

Myo-Inositol Reverses TGF-β1-Induced EMT in MCF-10A Non-Tumorigenic Breast Cells

 ,
,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Cell Model and Treatments
2.2. RNA Extraction and RT-qPCR
2.3. Western Blot Assay
2.4. Confocal Microscopy
2.5. Migration and Invasion Assays
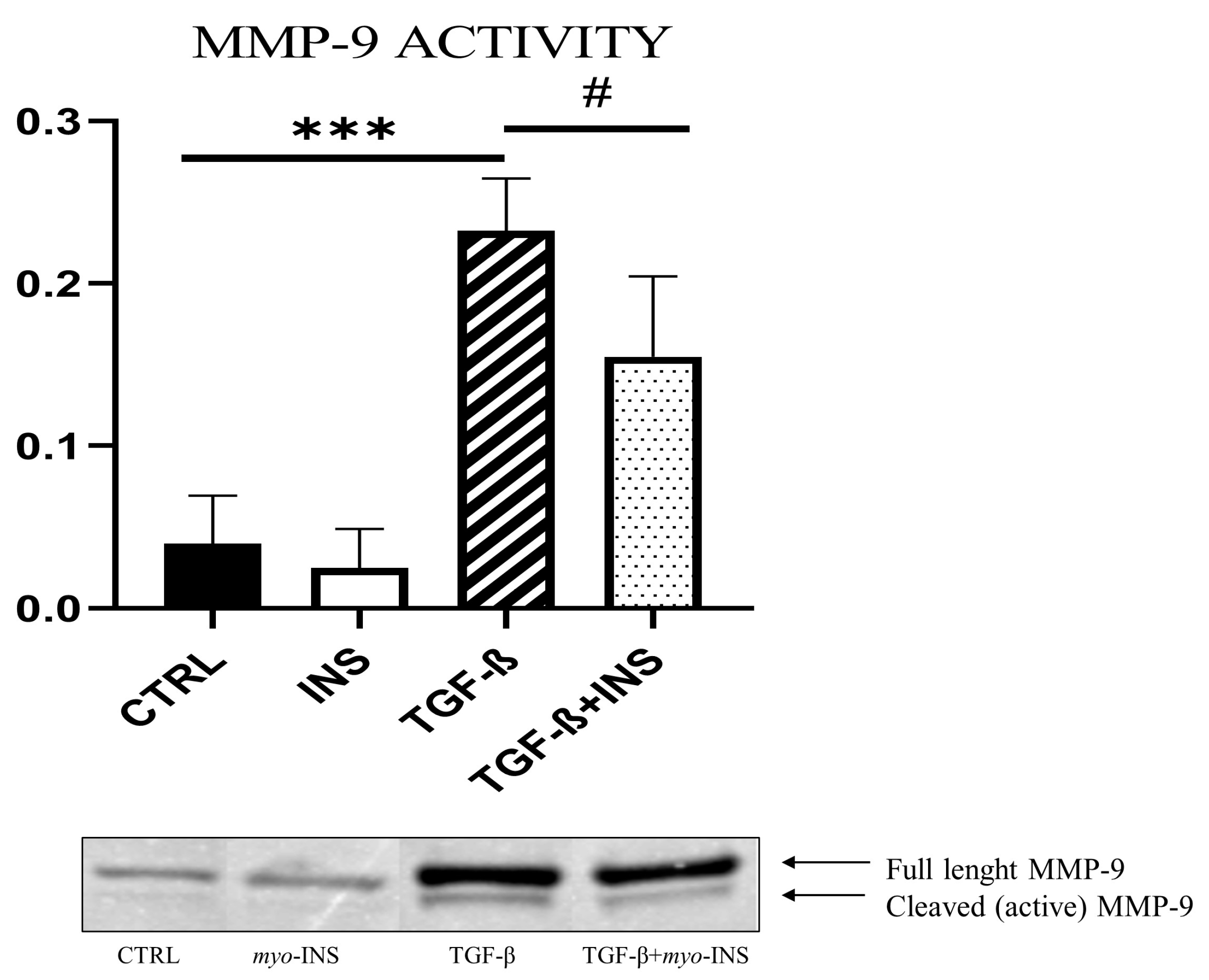
2.6. Zymography
2.7. E-Cadherin Interference
2.8. Statistical Analysis
3. Results
3.1. TGF-β1 Induces Morphological Changes in MCF-10A Cells
3.2. Modulation of Gene Expression during EMT
3.3. Myo-Ins Counteracts TGF-β1-Dependent Effects on EMT Marker Expressions
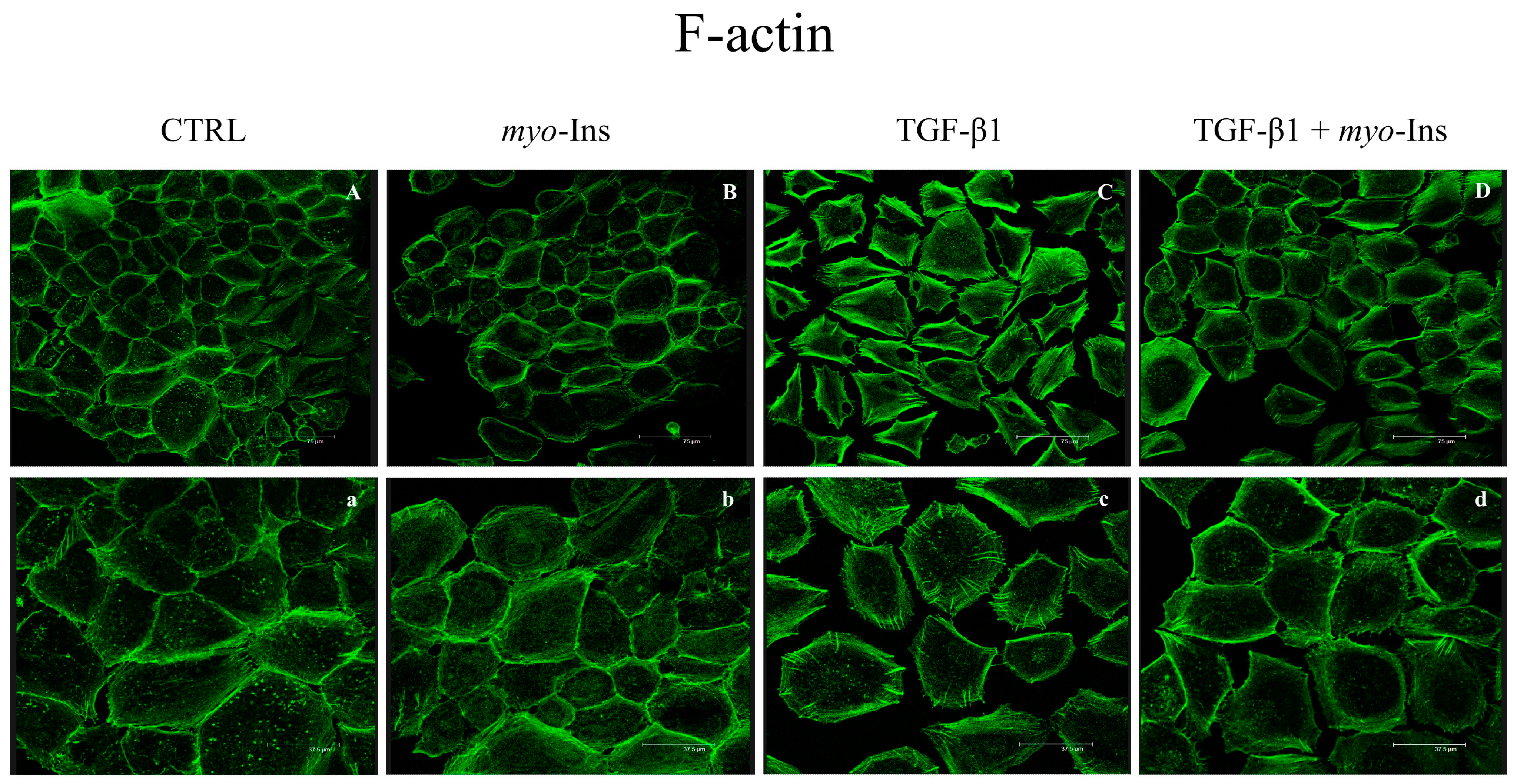
3.4. F-Actin Remodeling
3.5. Myo-Ins Almost Completely Inhibits the Migrating and Invasive Capabilities Induced by TGF-β1
3.6. Myo-Ins Antagonizes TGF-β1-Dependent Effects on CSK Rearrangement
3.7. Myo-Ins Restores Physiological E-Cadherin/β-Catenin Distribution
3.8. E-Cadherin Silencing
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [Green Version]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial–mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Jolly, M.K.; Ware, K.E.; Gilja, S.; Somarelli, J.A.; Levine, H. EMT and MET: Necessary or permissive for metastasis? Mol. Oncol. 2017, 11, 755–769. [Google Scholar] [CrossRef] [Green Version]
- Tian, X.; Liu, Z.; Niu, B.; Zhang, J.; Tan, T.K.; Lee, S.R.; Zhao, Y.; Harris, D.C.; Zheng, G. E-cadherin/β-catenin complex and the epithelial barrier. J. Biomed. Biotechnol. 2011, 2011, 567305. [Google Scholar] [CrossRef] [Green Version]
- Zavadil, J.; Böttinger, E.P. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene 2005, 24, 5764–5774. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial–mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinck, A.P.; Mueller, T.D.; Springer, T.A. Structural Biology and Evolution of the TGF-β Family. Cold Spring Harb. Perspect. Biol. 2016, 8, a022103. [Google Scholar]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Sistigu, A.; Di Modugno, F.; Manic, G.; Nisticò, P. Deciphering the loop of epithelial-mesenchymal transition, inflammatory cytokines and cancer immunoediting. Cytokine Growth Factor. Rev. 2017, 36, 67–77. [Google Scholar] [CrossRef]
- López-Novoa, J.M.; Nieto, M.A. Inflammation and EMT: An alliance towards organ fibrosis and cancer progression. EMBO Mol. Med. 2009, 1, 303–314. [Google Scholar] [CrossRef] [Green Version]
- Suarez-Carmona, M.; Lesage, J.; Cataldo, D.; Gilles, C. EMT and inflammation: Inseparable actors of cancer progression. Mol. Oncol. 2017, 11, 805–823. [Google Scholar] [CrossRef]
- Beach, J.R.; Hussey, G.S.; Miller, T.E.; Chaudhury, A.; Patel, P.; Monslow, J.; Zheng, Q.; Keri, R.A.; Reizes, O.; Bresnick, A.R.; et al. Myosin II isoform switching mediates invasiveness after TGF-beta-induced epithelial-mesenchymal transition. Proc. Natl. Acad. Sci. USA 2011, 108, 17991–17996. [Google Scholar] [CrossRef] [Green Version]
- Gunasinghe, N.P.; Wells, A.; Thompson, E.W.; Hugo, H.J. Mesenchymal-epithelial transition (MET) as a mechanism for metastatic colonisation in breast cancer. Cancer Metastasis. Rev. 2012, 31, 469–478. [Google Scholar] [CrossRef]
- Pei, D.; Shu, X.; Gassama-Diagne, A.; Thiery, J.P. Mesenchymal-epithelial transition in development and reprogramming. Nat. Cell Biol. 2019, 21, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Ascenção, K.; Dilek, N.; Augsburger, F.; Panagaki, T.; Zuhra, K.; Szabo, C. Pharmacological induction of mesenchymal-epithelial transition via inhibition of H2S biosynthesis and consequent suppression of ACLY activity in colon cancer cells. Pharmacol. Res. 2021, 165, 105393. [Google Scholar] [CrossRef] [PubMed]
- Proietti, S.; Cucina, A.; Pensotti, A.; Fuso, A.; Marchese, C.; Nicolini, A.; Bizzarri, M. Tumor reversion and embryo morphogenetic factors. Semin. Cancer Biol. 2022, 79, 83–90. [Google Scholar] [CrossRef]
- Bizzarri, M.; Cucina, A.; Proietti, S. Tumor Reversion: Mesenchymal-Epithelial Transition as a Critical Step in Managing the Tumor-Microenvironment Cross-Talk. Curr. Pharm. Des. 2017, 23, 4705–4715. [Google Scholar] [CrossRef]
- Bizzarri, M.; Fuso, A.; Dinicola, S.; Cucina, A.; Bevilacqua, A. Pharmacodynamics and pharmacokinetics of inositol(s) in health and disease. Expert Opin. Drug Metab. Toxicol. 2016, 12, 1181–1196. [Google Scholar] [CrossRef]
- Baldassarre, M.P.A.; Di Tomo, P.; Centorame, G.; Pandolfi, A.; Di Pietro, N.; Consoli, A.; Formoso, G. Myoinositol Reduces Inflammation and Oxidative Stress in Human Endothelial Cells Exposed In Vivo to Chronic Hyperglycemia. Nutrients 2021, 13, 2210. [Google Scholar] [CrossRef] [PubMed]
- Bizzarri, M.; Dinicola, S.; Bevilacqua, A.; Cucina, A. Broad Spectrum Anticancer Activity of Myo-Inositol and Inositol Hexakisphosphate. Int. J. Endocrinol. 2016, 2016, 5616807. [Google Scholar] [CrossRef] [Green Version]
- Bakin, A.V.; Tomlinson, A.K.; Bhowmick, N.A.; Moses, H.L.; Arteaga, C.L. Phosphatidylinositol 3-kinase function is required for transforming growth factor-β-mediated epithelial to mesenchymal transition and cell migration. J. Biol. Chem. 2000, 275, 36803–36810. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.; Zhang, Z.; Zhou, C.; Liu, L.; Huang, C. Epithelial-mesenchymal transition: The history, regulatory mechanism, and cancer therapeutic opportunities. MedComm 2022, 3, e144. [Google Scholar] [CrossRef]
- Vincent, T.; Neve, E.P.; Johnson, J.R.; Kukalev, A.; Rojo, F.; Albanell, J.; Pietras, K.; Virtanen, I.; Philipson, L.; Leopold, P.L.; et al. A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-beta mediated epithelial-mesenchymal transition. Nat. Cell Biol. 2009, 11, 943–950. [Google Scholar] [CrossRef] [Green Version]
- Poltavets, V.; Kochetkova, M.; Pitson, S.M.; Samuel, M.S. The role of the extracellular matrix and its molecular and cellular regulators in cancer cell plasticity. Front. Oncol. 2018, 8, 431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, H.; Kim, B.; Moon, B.I.; Oh, E.S. Cytokeratin 18 is necessary for initiation of TGF-β1-induced epithelial-mesenchymal transition in breast epithelial cells. Mol. Cell Biochem. 2016, 423, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [Green Version]
- Ngok, S.P.; Lin, W.H.; Anastasiadis, P.Z. Establishment of epithelial polarity—GEF who’s minding the GAP? J. Cell Sci. 2014, 127, 3205–3215. [Google Scholar] [CrossRef] [Green Version]
- Barrios-Rodiles, M.; Brown, K.R.; Ozdamar, B.; Bose, R.; Liu, Z.; Donovan, R.S.; Shinjo, F.; Liu, Y.; Dembowy, J.; Taylor, I.W.; et al. High-throughput mapping of a dynamic signaling network in mammalian cells. Science 2005, 307, 1621–1625. [Google Scholar] [CrossRef] [Green Version]
- Galvagni, F.; Baldari, C.T.; Oliviero, S.; Orlandini, M. An apical actin-rich domain drives the establishment of cell polarity during cell adhesion. Histochem. Cell Biol. 2012, 138, 419–433. [Google Scholar] [CrossRef] [Green Version]
- Slováková, J.; Sikora, M.; Arslan, F.N.; Caballero-Mancebo, S.; Krens, S.F.G.; Kaufmann, W.A.; Merrin, J.; Heisenberg, C.P. Tension-dependent stabilization of E-cadherin limits cell-cell contact expansion in zebrafish germ-layer progenitor cells. Proc. Natl. Acad. Sci. USA 2022, 119, e2122030119. [Google Scholar] [CrossRef] [PubMed]
- Igaki, T.; Pagliarini, R.A.; Xu, T. Loss of cell polarity drives tumor growth and invasion through JNK activation in Drosophila. Curr. Biol. 2006, 16, 1139–1146. [Google Scholar] [CrossRef] [Green Version]
- Loh, C.Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef] [Green Version]
- Sanghvi-Shah, R.; Weber, G.F. Intermediate Filaments at the Junction of Mechanotransduction, Migration, and Development. Front. Cell Dev. Biol. 2017, 5, 81. [Google Scholar] [CrossRef] [Green Version]
- Cano, A.; Pérez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; del Barrio, M.G.; Portillo, F.; Nieto, M.A. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Olmeda, D.; Jordá, M.; Peinado, H.; Fabra, A.; Cano, A. Snail silencing effectively suppresses tumour growth and invasiveness. Oncogene 2007, 26, 1862–1874. [Google Scholar] [CrossRef] [Green Version]
- Peinado, H.; Quintanilla, M.; Cano, A. Transforming growth factor beta-1 induces snail transcription factor in epithelial cell lines: Mechanisms for epithelial mesenchymal transitions. J. Biol. Chem. 2003, 278, 21113–21123. [Google Scholar] [CrossRef] [Green Version]
- Dumont, N.; Bakin, A.V.; Arteaga, C.L. Autocrine transforming growth factor-beta signaling mediates Smad-independent motility in human cancer cells. J. Biol. Chem. 2003, 278, 3275–3285. [Google Scholar] [CrossRef] [Green Version]
- Janda, E.; Lehmann, K.; Killisch, I.; Jechlinger, M.; Herzig, M.; Downward, J.; Beug, H.; Grünert, S. Ras and TGF-β cooperatively regulate epithelial cell plasticity and metastasis: Dissection of Ras signaling pathways. J. Cell Biol. 2002, 156, 299–313. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Yang, Z.; Lu, N. A new role for the PI3K/Akt signaling pathway in the epithelial-mesenchymal transition. Cell Adh. Migr. 2015, 9, 317–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radisky, E.S.; Radisky, D.C. Matrix metalloproteinase-induced epithelial-mesenchymal transition in breast cancer. J. Mammary Gland Biol. Neoplasia 2010, 15, 201–212. [Google Scholar] [CrossRef] [Green Version]
- Krstic, J.; Santibanez, J.F. Transforming growth factor-beta and matrix metalloproteinases: Functional interactions in tumor stroma-infiltrating myeloid cells. Sci. World J. 2014, 2014, 521754. [Google Scholar] [CrossRef] [Green Version]
- Ignotz, R.A.; Massagué, J. Transforming growth factor-beta stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J. Biol. Chem. 1986, 261, 4337–4345. [Google Scholar] [CrossRef]
- Hu, J.; Jo, M.; Eastman, B.M.; Gilder, A.S.; Bui, J.D.; Gonias, S.L. uPAR induces expression of transforming growth factor β and interleukin-4 in cancer cells to promote tumor-permissive conditioning of macrophages. Am. J. Pathol. 2014, 184, 3384–3393. [Google Scholar] [CrossRef] [Green Version]
- Chao, Y.L.; Shepard, C.R.; Wells, A. Breast carcinoma cells re-express E-cadherin during mesenchymal to epithelial reverting transition. Mol. Cancer 2010, 9, 179. [Google Scholar] [CrossRef] [Green Version]
- Perl, A.K.; Wilgenbus, P.; Dahl, U.; Semb, H.; Christofori, G. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature 1998, 392, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Vleminckx, K.; Vakaet, L., Jr.; Mareel, M.; Fiers, W.; van Roy, F. Genetic manipulation of E-cadherin expression by epithelial tumor cells reveals an invasion suppressor role. Cell 1991, 66, 107–119. [Google Scholar] [CrossRef]
- Dinicola, S.; Fabrizi, G.; Masiello, M.G.; Proietti, S.; Palombo, A.; Minini, M.; Harrath, A.H.; Alwasel, S.H.; Ricci, G.; Catizone, A.; et al. Inositol induces mesenchymal-epithelial reversion in breast cancer cells through cytoskeleton rearrangement. Exp. Cell Res. 2016, 345, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Yao, D.; Dai, C.; Peng, S. Mechanism of the mesenchymal-epithelial transition and its relationship with metastatic tumor formation. Mol. Cancer Res. 2011, 9, 1608–1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteban, M.A.; Bao, X.; Zhuang, Q.; Zhou, T.; Qin, B.; Pei, D. The mesenchymal-to-epithelial transition in somatic cell reprogramming. Curr. Opin. Genet. Dev. 2012, 22, 423–428. [Google Scholar] [CrossRef]
- Gandalovičová, A.; Rosel, D.; Fernandes, M.; Veselý, P.; Heneberg, P.; Čermák, V.; Petruželka, L.; Kumar, S.; Sanz-Moreno, V.; Brábek, J. Migrastatics-Anti-metastatic and Anti-invasion Drugs: Promises and Challenges. Trends. Cancer 2017, 3, 391–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solomon, J.; Raškova, M.; Rösel, D.; Brábek, J.; Gil-Henn, H. Are We Ready for Migrastatics? Cells 2021, 10, 1845. [Google Scholar] [CrossRef] [PubMed]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monti, N.; Dinicola, S.; Querqui, A.; Fabrizi, G.; Fedeli, V.; Gesualdi, L.; Catizone, A.; Unfer, V.; Bizzarri, M. Myo-Inositol Reverses TGF-β1-Induced EMT in MCF-10A Non-Tumorigenic Breast Cells. Cancers 2023, 15, 2317. https://doi.org/10.3390/cancers15082317
Monti N, Dinicola S, Querqui A, Fabrizi G, Fedeli V, Gesualdi L, Catizone A, Unfer V, Bizzarri M. Myo-Inositol Reverses TGF-β1-Induced EMT in MCF-10A Non-Tumorigenic Breast Cells. Cancers. 2023; 15(8):2317. https://doi.org/10.3390/cancers15082317
Chicago/Turabian StyleMonti, Noemi, Simona Dinicola, Alessandro Querqui, Gianmarco Fabrizi, Valeria Fedeli, Luisa Gesualdi, Angela Catizone, Vittorio Unfer, and Mariano Bizzarri. 2023. "Myo-Inositol Reverses TGF-β1-Induced EMT in MCF-10A Non-Tumorigenic Breast Cells" Cancers 15, no. 8: 2317. https://doi.org/10.3390/cancers15082317
APA StyleMonti, N., Dinicola, S., Querqui, A., Fabrizi, G., Fedeli, V., Gesualdi, L., Catizone, A., Unfer, V., & Bizzarri, M. (2023). Myo-Inositol Reverses TGF-β1-Induced EMT in MCF-10A Non-Tumorigenic Breast Cells. Cancers, 15(8), 2317. https://doi.org/10.3390/cancers15082317