Induction | Acute Lymphocytic Leukemia, Chronic Lymphocytic Leukemia | [204] |
| Enzyme Name | Enzyme Isoform | Type of Cancer | Molecules | Roles in Glycolysis and Autophagy | Ref. |
|---|---|---|---|---|---|
| HK | HK2 | NSCLC | 2-DG | Induce autophagy and apoptosis by increasing LC3II and cleaved caspase3 protein levels. | [373] |
| NSCLC | SiRNA | Inhibit cancer cell proliferation and tumorigenesis through decreasing glycolysis and HK2 expression at both the mRNA and protein levels. | [374] | ||
| NSCLC | miR-143 | Inhibit glycolysis and autophagy by the knockdown of HK2 and Atg2B. | [375] | ||
| PFKFBs | PFKFB3 | NSCLC | Erlotinib drives | Reduce the EGF-mediated increase in glycolysis. | [376] |
| PFKFB4 | SCLC | Ibrutinib | Inhibit autophagy by targeting Erk. | [377] | |
| GAPDH | - | NSCLC | SiRNA | Reduce glycolysis and cease tumor cell proliferation. | [131] |
| PK | PKM2 | NSCLC | pshRNA | Induce autophagy and apoptosis. | [378] |
| LDH | LDHA | Lung and Breast Cancer | WZB117 and ShRNA | Reduce glycolysis and the induction of apoptosis. | [379] |
| HIF | HIF-1 | NSCLC | SP600125 | Inhibit hypoxia-induced autophagy by targeting the BECLIN1 protein. | [379] |
| NSCLC | Chloramphenicol | Inhibit hypoxia-induced autophagy. | [380] | ||
| Lactate | - | NSCLC | SiRNA | Reduce pH-stimulated autophagy by the knockdown of the BECLIN1 protein and ER stress marker protein (GRP78). | [365] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alizadeh, J.; Kavoosi, M.; Singh, N.; Lorzadeh, S.; Ravandi, A.; Kidane, B.; Ahmed, N.; Mraiche, F.; Mowat, M.R.; Ghavami, S. Regulation of Autophagy via Carbohydrate and Lipid Metabolism in Cancer. Cancers 2023, 15, 2195. https://doi.org/10.3390/cancers15082195
Alizadeh J, Kavoosi M, Singh N, Lorzadeh S, Ravandi A, Kidane B, Ahmed N, Mraiche F, Mowat MR, Ghavami S. Regulation of Autophagy via Carbohydrate and Lipid Metabolism in Cancer. Cancers. 2023; 15(8):2195. https://doi.org/10.3390/cancers15082195
Chicago/Turabian StyleAlizadeh, Javad, Mahboubeh Kavoosi, Navjit Singh, Shahrokh Lorzadeh, Amir Ravandi, Biniam Kidane, Naseer Ahmed, Fatima Mraiche, Michael R. Mowat, and Saeid Ghavami. 2023. "Regulation of Autophagy via Carbohydrate and Lipid Metabolism in Cancer" Cancers 15, no. 8: 2195. https://doi.org/10.3390/cancers15082195
APA StyleAlizadeh, J., Kavoosi, M., Singh, N., Lorzadeh, S., Ravandi, A., Kidane, B., Ahmed, N., Mraiche, F., Mowat, M. R., & Ghavami, S. (2023). Regulation of Autophagy via Carbohydrate and Lipid Metabolism in Cancer. Cancers, 15(8), 2195. https://doi.org/10.3390/cancers15082195