


Chromatin Remodelling Molecule ARID1A Determines Metastatic Heterogeneity in Triple-Negative Breast Cancer by Competitively Binding to YAP

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Samples
2.2. Cell Lines and Cell Culture
2.3. Lentiviral shRNA Production, Infection and Knockdown of Genes
| Species | Gene | Sequences |
| mouse | ARID1AKO-1 | 5′-GGTCCCTGTTGTTGCGAGTA-3′ |
| mouse | ARID1AKO-2 | 5′-gACCCCATGACCATGCAGGGC-3′ |
| mouse | YAPKD-1 | 5′-AGGCCAGTACTGATGCAGGTA-3′ |
| mouse | YAPKD-2 | 5′-CAGGACCTCTTCCTGATGGAT-3′ |
| human | ARID1AKO-1 | 5′-CACCGATGGTCATCGGGTACCGCTG-3′ |
| human | ARID1AKO-2 | 5′-CACCGCCCCTCAATGACCTCCAGTA-3′ |
| human | ARID1AOE-1 | 5′-CACCGGGCGCTCTAGCCGCTCAGTC-3′ |
| human | ARID1AOE-2 | 5′-CACCGCTTGGGTCGAGGCTGCTGCG-3′ |
| human | YAPKD-1 | 5′-CCCAGTTAAATGTTCACCAAT-3′ |
| human | YAPKD-2 | 5′-CACCAAGCTAGATAAAGAA-3′ |
2.4. Cellular Fractionation, Western Blot and Co-IP Assay
2.5. Preparation of RNA and Quantitative Real-Time PCR
2.6. RNA-Seq Library Construction and Analysis
2.7. Wound Healing Assay
2.8. Transwell Migration and Invasion Assays
2.9. Immunofluorescent Staining
2.10. In Vivo Tumour Metastasis Model
2.11. Immunohistochemical Analysis
2.12. Hematoxylin/Eosin Staining
2.13. Statistical Analysis
3. Results
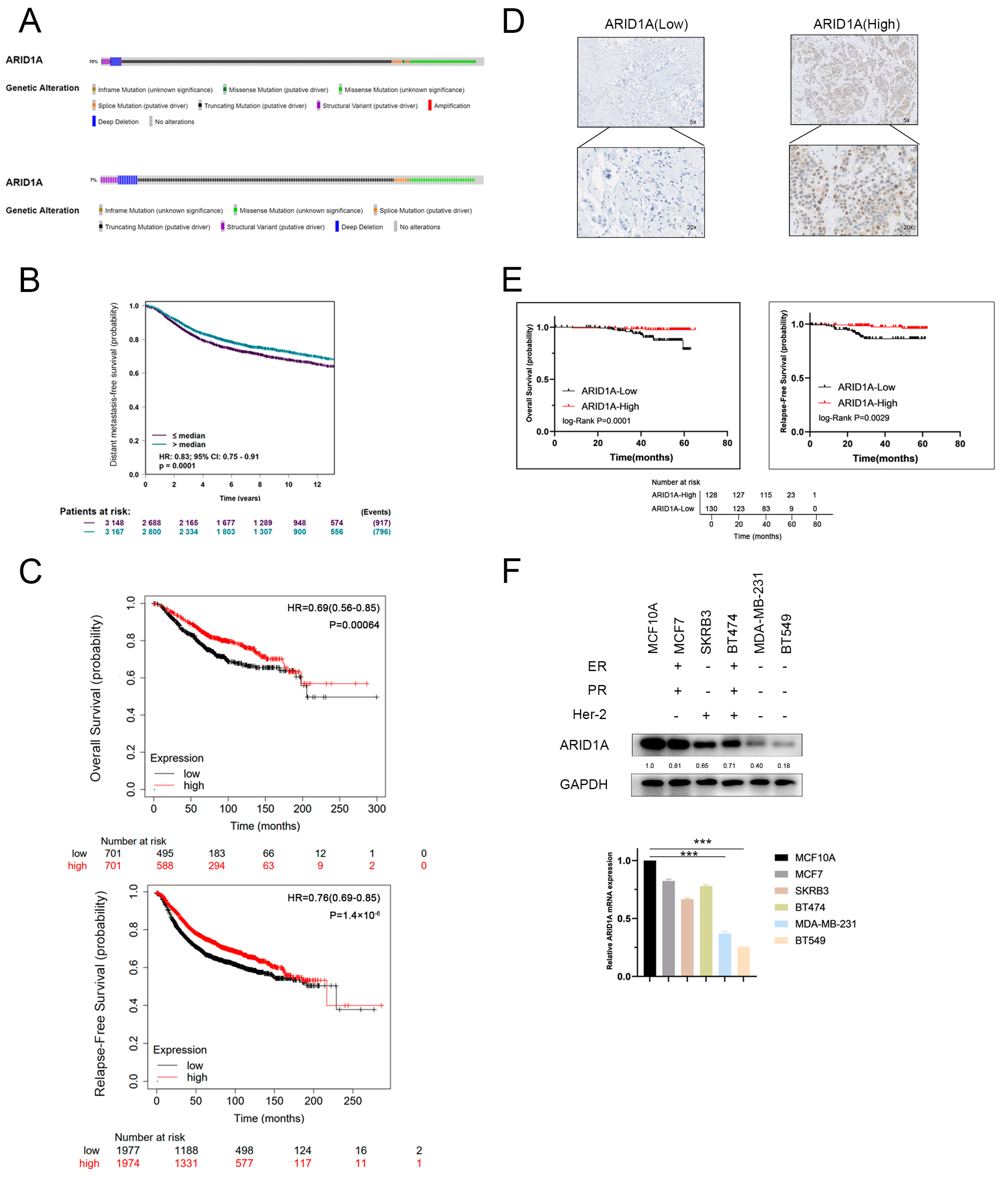
3.1. ARID1A-Low Expression Associated with Metastasis and Poor Prognosis in TNBC
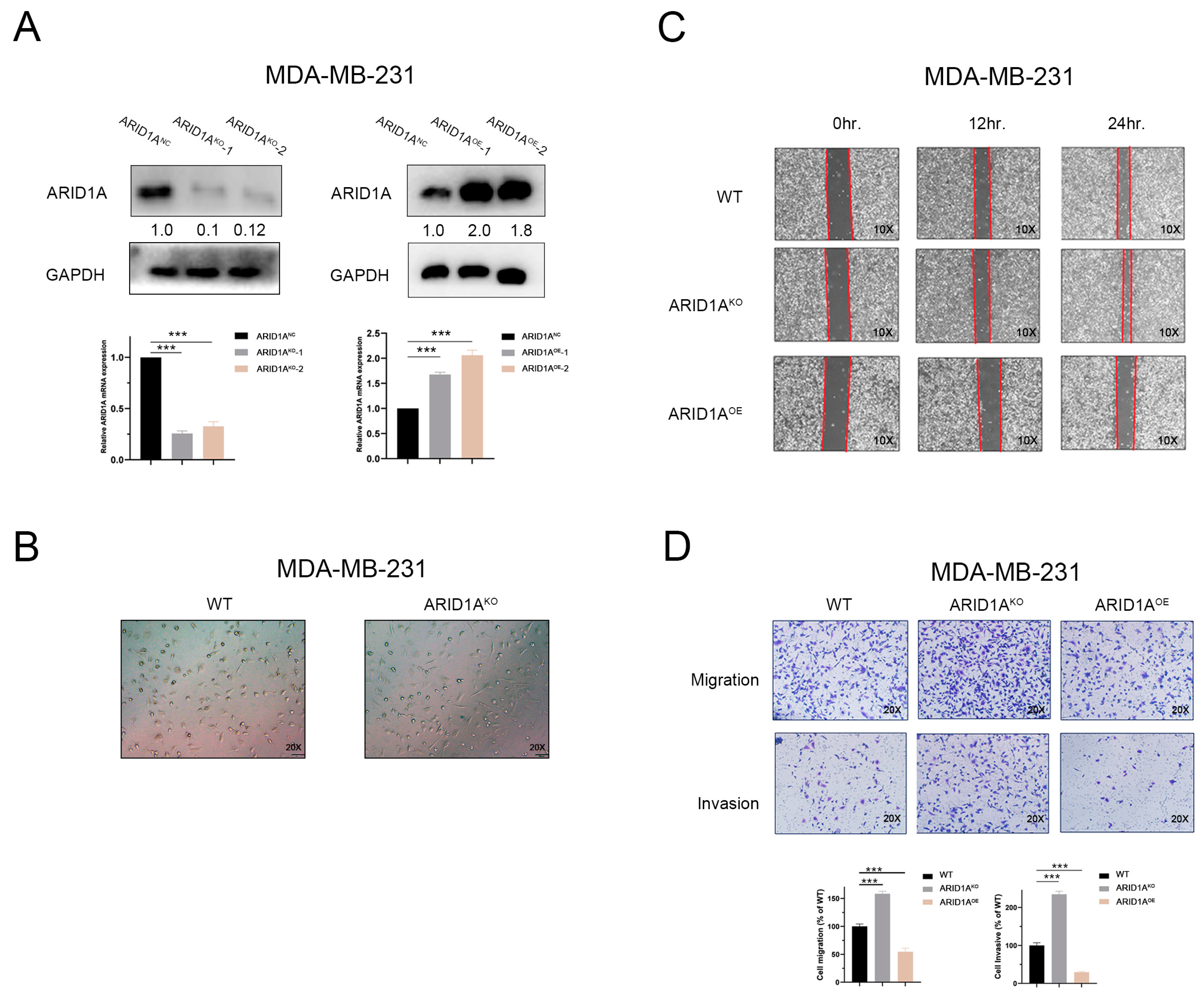
3.2. Inhibition of ARID1A Promotes Migration and Invasion in TNBC Cells
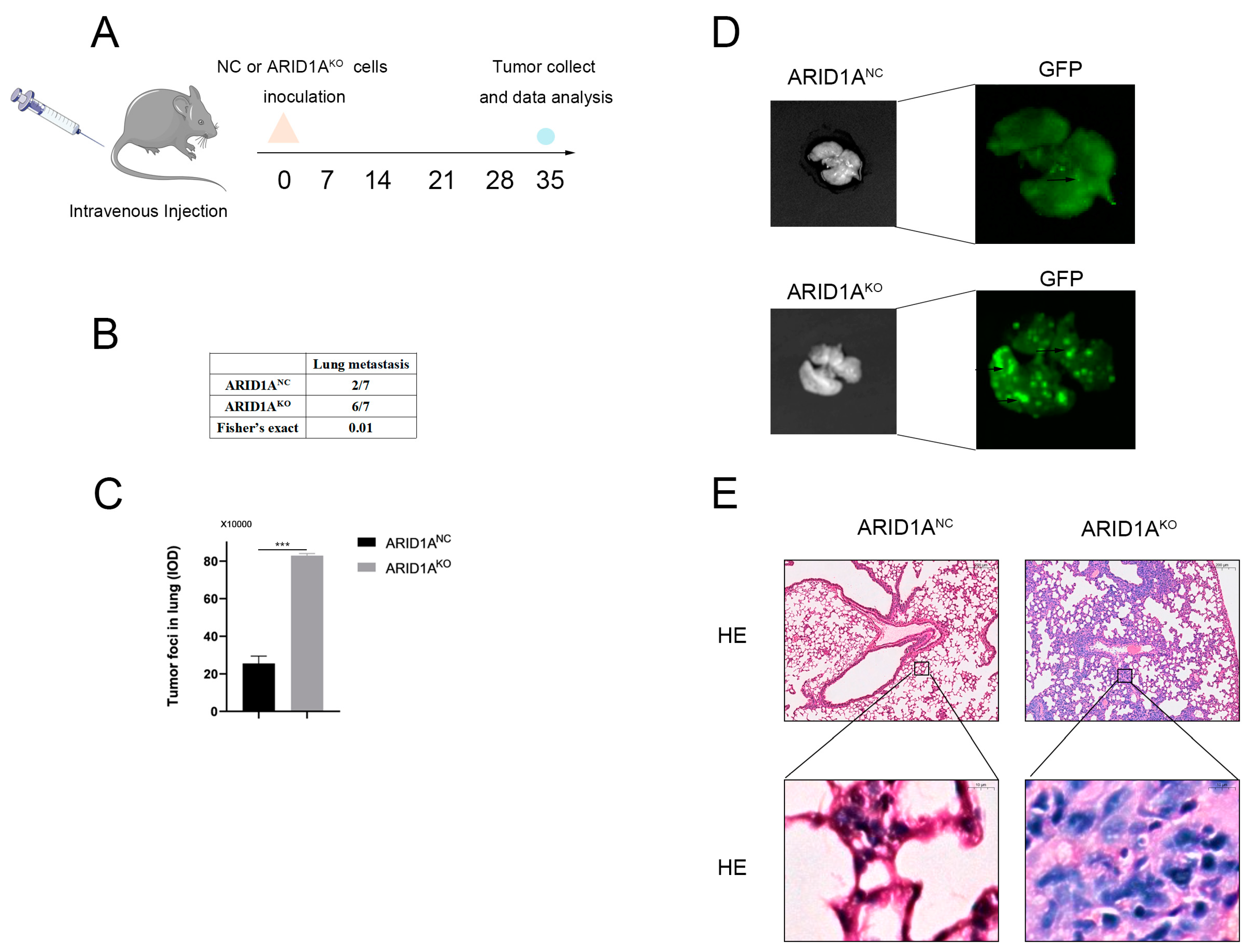
3.3. ARID1A Inhibits TNBC Metastasis In Vivo
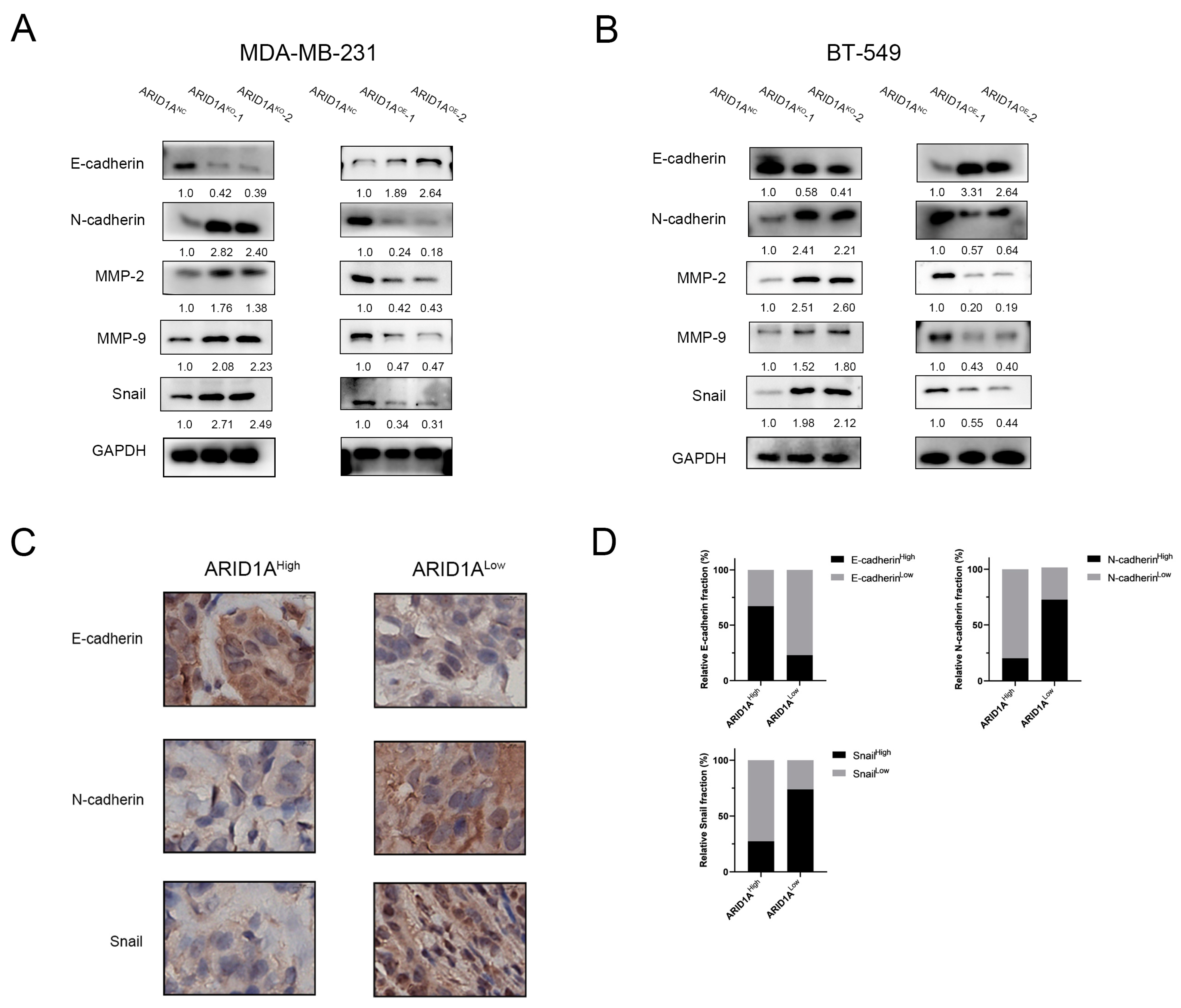
3.4. ARID1A Regulates EMT in TNBC
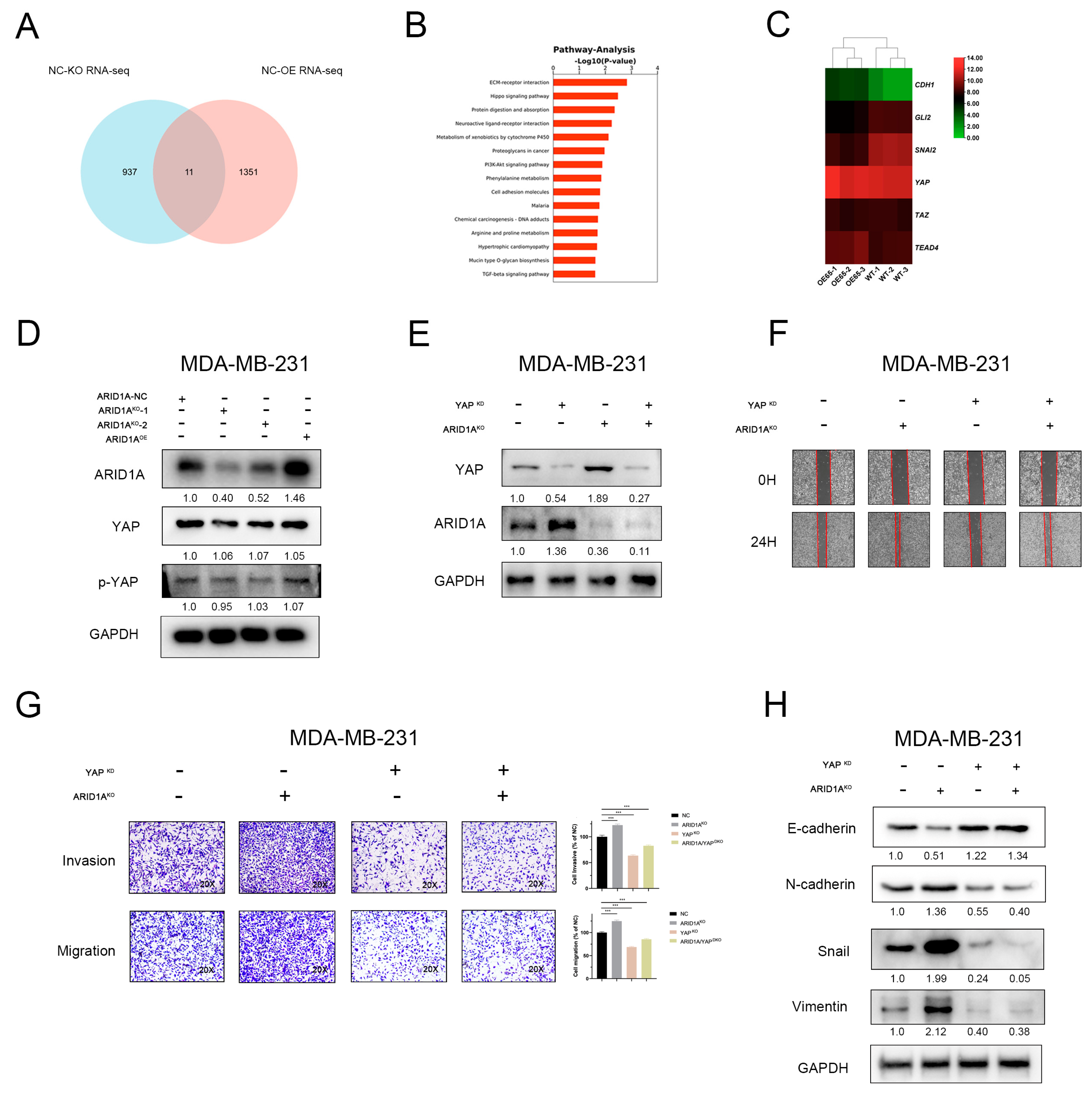
3.5. YAP and Hippo Pathway Involved in ARID1A-Regulated EMT and Tumour Metastasis
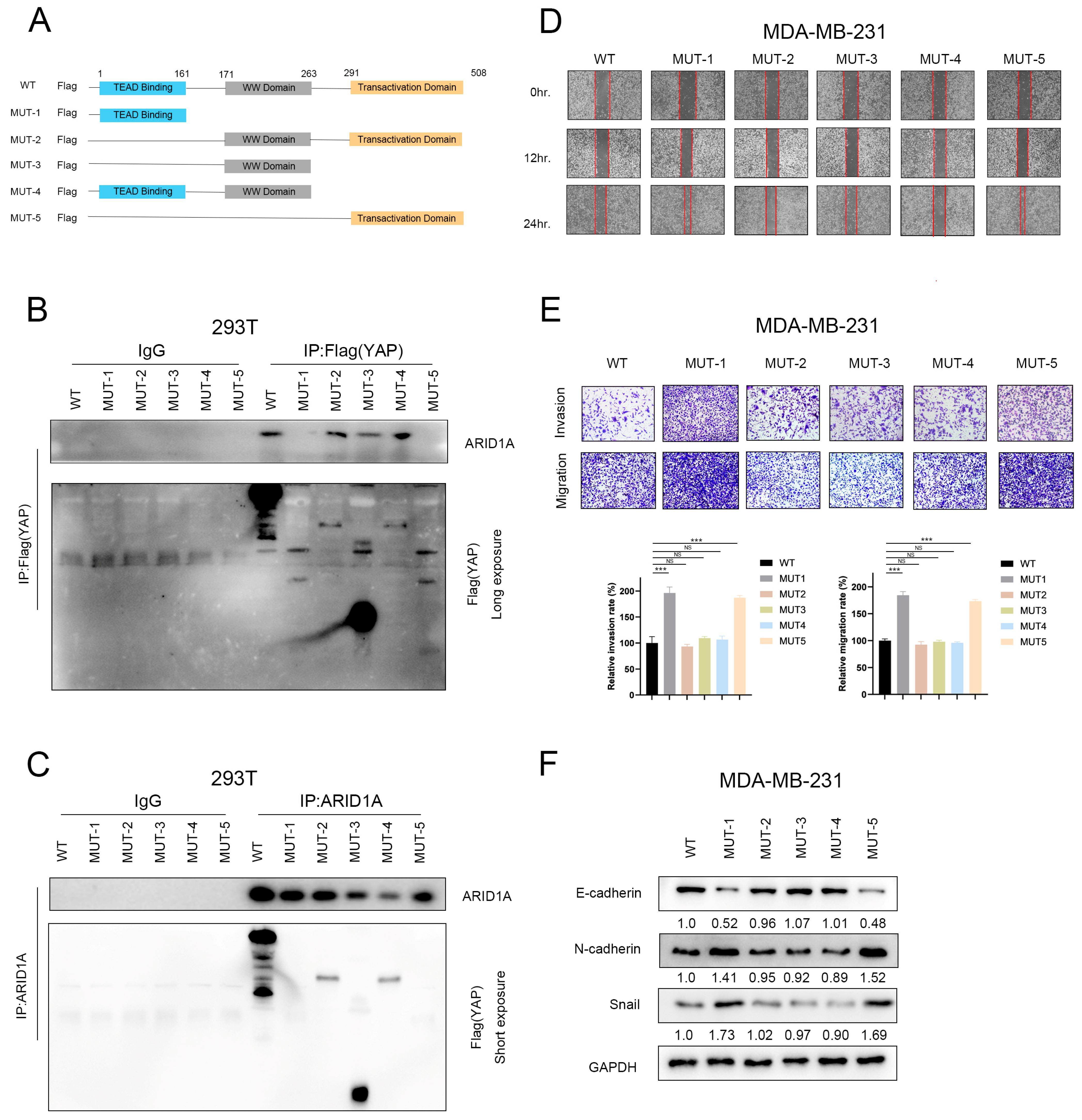
3.6. ARID1A Competitively Binds YAP to Form ARID1A/YAP Complex
3.7. ARID1A Combined with YAP via the WW Domain
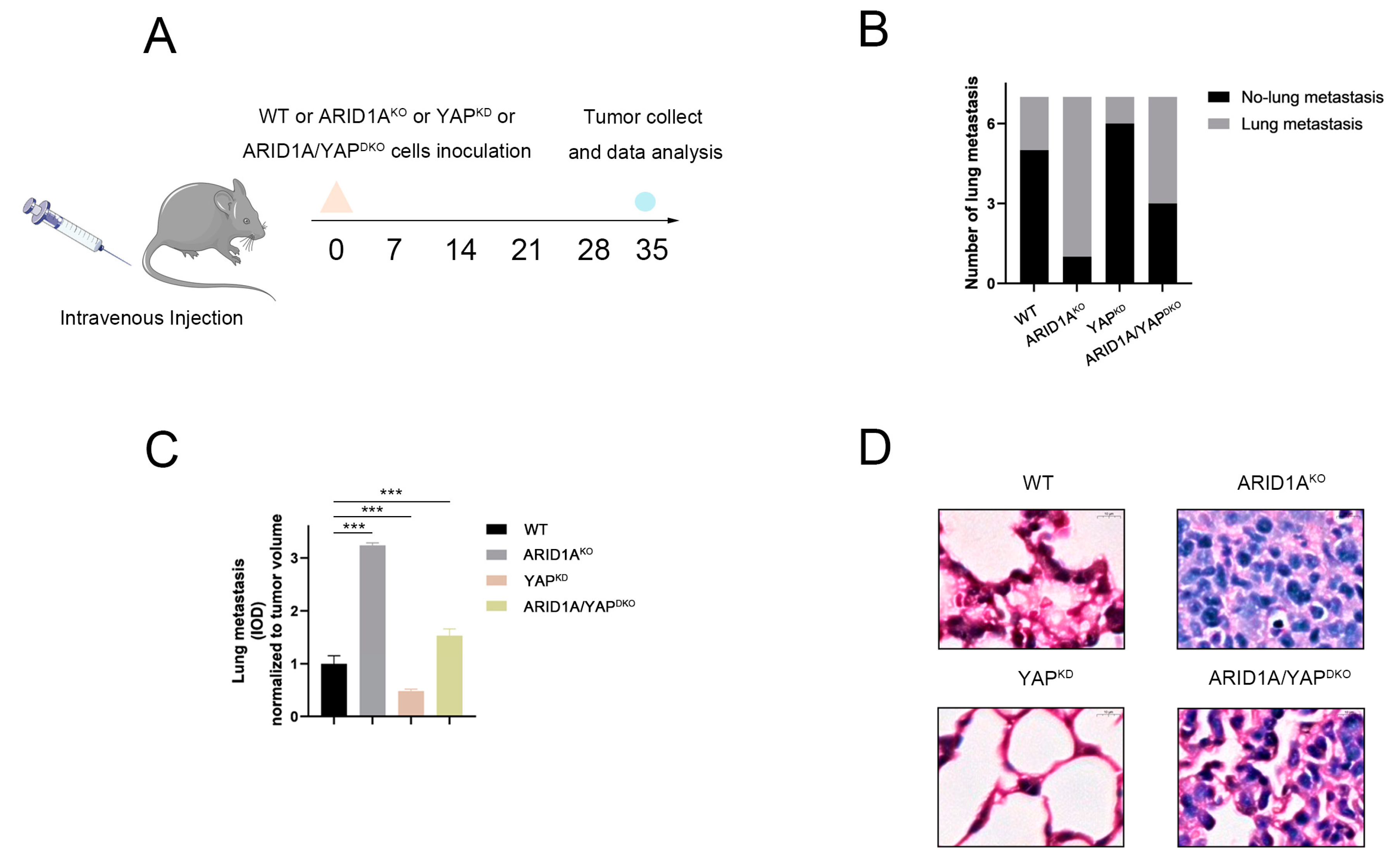
3.8. ARID1A and YAP Co-Regulate Tumour Metastasis In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Loibl, S.; Poortmans, P.; Morrow, M.; Denkert, C.; Curigliano, G. Breast cancer. Lancet 2021, 397, 1750–1769. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.; Miller, K.; Jemal, A. Cancer statistics, 2020. CA A Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Vandsemb, E.; Herbst, R.; Chen, L. Adaptive immune resistance at the tumour site: Mechanisms and therapeutic opportunities. Nat. Rev. Drug Discov. 2022, 21, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed]
- Mittal, P.; Roberts, C. The SWI/SNF complex in cancer—Biology, biomarkers and therapy. Nat. Rev. Clin. Oncol. 2020, 17, 435–448. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Roberts, C. ARID1A mutations in cancer: Another epigenetic tumor suppressor? Cancer Discov. 2013, 3, 35–43. [Google Scholar] [CrossRef]
- Kandoth, C.; Schultz, N.; Cherniack, A.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A.; Pashtan, I.; Shen, R.; Benz, C.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef]
- Shen, J.; Ju, Z.; Zhao, W.; Wang, L.; Peng, Y.; Ge, Z.; Nagel, Z.; Zou, J.; Wang, C.; Kapoor, P.; et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat. Med. 2018, 24, 556–562. [Google Scholar] [CrossRef]
- Nagarajan, S.; Rao, S.; Sutton, J.; Cheeseman, D.; Dunn, S.; Papachristou, E.; Prada, J.; Couturier, D.; Kumar, S.; Kishore, K.; et al. ARID1A influences HDAC1/BRD4 activity, intrinsic proliferative capacity and breast cancer treatment response. Nat. Genet. 2020, 52, 187–197. [Google Scholar] [CrossRef]
- Kim, M.; Park, J.; Bouhaddou, M.; Kim, K.; Rojc, A.; Modak, M.; Soucheray, M.; McGregor, M.; O’Leary, P.; Wolf, D.; et al. A protein interaction landscape of breast cancer. Science 2021, 374, eabf3066. [Google Scholar] [CrossRef]
- Tao, Z.; Li, T.; Feng, Z.; Liu, C.; Shao, Y.; Zhu, M.; Gong, C.; Wang, B.; Cao, J.; Wang, L.; et al. Characterizations of Cancer Gene Mutations in Chinese Metastatic Breast Cancer Patients. Front. Oncol. 2020, 10, 1023. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Jang, G.; Sim, S.; Park, I.; Kim, K.; Park, C. SMARCA4 Depletion Induces Cisplatin Resistance by Activating YAP1-Mediated Epithelial-to-Mesenchymal Transition in Triple-Negative Breast Cancer. Cancers 2021, 13, 5474. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Varelas, X.; Guan, K. Targeting the Hippo pathway in cancer, fibrosis, wound healing and regenerative medicine. Nat. Rev. Drug Discov. 2020, 19, 480–494. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Xie, S.; Yao, X.; Dey, A. Transcriptional Regulation of the Hippo Pathway: Current Understanding and Insights from Single-Cell Technologies. Cells 2022, 11, 2225. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Liu, Y.; Xiao, Y.; Hu, X.; Jiang, L.; Zuo, W.; Ma, D.; Ding, J.; Zhu, X.; Zou, J.; et al. Molecular subtyping and genomic profiling expand precision medicine in refractory metastatic triple-negative breast cancer: The future trial. Cell Res. 2021, 31, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; LaFramboise, W.; Liu, T.; Cao, D.; Luvison, A.; Miller, C.; Lyons, M.; O’Sullivan, R.; Zureikat, A.; Hogg, M.; et al. Loss of Chromatin-Remodeling Proteins and/or CDKN2A Associates with Metastasis of Pancreatic Neuroendocrine Tumors and Reduced Patient Survival Times. Gastroenterology 2018, 154, 2060–2063.e2068. [Google Scholar] [CrossRef]
- Xu, S.; Tang, C. ARID1AThe Role of in Tumors: Tumor Initiation or Tumor Suppression? Front. Oncol. 2021, 11, 745187. [Google Scholar] [CrossRef]
- Zafra, M.; Dow, L. Revealing ARID1A Function in gastric Cancer from the Bottom Up. Cancer Discov. 2021, 11, 1327–1329. [Google Scholar] [CrossRef]
- Chen, G.; Li, X.; Ji, C.; Liu, P.; Zhou, L.; Xu, D.; Wang, D.; Li, J.; Yu, J. Early myeloid-derived suppressor cells accelerate epithelial-mesenchymal transition by downregulating ARID1A in luminal A breast cancer. Front. Bioeng. Biotechnol. 2022, 10, 973731. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Ma, S.; Meng, Z.; Chen, R.; Guan, K. The Hippo Pathway: Biology and Pathophysiology. Annu. Rev. Biochem. 2019, 88, 577–604. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zhang, Y.; Yang, J.; Zhan, H.; Zhou, Z.; Jiang, Y.; Shi, X.; Fan, X.; Zhang, J.; Luo, W.; et al. Zinc-Dependent Regulation of ZEB1 and YAP1 Coactivation Promotes Epithelial-Mesenchymal Transition Plasticity and Metastasis in Pancreatic Cancer. Gastroenterology 2021, 160, 1771–1783.e1771. [Google Scholar] [CrossRef] [PubMed]
- Koo, J.; Guan, K. Interplay between YAP/TAZ and Metabolism. Cell Metab. 2018, 28, 196–206. [Google Scholar] [CrossRef]
- Li, T.; Liu, J.; Feng, J.; Liu, Z.; Liu, S.; Zhang, M.; Zhang, Y.; Hou, Y.; Wu, D.; Li, C.; et al. Variation in the life history strategy underlies functional diversity of tumors. Natl. Sci. Rev. 2021, 8, nwaa124. [Google Scholar] [CrossRef] [PubMed]
- Pocaterra, A.; Romani, P.; Dupont, S. YAP/TAZ functions and their regulation at a glance. J. Cell Sci. 2020, 133, jcs230425. [Google Scholar] [CrossRef]
- Totaro, A.; Panciera, T.; Piccolo, S. YAP/TAZ upstream signals and downstream responses. Nat. Cell Biol. 2018, 20, 888–899. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Chen, X.; Qiao, X.; Xie, Y.; Guo, D.; Li, B.; Cao, J.; Tao, Z.; Hu, X. Chromatin Remodelling Molecule ARID1A Determines Metastatic Heterogeneity in Triple-Negative Breast Cancer by Competitively Binding to YAP. Cancers 2023, 15, 2447. https://doi.org/10.3390/cancers15092447
Wang Y, Chen X, Qiao X, Xie Y, Guo D, Li B, Cao J, Tao Z, Hu X. Chromatin Remodelling Molecule ARID1A Determines Metastatic Heterogeneity in Triple-Negative Breast Cancer by Competitively Binding to YAP. Cancers. 2023; 15(9):2447. https://doi.org/10.3390/cancers15092447
Chicago/Turabian StyleWang, Ye, Xinyu Chen, Xiaosu Qiao, Yizhao Xie, Duancheng Guo, Bin Li, Jianing Cao, Zhonghua Tao, and Xichun Hu. 2023. "Chromatin Remodelling Molecule ARID1A Determines Metastatic Heterogeneity in Triple-Negative Breast Cancer by Competitively Binding to YAP" Cancers 15, no. 9: 2447. https://doi.org/10.3390/cancers15092447
APA StyleWang, Y., Chen, X., Qiao, X., Xie, Y., Guo, D., Li, B., Cao, J., Tao, Z., & Hu, X. (2023). Chromatin Remodelling Molecule ARID1A Determines Metastatic Heterogeneity in Triple-Negative Breast Cancer by Competitively Binding to YAP. Cancers, 15(9), 2447. https://doi.org/10.3390/cancers15092447