
Myeloid NGS Analyses of Paired Samples from Bone Marrow and Peripheral Blood Yield Concordant Results: A Prospective Cohort Analysis of the AGMT Study Group

 , , , , , and
, , , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
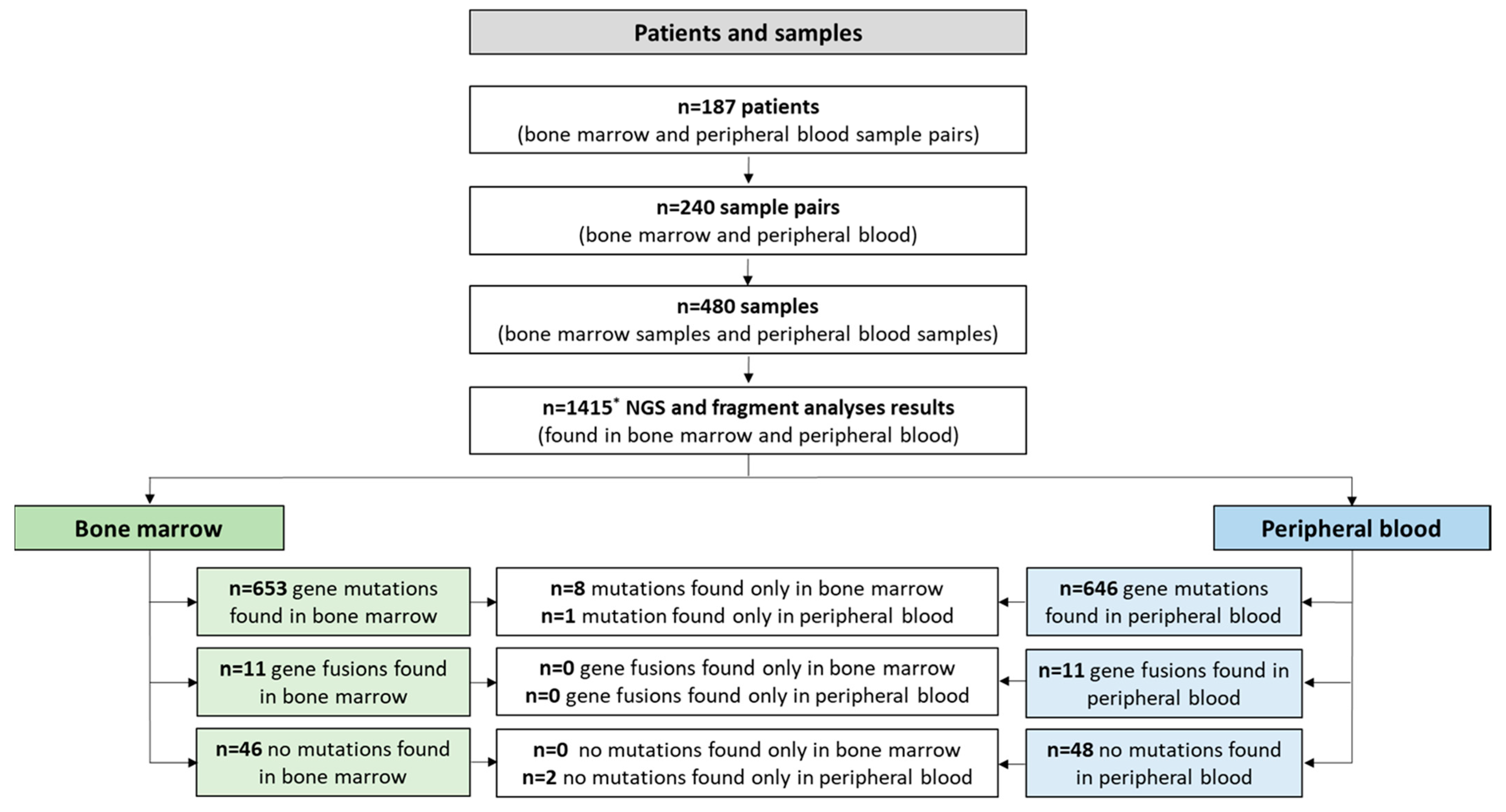
2.1. Patients and Cohort
2.2. Mutational Analyses
2.3. Bioinformatic Analyses
2.4. Statistical Analyses
3. Results
3.1. Patient Characteristics
3.2. Mutational Analyses—Overview

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patients with PB and BM Sample Pairs (n = 187) | Total no. of PB and BM Sample Pairs (n = 240) | |
|---|---|---|
| Mean days between PB and BM samples (SD) | 2.2 (10.5) | 2.2 (10.5) |
| Median (IQR) | 0.0 (0.0–0.0) | 0.0 (0.0–0.0) |
| Min–max | 0–118 | 0–118 |
| Unknown, n (%) | 0 (0.0) | 0 (0.0) |
| WHO 2016 Classification: MDS, n (%) | 43 (23.0) | 63 (26.3) |
| MDS/MPN | 15 (8.0) | 16 (6.7) |
| AML | 46 (24.6) | 72 (30) |
| MPN | 33 (17.6) | 38 (15.8) |
| Others 1 | 50 (26.7) | 51 (21.3) |
| Unknown | 0 (0.0) | 0 (0.0) |
| Mean age (SD), years | 66.1 (14.7) | 65.2 (14.8) |
| Median (IQR) | 70.0 (58.0–77.7) | 68.5 (57.5–76.3) |
| Min–max | 18–90 | 18–90 |
| Unknown | 0 (0.0) | 0 (0.0) |
| Sex: Female, n (%) | 87 (46.5) | 115 (47.9) |
| Male | 100 (53.5) | 125 (52.1) |
| Unknown | 0 (0.0) | 0 (0.0) |
| Treatment-related disease: No, n (%) | 170 (90.9) | 218 (90.8) |
| Yes | 17 (9.1) | 22 (9.2) |
| Unknown | 0 (0.0) | 0 (0.0) |
| Normal karyotype: No, n (%) | 53 (28.3) | 60 (25.0) |
| Yes | 108 (57.8) | 134 (55.8) |
| Unknown | 26 (13.9) | 46 (19.2) |
| Complex karyotype: No, n (%) | 148 (79.1) | 180 (75.0) |
| Yes | 13 (6.9) | 14 (5.8) |
| Unknown | 26 (13.9) | 46 (19.2) |
| Monosomal karyotype: No, n (%) | 155 (82.9) | 188 (78.3) |
| Yes | 6 (3.2) | 6 (2.5) |
| Unknown | 26 (13.9) | 46 (19.2) |
| Peripheral blood blasts, %: Mean (SD) | 5.8 (16.5) | 5.0 (14.9) |
| Median (IQR) | 0.0 (0.0–2.0) | 0.0 (0.0–1.0) |
| Min–max | 0.0–99.0 | 0.0–99.0 |
| Unknown, n (%) | 0 (0.0) | 3 (1.3) |
| Bone marrow blasts histology, %: Mean (SD) | 9.0 (18.9) | 10.1 (20.7) |
| Median (IQR) | 2.5 (2.5–2.5) | 2.5 (2.5–2.5) |
| Min–max | 0.0–95.0 | 0.0–95.0 |
| Unknown, n (%) | 21 (11.2) | 41 (17.2) |
| Bone marrow blasts aspirate, %: Mean (SD) | 12.7 (25.1) | 14.0 (25.9) |
| Median (IQR) | 2.0 (1.0–6.0) | 2.0 (1.0–8.0) |
| Min–max | 0.0–100.0 | 0.0–100.0 |
| Unknown, n (%) | 31 (16.6) | 43 (17.9) |
| White blood cell count, G/L: Mean (SD) | 13.9 (34.6) | 11.6 (30.9) |
| Median (IQR) | 4.8 (2.7–9.1) | 4.2 (2.2–8.7) |
| Min–max | 0.6–305.6 | 0.5–305.6 |
| Unknown, n (%) | 0 (0.0) | 0 (0.0) |
| Absolute neutrophil count, G/L: Mean (SD) | 7.9 (23.9) | 6.6 (21.3) |
| Median (IQR) | 2.7 (1.2–5.3) | 2.3 (0.8–4.8) |
| Min–max | 0.0–226.1 | 0.0–226.1 |
| Unknown, n (%) | 0 (0.0) | 0 (0.0) |
| Monocytes, %: Mean (SD) | 9.2 (9.6) | 9.6 (10.1) |
| Median (IQR) | 6.8 (3.0–12.0) | 7 (3.0–12.0) |
| Min–max | 0.0–72.0 | 0.0–72.0 |
| Unknown, n (%) | 0 (0.0) | 1 (0.4) |
| Lymphocytes, %: Mean (SD) | 27.7 (20.1) | 29.6 (21.18) |
| Median (IQR) | 23.0 (13.0–36.0) | 25.0 (13.8–40.0) |
| Min–max | 0.9–95.0 | 0.9–98.0 |
| Unknown, n (%) | 0 (0.0) | 1 (0.4) |
| Hemoglobin, g/dL: Mean (SD) | 106 (2.5) | 10.5 (2.4) |
| Median (IQR) | 10.3 (8.8–12.3) | 10.1 (8.8–12.2) |
| Min–max | 5.8–17.3 | 5.6–17.3 |
| Unknown, n (%) | 0 (0,0) | 0 (0.0) |
| Mean cell volume, fl: Mean (SD) | 92.7 (9.0) | 92.6 (9.1) |
| Median (IQR) | 91.4 (86.7–97.6) | 91.3 (86.3–97.6) |
| Min–max | 62.6–120.1 | 62.6–120.1 |
| Unknown, n (%) | 1 (0.5) | 1 (0.4) |
| Mean cell hemoglobin, pg: Mean (SD) | 31.7 (3.53) | 31.6 (3.5) |
| Median (IQR) | 31.3 (29.6–33.7) | 31.2 (29.5–33.6) |
| Min–max | 18.7–44.4 | 18.7–44.4 |
| Unknown, n (%) | 1 (0.5) | 1 (0.4) |
| Platelet count, G/L: Mean (SD) | 198.5 (234.1) | 191.3 (219.1) |
| Median (IQR) | 131.0 (58.0–223.0) | 132.0 (53.5–230.5) |
| Min–max | 6–1893 | 6–1893 |
| Unknown, n (%) | 0 (0,0) | 0 (0,0) |
| Ferritin, µg/L: Mean (SD) | 784.3 (1003.5) | 1030.7 (1607.6) |
| Median (IQR) | 412.0 (189.5–1001.5) | 467.5 (196.5–1382.0) |
| Min–max | 16–7212 | 11.0–1346 |
| Unknown, n (%) | 79 (42.2) | 116 (48.3) |
| Creatinine, mg/dL: Mean (SD) | 1.1 (0.83) | 1.0 (0.75) |
| Median (IQR) | 0.9 (0.7–1.1) | 0.9 (0.7–1.1) |
| Min–max | 0.3–9.5 | 0.3–9.5 |
| Unknown, n (%) | 8 (4.3) | 13 (5.4) |
| Bilirubin, mg/dL: Mean (SD) | 9.8 (0.9) | 0.7 (0.85) |
| Median (IQR) | 0.5 (0.4–0.8) | 0.5 (0.4–0.8) |
| Min–max | 0.1–8.0 | 0.1–8.0 |
| Unknown, n (%) | 9 (4.8) | 15 (6.3) |
3.3. Mutational Analyses—Concordance and Predictive Value
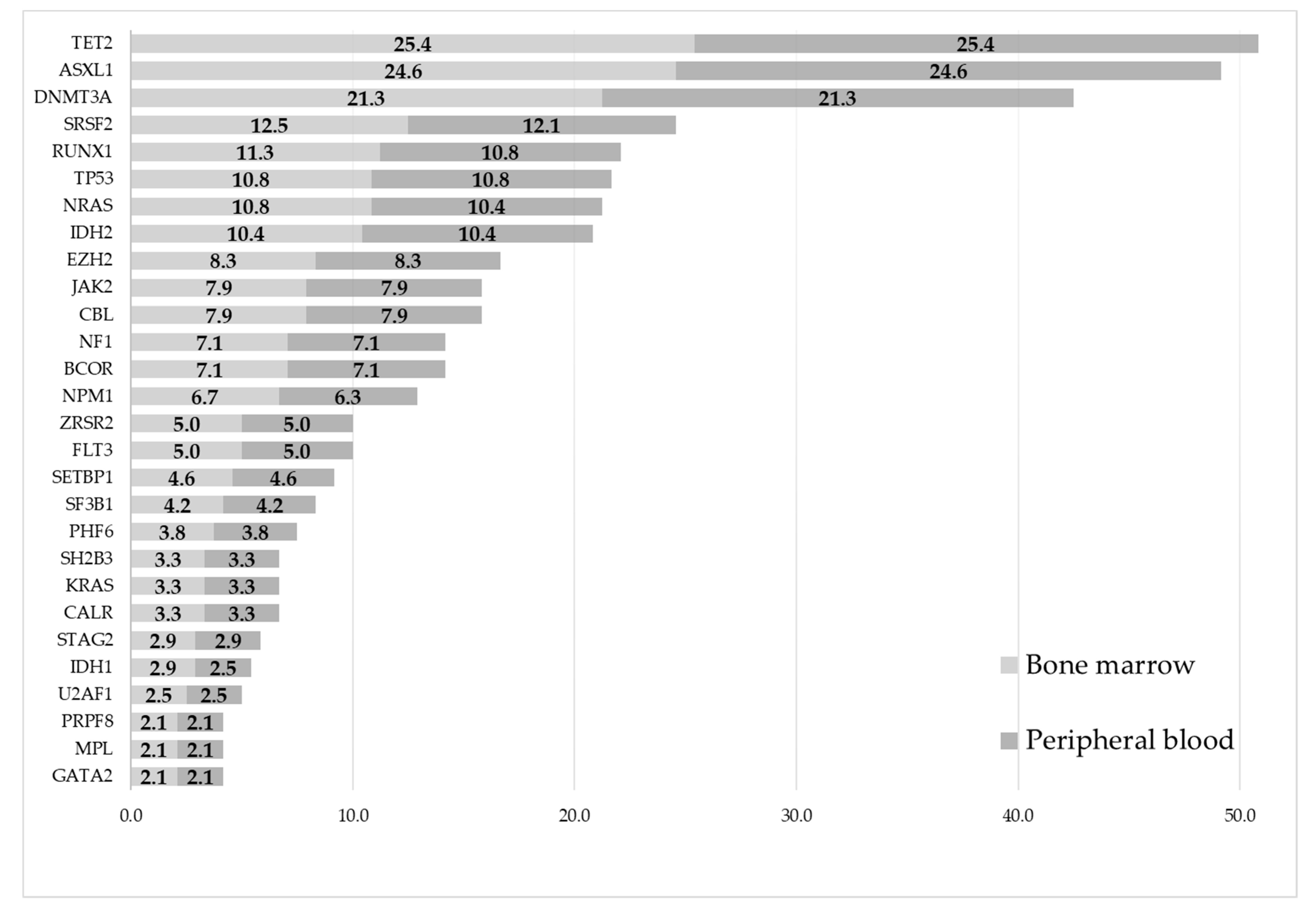
3.4. Mutational Analyses—Occurrence of Mutations
- Acute myeloid leukemia: DNMT3A (31.9% vs. 31.9%), NPM1 (19.4% vs. 18.1%), IDH2 (19.4% vs. 19.4%), TET2 (19.4% vs. 19.4%), and TP53 (15.3% vs. 15.3%) (Figure S1).
- Myelodysplastic neoplasms: TET2 (37.1% vs. 37.1%), ASXL1 (33.9% vs. 33.9%), DNMT3A (21.0% vs. 21.0%), TP53 (17.7% vs. 17.7%), and RUNX1 (17.7% vs. 17.7%) (Figure S2).
- Myelodysplastic/myeloproliferative overlap syndromes: ASXL1 (52.9% vs. 52.9%), TET2 (41.2% vs. 41.2%), SRSF2 (35.3% vs. 35.3%), NRAS (29.4% vs. 29.4%), and RUNX1 (23.5% vs. 23.5%) (Figure S3).
- Myeloproliferative neoplasms: ASXL1 (34.2% vs. 34.2%), TET2 (31.6% vs. 31.6%), JAK2 (31.6% vs. 31.6%), SRSF (23.7% vs. 21.1%), and CALR (21.1% vs. 21.1%) (Figure S4).
- Other (i.e., non-myeloid) diagnoses: DNMT3A (15.7% vs. 15.7%), ASXL1 (13.7% vs. 13.7%), TET2 (9.8% vs. 9.8%), MYD88 (7.8% vs. 7.8%), and CBL (7.8% vs. 7.8%) (Figure S5).

3.5. Mutational Analyses—Correlation of BMVAF vs. PBVAF

3.6. Mutational Analyses—Agreement
3.7. Discordant Mutations
3.8. Further Subanalyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 Revision of the World Health Organization Classification of Lymphoid Neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [Green Version]
- Pleyer, L.; Leisch, M.; Kourakli, A.; Padron, E.; Maciejewski, J.P.; Xicoy Cirici, B.; Kaivers, J.; Ungerstedt, J.; Heibl, S.; Patiou, P.; et al. Outcomes of Patients with Chronic Myelomonocytic Leukaemia Treated with Non-Curative Therapies: A Retrospective Cohort Study. Lancet Haematol. 2021, 8, e135–e148. [Google Scholar] [CrossRef] [PubMed]
- Pleyer, L.; Neureiter, D.; Faber, V.; Greil, R. Myelodysplastic Syndromes (MDS). In Chronic Myeloid Neoplasias and Clonal Overlap Syndromes; Greil, R., Pleyer, L., Faber, V., Neureiter, D., Eds.; Springer: Vienna, Austria, 2010; pp. 153–222. ISBN 978-3-211-79891-1. [Google Scholar]
- Valent, P.; Orazi, A.; Savona, M.R.; Patnaik, M.M.; Onida, F.; van de Loosdrecht, A.A.; Haase, D.; Haferlach, T.; Elena, C.; Pleyer, L.; et al. Proposed Diagnostic Criteria for Classical Chronic Myelomonocytic Leukemia (CMML), CMML Variants and Pre-CMML Conditions. Haematologica 2019, 104, 1935–1949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pleyer, L.; Döhner, H.; Dombret, H.; Seymour, J.; Schuh, A.; Beach, C.; Swern, A.; Burgstaller, S.; Stauder, R.; Girschikofsky, M.; et al. Azacitidine for Front-Line Therapy of Patients with AML: Reproducible Efficacy Established by Direct Comparison of International Phase 3 Trial Data with Registry Data from the Austrian Azacitidine Registry of the AGMT Study Group. IJMS 2017, 18, 415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pleyer, L.; Sekeres, M.A. An Early Glimpse at Azacitidine plus Venetoclax for Myelodysplastic Syndromes. Lancet Haematol. 2022, 9, e714–e716. [Google Scholar] [CrossRef]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th Edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.-M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating Morphologic, Clinical, and Genomic Data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef]
- Pleyer, L.; Neureiter, D.; Faber, V.; Greil, R. Chronic Myelomonocytic Leukemia (CMML). In Chronic Myeloid Neoplasias and Clonal Overlap Syndromes; Greil, R., Pleyer, L., Faber, V., Neureiter, D., Eds.; Springer: Vienna, Austria, 2010; pp. 223–233. ISBN 978-3-211-79891-1. [Google Scholar]
- Schuh, A.C.; Döhner, H.; Pleyer, L.; Seymour, J.F.; Fenaux, P.; Dombret, H. Azacitidine in Adult Patients with Acute Myeloid Leukemia. Crit. Rev. Oncol./Hematol. 2017, 116, 159–177. [Google Scholar] [CrossRef]
- Itzykson, R.; Fenaux, P.; Bowen, D.; Cross, N.C.P.; Cortes, J.; De Witte, T.; Germing, U.; Onida, F.; Padron, E.; Platzbecker, U.; et al. Diagnosis and Treatment of Chronic Myelomonocytic Leukemias in Adults: Recommendations From the European Hematology Association and the European LeukemiaNet. HemaSphere 2018, 2, e150. [Google Scholar] [CrossRef]
- Leisch, M.; Jansko, B.; Zaborsky, N.; Greil, R.; Pleyer, L. Next Generation Sequencing in AML—On the Way to Becoming a New Standard for Treatment Initiation and/or Modulation? Cancers 2019, 11, 252. [Google Scholar] [CrossRef] [Green Version]
- Gangat, N.; Mudireddy, M.; Lasho, T.L.; Finke, C.M.; Nicolosi, M.; Szuber, N.; Patnaik, M.M.; Pardanani, A.; Hanson, C.A.; Ketterling, R.P.; et al. Mutations and Prognosis in Myelodysplastic Syndromes: Karyotype-Adjusted Analysis of Targeted Sequencing in 300 Consecutive Cases and Development of a Genetic Risk Model. Am. J. Hematol. 2018, 93, 691–697. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Lasho, T.L.; Patnaik, M.M.; Saeed, L.; Mudireddy, M.; Idossa, D.; Finke, C.; Ketterling, R.P.; Pardanani, A.; Gangat, N. Targeted Next-Generation Sequencing in Myelodysplastic Syndromes and Prognostic Interaction between Mutations and IPSS-R. Am. J. Hematol. 2017, 92, 1311–1317. [Google Scholar] [CrossRef] [Green Version]
- Yun, S.; Geyer, S.M.; Komrokji, R.S.; Al Ali, N.H.; Song, J.; Hussaini, M.; Sweet, K.L.; Lancet, J.E.; List, A.F.; Padron, E.; et al. Prognostic Significance of Serial Molecular Annotation in Myelodysplastic Syndromes (MDS) and Secondary Acute Myeloid Leukemia (SAML). Leukemia 2021, 35, 1145–1155. [Google Scholar] [CrossRef] [PubMed]
- Reinig, E.; Yang, F.; Traer, E.; Arora, R.; Brown, S.; Rattray, R.; Braziel, R.; Fan, G.; Press, R.; Dunlap, J. Targeted Next-Generation Sequencing in Myelodysplastic Syndrome and Chronic Myelomonocytic Leukemia Aids Diagnosis in Challenging Cases and Identifies Frequent Spliceosome Mutations in Transformed Acute Myeloid Leukemia. Am. J. Clin. Pathol. 2016, 145, 497–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maggioni, G.; Della Porta, M.G. Molecular Landscape of Myelodysplastic Neoplasms in Disease Classification and Prognostication. Curr. Opin. Hematol. 2023, 30, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Elena, C.; Gallì, A.; Such, E.; Meggendorfer, M.; Germing, U.; Rizzo, E.; Cervera, J.; Molteni, E.; Fasan, A.; Schuler, E.; et al. Integrating Clinical Features and Genetic Lesions in the Risk Assessment of Patients with Chronic Myelomonocytic Leukemia. Blood 2016, 128, 1408–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Chen, E.C.; Stahl, M.; Zeidan, A.M. Prognostication in Myelodysplastic Syndromes (Neoplasms): Molecular Risk Stratification Finally Coming of Age. Blood Rev. 2022, 5, 101033. [Google Scholar] [CrossRef] [PubMed]
- Duncavage, E.J.; Bagg, A.; Hasserjian, R.P.; DiNardo, C.D.; Godley, L.A.; Iacobucci, I.; Jaiswal, S.; Malcovati, L.; Vannucchi, A.M.; Patel, K.P.; et al. Genomic Profiling for Clinical Decision Making in Myeloid Neoplasms and Acute Leukemia. Blood 2022, 140, 2228–2247. [Google Scholar] [CrossRef]
- Greenberg, P.L.; Tuechler, H.; Schanz, J.; Sanz, G.; Garcia-Manero, G.; Solé, F.; Bennett, J.M.; Bowen, D.; Fenaux, P.; Dreyfus, F.; et al. Revised International Prognostic Scoring System for Myelodysplastic Syndromes. Blood 2012, 120, 2454–2465. [Google Scholar] [CrossRef] [Green Version]
- Döhner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and Management of AML in Adults: 2022 Recommendations from an International Expert Panel on Behalf of the ELN. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and Management of AML in Adults: 2017 ELN Recommendations from an International Expert Panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheson, B.D. Clinical Application and Proposal for Modification of the International Working Group (IWG) Response Criteria in Myelodysplasia. Blood 2006, 108, 419–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheson, B.D.; Bennett, J.M.; Kopecky, K.J.; Büchner, T.; Willman, C.L.; Estey, E.H.; Schiffer, C.A.; Doehner, H.; Tallman, M.S.; Lister, T.A.; et al. Revised Recommendations of the International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia. JCO 2003, 21, 4642–4649. [Google Scholar] [CrossRef]
- Savona, M.R.; Malcovati, L.; Komrokji, R.; Tiu, R.V.; Mughal, T.I.; Orazi, A.; Kiladjian, J.-J.; Padron, E.; Solary, E.; Tibes, R.; et al. An International Consortium Proposal of Uniform Response Criteria for Myelodysplastic/Myeloproliferative Neoplasms (MDS/MPN) in Adults. Blood 2015, 125, 1857–1865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeidan, A.M.; Platzbecker, U.; Bewersdorf, J.P.; Stahl, M.; Adès, L.; Borate, U.; Bowen, D.T.; Buckstein, R.J.; Brunner, A.M.; Carraway, H.E.; et al. Consensus Proposal for Revised International Working Group Response Criteria for Higher Risk Myelodysplastic Syndromes. Blood, 2023; Online ahead of print. [Google Scholar] [CrossRef]
- Tazi, Y.; Arango-Ossa, J.E.; Zhou, Y.; Bernard, E.; Thomas, I.; Gilkes, A.; Freeman, S.; Pradat, Y.; Johnson, S.J.; Hills, R.; et al. Unified Classification and Risk-Stratification in Acute Myeloid Leukemia. Nat. Commun. 2022, 13, 4622. [Google Scholar] [CrossRef] [PubMed]
- Nannya, Y.; Tobiasson, M.; Sato, S.; Bernard, E.; Ohtake, S.; Takeda, J.; Creignou, M.; Zhao, L.; Kusakabe, M.; Shibata, Y.; et al. Post-Azacitidine Clone Size Predicts Outcome of Patients with Myelodysplastic Syndromes and Related Myeloid Neoplasms. Blood Adv. 2023; Online ahead of print. [Google Scholar] [CrossRef]
- Welch, J.S.; Petti, A.A.; Miller, C.A.; Fronick, C.C.; O’Laughlin, M.; Fulton, R.S.; Wilson, R.K.; Baty, J.D.; Duncavage, E.J.; Tandon, B.; et al. TP53 and Decitabine in Acute Myeloid Leukemia and Myelodysplastic Syndromes. N. Engl. J. Med. 2016, 375, 2023–2036. [Google Scholar] [CrossRef]
- Bernard, E.; Tuechler, H.; Greenberg, P.L.; Hasserjian, R.P.; Arango Ossa, J.E.; Nannya, Y.; Devlin, S.M.; Creignou, M.; Pinel, P.; Monnier, L.; et al. Molecular International Prognostic Scoring System for Myelodysplastic Syndromes. NEJM Evid. 2022, 1. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Dinmohamed, A.G.; van Norden, Y.; Visser, O.; Posthuma, E.F.M.; Huijgens, P.C.; Sonneveld, P.; van de Loosdrecht, A.A.; Jongen-Lavrencic, M. The Use of Medical Claims to Assess Incidence, Diagnostic Procedures and Initial Treatment of Myelodysplastic Syndromes and Chronic Myelomonocytic Leukemia in the Netherlands. Leuk. Res. 2015, 39, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Pleyer, L.; Pfeilstocker, M.; Stauder, R.; Heibl, S.; Sill, H.; Girschikofsky, M.; Stampfl-Mattesberger, M.; Tinchon, C.; Hartmann, B.; Petzer, A.; et al. Peripheral Blood Complete Remission Provides Added Value to the Classical Definition of Morphologic Complete Remission—A Prospective Cohort Study of 1441 Patients with MDS, CMML and AML Treated within the Austrian Azacitidine Registry. Blood 2021, 138, 3387. [Google Scholar] [CrossRef]
- Pleyer, L.; Pfeilstocker, M.; Stauder, R.; Heibl, S.; Sill, H.; Girschikofsky, M.; Stampfl-Mattersberger, M.; Tinchon, C.; Petzer, A.; Schmitt, C.A.; et al. Expanding on Current Definitions of Hematologic Improvement in MDS, CMML and AML: Landmark Analyses of 1301 Patients Treated with Azacitidine in the Austrian Registry of Hypomethylating Agents By the AGMT-Study Group. Blood 2019, 134, 3821. [Google Scholar] [CrossRef]
- van Lom, K.; Hagemeijer, A.; Smit, E.; Hählen, K.; Groeneveld, K.; Löwenberg, B. Cytogenetic Clonality Analysis in Myelodysplastic Syndrome: Monosomy 7 Can Be Demonstrated in the Myeloid and in the Lymphoid Lineage. Leukemia 1995, 9, 1818–1821. [Google Scholar] [PubMed]
- Asadi Fakhr, Z.; Mehrzad, V.; Izaditabar, A.; Salehi, M. Evaluation of the Utility of Peripheral Blood vs Bone Marrow in Karyotype and Fluorescence in Situ Hybridization for Myelodysplastic Syndrome Diagnosis. J. Clin. Lab. Anal. 2018, 32, e22586. [Google Scholar] [CrossRef] [Green Version]
- Braulke, F.; Jung, K.; Schanz, J.; Götze, K.; Müller-Thomas, C.; Platzbecker, U.; Germing, U.; Brümmendorf, T.H.; Bug, G.; Ottmann, O.; et al. Molecular Cytogenetic Monitoring from CD34+ Peripheral Blood Cells in Myelodysplastic Syndromes: First Results from a Prospective Multicenter German Diagnostic Study. Leuk. Res. 2013, 37, 900–906. [Google Scholar] [CrossRef]
- Cherry, A.M.; Slovak, M.L.; Campbell, L.J.; Chun, K.; Eclache, V.; Haase, D.; Haferlach, C.; Hildebrandt, B.; Iqbal, A.M.; Jhanwar, S.C.; et al. Will a Peripheral Blood (PB) Sample Yield the Same Diagnostic and Prognostic Cytogenetic Data as the Concomitant Bone Marrow (BM) in Myelodysplasia? Leuk. Res. 2012, 36, 832–840. [Google Scholar] [CrossRef]
- Lucas, F.; Michaels, P.D.; Wang, D.; Kim, A.S. Mutational Analysis of Hematologic Neoplasms in 164 Paired Peripheral Blood and Bone Marrow Samples by Next-Generation Sequencing. Blood Adv. 2020, 4, 4362–4365. [Google Scholar] [CrossRef]
- Ruan, M.; Liu, L.; Qi, B.; Chen, X.; Chang, L.; Zhang, A.; Liu, F.; Wang, S.; Liu, X.; Chen, X.; et al. Targeted Next-Generation Sequencing of Circulating Tumor DNA, Bone Marrow, and Peripheral Blood Mononuclear Cells in Pediatric AML. Front. Oncol. 2021, 11, 666470. [Google Scholar] [CrossRef]
- Jumniensuk, C.; Nobori, A.; Lee, T.; Senaratne, T.N.; Rao, D.; Pullarkat, S. Concordance of Peripheral Blood and Bone Marrow Next-Generation Sequencing in Hematologic Neoplasms. Adv. Hematol. 2022, 2022, 8091746. [Google Scholar] [CrossRef]
- Stasik, S.; Burkhard-Meier, C.; Kramer, M.; Middeke, J.M.; Oelschlaegel, U.; Sockel, K.; Ehninger, G.; Serve, H.; Müller-Tidow, C.; Baldus, C.D.; et al. Deep Sequencing in CD34+ Cells from Peripheral Blood Enables Sensitive Detection of Measurable Residual Disease in AML. Blood Adv. 2022, 6, 3294–3303. [Google Scholar] [CrossRef] [PubMed]
- Muffly, L.; Sundaram, V.; Chen, C.; Yurkiewicz, I.; Kuo, E.; Burnash, S.; Spiegel, J.Y.; Arai, S.; Frank, M.J.; Johnston, L.J.; et al. Concordance of Peripheral Blood and Bone Marrow Measurable Residual Disease in Adult Acute Lymphoblastic Leukemia. Blood Adv. 2021, 5, 3147–3151. [Google Scholar] [CrossRef] [PubMed]
- Fries, C.; Adlowitz, D.G.; Spence, J.M.; Spence, J.P.; Rock, P.J.; Burack, W.R. Acute Lymphoblastic Leukemia Clonal Distribution between Bone Marrow and Peripheral Blood. Pediatr. Blood Cancer 2020, 67, e28280. [Google Scholar] [CrossRef] [PubMed]
- Mohamedali, A.M.; Gäken, J.; Ahmed, M.; Malik, F.; Smith, A.E.; Best, S.; Mian, S.; Gaymes, T.; Ireland, R.; Kulasekararaj, A.G.; et al. High Concordance of Genomic and Cytogenetic Aberrations between Peripheral Blood and Bone Marrow in Myelodysplastic Syndrome (MDS). Leukemia 2015, 29, 1928–1938. [Google Scholar] [CrossRef] [PubMed]
- Godwin, C.D.; Zhou, Y.; Othus, M.; Asmuth, M.M.; Shaw, C.M.; Gardner, K.M.; Wood, B.L.; Walter, R.B.; Estey, E.H. Acute Myeloid Leukemia Measurable Residual Disease Detection by Flow Cytometry in Peripheral Blood vs Bone Marrow. Blood 2021, 137, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Coleman, J.F.; Theil, K.S.; Tubbs, R.R.; Cook, J.R. Diagnostic Yield of Bone Marrow and Peripheral Blood FISH Panel Testing in Clinically Suspected Myelodysplastic Syndromes and/or Acute Myeloid Leukemia: A Prospective Analysis of 433 Cases. Am. J. Clin. Pathol. 2011, 135, 915–920. [Google Scholar] [CrossRef] [PubMed]
- Leisch, M.; Pfeilstöcker, M.; Stauder, R.; Heibl, S.; Sill, H.; Girschikofsky, M.; Stampfl-Mattersberger, M.; Tinchon, C.; Hartmann, B.; Petzer, A.; et al. Adverse Events in 1406 Patients Receiving 13,780 Cycles of Azacitidine within the Austrian Registry of Hypomethylating Agents—A Prospective Cohort Study of the AGMT Study-Group. Cancers 2022, 14, 2459. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows–Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and Accurate Long-Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve Years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative Genomics Viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schober, P.; Boer, C.; Schwarte, L.A. Correlation Coefficients: Appropriate Use and Interpretation. Anesth. Analg. 2018, 126, 1763–1768. [Google Scholar] [CrossRef]
- Bland, J.M.; Altman, D.G. Statistical Methods for Assessing Agreement between Two Methods of Clinical Measurement. Lancet 1986, 1, 307–310. [Google Scholar] [CrossRef]
- Pleyer, L.; Vaisband, M.; Pfeilstocker, M.; Stauder, R.; Heibl, S.; Sill, H.; Girschikofsky, M.; Stampf-Mattersberger, M.; Tinchon, C.; Hartmann, B.; et al. Cox Proportional Hazards Deep Neural Network Identifies Peripheral Blood Complete Remission (PB-CR) to Be at Least Equivalent to Morphologic CR in Predicting Outcomes of Patients Treated with Azacitidine—A Prospective Cohort Study by the AGMT; ASH Oral: New Orleans, LA, USA, 2022. [Google Scholar]
- Sargas, C.; Ayala, R.; Larráyoz, M.J.; Chillón, M.C.; Carrillo-Cruz, E.; Bilbao-Sieyro, C.; Prados de la Torre, E.; Martínez-Cuadrón, D.; Rodríguez-Veiga, R.; Boluda, B.; et al. Molecular Landscape and Validation of New Genomic Classification in 2668 Adult AML Patients: Real Life Data from the PETHEMA Registry. Cancers 2023, 15, 438. [Google Scholar] [CrossRef] [PubMed]
- Kayser, S.; Levis, M.J. The Clinical Impact of the Molecular Landscape of Acute Myeloid Leukemia. Haematologica 2023, 108, 308–320. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S. Genetics of MDS. Blood 2019, 133, 1049–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiereghin, C.; Travaglino, E.; Zampini, M.; Saba, E.; Saitta, C.; Riva, E.; Bersanelli, M.; Della Porta, M.G. The Genetics of Myelodysplastic Syndromes: Clinical Relevance. Genes 2021, 12, 1144. [Google Scholar] [CrossRef]
- Prassek, V.V.; Rothenberg-Thurley, M.; Sauerland, M.C.; Herold, T.; Janke, H.; Ksienzyk, B.; Konstandin, N.P.; Goerlich, D.; Krug, U.; Faldum, A.; et al. Genetics of Acute Myeloid Leukemia in the Elderly: Mutation Spectrum and Clinical Impact in Intensively Treated Patients Aged 75 Years or Older. Haematologica 2018, 103, 1853–1861. [Google Scholar] [CrossRef] [Green Version]
- Sexauer, A.; Perl, A.; Yang, X.; Borowitz, M.; Gocke, C.; Rajkhowa, T.; Thiede, C.; Frattini, M.; Nybakken, G.E.; Pratz, K.; et al. Terminal Myeloid Differentiation in Vivo Is Induced by FLT3 Inhibition in FLT3/ITD AML. Blood 2012, 120, 4205–4214. [Google Scholar] [CrossRef] [Green Version]
- Kiladjian, J.-J.; Bourgeois, E.; Lobe, I.; Braun, T.; Visentin, G.; Bourhis, J.-H.; Fenaux, P.; Chouaib, S.; Caignard, A. Cytolytic Function and Survival of Natural Killer Cells Are Severely Altered in Myelodysplastic Syndromes. Leukemia 2006, 20, 463–470. [Google Scholar] [CrossRef] [Green Version]
- Miura, I.; Kobayashi, Y.; Takahashi, N.; Saitoh, K.; Miura, A.B. Involvement of Natural Killer Cells in Patients with Myelodysplastic Syndrome Carrying Monosomy 7 Revealed by the Application of Fluorescence in Situ Hybridization to Cells Collected by Means of Fluorescence-Activated Cell Sorting: Short Report. Br. J. Haematol. 2000, 110, 876–879. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, T.; Koike, K.; Agematsu, K.; Itoh, S.; Hagimoto, R.; Kitazawa, Y.; Higuchi, T.; Sawai, N.; Matsui, H.; Komiyama, A. Cytogenetic Clonality Analysis in Monosomy 7 Associated with Juvenile Myelomonocytic Leukemia: Clonality in B and NK Cells, but Not in T Cells. Leuk. Res. 1998, 22, 887–892. [Google Scholar] [CrossRef] [PubMed]
- Meers, S.; Vandenberghe, P.; Boogaerts, M.; Verhoef, G.; Delforge, M. The Clinical Significance of Activated Lymphocytes in Patients with Myelodysplastic Syndromes: A Single Centre Study of 131 Patients. Leuk. Res. 2008, 32, 1026–1035. [Google Scholar] [CrossRef]
- Ma, L.; Delforge, M.; Van Duppen, V.; Verhoef, G.; Emanuel, B.; Boogaerts, M.; Hagemeijer, A.; Vandenberghe, P. Circulating Myeloid and Lymphoid Precursor Dendritic Cells Are Clonally Involved in Myelodysplastic Syndromes. Leukemia 2004, 18, 1451–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohty, M.; Jarrossay, D.; Lafage-Pochitaloff, M.; Zandotti, C.; Brière, F.; de Lamballeri, X.-N.; Isnardon, D.; Sainty, D.; Olive, D.; Gaugler, B. Circulating Blood Dendritic Cells from Myeloid Leukemia Patients Display Quantitative and Cytogenetic Abnormalities as Well as Functional Impairment. Blood 2001, 98, 3750–3756. [Google Scholar] [CrossRef] [Green Version]
- Matteo Rigolin, G.; Howard, J.; Buggins, A.; Sneddon, C.; Castoldi, G.; Hirst, W.J.R.; Mufti, G.J. Phenotypic and Functional Characteristics of Monocyte-Derived Dendritic Cells from Patients with Myelodysplastic Syndromes: Monocyte-Derived Dendritic Cells in Myelodysplastic Syndromes. Br. J. Haematol. 1999, 107, 844–850. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, L.; Astrand-Grundström, I.; Arvidsson, I.; Jacobsson, B.; Hellström-Lindberg, E.; Hast, R.; Jacobsen, S.E. Isolation and Characterization of Hematopoietic Progenitor/Stem Cells in 5q-Deleted Myelodysplastic Syndromes: Evidence for Involvement at the Hematopoietic Stem Cell Level. Blood 2000, 96, 2012–2021. [Google Scholar] [CrossRef] [PubMed]
- Thanopoulou, E.; Cashman, J.; Kakagianne, T.; Eaves, A.; Zoumbos, N.; Eaves, C. Engraftment of NOD/SCID-Beta2 Microglobulin Null Mice with Multilineage Neoplastic Cells from Patients with Myelodysplastic Syndrome. Blood 2004, 103, 4285–4293. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, H.J.; Broudy, V.C.; Magenis, R.E.; Olson, S.; Tomar, D.; Barton, S.; Fitchen, J.H.; Bagby, G.C. Cytogenetic Evidence for Involvement of B Lymphocytes in Acquired Idiopathic Sideroblastic Anemias. Blood 1987, 70, 1003–1005. [Google Scholar] [CrossRef] [Green Version]
- White, N.J.; Nacheva, E.; Asimakopoulos, F.A.; Bloxham, D.; Paul, B.; Green, A.R. Deletion of Chromosome 20q in Myelodysplasia Can Occur in a Multipotent Precursor of Both Myeloid Cells and B Cells. Blood 1994, 83, 2809–2816. [Google Scholar] [CrossRef]
- Sasaki, K.; Kanagal-Shamanna, R.; Montalban-Bravo, G.; Assi, R.; Jabbour, E.; Ravandi, F.; Kadia, T.; Pierce, S.; Takahashi, K.; Nogueras Gonzalez, G.; et al. Impact of the Variant Allele Frequency of ASXL1, DNMT3A, JAK2, TET2, TP53, and NPM1 on the Outcomes of Patients with Newly Diagnosed Acute Myeloid Leukemia. Cancer 2020, 126, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal Hematopoiesis of Indeterminate Potential and Its Distinction from Myelodysplastic Syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]




| First Author | Pts, n | Paired Samples, n | Disease, n/n (%) | Method | Company, kit | Days between BM-PB Analyses, Mean (min–max) | Concordance between Paired Samples (BM and PB) | Concordance between Paired Mutations (BM and PB) | Coefficient |
|---|---|---|---|---|---|---|---|---|---|
| Jansko-Gadermeir B. [current manuscript] | 187 | 240 | AML, 46/187 (24.6%) MDS, 43/187 (23.0%) MDS/MPN, 15/187 (8.0%) MPN, 33/187 (17.6%) Others, 50/187 (26.7%) | NGS | Illumina® AmpliSeq™ myeloid panel (40 genes, 29 driver fusion genes) Leukostrat Invivoscribe 2.0 (FLT3-ITD/TKD) | 2 (0–118) | Complete concordance: 231/240 (96%) Partial concordance: 7/240 (3%) | Concordance: 702/711 (99.7%) | r = 0.93 p < 0.0001 |
| Jumniensuk C. [42] | 163 | 163 | Cytopenia, 54/163 (33%) NHL, 31/163 (19%) AML, 23/163 (14%) MDS, 53/163 (13%) MPN, 21/163 (13%) MDS/MPN, 11/163 (7%) Others, 2/163 (1%) | NGS | Illumina® TruSight (54 genes) | 63 (0–334) | Complete concordance: 124/163 (76%) Partial concordance: 26/163 (16%) | Concordance: not given | κ = 0·79 p < 0·0001 |
| Stasik S. [43] | 29 | 35 | MDS, 2/40 (5%) AML, 38/40 (95%) | NGS (CD34+ MRD) | Life Technologies custom panel (4 genes) | Not reported | Complete concordance: not given Partial concordance: not given | Concordance: not given | r = 0·90 p <0·0001 |
| Muffly L. [44] | 62 | 126 | T-ALL, 8/62 (13%) B-ALL, 54/62 (87%) | NGS (MRD) | Adaptive Biotechnologies clonoSEQ assay (TCR rearrangement) | Not reported | Complete concordance: 112/126 (89%) Partial concordance: not applicable | Concordance: not given | r = 0·87 p <0·0001 |
| Ruan M. [41] | 20 | 20 | Pediatric AML, 20/20 (100%) | NGS | AcornMed Biotechnology customized Gene Panel (137 genes) | Not reported | Complete concordance: 155/209 (74%) Partial concordance: not given | Concordance: 155/239 (74%) | r = 0·95 p < 0·001 |
| Lucas F. [40] | 164 | 164 | Myeloid neoplasias, 129/164 (79%) Lymphoid neoplasm, 32/164 (20%) MPAL, 3/126 (1.8%) | NGS | Rapid Heme Panel (95 genes) | 2 (0–14) | Complete concordance: 130/164 (79%) Partial concordance: not given | Concordance: 278/329 (84.5%) | Not given |
| Fries C. [45] | 16 | 16 | B-ALL, 16/16 (100%) | NGS | IGH Vh-DJh rearrangement | Not reported | Complete concordance: 11/16 (69%) Partial concordance: 4/16 (25%) | Concordance: 23/28 (82.1%) 2 | Not given |
| Mohmedali A.M. [46] | 183 | 183 | MDS, 183/183 (100%) | NGS | Illumina custom panel (24 genes) | Not reported | Complete concordance: 177/183 (97%) Partial concordance: not given | Concordance: 234/240 (97.5%) | Not given |
| Peripheral blood | ||||
|---|---|---|---|---|
| Positive | Negative | Total | ||
| Bone marrow | Positive | 656 | 8 | 664 |
| Negative | 1 | 1840 | 1841 | |
| Total | 657 | 1848 | 2505 | |
| ID | Sex | Age at Initial Diagnosis | Initial Diagnosis | BM Blasts, % | PB Blasts, % | WBC, G/L | Mutations Detected in BM, n | Mutations Detected in PB, n | Discordant Mutation | Pathway | VAF in BM, % | VAF in PB, % |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | f | 55 | AML | 1 | 0 | 4.6 | 2 | 1 | NPM1 | Nucleolar multifunctional protein | 0.6 | Not found |
| 2 | f | 84 | AML | 87 | 1 | 12.5 | 6 | 7 | ASLX1 | DNA methylation related | Not found | 1.0 |
| 3 | f | 83 | MDS | 2.5 | 0 | 5.2 | 6 | 5 | SETBP1 | DNA replication | 1.1 | Not found |
| 4 | f | 81 | AML | 19 | 2 | 1.7 | 5 | 4 | NRAS | RAS pathway | 1.3 | Not found |
| 5 | f | 79 | MDS | 3 | not done | 1.5 | 2 | 1 | RB1 | Tumor suppressor | 1.9 | Not found |
| 6 | f | 75 | MPN | 2.5 | 2 | 3.6 | 4 | 3 | SRSF2 | Splicing factor | 2.2 | Not found |
| 7 | f | 70 | MDS | 8 | 0 | 2.5 | 1 | 0 | ASLX1 | DNA methylation related | 2.5 | Not found |
| 8 | f | 76 | AML | 40 | 0 | 3.1 | 9 | 8 | RUNX1 | Transcription factor | 3.7 | Not found |
| 9 | f | 70 | Waldenstrom’s disease | 2.5 | 0 | 6.0 | 1 | 0 | IDH1 | DNA methylation related | 9.1 | Not found |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jansko-Gadermeir, B.; Leisch, M.; Gassner, F.J.; Zaborsky, N.; Dillinger, T.; Hutter, S.; Risch, A.; Melchardt, T.; Egle, A.; Drost, M.; et al. Myeloid NGS Analyses of Paired Samples from Bone Marrow and Peripheral Blood Yield Concordant Results: A Prospective Cohort Analysis of the AGMT Study Group. Cancers 2023, 15, 2305. https://doi.org/10.3390/cancers15082305
Jansko-Gadermeir B, Leisch M, Gassner FJ, Zaborsky N, Dillinger T, Hutter S, Risch A, Melchardt T, Egle A, Drost M, et al. Myeloid NGS Analyses of Paired Samples from Bone Marrow and Peripheral Blood Yield Concordant Results: A Prospective Cohort Analysis of the AGMT Study Group. Cancers. 2023; 15(8):2305. https://doi.org/10.3390/cancers15082305
Chicago/Turabian StyleJansko-Gadermeir, Bettina, Michael Leisch, Franz J. Gassner, Nadja Zaborsky, Thomas Dillinger, Sonja Hutter, Angela Risch, Thomas Melchardt, Alexander Egle, Manuel Drost, and et al. 2023. "Myeloid NGS Analyses of Paired Samples from Bone Marrow and Peripheral Blood Yield Concordant Results: A Prospective Cohort Analysis of the AGMT Study Group" Cancers 15, no. 8: 2305. https://doi.org/10.3390/cancers15082305
APA StyleJansko-Gadermeir, B., Leisch, M., Gassner, F. J., Zaborsky, N., Dillinger, T., Hutter, S., Risch, A., Melchardt, T., Egle, A., Drost, M., Larcher-Senn, J., Greil, R., & Pleyer, L. (2023). Myeloid NGS Analyses of Paired Samples from Bone Marrow and Peripheral Blood Yield Concordant Results: A Prospective Cohort Analysis of the AGMT Study Group. Cancers, 15(8), 2305. https://doi.org/10.3390/cancers15082305