1. Introduction
Lung cancer is the leading cause of cancer-related deaths among both men and women in the United States with 5-year survival rate of just 19%. Advances in genetic profiling of lung adenocarcinoma, the most common subtype of non-small-cell lung carcinoma, has led to the identification of several oncogenic drivers. Most prominent among them are
EGFR,
KRAS, and
MET [
1]. Specific mutations in the EGFR tyrosine kinase domain sensitize lung adenocarcinoma tumors to respond differentially to EGFR tyrosine kinase inhibitors (TKIs). Osimertinib, a third-generation EGFR TKI, targets mutant EGFR harboring both TKI-sensitizing and first/second-generation EGFR TKI-resistant T790M mutation in lung adenocarcinoma patients [
2,
3]. Unfortunately, all patients treated with osimertinib eventually develop acquired resistance. Recent studies have revealed various mechanisms of acquired resistance to osimertinib [
4,
5,
6,
7,
8,
9,
10]. However, no resistance mechanism can be identified in a significant number of patients. Most importantly, a large number of identified resistance mechanisms do not reveal an actionable target. Therefore, further studies are necessary to develop new therapies that are agnostic to specific resistance mechanisms; therapies that can target resistant tumors with either an unidentified resistance mechanism or with an identified, hitherto unactionable target of resistance. It is worth exploring the pathways that are not apparently linked to the EGFR pathway or compensatory parallel signaling pathways for targeted therapy. This may include pathways essential for cell survival and resistance in tumors, such as DNA repair or the cell cycle. Inhibition of DNA repair mechanisms or the cell cycle may be exploited to provide an additive or synergistic effect to osimertinib treatment of EGFR mutant tumors.
The cell cycle plays an important role in mammalian cell proliferation [
11]. The cell cycle can be regulated either by cyclin-dependent kinases (CDKs, catalytic subunit) in association with cyclins (regulatory subunit) or by cell-cycle checkpoint sensors to correct deficiency in replication or chromosome separations during cell divisions [
12,
13]. Cell-cycle checkpoint inhibitors that target cell-cycle components, specifically CDKs, could serve as potential therapeutic agents to treat human cancer [
12]. CDK1-6 bind multiple cyclins and are directly involved in cell-cycle progression, whereas CDK7-20 bind to single cyclins and are involved in transcriptional regulation [
12,
14]. CDK7, CDK12, and CDK13 regulate transcription by phosphorylating the C-terminal domain (CTD) of RNA polymerase II. CDK7 is involved in phosphorylation of serine-5 and serine-7 residues of RNA polymerase II CTD to initiate transcription, whereas CDK12 and 13 are involved in phosphorylation of serine-2 residue to promote elongation of the RNA polymerase II transcription complex. CDK12 and CDK13 inhibitors are known to inhibit transcription of DNA repair genes leading to inhibition of DNA repair mechanisms and causing cell death [
15]. CDK12 function and its role as a target for cancer treatment has been reviewed recently [
16]. We have also shown that non-functional G879V mutation in CDK12 results in reduced expression of long transcript genes that include DNA repair pathway genes, demonstrating that this mutation affects CDK12 DNA damage repair function [
17,
18]. We hypothesized that CDK12/13 inhibition will be able to circumvent both known and unknown mechanisms of resistance to osimertinib by virtue of targeting the DNA repair pathway that is independent of the hitherto known mechanisms of resistance to osimertinib. In this study, we sought to examine the role of CDK12/13 inhibitors in overcoming osimertinib resistance in human tumors. First, we developed osimertinib-resistant cells by long-term culture of sensitive cells in the presence of osimertinib to replicate in vivo selection of resistant tumors in patients treated with osimertinib. These resistant human lung adenocarcinoma cell lines were tested for growth inhibition in vitro by two novel CDK12/13 inhibitors. We further assessed the potency of these drugs in an osimertinib-resistant mouse xenograft model and demonstrated that these CDK12/13 inhibitors, in combination with osimertinib, can overcome osimertinib resistance in vivo. These results raise the possibility of the evaluation of CDK12/13 inhibitors in clinical studies in combination with osimertinib to overcome resistance.
3. Discussion
In this study, we leveraged the availability of CDK12/13 inhibitor AU-15606 and AU-16770 to test if these compounds could overcome osimertinib resistance in lung adenocarcinoma cells. CDK12 plays an important role as a regulator of the expression of DDR genes that happen to have long transcripts. A recent study showed that loss of CDK12 in cancer cells results in premature cleavage and polyadenylation of DDR gene transcripts, leading to decreased DNA damage response and lower recovery of affected cells [
15]. We anticipated similar outcomes in response to AU-15606 and AU-16770 CDK12/13 inhibitors.
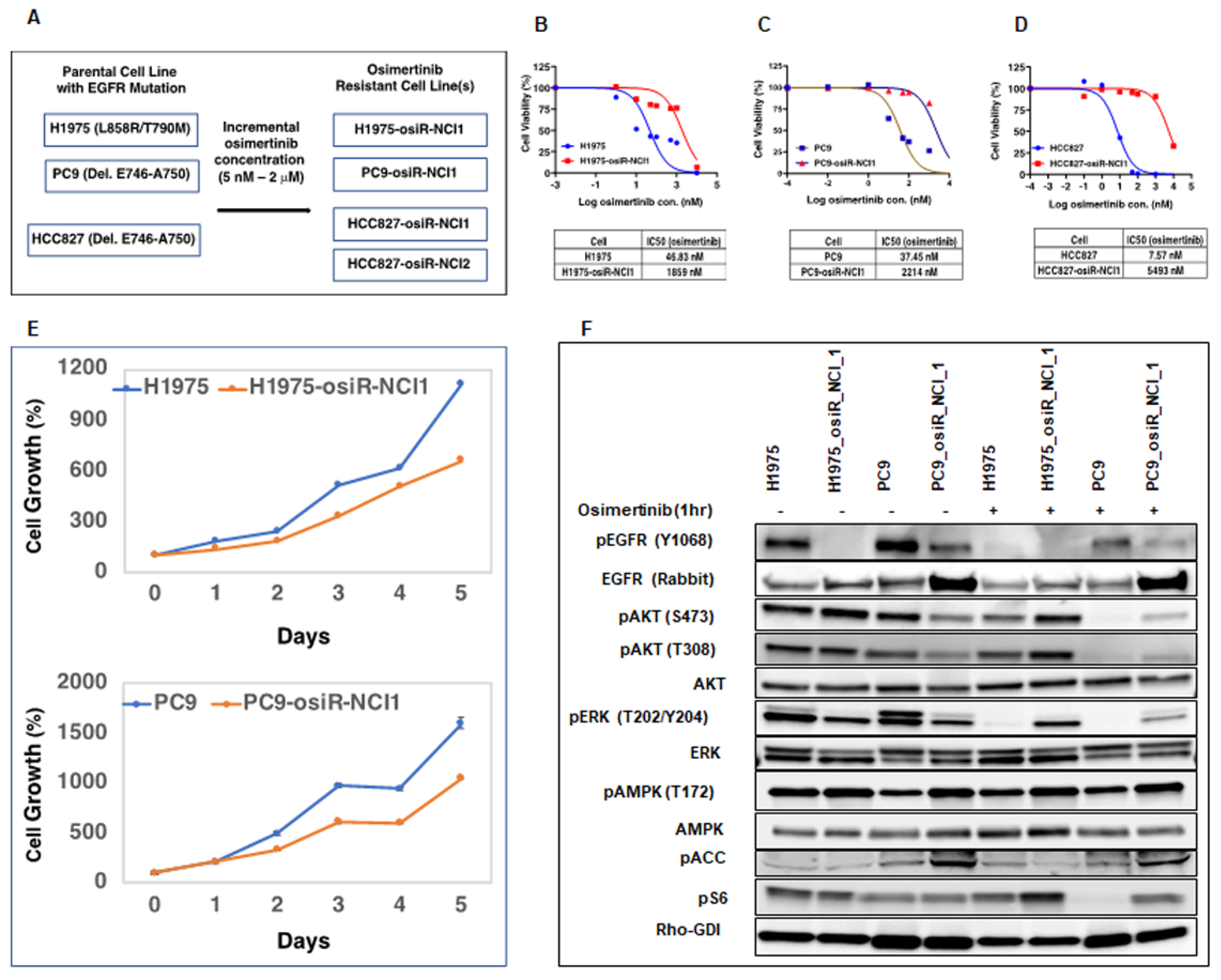
Initially, we generated isogenic drug-resistant EGFR mutant lung adenocarcinoma cell lines by osimertinib dose escalation. We utilized this model system to study the mechanism of osimertinib resistance with the intention of developing more effective anti-cancer therapies. It is well documented that cancer cells develop drug resistance due to several mechanisms, including tumor heterogeneity, EMT, and inhibition of cell death [
24]. Here, we showed that these osimertinib-resistant cells (H1975-osiR-NCI1, PC9-osiR-NCI1, and HCC827-osiR-NCI1/2) grow slower than the parental cells, suggesting that altered proliferation may be associated with drug resistance (
Figure 1E). We further demonstrated that both the AKT survival and MAPK (ERK) cell proliferation pathways remain active even after osimertinib treatment of resistant cells (
Figure 1F). Interestingly, the basal pERK level was lower in the osimertinib-resistant cells compared to the sensitive cells. This is related to the significantly lower level of pEGFR in the resistant cells (
Figure 1F, pEGFR, lanes 2 and 4) and phenotypically correlates with the reduced growth observed in the resistant cells compared to the sensitive cells (
Figure 1E). However, while osimertinib treatment of parental H1975 and PC9 cells almost completely abrogates pERK levels, there is incomplete inhibition of ERK phosphorylation in the osimertinib-resistant cells, suggesting that residual pERK levels in the presence of osimertinib continue to promote growth signals and hence resistance to osimertinib (
Figure 1F, pERK lanes 6 and 8). This provides the resistant cells with a growth advantage compared to parental cells.
Under normal cell growth conditions, pERK translocates to the nucleus and activates transcription factors such as FOS, ELK, and other proteins resulting in cell-proliferation-specific gene expression. Unphosphorylated ERK is retained in the cytoplasm and cannot activate the above genes. Furthermore, cytoplasmic ERK activates proapoptotic genes, such as
DAPK, which inhibits growth and promotes apoptosis [
25]. It is still possible that a parallel receptor tyrosine kinase pathway is activated in these resistant cells. We have previously demonstrated that pAKT is activated in PC9-osiR and HCC827-osiR cells harboring EGFR Del746-750 mutant. Furthermore, dactolisib, a dual PI3K/AKT inhibitor, inhibits activation of AKT and circumvents osimertinib resistance [
26].
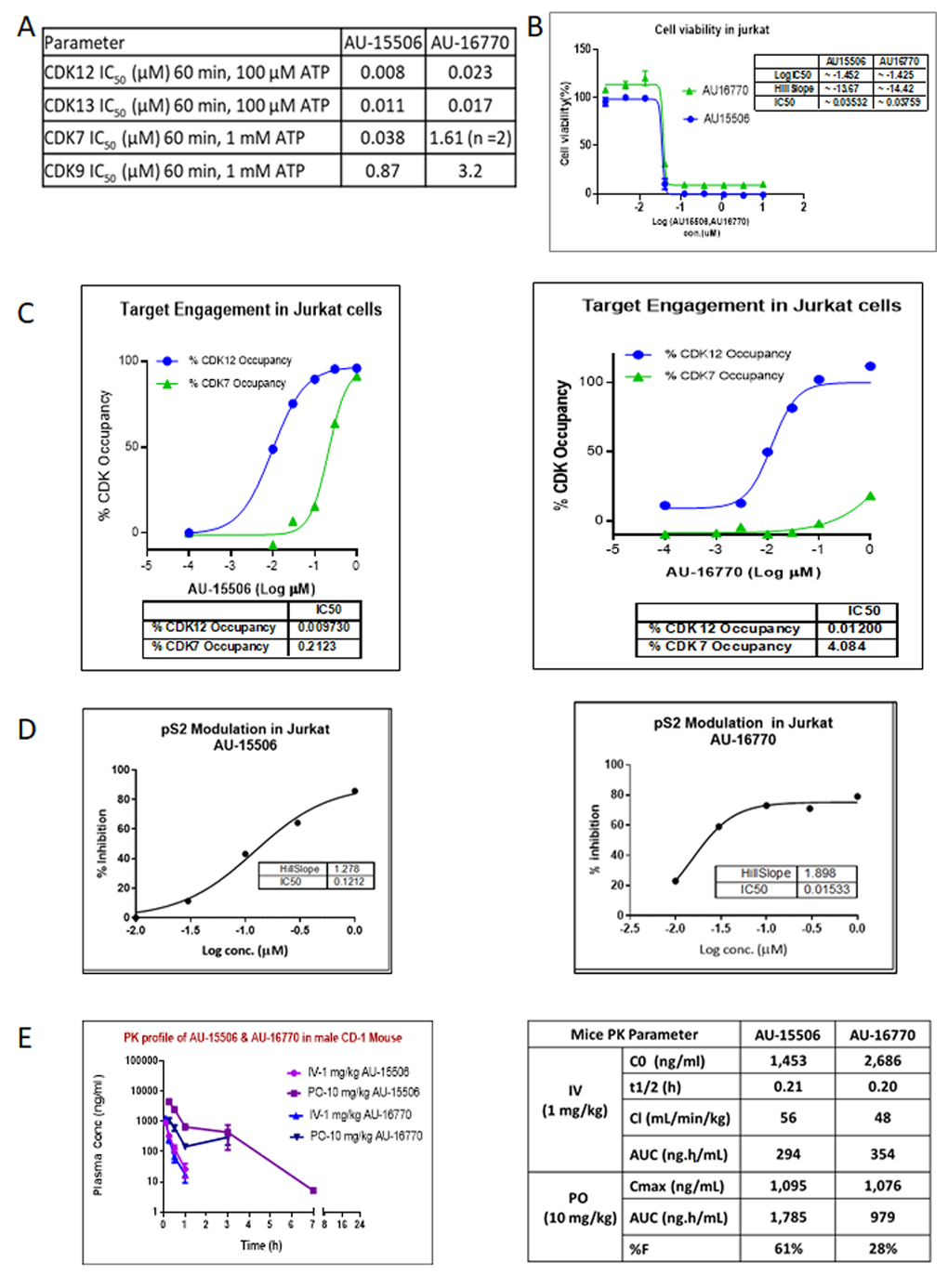
In order to target pathways other than EGFR signaling to develop successful combination therapies for overcoming osimertinib resistance, we assessed the potency of two novel CDK12/13 inhibitors, AU-15506 and AU-16770. Initially, we characterized these lead compounds for their effectiveness in inhibiting CDKs, particularly CDK12, CDK13, CDK7, and CDK9, in an in vitro kinase assay. We observed that these compounds inhibit CDK12 and 13 more efficiently than CDK7 and CDK9 (
Figure 2A). These compounds were found to have a minimal off-target effect at micromolar concentrations when tested in a panel of 27 other kinases (
Supplementary Figure S2). In our study, the compounds AU-16770 and AU-15506 tested at 1 μM and 10 μM showed high selectivity in inhibiting CDK12/13, while some marginal activity was observed for SGK1, GLK, and FGFR1 kinases at micromolar concentrations of the drugs (
Supplementary Figure S2). High selectivity is expected to increase the therapeutic index of these inhibitors. There are not well explored correlations for activities of SGK1 and GLK with CDK12/13. However, CDK12 inhibition may downregulate the expression of FGFR1 and other FGF receptors, potentially inhibiting the signaling pathways that promote cancer cell growth and survival. The CDK12 inhibitor reduced the expression of FGFR1 and other FGF receptors in ovarian cancer cells, leading to decreased cell proliferation and increased apoptosis. The reference molecule CDK12/13 inhibitor, THZ531 [
27], also showed 82% inhibition of FGFR1 at 1 μM concentration of the drug. However, AU-16770 and AU-15506 inhibits <50% at 1 μM and this inhibition may not result in additional toxicities.
Next, in the Jurkat cell viability assay, these compounds showed high inhibitory activity as determined by their very low IC
50 (
Figure 2B). When we performed target engagement assay in Jurkat cells, we observed that these compounds bind irreversibly to the intended target, CDK12, more efficiently than CDK7, another member of CDK family with high kinase domain similarity (
Figure 2C). We further validated the effectiveness of these compounds by examining inhibition of phospho-serine modification of RNA pol II, a substrate of CDK12 (
Figure 2D). Therefore, we concluded that AU15606 and AU-16770 are CDK12/13-specific highly potent anti-cancer agents in vitro. Finally, we performed pharmacokinetic assays of AU-15506 and AU-16770 in mice and analyzed the pharmacokinetic parameters. When administered through intravenous injection, these compounds reached the highest concentration in the blood stream in ~0.2 h, and drug clearance appeared to be similar between the two compounds (56 mL/min/kg vs. 48 mL/min/kg). When these drugs were administered through oral gavage (PO), the bioavailability of these compounds appeared to vary significantly (61% vs. 28%). However, despite bioavailability nonequivalence, AU-16770 had better therapeutic equivalence. Furthermore, these compounds appeared to be more specific and effective (IC50s: 0.035 nM and 0.038 nM) in Jurkat cells compared to THZ531 (IC50 = 50 nM), a first-in-class covalent inhibitor of CDK12/13 [
18]. Therefore, these compounds need to be validated further in vitro and in vivo as effective anti-cancer agents to inhibit osimertinib-resistant tumor cell growth.
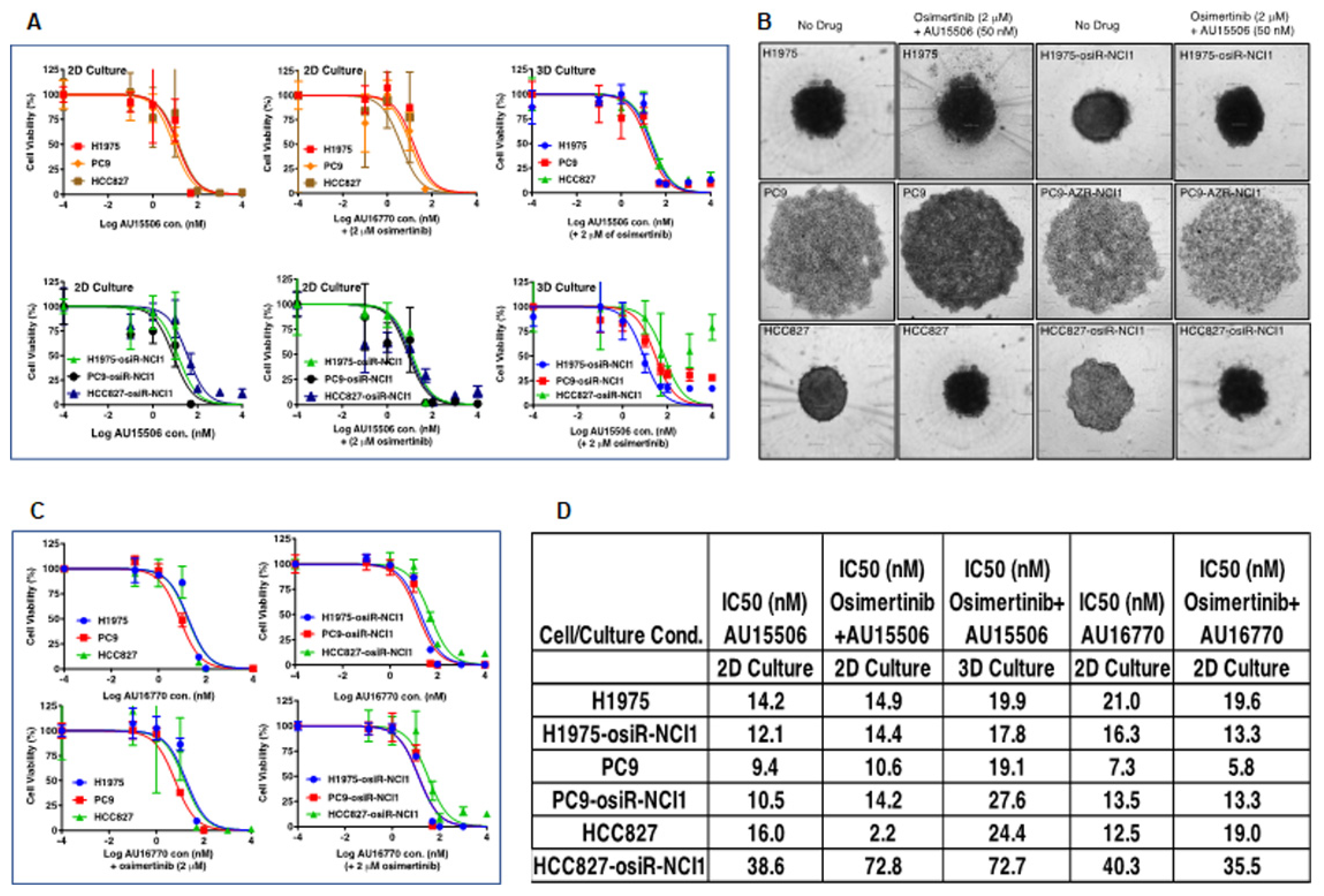
We found that CDK12/13 inhibitors, AU-15506 and AU-16770, are very effective in inhibiting osimertinib-resistant H1975-osiR-NCI1, PC9-osiR-NCI1, and HCC827-osiR-NCI1/2 cancer cell growth in vitro, either alone or in combination with osimertinib (
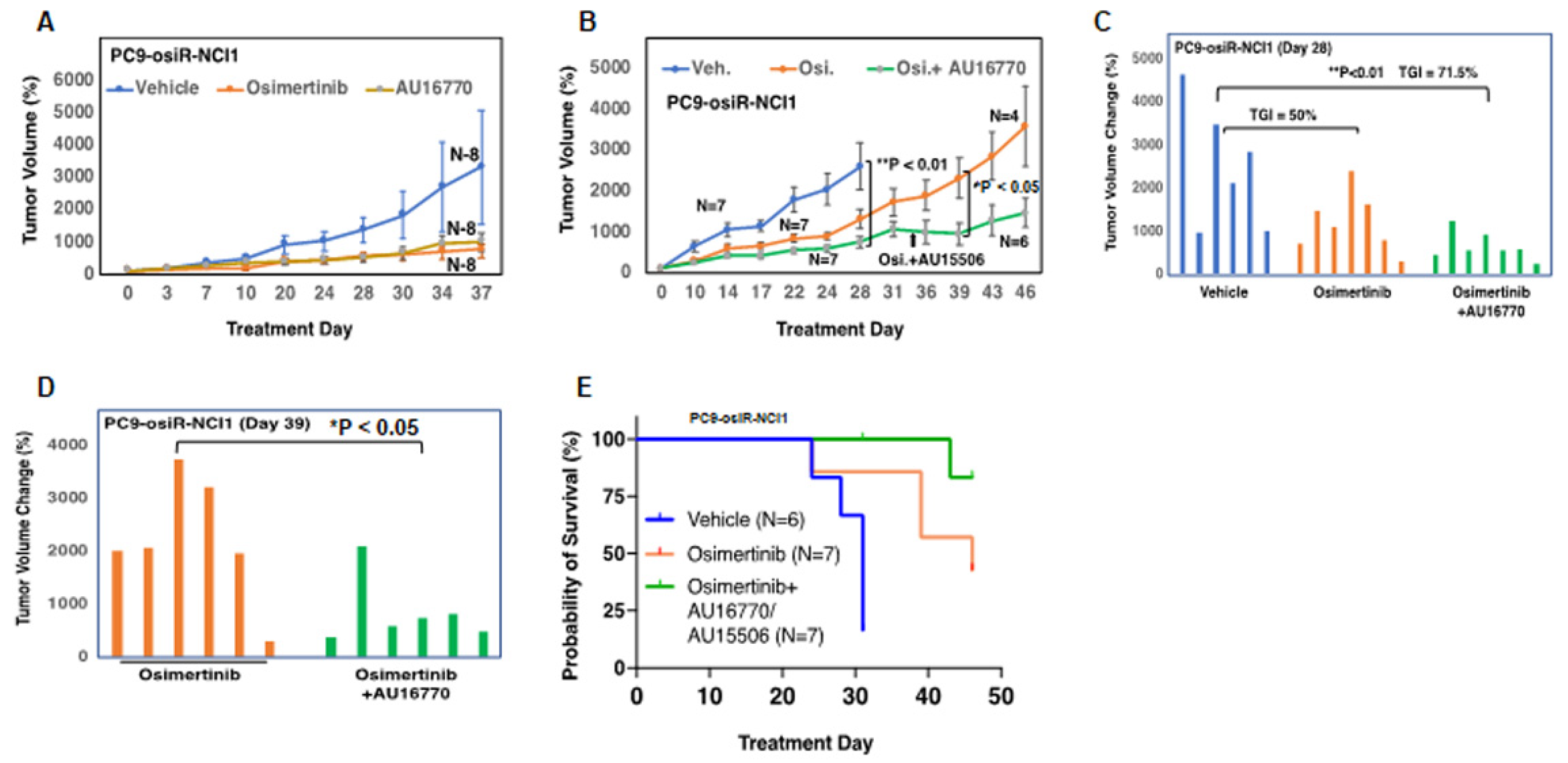
Figure 3A–C). However, AU16770 was only effective in reducing xenograft tumor growth when combined with osimertinib (
Figure 4A–H). This suggests that osimertinib-resistant patients may still need to take osimertinib and other novel drugs to prevent disease recurrence and survive longer. Indeed, we demonstrated that a lung adenocarcinoma patient with
ERBB2 amplification and harboring CDK12-G879V mutation responded to HER2-directed targeted therapy in combination with standard chemotherapy that explored the vulnerability of impaired DNA damage repair caused by the CDK12-G879V mutation [
18].
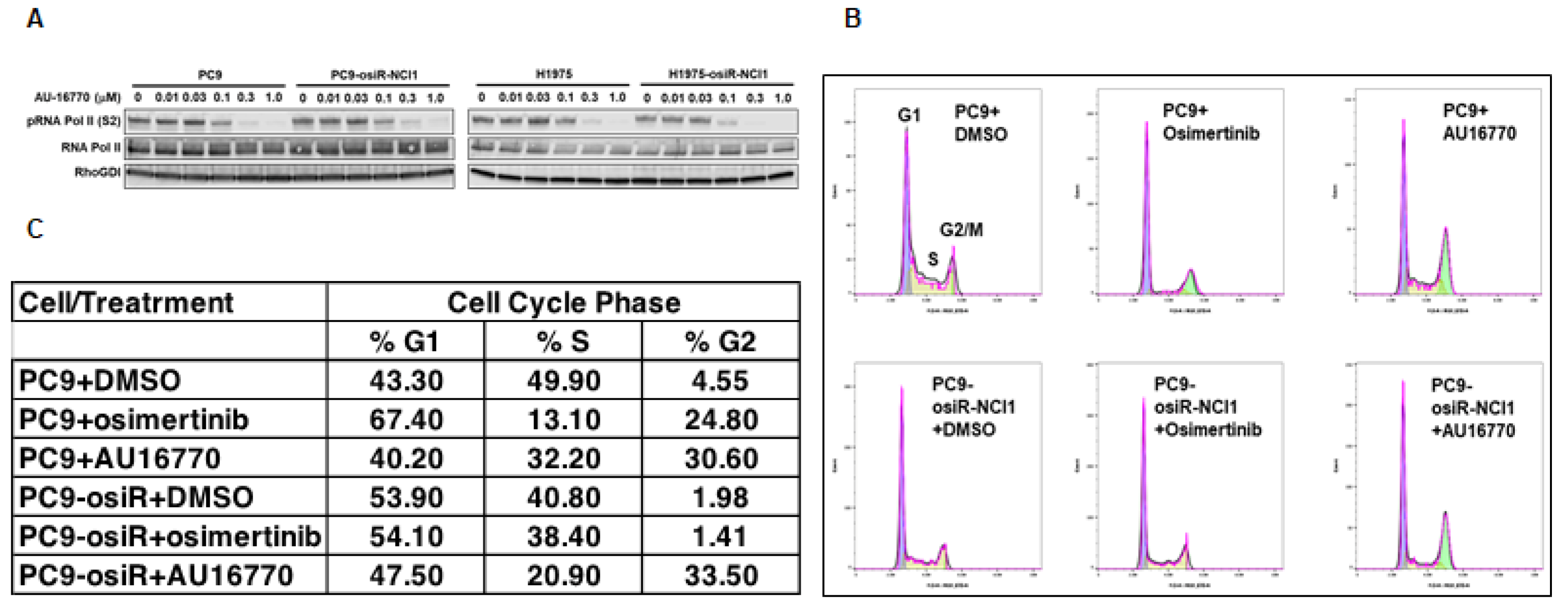
Proliferation of cells depend on normal duplication of genetic materials and cell divisions using the highly regulated cell-cycle process, and aberration may lead to cancer development. This process is controlled by CDKs (CDK4-6) in association with cyclins. Therefore, inhibitors that block CDK activities are potential anti-cancer agents [
28]. Here, we demonstrated that the CDK12/13 inhibitor AU-16770 prevented the transition of osimertinib-resistant cells from the G2 to M phase, consistent with other studies (
Figure 5B,C). It is interesting to note that because CDK12 is required during DNA transcription, depletion or inhibition of CDK12 activity is expected to arrest cells at the G1/S phase [
29]. Further studies are necessary to understand how AU-16770 is responsible for growth arrest of PC9-osiR-NCI1 cells at the G2/M phase.
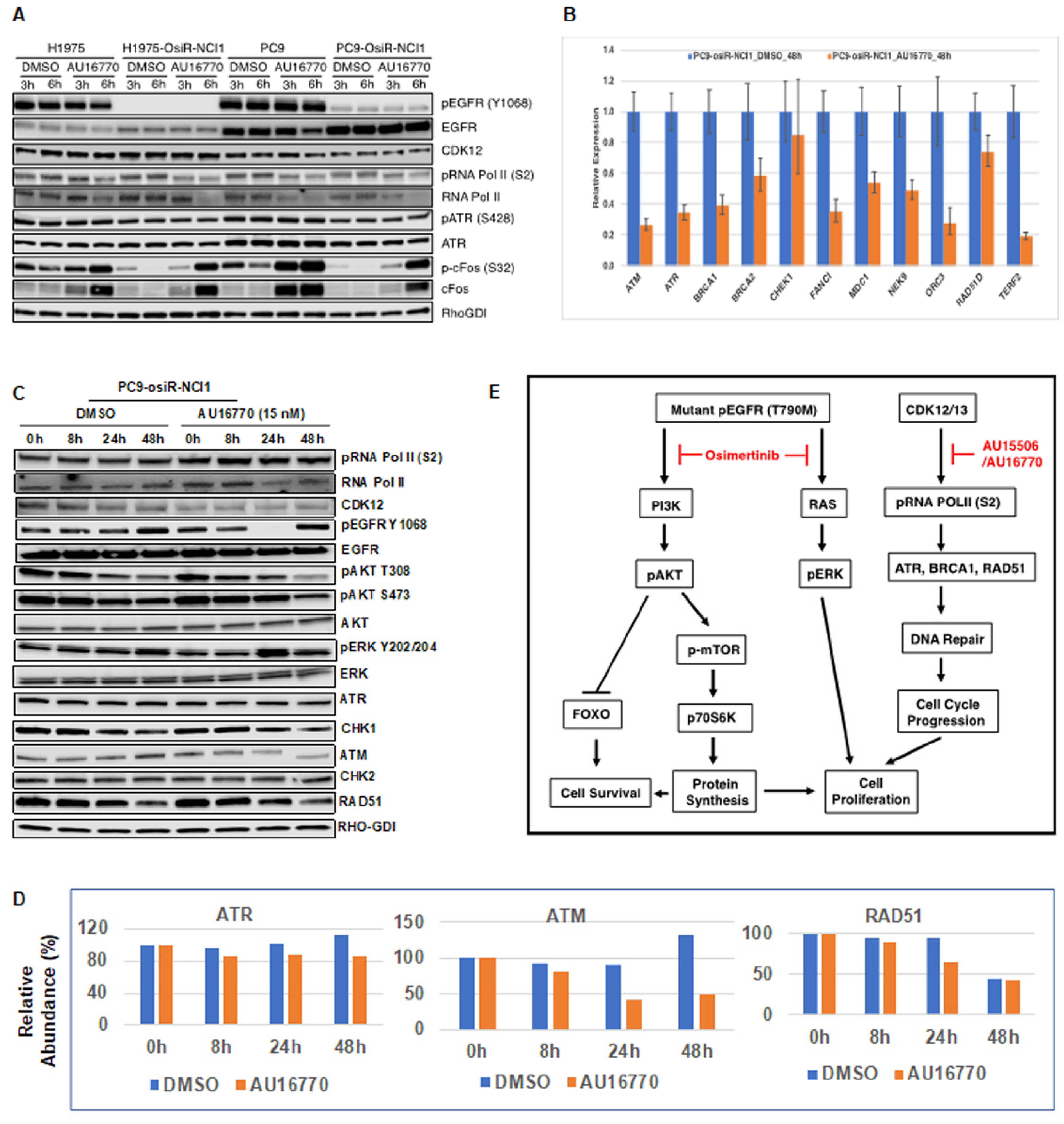
Phosphorylation and dephosphorylation of intracellular signaling components play an important role in modulating cell survival and cell proliferation. We performed immunoblot analyses of several intracellular proteins to determine if their phosphorylation status correlated with cellular AU-16770 sensitivity (
Figure 6). To our surprise, we observed that AU-16770 treatment increases the synthesis of cFOS, leading to higher phospho cFOS proteins both in osimertinib-sensitive and -resistant cells (
Figure 6A). Further experiments are clearly necessary to understand this effect. Quantitative PCR analysis of long transcripts associated with DNA damage response (DDR) genes indicated that, with the exception of
CHEK1, expression of
ATR,
ATM,
BRCA1,
BRCA2,
FANC1,
MDC1,
NEK9,
ORC3,
RAD51, and
TERF2 were significantly reduced in PC9-osiR cells upon treatment with AU-16770 for 48 h (
Figure 6B), supporting impaired DDR and cell-cycle arrest. Previously, we have also shown that the G879V mutation in CDK12 affects its DNA repair function and makes cells susceptible to chemotherapy [
18]. Interestingly, in our Western blot analyses (
Figure 6C) of cell extracts of PC9-osiR-NCI cells treated with AU-16770, we did not demonstrate a reduction in RNA Pol II serine 2 phosphorylation as shown in
Figure 5. This is presumably due to usage of a much lower (15 nM) concentration of AU-16770 compared to 100–300 nM that was used in the experiment shown in
Figure 5. However, there was a reduction in protein expression of some DDR proteins, such as ATM, ATR, and RAD51 at 24–48 h upon AU-16770 treatment (
Figure 6C,D).
Recently, it has been reported that inhibition of RAD51 is associated with decreased cervical cancer cell proliferation in vitro and in cervical cancer xenografts by attenuating cell-cycle transition [
30]. Therefore, it is possible that osimertinib and AU-16770 combination treatment results in inhibition of RAD51 expression and ATR and cFOS phosphorylation, leading to the suppression of proliferation-specific gene expression and inhibition of cell-cycle transition of osimertinib-resistant cells. Based on our results, we have modeled the activation of different pathways in osimertinib-resistant cells and suppression of these pathways in the presence of AU-15506/AU-16770 (
Figure 6E). We conclude that osimertinib and AU-16770 work synergistically to attenuate gene expression and protein synthesis of a specific set of genes that are critical for cell survival and cell proliferation [
23,
30].
In this study, we demonstrated that osimertinib-resistant cells generated in culture may serve as an excellent model system for osimertinib resistance in patients. This system allowed us to identify new anti-cancer agents to circumvent resistance. The role of CDK12/13 inhibitors as anti-cancer therapeutic agents is well known, but this is the first study to explore the use of CDK12/13 inhibitors in overcoming osimertinib resistance. Interestingly, the CDK12/13 inhibitors used in this study were equally effective in inhibiting the growth of the parental osimertinib-sensitive cells in culture as a monotherapy, underscoring the DDR and cell-cycle progression pathway inhibition that is parallel and non-overlapping to EGFR pathway inhibition by osimertinib.
4. Materials and Methods
4.1. Reagents and Antibodies
RPMI-1640 tissue culture medium was obtained from Millipore-Sigma (Burlington, MA, USA) and FBS was obtained from GeminiBio (West Sacramento, CA, USA). All chemicals were obtained from Millipore-Sigma, unless stated otherwise. Complete minitab protease inhibitor (#11836170001), and PhosStop phosphatase inhibitor (#49068450001) were obtained from Millipore-Sigma. Nitrocellulose Western transfer sandwich kit (#1704271) was obtained from Bio-Rad laboratories, (Hong Kong, China). Tyrosine kinase inhibitor osimertinib was obtained from ChemieTek (#CT-A9291) and CDK12 inhibitors AU-15506 (AU-CBB-15506) and AU-16770 (AU-CBB-16770) were provided by Aurigene Discovery Technologies Ltd. (Bengaluru, India). Mouse anti-EGFR mAb was obtained from BD Biosciences (#610017, East Rutherford. NJ, USA). Polyclonal or monoclonal antibodies to pEGFR-Y1068 (#2234), AKT (#4691), ERK (#4370), AMPK-alpha (#5831), ATR (#13934), ATM (#2873), RAD51 (#8875), CHK1 (#2360), CHK2 (#6334), pAKT (#4370, #4370), pERK (#4370), pACC (#11818), and pS6 (#4858) were obtained from Cell Signaling Technology (Danvers, MA, USA). Polyclonal or monoclonal antibodies to CDK12 (ABE1861), RNA Pol II [630849], pSer 2 RNA Pol II (MABE953), and pSer 5 RNA Pol II (MABE954) were obtained from Millipore-Sigma.
4.2. Cell Lines
Parental H1975 (#CRL-5908) and HCC827 (#CRL-5908) cell lines were purchased from the ATCC (Manassas, VA, USA), and the PC9 cell line was obtained from the Varmus Laboratory (MSKCC, New York, NY, USA). All human lung adenocarcinoma cells were maintained in RPMI supplemented with 10% FBS, 100 units/mL penicillin, and 100 μg/mL streptomycin. Jurkat cells procured from ATCC (#TIB-152) were cultured in RPMI with 10% FBS. Cells were authenticated by short tandem repeat (STR) profiling using the AmpFLSTR Identifiler kit (ThermoFisher, Waltham, MA, USA, #4322288) at the Protein Expression Laboratory (NCI, Frederick, Bethesda, MD, USA).
4.3. Generation of Osimertinib-Resistant Cell Lines
Initially, H1975, PC9, and HCC827 parental cells were seeded at low density (5 × 105/10 mL) in 10 cm Petri dishes containing RPMI-1640 medium, 10% FBS, and 1% penicillin/streptomycin. The following day, the medium was replaced with a fresh medium, supplemented with either 5 nM (HCC827) or 25 nM (H1975 and PC9) osimertinib, which led to a significant loss in viability within 2 days. After 3–4 days of growth, surviving cells were trypsinized and plated again at the same initial density and the next day, the medium was replaced with a fresh growth medium, supplemented with 2× (relative to the previous concentration) of osimertinib. This process continued until osimertinib concentration reached 2 mM. The newly generated osimertinib-resistant cells (H1975-osiR-NCI1, PC9-osiR-NCI1, HCC827-osiR-NCI1, and HCC827-osiR-NCI2) were then maintained at 2 mM osimertinib.
4.4. Cell Extract and Mouse Tissue Extract Preparation, Immunoprecipitation, and Immunoblot Analysis
Cells were washed with ice-cold PBS and lysed in RIPA buffer (150 mmol/L NaCl, 1.0% IGEPAL CA-630, 0.5% sodium deoxycholate, 0.1% SDS, and 50 mmol/L Tris, pH 8.0) supplemented with protease and phosphatase inhibitors. Tissue extracts were prepared using a TissueLyser (Qiagen, Hilden, Germany) following the manufacturer’s protocol. Protein concentrations were quantified using the BCA method (ThermoFisher). Cell lysates were combined with 4× SDS sample buffer (Millipore Sigma, Burlington, MA, USA), incubated at 95 °C for 5 min and fractionated by 4–15% polyacrylamide-SDS PAGE. Proteins were transferred to nitrocellulose membranes using a semidry transfer method (Bio-Rad) and probed with the specific antibodies as described in the figures.
4.5. RNA POL II Ser 2 (pS2) Phosphorylation Detection by Western Blot
For pS2 detection by Western blot, Jurkat cells were seeded at a density of 1 × 106 cells/mL in 5 mL of complete medium in a T25 flask. Cells were treated with the compounds for 6 h. After 6 h, cells were harvested and lysed with cell lysis buffer containing 50 mM HEPES (pH 7.4), 150 mM of sodium chloride, 5 mM of EDTA, and 1% of Triton ×100 (Sigma, Livonia, MI, USA), supplemented with protease and phosphatase inhibitor cocktails (Sigma, USA). Protein quantification was conducted using the BCA method (Thermo Scientific, Waltham, MA, USA, #23277). Total protein of 40 µg was resolved in 8% SDS-PAGE gel. Proteins were transferred onto PVDF-FL (Millipore # IPFL00010) membranes using the wet transfer method (30 V, 16 h). After protein transfer, PVDF-FL membranes were blocked using the Odyssey blocking buffer (Licor #927-50010) at room temperature for 1 h. pS2 RNA Pol II antibody at a dilution of 1:4000 (Bethyl # A300-654A) and β-actin primary antibody at a dilution of 1:20,000 (Santa Cruz #Ss-69879) were diluted in the Odyssey blocking buffer and added to the membrane and incubated over night at 4 °C on a rocker. The membrane was washed 3× times with TBST for 10 min each and incubated with Licor anti-rabbit (Licor #926-32211) and anti-mouse (Licor #926-68070) secondary antibodies at 1:10,000 dilution in the Odyssey blocking buffer at room temperature for 1 h. Membranes were washed 3× times with TBST and scanned using an LICOR Odyssey™ infrared scanner in the 800 and 680 channels. The raw data image file was stored in an LICOR proprietary format. The percent inhibition of pS2 in treated samples was calculated relative to the untreated controls. The data were analyzed using GraphPad Prism 6.0 (San Diego, CA, USA).
4.6. Cell Growth and Growth Inhibition Assays
Osimertinib-sensitive (H1975, PC9, and HCC827) or osimertinib-resistant (H1975-osiR-NCI1, PC9-osiR-NCI1, and HCC827-osiR-NCI1) cells (1 × 106) were seeded into 10 cm Petri dishes and grown in RPMI-1640 medium supplemented with 10% FBS, 1% penicillin/streptomycin, and 2 mM osimertinib for 3–4 days. Cells were trypsinized, replated in 10 cm Petri dishes, and grown in RPMI-1640, 10% FBS and 1% penicillin/streptomycin but without osimertinib prior to the assay. Approximately 1500 cells/90 mL were plated in 96-well plates with 4 wells for each treatment condition. Plates were incubated at 37 °C. For the cell growth assay, one set of cells was treated each day for 5 days with cell titer glow reagent (Promega), and luciferase luminescence (indicator of cell viability) was measured according to the manufacturer’s protocol. For the growth inhibition end-point assay, cells in 96-well plates were treated with target drugs as follows. Drugs were serially diluted from 100 mM to 0.001 mM in RPMI-1640 medium containing 1% DMSO, and 10 mL was added to cells (90 mL) for a final concentration of 10 mM to 0.0001 mM. Cells were then allowed to grow for 72 h before the cell viability assay using cell titer glow reagent (Promega, Madison, WI, USA) according to manufacturer’s protocol. Data were plotted in GraphPad Prism to generate the growth inhibition curves and to determine the inhibitory concentrations of drug (IC50) for 50% growth inhibition.
4.7. General Method for Kinase Screening
Screening of the compounds in the in-house kinome panel was conducted using time-resolved (TR) measurements of fluorescence with fluorescence resolution energy transfer (TR-FRET) assay and Kinase Glo assay formats. Out of 26 kinases, 20 kinases (ALK, c-SRC, FLT3, FLT4, JAK2, KDR, ZAP70, TRKA, Aurora A, INSR, PDGFR-β, AXL, MUSK, EGFR, FAK, c-MET, FGFR1, RON, ACK1, TIE2) were performed in TR-FRET assay and 6 kinases (IRAK4, GLK, PI3Kγ, GSK3β, PKCα, SGK1) were performed in Kinase Glo assay format. All the kinase assays were performed at their respective ATP Km concentration. Screening of the compounds was conducted at 0.1 µM and 1 µM and the percent inhibition was calculated.
4.8. Jurkat Cell Viability Assay
For Jurkat cell viability assay, cells were seeded at a density of 10,000 cell/well in 90 µL of complete RPMI media in 96-well U-bottom plate (Corning #CLS 3799) and incubated at 37 °C in a CO2 incubator until compound addition. The compounds AU-15506 and AU-16770 were diluted using half-log dilutions in DMSO starting from 5 mM, which served as 500× stock. Serial dilution was performed 9 times. From the 500× DMSO stock, 2 µL was diluted into 98 µL of media to obtain intermediate dilution (10× stock of desired concentration). From the intermediate dilution, 10 µL was added to respective wells in triplicate. The control wells were treated with 0.2% DMSO. The assay plate was incubated for 72 h at 37 °C in a CO2 incubator. After 72 h of incubation, the assay plate was centrifuged at 1500 rpm for 5 min at room temperature and 50 μL of media was removed from each well with a multichannel pipette, followed by the addition of 50 μL of XTT (Thermo Fisher Scientific #X6473) working solution. The assay plates were incubated in a CO2 incubator at 37 °C until color development. The assay plates were read at 465 nM in a plate reader (Molecular Devices, Spectromax M3). Percent inhibition of proliferation of compound-treated wells was compared to that in DMSO-treated wells. The data were analyzed using GraphPad Prism 6.0 (San Diego, CA, USA).
4.9. CDK12 and CDK7 Target Engagement Assay
Jurkat cells were cultured in RPMI media (Lonza 12-115F) with 10% FBS. For the target engagement assay, cells were seeded at a density of 1 × 106 cells/mL in 7 mL of complete medium in T25 flask. Cells were treated with the compounds for 6 h. After 6 h, cells were harvested and lysed with a cell lysis buffer containing 50 mM HEPES (pH 7.4), 150 mM of sodium chloride, 5 mM of EDTA, and 1% of Triton ×100 (Sigma, USA), supplemented with protease and phosphatase inhibitor cocktails (Sigma, USA). Protein quantification was conducted using the Pierce BCA assay kit (Thermo Scientific #23277) following the supplier’s protocol.
4.9.1. Plate Based CDK12 Target Engagement
In 96-well plates (Sigma #CLS-9018), 200 µg protein sample for each treatment was added to 100 µL volume of 1× HEPES lysis buffer. A total of 100 µL lysis buffer was used as a blank. To each sample, 1 µL of 100 mM DTT and 1 µM of Bio-THZ531 in DMSO were added. The samples were transferred to a plate shaker and incubated overnight at 4 °C. On the following day, the samples were incubated in shaker at RT for 4 h. Precoated streptavidin microplates (Thermo Fisher #15501) were equilibrated at RT for 15 min and washed 4 times with 200 µL of reagent diluent each time (Tris-buffered saline with 0.05% tween and 0.5% BSA). A total of 100 μL of each of the Bio-THZ531 incubated samples was transferred to the streptavidin-coated microplate and incubated for 2 h at RT on a plate shaker. The sample from the microplate was discarded and the plate was washed 4 times with 200 µL of reagent diluent each time. CDK12 primary antibody (CST #11973s) was diluted in reagent dilution 1:1000, and 100 µL of the antibody dilution was added to each well in the microplate. The plate was incubated overnight at 4 °C on a plate shaker. The diluted antibody from the microplate was discarded and the plate was washed 4 times with 200 µL of reagent diluent each time. A total 100 μL/well of HRP-labelled anti-rabbit antibody (Invitrogen #656120) (1:3000 dilution in reagent diluent) was added to the microplate and incubated for 2 h at room temperature on a plate shaker. The diluted antibody from the microplate was discarded and the plate was washed 5 times with 200 µL of reagent diluent each time. A total of 100 μL of TMB substrate (ThermoFisher #34028) was added to each well in the microplate and incubated at RT. The development of blue color was monitored, and the assay was stopped by adding 50 µL of 2N Sulfuric acid in each well. Absorbance was measured at 450 nm and 570 nm wave lengths on a plate reader (Molecular Devices Spectromax M3). The percentage of CDK12 occupancy was calculated for compound treatment samples with reference to the untreated samples. The untreated sample was considered to have an occupancy of 100%. The data were analyzed using GraphPad Prism 6.0 (San Diego, CA, USA).
4.9.2. Plate-Based CDK7 Target Engagement
A high-binding microplate (ThermoFisher #15501) was coated with 100 µL CDK7 antibody (Bethyl Labs #A300-405A) (1:500 dilution in PBS) in each well and incubated overnight at 4 °C. The following day, coating buffer was discarded, and the plate was washed 4 times with 300 µL reagent diluent (Tris-buffered saline with 0.05% tween and 0.5% BSA). A total of 200 µL of blocking buffer (Tris-buffered saline with 0.05% tween and 2.5% BSA) was added to the wells in the microplate and incubated at RT for 2 h on a plate shaker. The blocking solution was discarded, and the microplate was washed 4 times with 300 µL reagent diluent. A total of 200 µg of cell lysate from each treatment in 1× HEPES lysis buffer was added to the wells in the microplate. The microplate was incubated for 2 h at RT on a plate shaker. The samples in the plates were discarded and the microplate was washed 4 times with 300 µL reagent diluent. A total of 100 µL of 1 µM Bio-THZ1 was added to the wells in the microplate and incubated overnight at 4 °C. The next day, the microplate was incubated at RT for 4 h in a shaker. The Bio-THZ1 solution was discarded, and the microplate was washed 5 times with 300 µL reagent diluent. A total of 100 µL 1× streptavidin–HRP was added to the wells in the microplate and incubated at room temperature for 1 h on a plate shaker. After 1 h of incubation, the solution in the microplates was discarded and the wells were washed 7 times with 300 µL of reagent diluent and 100 µL of TMB substrate was added to the well and incubated at RT. The development of blue color was monitored, and the assay was stopped by adding 50 µL of 2N Sulfuric acid in each well. Absorbance was measured at 450 nm and 570 nm wave lengths on plate reader (Molecular Devices Spectromax M3). The percentage of CDK7 occupancy was calculated for compound treatment samples with reference to the untreated samples. The untreated sample was considered to have an occupancy of 100%. The data were analyzed using GraphPad Prism 6.0 (San Diego, CA, USA).
4.10. Cell-Cycle Analysis
PC9 and PC9-osiR-NCI1 cells were initially grown in an RPMI-1640 medium supplemented with 10% FBS, 1% penicillin/streptomycin, and 2 mM osimertinib. Following 3–4 days of growth, cells were trypsinized, replated in 10 cm Petri dishes and grown in drug-free RPMI-1640, 10% FBS, and 1% penicillin/streptomycin. Both groups of cells were treated with DMSO (vehicle), osimertinib (50 nM), or AU-16770 (100 nM) for 18 h. Cells were trypsinized, washed with PBS, fixed in 70% ethanol, washed twice in PBS, and approximately 5 × 105 cells/mL were incubated in 50 mg/mL propidium iodide/PBS and 100 units/mL RNase to label the DNA for 1 h before it was analyzed by FACS.
4.11. Quantitative PCR
PC9-osiR cells were grown in 60 cm plates (in triplicate) in a drug-free medium as described above. Cells were then treated with either DMSO (vehicle) or AU-16770 for 0 h, 8 h, 24 h, and 48 h, then lysed in an RLT buffer (Qiagen) containing 2-Mercaptoethanol. Total RNA was then purified using the RNeasy Mini kit (Qiagen) following the manufacturer’s protocol. A high-capacity RNA to cDNA kit (ThermoFisher) was used to synthesize random primed cDNA from 1.5 ug DNAse-treated RNA. Real-time PCR was conducted in 384-well plates using a ViiA7 Real-time PCR system (Applied Biosystems, Waltham, MA, USA). Singleplex reactions (10 μL) containing an FAM-MGB expression assay for the gene of interest (ATM Hs01112326_m1, ATR Hs00992138_m1, BRCA1 Hs00212914_m1, BRCA2 Hs01037421_m1, CHEK1 Hs00967510_g1, FANCI Hs01105312_m1, MDC1 Hs01029034_m1, NEK9 Hs00929599_m1, ORC3 Hs01031861_g1, RAD51D Hs00979545_g1, TERF2 Hs01030573_m1) or GAPDH endogenous control (ThermoFisher) were performed using cDNA synthesized from 20 ng of RNA and 1× Universal Master Mix (ThermoFisher—without Amp Erase UNG). The comparative Ct method (delta, delta Ct) was used to determine the relative expression normalized to GAPDH (Applied Biosystems® ViiA™ 7Real-Time PCR System Getting Started Guides). Samples were analyzed in quadruplicate, and values were expressed as the mean ± SE.
4.12. Copy Number Assay
Genomic DNA was isolated using the AllPrep DNA/RNA Mini kit (Qiagen) following the manufacturer’s protocol. Real-time PCR was conducted in 384-well plates using a ViiA7 Real-time PCR system (Applied Biosystems). Multiplex reactions (10 μL) containing an FAM-MGB copy number assay for ERBB2- (Hs00159103_cn) or MET (Hs01602615_cn) and a VIC TAMRA TERT copy number reference assay (ThermoFisher) were performed using 10 ng genomic DNA and 1x Universal Master Mix (ThermoFisher—without Amp Erase UNG). The comparative Ct method (delta, delta Ct) was used to determine the relative copy number normalized to TERT, relative to the parental cell line (Applied Biosystems® ViiA™ 7Real-Time PCR System Getting Started Guides). Samples were analyzed in quadruplicate, and values were expressed as the mean ± SD.
4.13. Xenograft Studies
All mice xenograft studies were conducted on 5–6-week-old female athymic nude mice (NSG) with approval from the NCI Animal Care and Use Committee (ACUC). Mice were obtained from NCI-Frederick and maintained in a pathogen-free facility at the NCI. H1975-osiR-NCI1 or PC9-osiR-NCI1 cells were cultured in RPMI (Millipore-Sigma) supplemented with 10% fetal bovine serum (Gemini) and 1% penicillin/streptomycin. All cell lines were then cultured in a humidified incubator with 5% CO2 at 37 °C. Cells were detached using 0.25% trypsin (ThermoFisher) and resuspended in PBS prior to implantation. Approximately 2 × 106 cells/0.1 mL/mouse were implanted subcutaneously. Between 7–10 days post-injection, mice were visually checked for pulpable tumor formation and treated (oral gavage) with 5 mg/day/kg body weight for another 12–14 days. Tumor growth was monitored twice weekly by bilateral caliper measurement and tumor volume was calculated using the length × diameter2 formula. Mice with tumor volume <200 mm3 were randomized into vehicle or treatment groups to ensure equal distribution across groups. However, for tumor tissue collection and lysate preparation, mice with xenografts were maintained with osimertinib until tumor size reached approximately 900 mm3 prior to drug treatment for 5 days. Vehicle (0.5% methyl cellulose), osimertinib (5 mg/kg/day), AU-15506 (5 mg/kg/day), or AU-16770 (5 mg/kg/day) were administered by oral gavage. AU-15506 or AU-16770 were prepared in a solution containing 10% PEG400, 10% D-ɑ-tocopheryl polyethylene glycol succinate (TPGS), and 0.4% Tween 80. Tumor size was measured twice weekly. Tumor volume for each mouse was converted to percent change based on a baseline volume of 100% on treatment day 0. Tumor growth inhibition was assessed by comparison of either the average tumor volume or mean changes in tumor volume for the control and treatment groups. Statistical significance was evaluated using a two-tailed Student’s t test.
4.14. In Vivo Preclinical Pharmacokinetic Analyses
Animal experimental procedures used in this study were approved by the Institutional Animal Ethical Committee based on the Committee for the Purpose of Control and Supervision on Experiments on Animals guidelines. Mice were used for the experiment after one week of acclimatization to standard laboratory conditions. Mice were fed with standard diet and water ad libitum.
Pharmacokinetic profiling was carried out using oral and intravenous route (IV) dosing. Three mice per route were administered with the test compound at indicated dose levels dissolved in specified vehicle (intravenous arm contains 2% v/v NMP + 30% v/v PEG + QS saline, oral arm contains 0.2% w/v tween 80 and 0.5% w/v methyl cellulose). Bolus IV dosing was performed in male CD1 mice, whereas oral dosing was conducted through oral gavage. The plasma samples were collected at designated time points and were frozen and stored at below −70 °C until analysis. The plasma samples were deproteinized with acetonitrile containing the internal standard, followed by centrifugation, and the supernatants were used for analysis. Quantitative bioanalysis of the AU-15506 and AU-16770 in the plasma samples was conducted using the fit-for-purpose LC-MS/MS method.
The PK data were expressed as mean ± standard deviation and PK parameters were determined using WinNonlin 8.1 software. The plasma concentration after injection (C0 min), the area under the concentration–time curve from time zero to 24 h (AUC0-t), Vdss, and CLtotal for after IV administration were obtained. The maximum plasma concentration (Cmax), time to maximum plasma concentration (Tmax), AUC0-t, and %F after oral administration were also obtained.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





