



Versatile Platinum(IV) Prodrugs of Naproxen and Acemetacin as Chemo-Anti-Inflammatory Agents

 , , , , , and
, , , , , and 
Abstract
:Simple Summary
Abstract
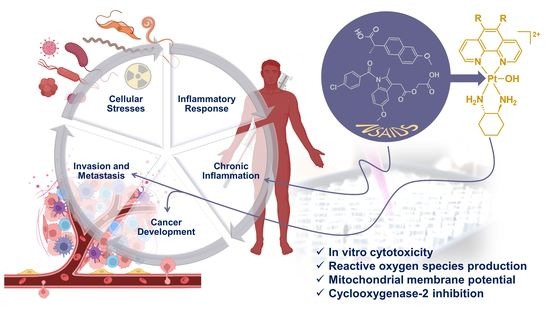
1. Introduction
2. Materials and Methods
2.1. Materials
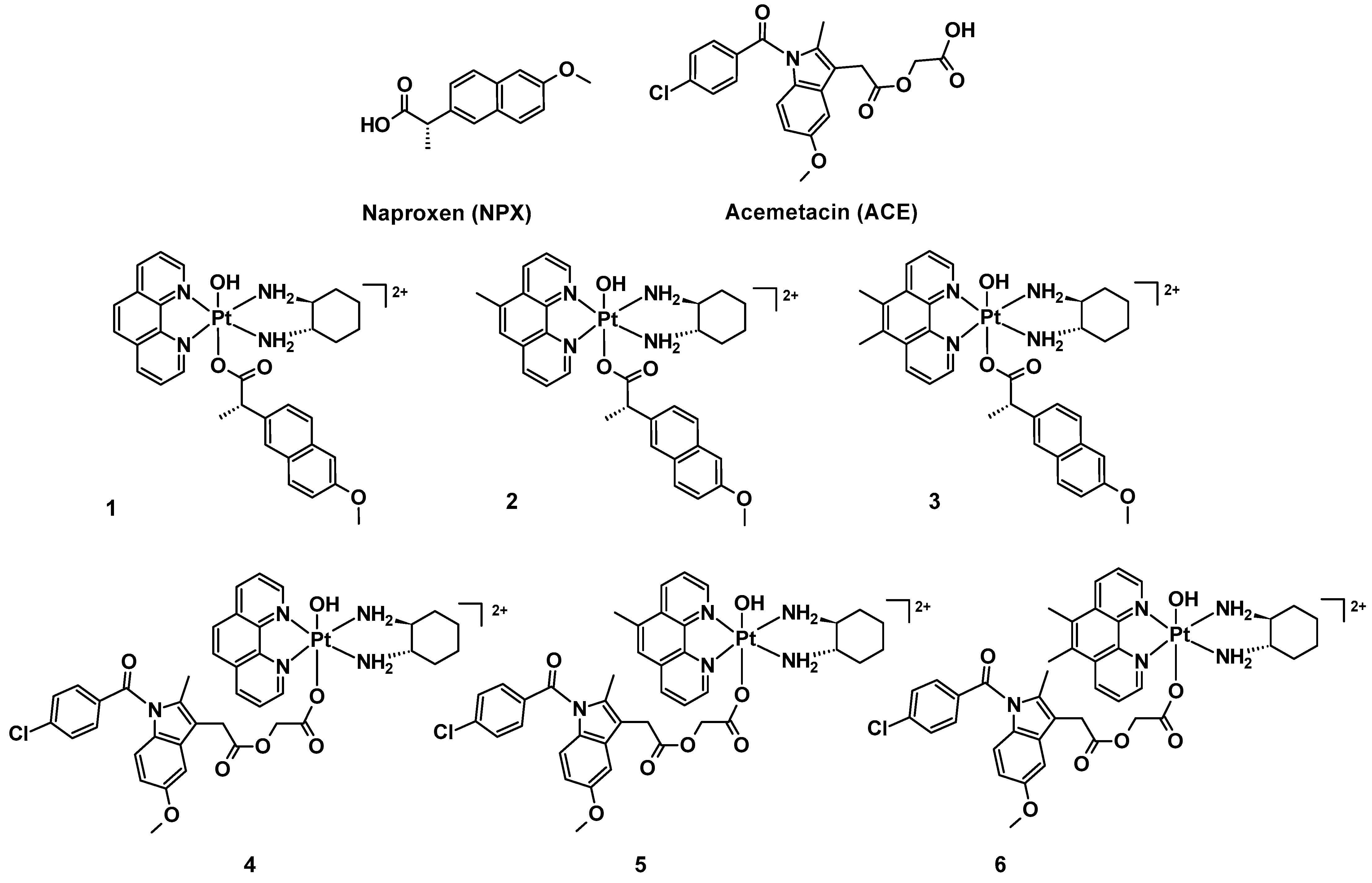
2.2. Chemistry
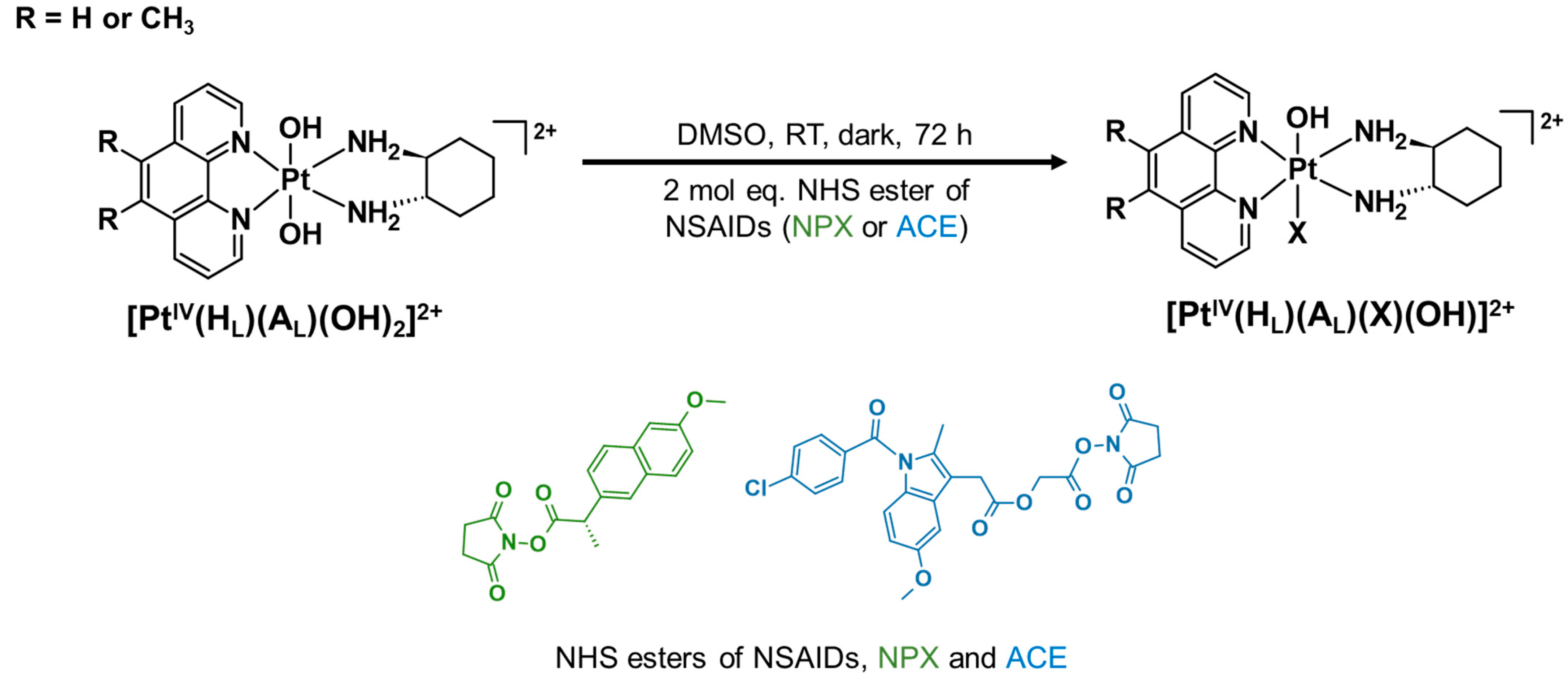
2.2.1. Synthesis Route of NHS Esters
2.2.2. Synthesis Route of Platinum(II) Precursors and Platinum(IV) Scaffolds
2.2.3. Synthesis Route of Platinum(IV) Derivatives Incorporating NPX and ACE (1–6)
2.3. Instrumentation
2.4. Physicochemical and Biological Investigations
2.4.1. Solubility Measurements
2.4.2. Stability Studies
2.4.3. Lipophilicity Studies
2.4.4. Reduction Studies
2.4.5. Cell Viability Assays
2.4.6. Reactive Oxygen Species (ROS) Potential
2.4.7. Mitochondrial Membrane Potential (MtMP)
2.4.8. Cyclooxygenase-2 (COX-2) Inhibition
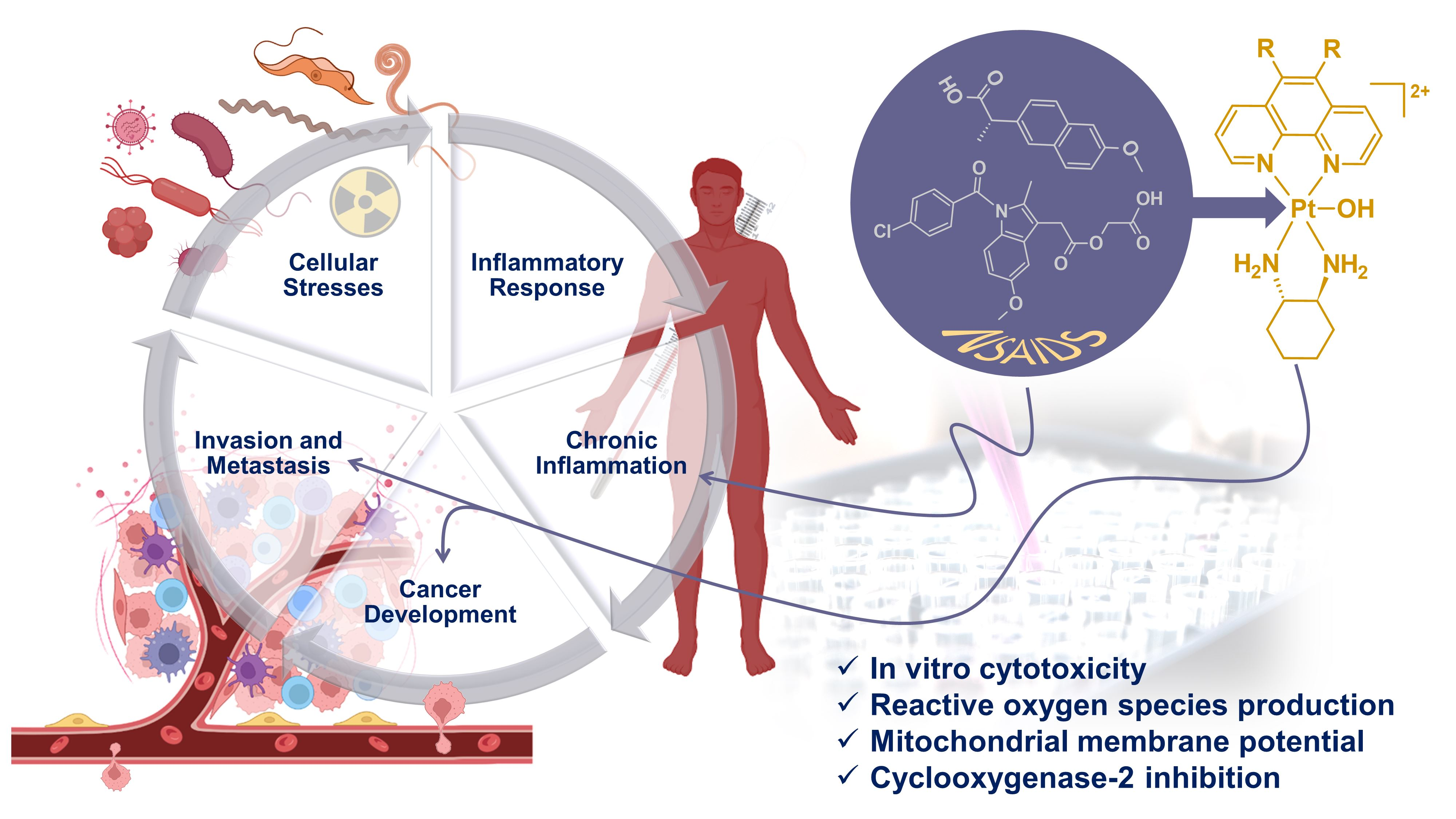
3. Results and Discussion
3.1. Synthesis and Characterisation
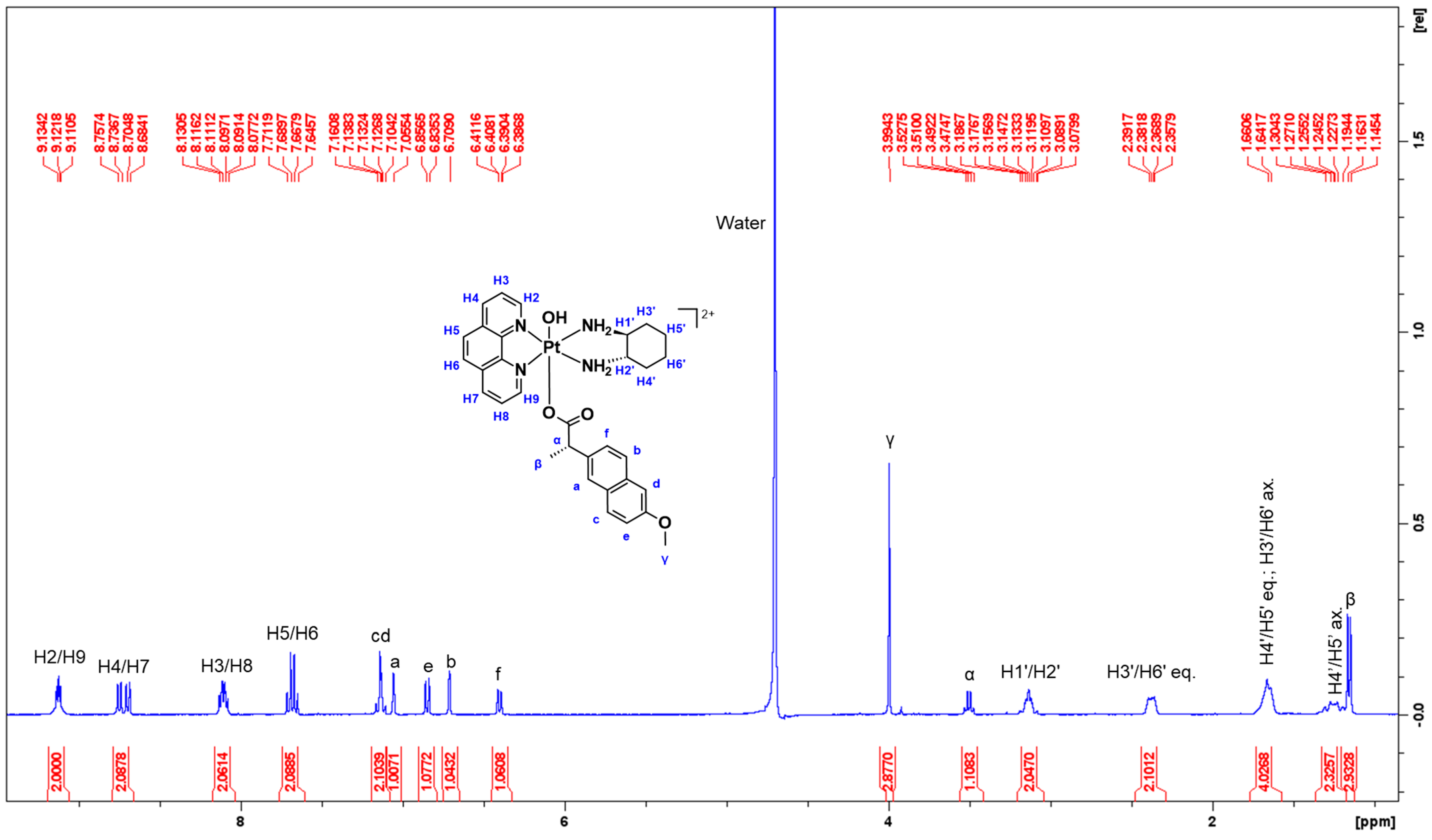
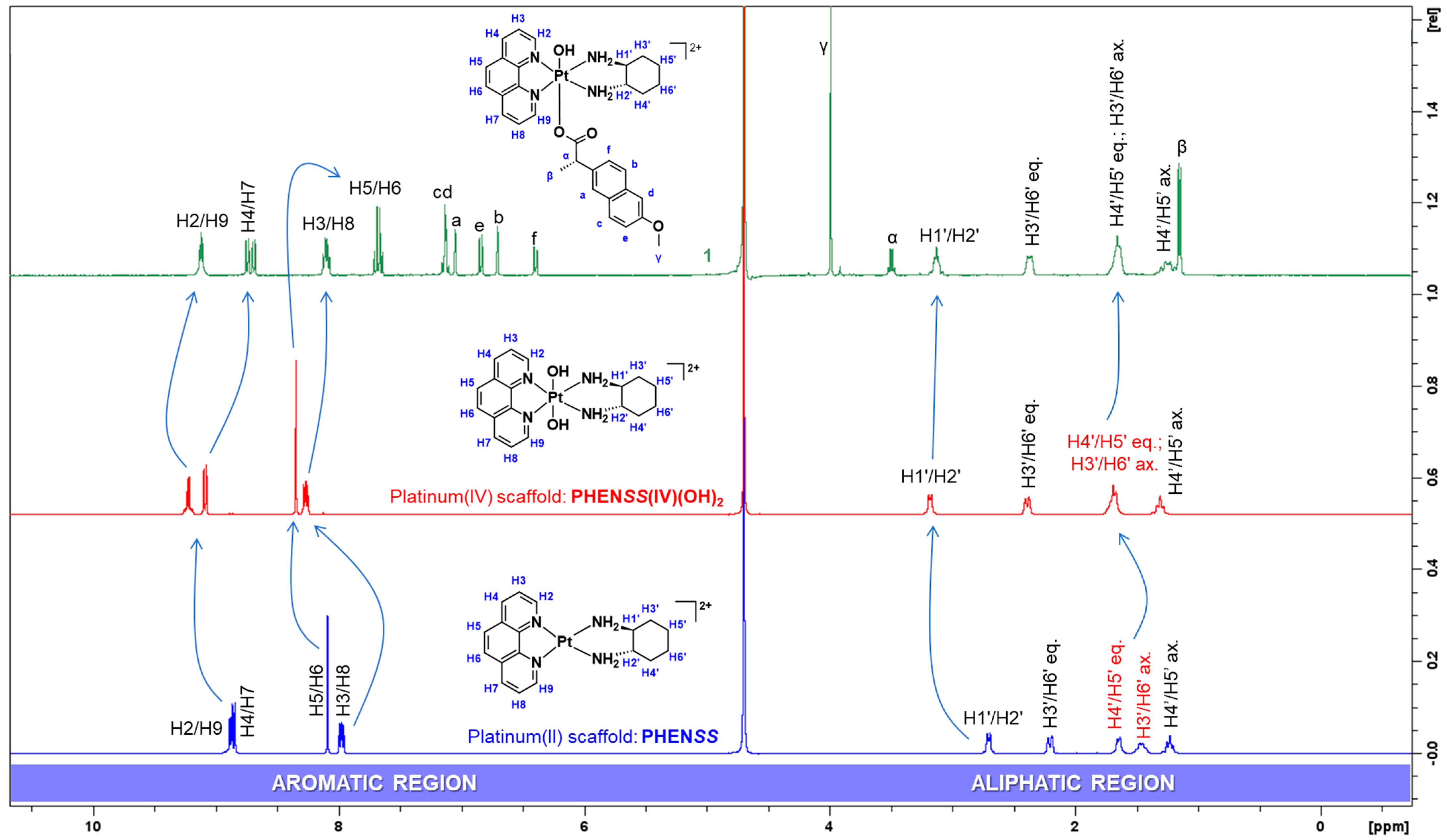
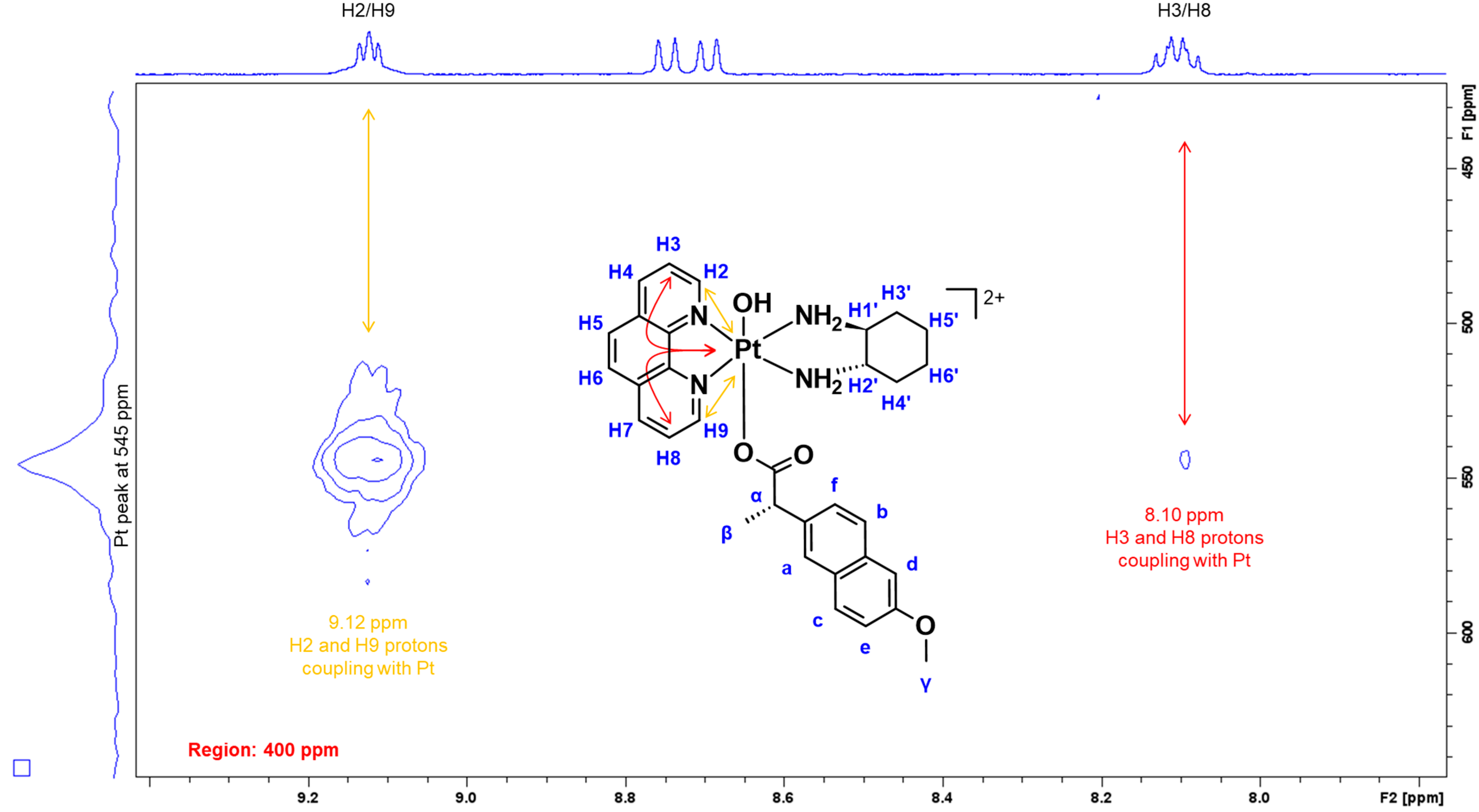
3.1.1. NMR Spectra Assignment
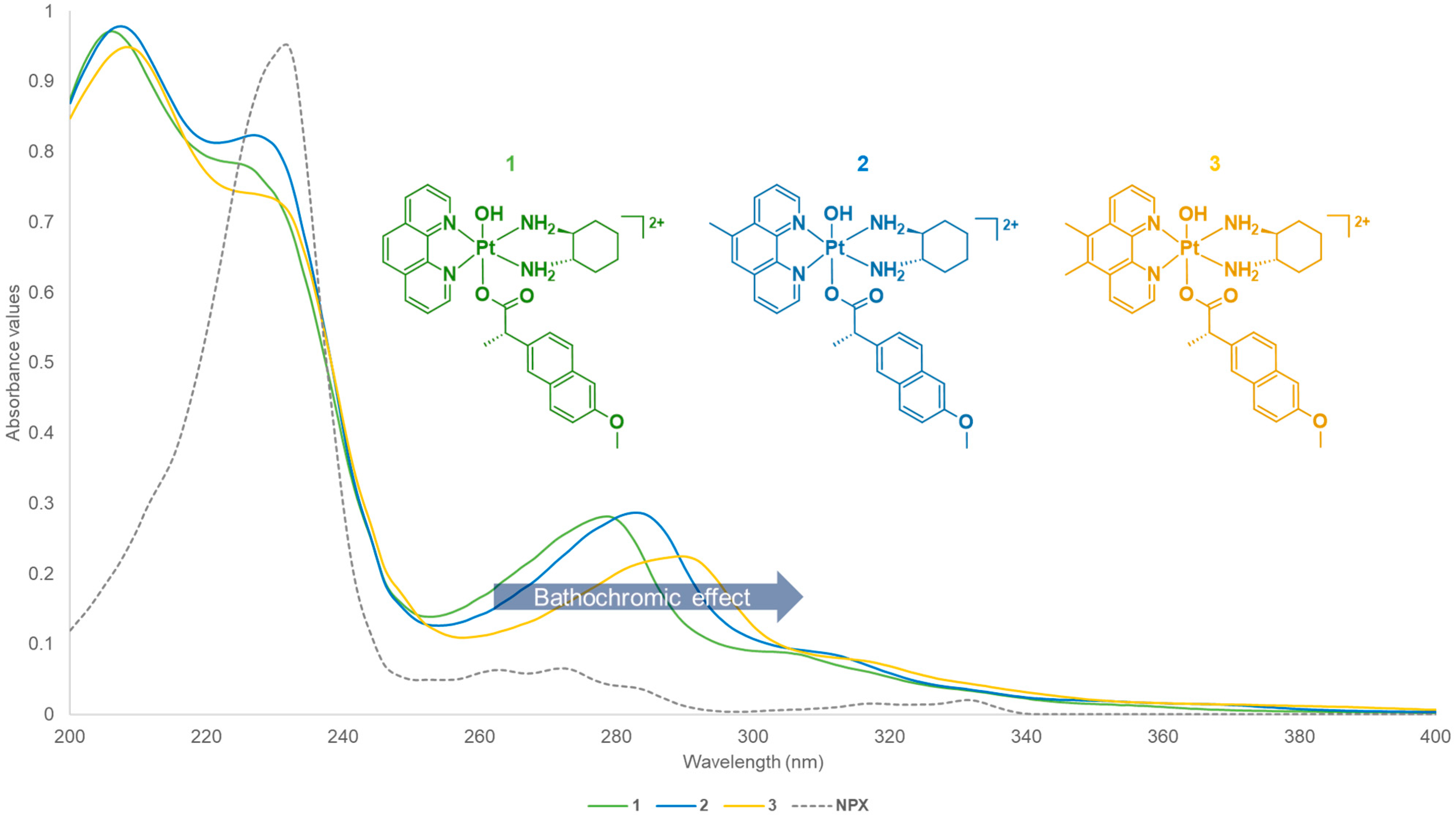
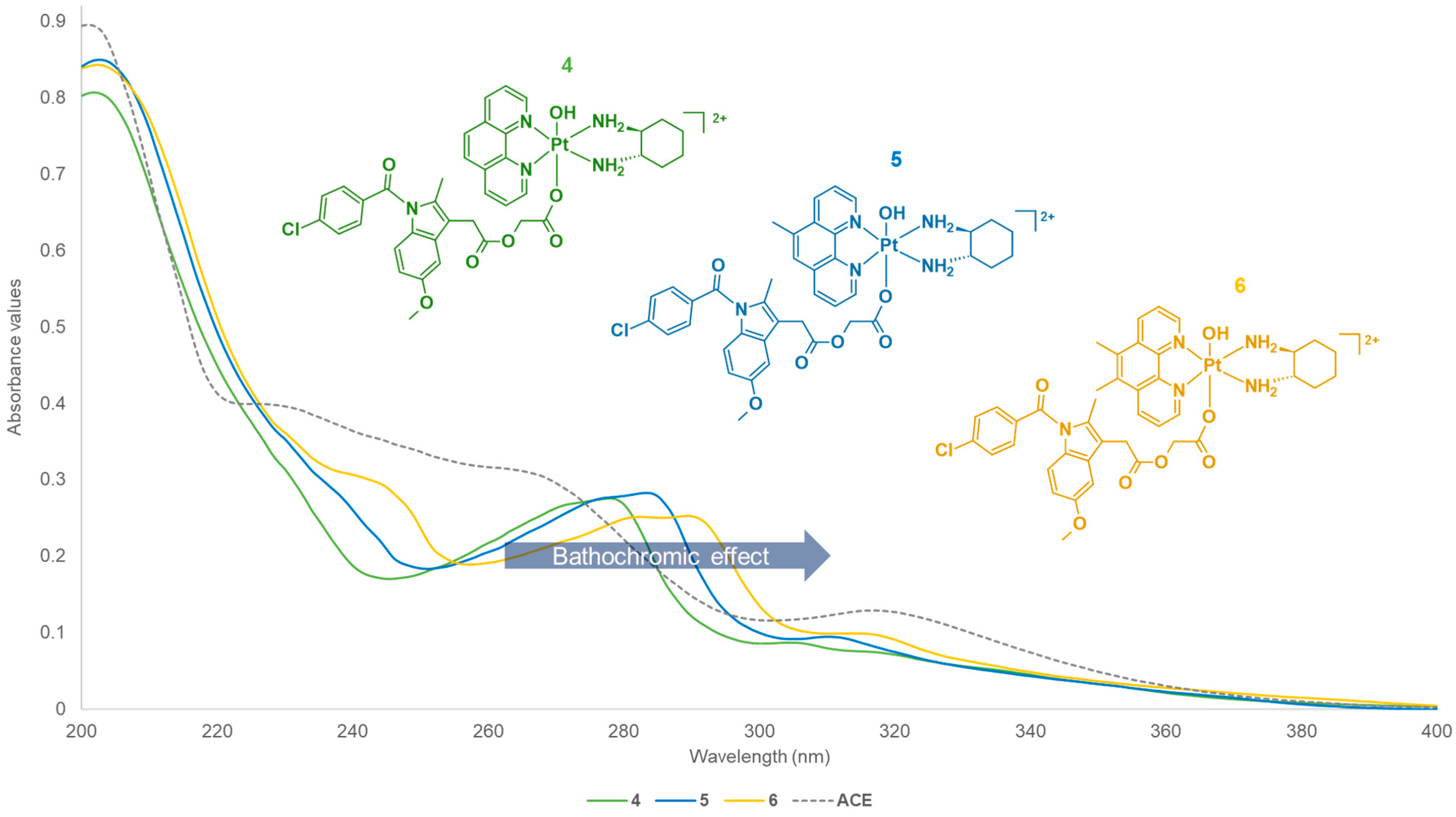
3.1.2. Electronic Spectra Analysis
3.1.3. Solubility Measurements
3.1.4. Lipophilicity Measurements
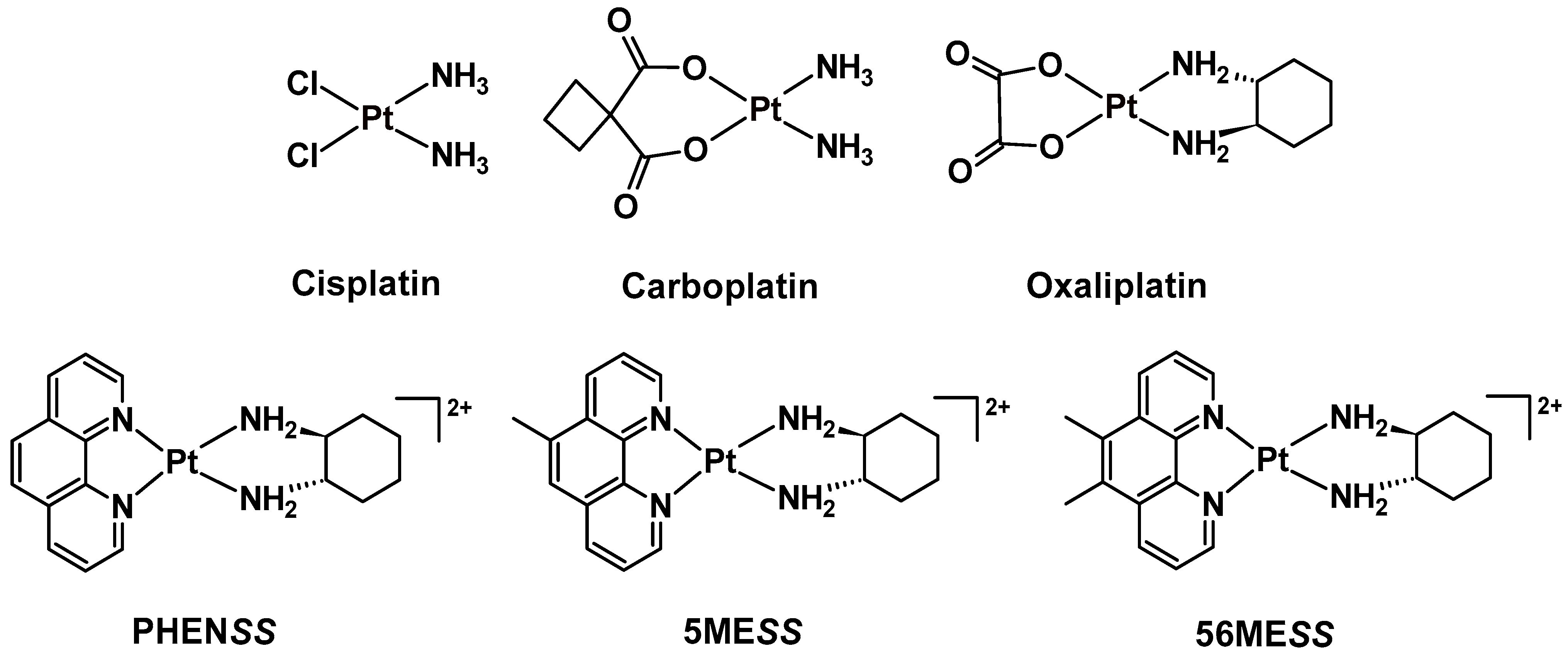
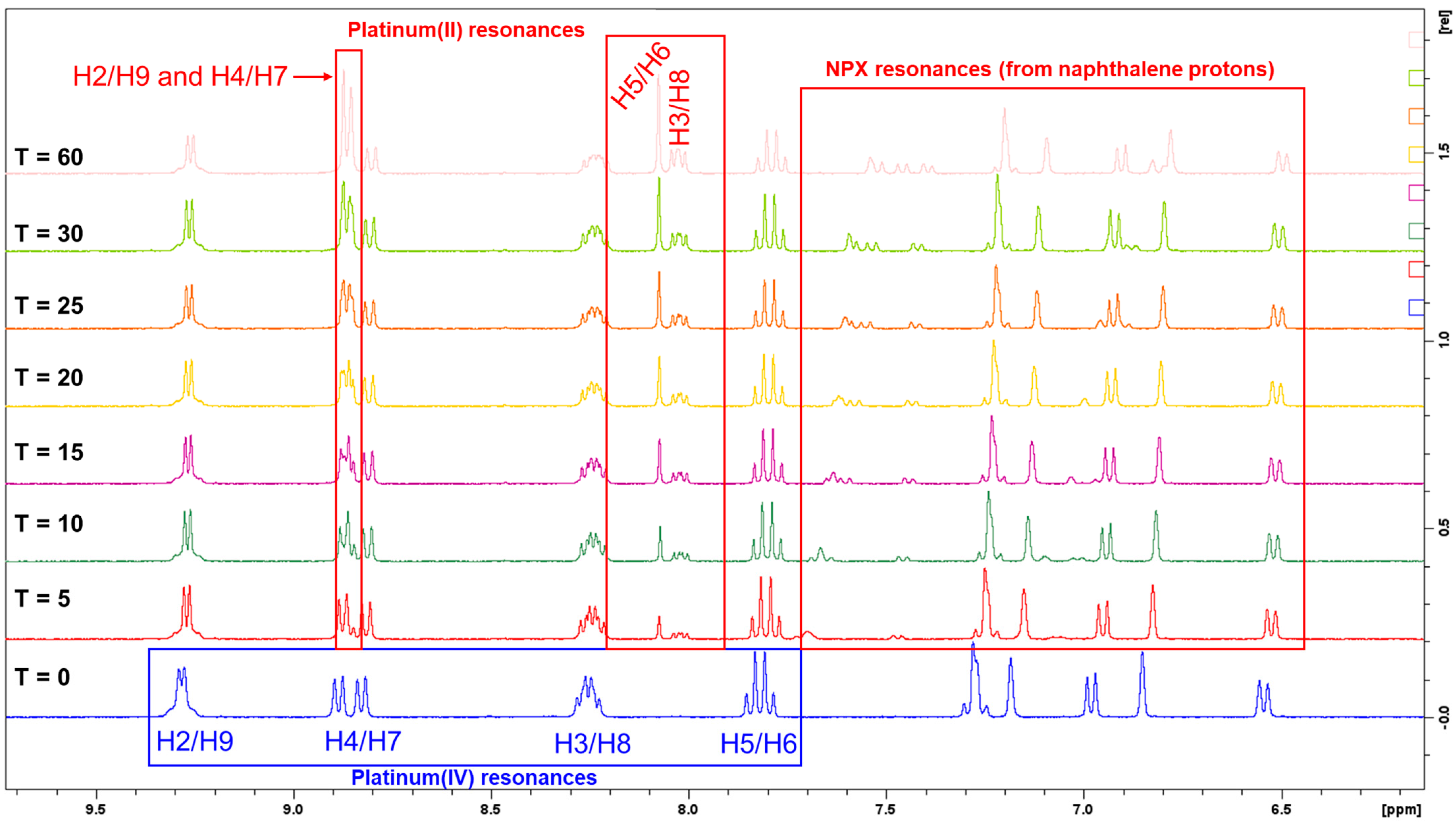
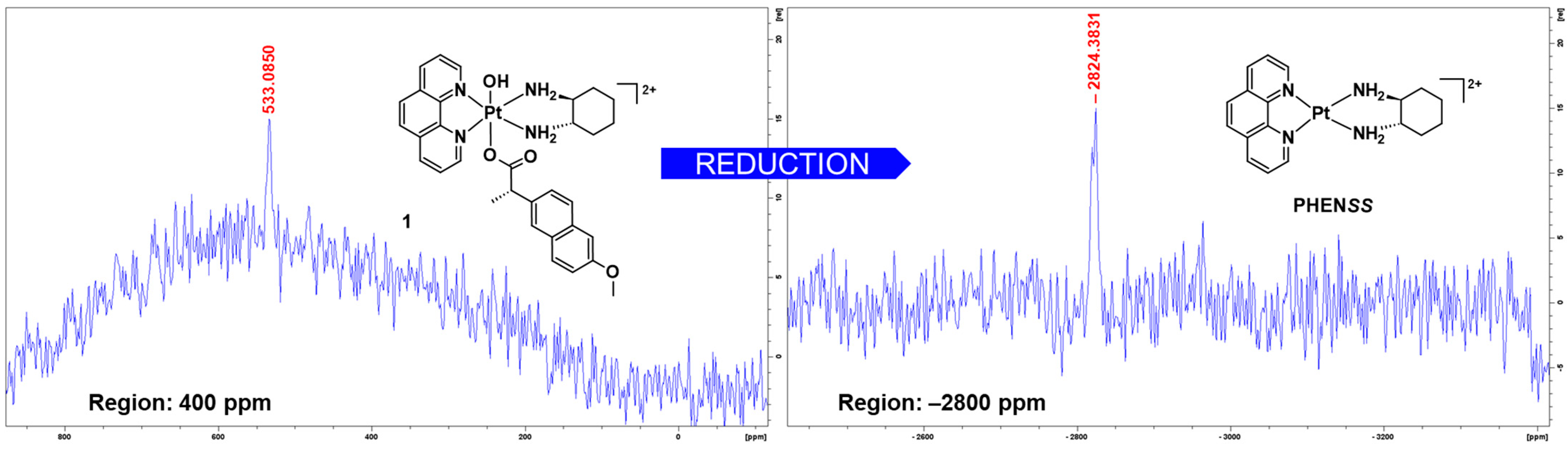
3.1.5. Preliminary Reduction Studies
3.1.6. Stability Studies
3.2. Biological Investigations
3.2.1. Growth Inhibition Studies
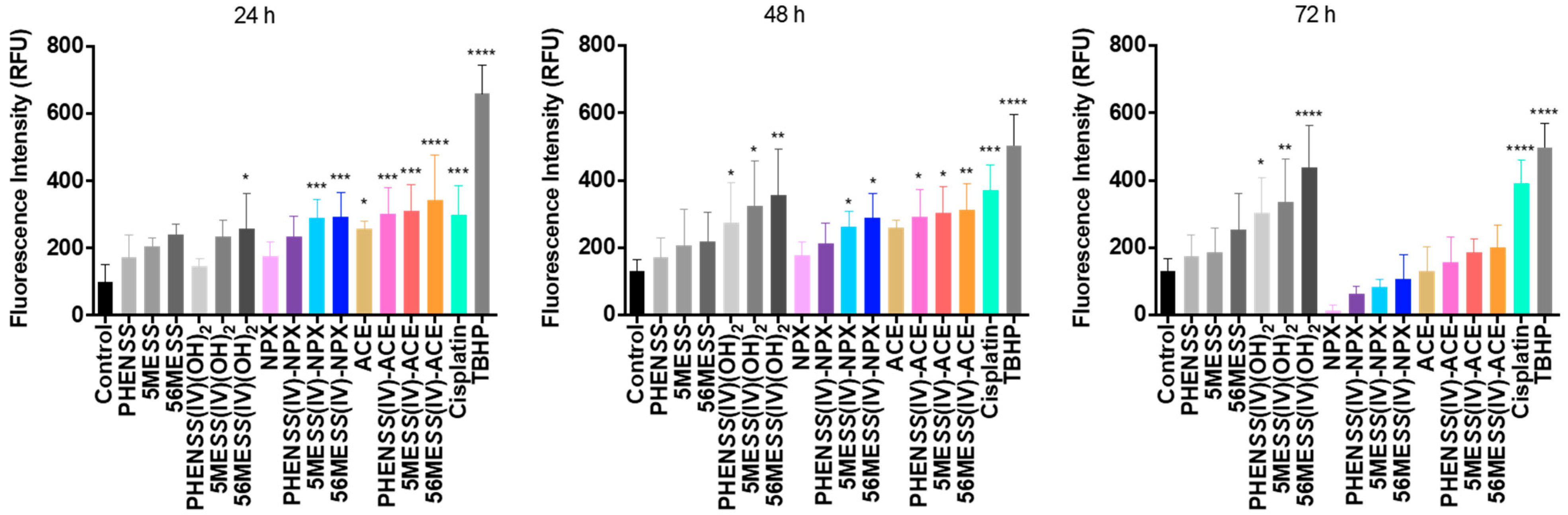
3.2.2. ROS Potential
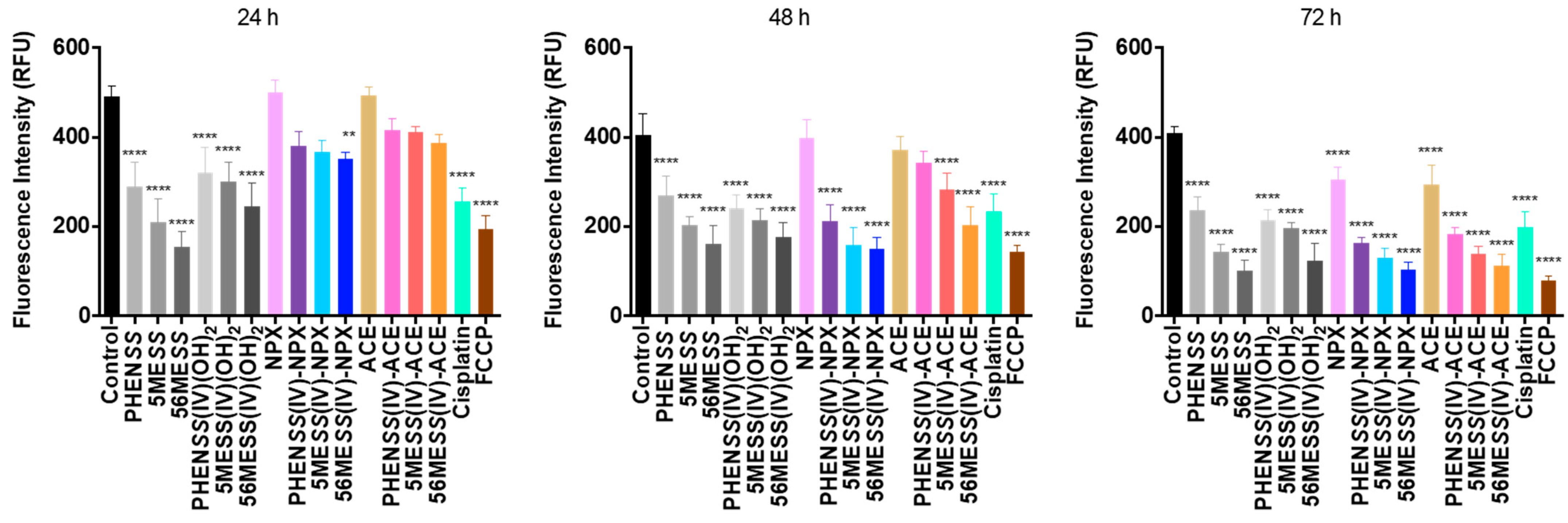
3.2.3. Mitochondrial Membrane Potential (MtMP)
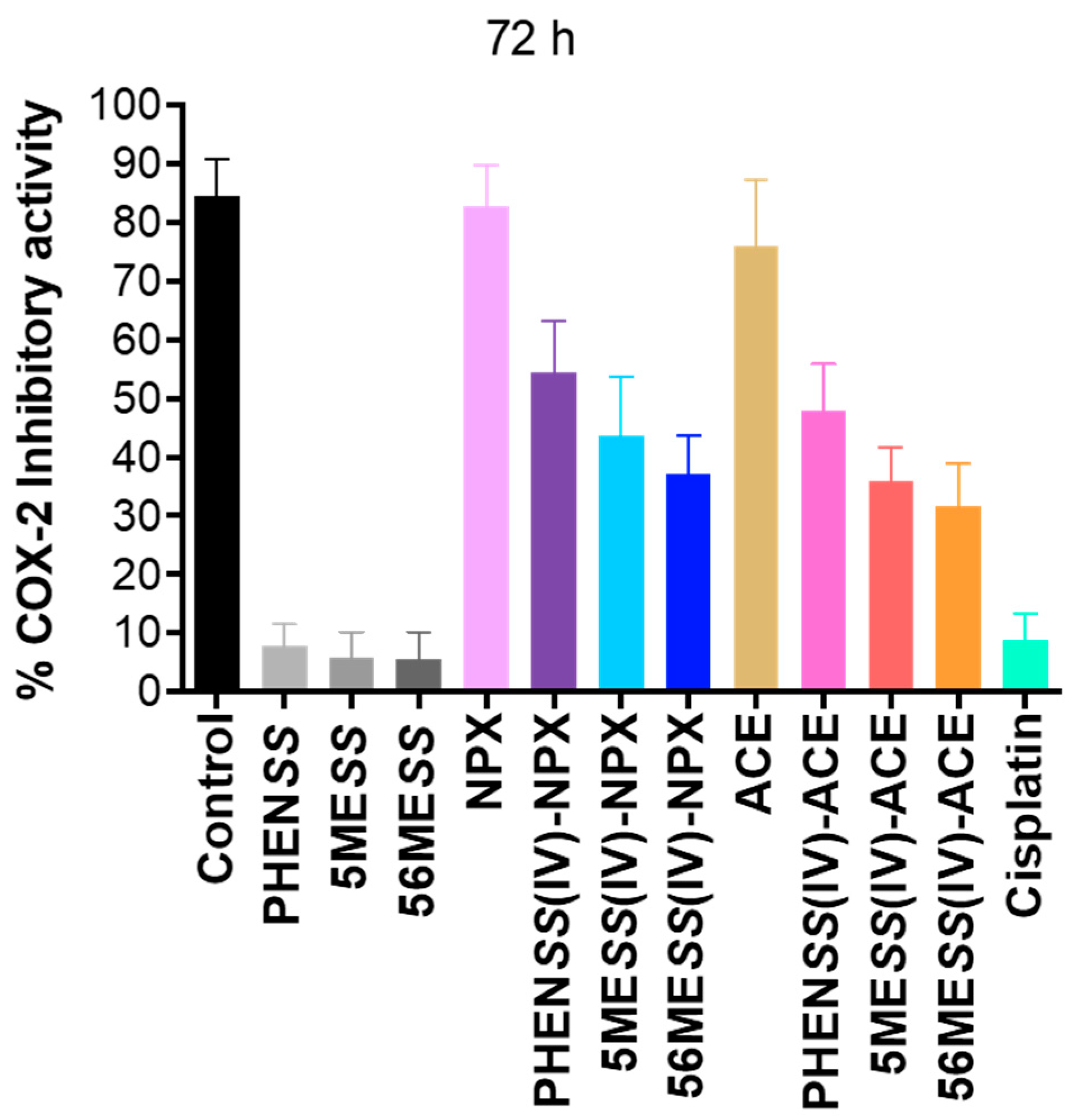
3.2.4. Cyclooxygenase-2 (COX) Inhibition
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Paprocka, R.; Wiese-Szadkowska, M.; Janciauskiene, S.; Kosmalski, T.; Kulik, M.; Helmin-Basa, A. Latest developments in metal complexes as anticancer agents. Coord. Chem. Rev. 2022, 452, 214307. [Google Scholar] [CrossRef]
- Pucci, C.; Martinelli, C.; Ciofani, G. Innovative approaches for cancer treatment: Current perspectives and new challenges. Ecancermedicalscience 2019, 13, 961. [Google Scholar] [CrossRef] [PubMed]
- Siddik, Z.H.; Al-Baker, S.; Thai, G.; Khokhar, A.R. Antitumor activity of isomeric 1,2-diaminocyclohexane platinum(IV) complexes. J. Cancer Res. Clin. Oncol. 1994, 120, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Kelland, L.R.; Sharp, S.Y.; O’Neill, C.F.; Raynaud, F.I.; Beale, P.J.; Judson, I.R. Mini-review: Discovery and development of platinum complexes designed to circumvent cisplatin resistance. J. Inorg. Biochem. 1999, 77, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.; Kumar, S.; Tchounwou, P.B. Cisplatin-based chemotherapy of human cancers. J. Cancer Sci. Ther. 2019, 11, 97. [Google Scholar]
- Brabec, V.; Kasparkova, J. Modifications of DNA by platinum complexes. Relation to resistance of tumors to platinum antitumor drugs. Drug Resist. Updat. 2005, 8, 131–146. [Google Scholar] [CrossRef]
- Florea, A.M.; Busselberg, D. Cisplatin as an anti-tumor drug: Cellular mechanisms of activity, drug resistance and induced side effects. Cancers 2011, 3, 1351–1371. [Google Scholar] [CrossRef]
- Gomez-Ruiz, S.; Maksimovic-Ivanic, D.; Mijatovic, S.; Kaluderovic, G.N. On the discovery, biological effects, and use of cisplatin and metallocenes in anticancer chemotherapy. Bioinorg. Chem. Appl. 2012, 2012, 140284. [Google Scholar] [CrossRef]
- Rocha, C.R.R.; Silva, M.M.; Quinet, A.; Cabral-Neto, J.B.; Menck, C.F.M. DNA repair pathways and cisplatin resistance: An intimate relationship. Clinics 2018, 73 (Suppl. S1), e478s. [Google Scholar] [CrossRef]
- Johnson, S.W.; Shen, D.W.; Pastan, I.; Gottesman, M.M.; Hamilton, T.C. Cross-resistance, cisplatin accumulation, and platinum–DNA adduct formation and removal in cisplatin-sensitive and -resistant human hepatoma cell lines. Exp. Cell Res. 1996, 226, 133–139. [Google Scholar] [CrossRef]
- Wheate, N.J.; Walker, S.; Craig, G.E.; Oun, R. The status of platinum anticancer drugs in the clinic and in clinical trials. Dalton Trans. 2010, 39, 8113–8127. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, J.T.; Lipp, H.P. Toxicity of platinum compounds. Expert. Opin. Pharmacother. 2003, 4, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Alassadi, S.; Pisani, M.J.; Wheate, N.J. A chemical perspective on the clinical use of platinum-based anticancer drugs. Dalton Trans. 2022, 51, 10835–10846. [Google Scholar] [CrossRef]
- Brodie, C.R.; Collins, J.G.; Aldrich-Wright, J.R. DNA binding and biological activity of some platinum(II) intercalating compounds containing methyl-substituted 1,10-phenanthrolines. Dalton Trans. 2004, 8, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Wheate, N.J.; Taleb, R.I.; Krause-Heuer, A.M.; Cook, R.L.; Wang, S.; Higgins, V.J.; Aldrich-Wright, J.R. Novel platinum(II)-based anticancer complexes and molecular hosts as their drug delivery vehicles. Dalton Trans. 2007, 43, 5055–5064. [Google Scholar] [CrossRef]
- Fisher, D.M.; Bednarski, P.J.; Grünert, R.; Turner, P.; Fenton, R.R.; Aldrich-Wright, J.R. Chiral platinum(II) metallointercalators with potent in vitro cytotoxic activity. Chem. Med. Chem. 2007, 2, 488–495. [Google Scholar] [CrossRef]
- Kemp, S.; Wheate, N.J.; Buck, D.P.; Nikac, M.; Collins, J.G.; Aldrich-Wright, J.R. The effect of ancillary ligand chirality and phenanthroline functional group substitution on the cytotoxicity of platinum(II)-based metallointercalators. J. Inorg. Biochem. 2007, 101, 1049–1058. [Google Scholar] [CrossRef]
- Fisher, D.M.; Fenton, R.; Aldrich-Wright, J. In Vivo studies of a platinum(II) metallointercalator. Chem. Comm. 2008, 43, 5613–5615. [Google Scholar] [CrossRef]
- Krause-Heuer, A.M.; Grunert, R.; Kuhne, S.; Buczkowska, M.; Wheate, N.J.; Le Pevelen, D.D.; Boag, L.R.; Fisher, D.M.; Kasparkova, J.; Malina, J.; et al. Studies of the mechanism of action of platinum(II) complexes with potent cytotoxicity in human cancer cells. J. Med. Chem. 2009, 52, 5474–5484. [Google Scholar] [CrossRef]
- Kostrhunova, H.; Zajac, J.; Novohradsky, V.; Kasparkova, J.; Malina, J.; Aldrich-Wright, J.R.; Petruzzella, E.; Sirota, R.; Gibson, D.; Brabec, V. A subset of new platinum antitumor agents kills cells by a multimodal mechanism of action also involving changes in the organization of the microtubule cytoskeleton. J. Med. Chem. 2019, 62, 5176–5190. [Google Scholar] [CrossRef] [PubMed]
- Macias, F.J.; Deo, K.M.; Pages, B.J.; Wormell, P.; Clegg, J.K.; Zhang, Y.; Li, F.; Zheng, G.; Sakoff, J.; Gilbert, J.; et al. Synthesis and analysis of the structure, diffusion and cytotoxicity of heterocyclic platinum(iv) complexes. Eur. J. Chem. 2015, 21, 16990–17001. [Google Scholar] [CrossRef] [PubMed]
- Garbutcheon-Singh, K.B.; Myers, S.; Harper, B.W.; Ng, N.S.; Dong, Q.; Xie, C.; Aldrich-Wright, J.R. The effects of 56MESS on mitochondrial and cytoskeletal proteins and the cell cycle in MDCK cells. Metallomics 2013, 5, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Moretto, J.; Chauffert, B.; Ghiringhelli, F.; Aldrich-Wright, J.R.; Bouyer, F. Discrepancy between in vitro and in vivo antitumor effect of a new platinum(II) metallointercalator. Investig. New Drugs 2011, 29, 1164–1176. [Google Scholar] [CrossRef]
- Harper, B.W.J.; Petruzzella, E.; Sirota, R.; Faccioli, F.F.; Aldrich-Wright, J.R.; Gandin, V.; Gibson, D. Synthesis, characterization and in vitro and in vivo anticancer activity of Pt(iv) derivatives of [Pt(1S,2S-DACH)(5,6-dimethyl-1,10-phenanthroline)]. Dalton Trans. 2017, 46, 7005–7019. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.D.; Hambley, T.W. Platinum(IV) antitumour compounds: Their bioinorganic chemistry. Coord. Chem. Rev. 2002, 232, 49–67. [Google Scholar] [CrossRef]
- Wong, D.Y.Q.; Ang, W.H. Development of platinum(IV) complexes as anticancer prodrugs: The story so far. Cosmos 2012, 8, 121–135. [Google Scholar] [CrossRef]
- Wang, Z.; Deng, Z.; Zhu, G. Emerging platinum(iv) prodrugs to combat cisplatin resistance: From isolated cancer cells to tumor microenvironment. Dalton Trans. 2019, 48, 2536–2544. [Google Scholar] [CrossRef]
- Jia, C.; Deacon, G.B.; Zhang, Y.; Gao, C. Platinum(IV) antitumor complexes and their nano-drug delivery. Coord. Chem. Rev. 2021, 429, 213640–213675. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, Z.; Deng, Z.; Zhu, G. Recent advances in the synthesis, stability, and activation of platinum(IV) anticancer prodrugs. Coord. Chem. Rev. 2021, 442, 213991–214023. [Google Scholar] [CrossRef]
- Ravera, M.; Gabano, E.; McGlinchey, M.J.; Osella, D. Pt(IV) antitumor prodrugs: Dogmas, paradigms, and realities. Dalton Trans. 2022, 51, 2121–2134. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.D.; Mellor, H.R.; Callaghan, R.; Hambley, T.W. Basis for design and development of platinum(IV) anticancer complexes. J. Med. Chem. 2007, 50, 3403–3411. [Google Scholar] [CrossRef]
- Coffetti, G.; Moraschi, M.; Facchetti, G.; Rimoldi, I. The challenging treatment of cisplatin-resistant tumors: State of the art and future perspectives. Molecules 2023, 28, 3407. [Google Scholar] [CrossRef]
- Wexselblatt, E.; Gibson, D. What do we know about the reduction of Pt(IV) pro-drugs? J. Inorg. Biochem. 2012, 117, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Bayat Mokhtari, R.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef]
- Johnstone, T.C.; Suntharalingam, K.; Lippard, S.J. The next generation of platinum drugs: Targeted Pt(ii) agents, nanoparticle delivery, and Pt(iv) prodrugs. Chem. Rev. 2016, 116, 3436–3486. [Google Scholar] [CrossRef]
- Spector, D.; Krasnovskaya, O.; Pavlov, K.; Erofeev, A.; Gorelkin, P.; Beloglazkina, E.; Majouga, A. Pt(IV) prodrugs with NSAIDs as axial ligands. Int. J. Mol. Sci. 2021, 22, 3817. [Google Scholar] [CrossRef]
- Barth, M.-C.; Häfner, N.; Runnebaum, I.B.; Weigand, W. Synthesis, characterization and biological investigation of the platinum(iv) tolfenamato prodrug-resolving cisplatin-resistance in ovarian carcinoma cell lines. Int. J. Mol. Sci. 2023, 24, 5718. [Google Scholar] [CrossRef]
- Spector, D.V.; Pavlov, K.G.; Akasov, R.A.; Vaneev, A.N.; Erofeev, A.S.; Gorelkin, P.V.; Nikitina, V.N.; Lopatukhina, E.V.; Semkina, A.S.; Vlasova, K.Y.; et al. Pt(IV) prodrugs with non-steroidal anti-inflammatory drugs in the axial position. J. Med. Chem. 2022, 65, 8227–8244. [Google Scholar] [CrossRef]
- Li, Z.; Wang, Q.; Li, L.; Chen, Y.; Cui, J.; Liu, M.; Zhang, N.; Liu, Z.; Han, J.; Wang, Z. Ketoprofen and loxoprofen platinum(iv) complexes displaying antimetastatic activities by inducing DNA damage, inflammation suppression, and enhanced immune response. J. Med. Chem. 2021, 64, 17920–17935. [Google Scholar] [CrossRef]
- Yasir Khan, H.; Parveen, S.; Yousuf, I.; Tabassum, S.; Arjmand, F. Metal complexes of NSAIDs as potent anti-tumor chemotherapeutics: Mechanistic insights into cytotoxic activity via multiple pathways primarily by inhibition of COX–1 and COX–2 enzymes. Coord. Chem. Rev. 2022, 453, 214316. [Google Scholar] [CrossRef]
- Mazhar, D.; Gillmore, R.; Waxman, J. COX and cancer. Int. J. Med. 2005, 98, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Sangha, S.; Yao, M.; Wolfe, M.M. Non-steroidal anti-inflammatory drugs and colorectal cancer prevention. Postgrad. Med. J. 2005, 81, 223–227. [Google Scholar] [CrossRef]
- Thun, M.J.; Henley, S.J.; Patrono, C. Nonsteroidal anti-inflammatory drugs as anticancer agents: Mechanistic, pharmacologic, and clinical issues. J. Natl. Cancer Ins. 2002, 94, 252–266. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Inza, S.; Ruberte, A.C.; Sanmartín, C.; Sharma, A.K.; Plano, D. NSAIDs: Old acquaintance in the pipeline for cancer treatment and prevention—Structural modulation, mechanisms of action, and bright future. J. Med. Chem. 2021, 64, 16380–16421. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Angulo, A.M.; Fuloria, J.; Prakash, O. Cyclooxygenase 2 inhibitors and colon cancer. Ochsner J. 2002, 4, 176–179. [Google Scholar]
- Ricciotti, E.; FitzGerald, G.A. Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 986–1000. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Cao, L.; Li, Z.; Qu, D. Metabolic reprogramming links chronic intestinal inflammation and the oncogenic transformation in colorectal tumorigenesis. Cancer Lett. 2019, 450, 123–131. [Google Scholar] [CrossRef]
- Pannunzio, A.; Coluccia, M. Cyclooxygenase-1 (COX-1) and COX-1 inhibitors in cancer: A review of oncology and medicinal chemistry literature. Pharmaceuticals 2018, 11, 101. [Google Scholar] [CrossRef]
- Szweda, M.; Rychlik, A.; Babińska, I.; Pomianowski, A. Significance of cyclooxygenase-2 in oncogenesis. J. Vet. Res. 2019, 63, 215–224. [Google Scholar] [CrossRef]
- Liu, B.; Qu, L.; Yan, S. Cyclooxygenase-2 promotes tumor growth and suppresses tumor immunity. Cancer Cell Int. 2015, 15, 106. [Google Scholar] [CrossRef] [PubMed]
- Aputen, A. Novel Platinum(IV) Prodrugs. Master’s Thesis, Western Sydney University, Campbelltown, Australia, 2019. [Google Scholar]
- Aputen, A.D.; Elias, M.G.; Gilbert, J.; Sakoff, J.A.; Gordon, C.P.; Scott, K.F.; Aldrich-Wright, J.R. Bioactive platinum(iv) complexes incorporating halogenated phenylacetates. Molecules 2022, 27, 7120. [Google Scholar] [CrossRef] [PubMed]
- Barré, A.; Tintas, M.; Levacher, V.; Papamicael, C.; Vincent, G. An overview of the synthesis of highly versatile n-hydroxysuccinimide esters. Synthesis 2017, 49, 472–483. [Google Scholar]
- Aputen, A.; Elias, M.G.; Gilbert, J.; Sakoff, J.A.; Gordon, C.P.; Scott, K.F.; Aldrich-Wright, J.R. Potent chlorambucil-platinum(IV) prodrugs. Int. J. Mol. Sci. 2022, 23, 10471. [Google Scholar] [CrossRef] [PubMed]
- Deo, K.M.; Sakoff, J.; Gilbert, J.; Zhang, Y.; Aldrich Wright, J.R. Synthesis, characterisation and influence of lipophilicity on cellular accumulation and cytotoxicity of unconventional platinum(iv) prodrugs as potent anticancer agents. Dalton Trans. 2019, 48, 17228–17240. [Google Scholar] [CrossRef]
- Deo, K.M.; Sakoff, J.; Gilbert, J.; Zhang, Y.; Aldrich Wright, J.R. Synthesis, characterisation and potent cytotoxicity of unconventional platinum(iv) complexes with modified lipophilicity. Dalton Trans. 2019, 48, 17217–17227. [Google Scholar] [CrossRef]
- Khoury, A.; Sakoff, J.A.; Gilbert, J.; Scott, K.F.; Karan, S.; Gordon, C.P.; Aldrich-Wright, J.R. Cyclooxygenase-inhibiting platinum(iv) prodrugs with potent anticancer activity. Pharmaceutics 2022, 14, 787. [Google Scholar] [CrossRef]
- Khoury, A.; Sakoff, J.A.; Gilbert, J.; Karan, S.; Gordon, C.P.; Aldrich-Wright, J.R. Potent platinum(iv) prodrugs that incorporate a biotin moiety to selectively target cancer cells. Pharmaceutics 2022, 14, 2780. [Google Scholar] [CrossRef]
- Tarleton, M.; Gilbert, J.; Robertson, M.J.; McCluskey, A.; Sakoff, J.A. Library synthesis and cytotoxicity of a family of 2-phenylacrylonitriles and discovery of an estrogen dependent breast cancer lead compound. Med. Chem. Comm. 2011, 2, 31–37. [Google Scholar] [CrossRef]
- López-Lázaro, M. A simple and reliable approach for assessing anticancer activity in vitro. Curr. Med. Chem. 2015, 22, 1324–1334. [Google Scholar] [CrossRef]
- López-Lázaro, M. Two preclinical tests to evaluate anticancer activity and to help validate drug candidates for clinical trials. Oncoscience 2015, 2, 91–98. [Google Scholar] [CrossRef]
- Elias, M.G.; Mehanna, S.; Elias, E.; Khnayzer, R.S.; Daher, C.F. A photoactivatable chemotherapeutic Ru(II) complex bearing bathocuproine ligand efficiently induces cell death in human malignant melanoma cells through a multi-mechanistic pathway. Chem. Biol. Interact. 2021, 348, 109644. [Google Scholar] [CrossRef]
- Mehanna, S.; Mansour, N.; Daher, C.F.; Elias, M.G.; Dagher, C.; Khnayzer, R.S. Drug-free phototherapy of superficial tumors: White light at the end of the tunnel. J. Photochem. Photobiol. B 2021, 224, 112324. [Google Scholar] [CrossRef] [PubMed]
- Garbutcheon-Singh, K.B.; Leverett, P.; Myers, S.; Aldrich-Wright, J.R. Cytotoxic platinum(II) intercalators that incorporate 1R,2R-diaminocyclopentane. Dalton Trans. 2013, 42, 918–926. [Google Scholar] [CrossRef] [PubMed]
- McGhie, B.S.; Sakoff, J.; Gilbert, J.; Aldrich-Wright, J.R. Synthesis and characterisation of platinum(IV) polypyridyl complexes with halide axial ligands. Inorg. Chim. Acta 2019, 495, 118964–118974. [Google Scholar] [CrossRef]
- McGhie, B.S.; Sakoff, J.; Gilbert, J.; Gordon, C.P.; Aldrich-Wright, J.R. Novel planar Pt(ii) cyclometallated cytotoxic complexes with g-quadruplex stabilisation and luminescent properties. Int. J. Mol. Sci. 2022, 23, 10469. [Google Scholar] [CrossRef]
- Schilt, A.A.; Taylor, R.C. Infra-red spectra of 1:10-phenanthroline metal complexes in the rock salt region below 2000 cm−1. J. Inorg. Nucl. Chem. 1959, 9, 211–221. [Google Scholar] [CrossRef]
- Wang, X.; Jia, G.; Yu, Y.; Gao, Y.; Zhang, W.; Wang, H.; Cao, Z.; Liu, J. A new homogeneous electrocatalyst for electrochemical carbonylation methanol to dimethyl carbonate. Quím. Nova 2015, 38, 298–302. [Google Scholar] [CrossRef]
- Reiher, M.; Brehm, G.; Schneider, S. Assignment of vibrational spectra of 1,10-phenanthroline by comparison with frequencies and Raman intensities from density functional calculations. J. Phys. Chem. A 2004, 108, 734–742. [Google Scholar] [CrossRef]
- Sun, Y.; Wei, H.; Zhang, Q.; Zhao, X. Platinum(II) complexes with 1,10-phenanthroline and hydrophilic alkoxyacetate ligands as potential antitumor agents. Chem. Biodivers. 2019, 16, e1800373. [Google Scholar] [CrossRef]
- Tolan, D.; Abdel-Monem, Y.K.; El-Nagar, M.A. Anti-tumor platinum (IV) complexes bearing the anti-inflammatory drug naproxen in the axial position. Appl. Organomet. Chem. 2019, 33, e4763. [Google Scholar] [CrossRef]
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug Solubility: Importance and Enhancement Techniques. ISRN Pharm. 2012, 2012, 195727. [Google Scholar] [CrossRef] [PubMed]
- Tsang, R.Y.; Al-Fayea, T.; Au, H.J. Cisplatin overdose: Toxicities and management. Drug Saf. 2009, 32, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, A.; Vaishampayan, U.N. Satraplatin: Leading the new generation of oral platinum agents. Expert Opin. Investig. Drugs 2009, 18, 1787–1797. [Google Scholar] [CrossRef]
- Vyas, N.; Turner, A.; Sewell, G. Platinum-based anticancer drugs in waste waters of a major UK hospital and predicted concentrations in recipient surface waters. Sci. Total Environ. 2014, 493, 324–329. [Google Scholar] [CrossRef]
- Mälkiä, A.; Murtomäki, L.; Urtti, A.; Kontturi, K. Drug permeation in biomembranes: In Vitro and In Silico prediction and influence of physicochemical properties. Eur. J. Pharm. Sci. 2004, 23, 13–47. [Google Scholar] [CrossRef]
- Starek, M.; Plenis, A.; Zagrobelna, M.; Dąbrowska, M. Assessment of lipophilicity descriptors of selected NSAIDs obtained at different tlc stationary phases. Pharmaceutics 2021, 13, 440. [Google Scholar] [CrossRef]
- Asirvatham, S.; Dhokchawle, B.V.; Tauro, S.J. Quantitative structure activity relationships studies of non-steroidal anti-inflammatory drugs: A review. Arab. J. Chem. 2019, 12, 3948–3962. [Google Scholar] [CrossRef]
- Dorwald, F.Z. Lead Optimization for Medicinal Chemists: Pharmacokinetic Properties of Functional Groups and Organic Compounds; Wiley-VCH: Weinheim, Germany, 2012. [Google Scholar]
- Chen, C.K.J.; Gui, X.; Kappen, P.; Renfrew, A.K.; Hambley, T.W. The effect of charge on the uptake and resistance to reduction of platinum(IV) complexes in human serum and whole blood models. Metallomics 2020, 12, 1599–1615. [Google Scholar] [CrossRef]
- Chen, C.K.J.; Kappen, P.; Gibson, D.; Hambley, T.W. trans-Platinum(iv) pro-drugs that exhibit unusual resistance to reduction by endogenous reductants and blood serum but are rapidly activated inside cells: 1H NMR and XANES spectroscopy study. Dalton Trans. 2020, 49, 7722–7736. [Google Scholar] [CrossRef]
- Chen, C.K.J.; Kappen, P.; Hambley, T.W. The reduction of cis-platinum(IV) complexes by ascorbate and in whole human blood models using 1H NMR and XANES spectroscopy. Metallomics 2019, 11, 686–695. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.-Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef]
- Jia, P.; Dai, C.; Cao, P.; Sun, D.; Ouyang, R.; Miao, Y. The role of reactive oxygen species in tumor treatment. RSC Adv. 2020, 10, 7740–7750. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of reactive oxygen species in cancer progression: Molecular mechanisms and recent advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Gupta, S.C.; Tyagi, A.K. Reactive oxygen species (ROS) and cancer: Role of antioxidative nutraceuticals. Cancer Lett. 2017, 387, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Snezhkina, A.V.; Kudryavtseva, A.V.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS Generation and antioxidant defense systems in normal and malignant cells. Oxid. Med. Cell Longev. 2019, 2019, 6175804. [Google Scholar] [CrossRef]
- Kumari, S.; Badana, A.K.; Murali, M.G.; Shailender, G.; Malla, R. Reactive oxygen species: A key constituent in cancer survival. Biomark. Insights 2018, 13, 1177271918755391. [Google Scholar] [CrossRef]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Reactive oxygen species and cancer paradox: To promote or to suppress? Free Radic. Biol. Med. 2017, 104, 144–164. [Google Scholar] [CrossRef]
- Mahmood, K.-A.S.; Ahmed, J.H.; Jawad, A.M. Non-steroidal anti-inflammatory drugs (NSAIDs), free radicals and reactive oxygen species (ROS): A review of literature. Med. J. Basrah 2009, 27, 46–53. [Google Scholar] [CrossRef]
- Ralph, S.J.; Pritchard, R.; Rodríguez-Enríquez, S.; Moreno-Sánchez, R.; Ralph, R.K. Hitting the bull’s-eye in metastatic cancers-nsaids elevate ros in mitochondria, inducing malignant cell death. Pharmaceuticals 2015, 8, 62–106. [Google Scholar] [CrossRef]
- Ramazani, E.; Tayarani-Najaran, Z.; Fereidoni, M. Celecoxib, indomethacin, and ibuprofen prevent 6-hydroxydopamine-induced PC12 cell death through the inhibition of NFκB and SAPK/JNK pathways. Iran J. Basic Med. Sci. 2019, 22, 477–484. [Google Scholar] [PubMed]
- Koncic, M.Z.; Rajic, Z.; Petric, N.; Zorc, B. Antioxidant activity of NSAID hydroxamic acids. Acta Pharm. 2009, 59, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Pistritto, G.; Trisciuoglio, D.; Ceci, C.; Garufi, A.; D’Orazi, G. Apoptosis as anticancer mechanism: Function and dysfunction of its modulators and targeted therapeutic strategies. Aging 2016, 8, 603–619. [Google Scholar] [CrossRef] [PubMed]
- Suski, J.; Lebiedzinska, M.; Bonora, M.; Pinton, P.; Duszynski, J.; Wieckowski, M.R. Relation between mitochondrial membrane potential and ROS formation. In Mitochondrial Bioenergetics: Methods and Protocols, 2nd ed.; Palmeira, C.M., Moreno, A.J., Eds.; Springer: New York, NY, USA, 2018; pp. 357–381. [Google Scholar]
- Joshi, D.; Bakowska, J. Determination of mitochondrial membrane potential and reactive oxygen species in live rat cortical neurons. J. Vis. Exp. 2011, 51, 2704. [Google Scholar]
- Zajac, J.; Kostrhunova, H.; Novohradsky, V.; Vrána, O.; Raveendran, R.; Gibson, D.; Kasparkova, J.; Brabec, V. Potentiation of mitochondrial dysfunction in tumor cells by conjugates of metabolic modulator dichloroacetate with a Pt(IV) derivative of oxaliplatin. J. Inorg. Biochem. 2016, 156, 89–97. [Google Scholar] [CrossRef]
- Suntharalingam, K.; Awuah, S.G.; Bruno, P.M.; Johnstone, T.C.; Wang, F.; Lin, W.; Zheng, Y.R.; Page, J.E.; Hemann, M.T.; Lippard, S.J. Necroptosis-inducing rhenium(V) oxo complexes. J. Am. Chem. Soc. 2015, 137, 2967–2974. [Google Scholar] [CrossRef]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Margreiter, R.; Ausserlechner, M.J.; Hagenbuchner, J. The complex interplay between mitochondria, ROS and entire cellular metabolism. Antioxidants 2022, 11, 1995. [Google Scholar] [CrossRef]
- Zhang, B.-b.; Wang, D.-g.; Guo, F.-f.; Xuan, C. Mitochondrial membrane potential and reactive oxygen species in cancer stem cells. Fam. Cancer 2015, 14, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Hawkey, C.J. COX-1 and COX-2 inhibitors. Best Pract. Res. Clin. Gastroenterol. 2001, 15, 801–820. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Wei, Y.; Chen, Q.; Xing, D. Cyclooxygenase 2-mediated apoptotic and inflammatory responses in photodynamic therapy treated breast adenocarcinoma cells and xenografts. J. Photochem. Photobiol. 2014, 134, 27–36. [Google Scholar] [CrossRef] [PubMed]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Platinum(IV) Complexes | Experimental Yields (%) | TR (min) | HPLC Peak Areas (%) | Mass-to-Charge Ratios (m/z) | |
|---|---|---|---|---|---|
| Calc. | Exp. | ||||
| 1 | 60.0 | 7.78 | 97 | 734.23 | 734.23 |
| 2 | 69.0 | 7.98 | 98 | 748.25 | 748.25 |
| 3 | 65.0 | 8.19 | 99 | 762.26 | 762.26 |
| 4 | 62.0 | 9.62 | 97 | 919.22 | 920.22 |
| 5 | 68.0 | 9.83 | 96 | 933.23 | 934.22 |
| 6 | 58.0 | 10.2 | 97 | 947.25 | 948.25 |
| Proton Labels | 1 | 2 | 3 |
|---|---|---|---|
| H2 | 9.12 (t, 2H, J = 5.0 Hz) (signals overlapping) | 9.12 (t, 1H, J = 5.6 Hz) | 9.06 (d, 2H, J = 5.5 Hz) (signals overlapping) |
| H9 | 9.04 (d, 1H, J = 5.6 Hz) | ||
| H4 | 8.74 (d, 1H, J = 8.3 Hz) | 8.84 (q, 1H, J = 8.5 Hz) | 8.84 (t, 2H, J = 9.0 Hz) (signals overlapping) |
| H7 | 8.70 (d, 1H, J = 8.3 Hz) | 8.60 (q, 1H, J = 8.3 Hz) | |
| H5 and H6 | 7.67 (q, 2H, J = 8.9 Hz) | - | - |
| H6 | - | 7.40 (d, 1H, J = 19 Hz) | - |
| H3 | 8.10 (m, 2H) (signals overlapping) | 8.10 (m, 2H) (signals overlapping) | 8.09 (m, 2H) (signals overlapping) |
| H8 | |||
| a | 7.05 (s, 1H) | 6.67 (d, 1H, J = 7.8 Hz) | 6.94 (s, 1H) |
| b | 6.71 (s, 1H) | 6.40 (d, 1H, J = 8.4 Hz) | 6.62 (s, 1H) |
| c | 7.12 (m, 2H) (signals overlapping) | 7.10 (d, 2H, J = 3.2 Hz) (signals overlapping) | 7.01 (s, 2H) (signals overlapping) |
| d | |||
| e | 6.84 (d, 1H, J = 8.5 Hz) | 7.03 (d, 1H, J = 4.5 Hz) | 6.77 (d, 1H, J = 8.5 Hz) |
| f | 6.40 (d, 1H, J1 = 1.4, J2 = 8.5 Hz) | 6.84 (dd, 1H, J1 = 2.4, J2 = 8.5 Hz) | 6.38 (d, 1H, J = 8.4 Hz) |
| γ | 3.99 (s, 3H) | 2.56 (s, 3H) | 3.92 (s, 3H) |
| α | 3.50 (m, 1H, J = 7.0 Hz) | 3.50 (m, 1H) | 3.48 (q, 1H, J = 7.0 Hz) |
| H1′ and H2′ | 3.12 (m, 2H) | 3.14 (d, 2H) | 3.16 (t, 2H) |
| CH3 (5 position) | - | 3.97 (s, 3H) | 3.92 (s, 3H) |
| 2 × CH3 (5 and 6 positions) | - | - | 2.42 (s, 8H) (signals overlapping) |
| H3′ and H6′ eq. | 2.37 (m, 2H) | 2.38 (d, 2H) | |
| H4′ and H5′ eq.; H3′ and H6′ ax. | 1.65 (m, 4H) | 1.66 (m, 4H) | 1.67 (m, 4H) |
| H4′ and H5′ ax. | 1.25 (m, 2H) | 1.27 (m, 2H) | 1.28 (m, 2H) |
| β | 1.15 (d, 3H, J = 7.1 Hz) | 1.15 (q, 3H, J = 4.6 Hz) | 1.15 (d, 3H, J = 7.0 Hz) |
| 1H/195Pt | 9.12, 8.10/545 | 9.12, 9.04/544 | 9.06, 8.09/534 |
| Proton Labels | 4 | 5 | 6 |
|---|---|---|---|
| H2 | 9.16 (d, 1H, J = 5.4 Hz) | 9.18 (dd, 1H, J = 5.5 Hz) | 9.12 (d, 1H, J = 5.4 Hz) |
| H9 | 9.14 (d, 1H, J = 5.5 Hz) | 9.06 (dd, 1H, J = 5.4 Hz) | 9.06 (d, 1H, J = 5.4 Hz) |
| H4 | 8.68 (q, 2H, J = 5.6 Hz) | 8.79 (q, 1H, J = 5.0 Hz) | 8.76 (q, 2H, J = 4.9 Hz) |
| H7 | 8.41 (q, 1H, J = 8.5 Hz) | ||
| H5 and H6 | 7.91 (s, 2H) | - | - |
| H6 | - | 7.58 (d, 1H, J = 6.4 Hz) | - |
| H3 | 8.03 (q, 2H, J = 5.8 Hz) (signals overlapping) | 8.08 (t, 1H, J = 6.8 Hz) | 7.98 (m, 1H) |
| H8 | 7.92 (m, 1H) | ||
| a and b | 7.21 (s, 4H) (signals overlapping) | 7.05 (q, 4H, J = 8.0 Hz) (signals overlapping) | 6.95 (s, 4H) (signals overlapping) |
| c and d | |||
| a1 | 6.53 (s, 1H) | 6.52 (s, 1H) | 6.53 (s, 2H) (signals overlapping) |
| b1 | 6.69 (d, 1H, J = 8.4 Hz) | 6.58 (d, 1H, J = 8.7 Hz) | |
| c1 | 6.42 (d, 1H, J = 8.4 Hz) | 6.27 (d, 1H, J = 8.6 Hz) | 6.18 (d, 1H, J = 7.6 Hz) |
| f | 3.55 (s, 3H) | 3.48 (s, 3H) | 3.44 (s, 3H) |
| α2 | 4.27 (s, 2H) | 4.28 (m, 2H) | 4.29 (s, 2H) |
| α1 | 3.37 (s, 2H) | 3.34 (s, 2H) | 3.33 (s, 2H) |
| H1′ and H2′ | 3.15 (s, 2H) | 3.15 (d, 2H) | 3.14 (m, 2H) |
| CH3 (5 position) | - | 2.50 (s, 3H) | - |
| 2 × CH3 (5 and 6 positions) | - | - | 2.38 (s, 8H) (signals overlapping) |
| H3′ and H6′ eq. | 2.33 (s, 2H) | 2.34 (d, 2H) | |
| e | 1.76 (s, 3H) | 1.65 (m, 7H) (signals overlapping) | 1.62 (m, 7H) (signals overlapping) |
| H4′ and H5′ eq.; H3′ and H6′ ax. | 1.65 (m, 4H) | ||
| H4′ and H5′ ax. | 1.21 (m, 2H) | 1.23 (m, 2H) | 1.23 (m, 2H) |
| β | - | - | - |
| 1H/195Pt | 9.16, 9.14, 8.03/547 | 9.18, 9.06, 8.08, 7.92/545 | 9.12, 9.09, 7.98/531 |
| Platinum (IV) Complexes | UV λmax nm (ε/M.cm−1 ± SD × 104) | CD λmax nm (Δε/M.cm−1 × 101) |
|---|---|---|
| 1 | 206 (8.92 ± 5.54), 227 (7.08 ± 3.59), 279 (2.56 ± 0.25), 306 (0.77 ± 0.88) | 202 (−1263), 224 (+178), 239 (+329), 281 (−137) |
| 2 | 207 (9.95 ± 6.78), 228 (8.39 ± 4.98), 283 (2.92 ± 0.12), 312 (0.86 ± 0.85) | 201 (−1061), 223 (+120), 232 (+48.1), 243 (+309), 287 (−109) |
| 3 | 208 (8.73 ± 5.35), 231 (6.74 ± 3.23), 289 (2.08 ± 0.42), 318 (0.67 ± 0.91) | 204 (−1007), 223 (+9.32), 230 (−9.99), 244 (+316), 293 (−93.2) |
| 4 | 203 (4.43 ± 0.42), 278 (1.15 ± 2.70), 305 (0.49 ± 3.40) | 206 (−447), 249 (+8.15) |
| 5 | 204 (18.6 ± 5.44), 284 (6.16 ± 2.63), 311 (2.12 ± 1.68) | 205 (−373), 209 (−419), 234 (−144) |
| 6 | 204 (16.7 ± 4.75), 244 (5.90 ± 2.34), 290 (5 ± 1.40), 316 (1.94 ± 0.47) | 214 (−175), 241 (−29.2), 285 (+11.9) |
| Platinum(IV) Complexes | Type of Bond and Frequency (cm−1) | |||||
|---|---|---|---|---|---|---|
| O-H Stretch (Alcohol) | C-H Stretch (Aromatic) | C-H Stretch (Alkyl) | C-C Stretch (Aromatic) | C-O Stretch (Alkyl Aryl Ether) | C-Cl Stretch (Halogen) | |
| 1 | 3380 | 3062 | 2936 | 1603 | 1220 | - |
| 2 | 3471 | 3076 | 2940 | 1605 | 1220 | - |
| 3 | 3466 | 3076 | 2937 | 1603 | 1218 | - |
| 4 | 3410 | 3063 | 2942 | 1609 | 1262 | 845 |
| 5 | 3385 | 3066 | 2943 | 1608 | 1265 | 846 |
| 6 | 3418 | 3071 | 2941 | 1609 | 1263 | 846 |
| Complexes | Solubility | |
|---|---|---|
| mol/L | mg/mL | |
| 1 | 3.7 × 10−2 | 32 |
| 2 | 3.2 × 10−2 | 28 |
| 3 | 4.3 × 10−2 | 38 |
| 4 | 1.5 × 10−2 | 26 |
| 5 | 2.9 × 10−2 | 31 |
| 6 | 3.3 × 10−2 | 36 |
| Cisplatin | - | 2.5 * |
| Oxaliplatin | - | 7.9 * |
| Carboplatin | - | 11.7 * |
| Complexes | Heterocyclic Ligands (or HL) | NSAID Axial Ligands | log kw Values |
|---|---|---|---|
| 1 | Phen | NPX | 1.44 |
| 2 | 5-Mephen | NPX | 1.46 |
| 3 | 5,6-Me2phen | NPX | 1.48 |
| 4 | Phen | ACE | 1.93 |
| 5 | 5-Mephen | ACE | 2.16 |
| 6 | 5,6-Me2phen | ACE | 2.29 |
| Platinum(IV) Complexes | Heterocyclic Ligands (or HL) | NSAID Axial Ligands | Reduction Times (min) | |
|---|---|---|---|---|
| T50% | T100% | |||
| 1 | Phen | NPX | 20–25 | post 60 |
| 2 | 5-Mephen | NPX | 10–15 | post 60 |
| 3 | 5,6-Me2phen | NPX | 10–15 | post 60 |
| 4 | Phen | ACE | 0–5 | 10–15 |
| 5 | 5-Mephen | ACE | 0–5 | 10–15 |
| 6 | 5,6-Me2phen | ACE | 0–5 | 5 |
| Platinum(IV) Prodrugs | HT29 | U87 | MCF7 | A2780 | H460 | A431 | Du145 | BE2C | SJG2 | MIA | MCF10A | ADDP | Mean GI50 Values | Selectivity Cytotoxicity Index (SCI) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GI50 Values (nM) | ||||||||||||||
| 1 | 400 ± 23 | 2570 ± 440 | 1120 ± 342 | 420 ± 50 | 860 ± 124 | 1020 ± 390 | 230 ± 17 | 1470 ± 67 | 1570 ± 67 | 660 ± 100 | 450 ± 24 | 400 ± 19 | 931 ± 139 | 1.96 |
| 2 | 210 ± 19 | 1330 ± 190 | 720 ± 240 | 290 ± 56 | 410 ± 37 | 530 ± 180 | 130 ± 27 | 680 ± 17 | 620 ± 43 | 370 ± 54 | 310 ± 12 | 280 ± 27 | 490 ± 75 | 2.38 |
| 3 | 28 ± 6 | 180 ± 23 | 60 ± 22 | 39 ± 9 | 56 ± 5 | 75 ± 33 | 10 ± 2 | 220 ± 12 | 230 ± 18 | 47 ± 7 | 36 ± 4 | 25 ± 2 | 84 ± 12 | 3.60 |
| 4 | 290 ± 30 | 1720 ± 140 | 840 ± 390 | 350 ± 47 | 610 ± 80 | 930 ± 490 | 150 ± 40 | 1300 ± 150 | 1450 ± 104 | 490 ± 36 | 360 ± 29 | 330 ± 27 | 735 ± 58 | 2.40 |
| 5 | 13 ± 8 | 140 ± 8 | 39 ± 18 | 30 ± 12 | 43 ± 8 | 60 ± 45 | 1.3 ± 0.4 | 250 ± 33 | 200 ± 21 | 29 ± 14 | 20 ± 5 | 9.3 ± 2 | 70 ± 15 | 15.4 |
| 6 | 4.7 ± 4 | 28 ± 3 | 12 ± 6 | 10 ± 4 | 16 ± 2 | 16 ± 12 | 0.22 ± 0.1 | 160 ± 19 | 130 ± 25 | 7.8 ± 4 | 3.4 ± 1 | 1.3 ± 0.5 | 32 ± 7 | 15.5 |
| Unconventional Platinum(II) Complexes | ||||||||||||||
| PHENSS | 160 ± 45 | 980 ± 270 | 1500 ± 500 | 230 ± 30 | 360 ± 35 | 480 ± 170 | 100 ± 38 | 380 ± 46 | 330 ± 66 | 200 ± 57 | 300 ± 58 | 190 ± 47 | 434 ± 110 | 3.00 |
| 5MESS | 33 ± 4 | 320 ± 26 | 200 ± 12 | 61 ± 10 | 41 ± 5 | 120 ± 25 | 22 ± 3 | 270 ± 38 | 220 ± 10 | 48 ± 2 | 30 ± 2 | 34 ± 2 | 117 ± 12 | 1.36 |
| 56MESS | 10 ± 1.6 | 35 ± 6.4 | 93 ± 44 | 76 ± 57 | 21 ± 2 | 29 ± 1 | 4.6 ± 0.4 | 59 ± 4 | 66 ± 22 | 13 ± 2 | 16 ± 1 | 13 ± 2 | 36 ± 10 | 3.48 |
| Unconventional Platinum(IV) Scaffolds | ||||||||||||||
| PHENSS(IV)(OH)2 | 710 ± 300 | 4900 ± 610 | 16,000 ± 4500 | 800 ± 84 | 1700 ± 200 | 4300 ± 530 | 310 ± 92 | 3000 ± 530 | 1700 ± 350 | 3400 ± 2200 | 1700 ± 200 | 1300 ± 350 | 3318 ± 880 | 5.48 |
| 5MESS(IV)(OH)2 | 60 ± 6 | 900 ± 58 | 1200 ± 390 | 240 ± 9 | 60 ± 5 | 360 ± 58 | 41 ± 5 | 1400 ± 300 | 640 ± 70 | 160 ± 29 | 130 ± 19 | 130 ± 22 | 443 ± 81 | 3.17 |
| 56MESS(IV)(OH)2 | 36 ± 7 | 190 ± 23 | 480 ± 140 | 59 ± 7 | 190 ± 150 | 120 ± 22 | 15 ± 2.6 | 240 ± 22 | 210 ± 45 | 43 ± 2.5 | 61 ± 7 | 170 ± 120 | 151 ± 50 | 4.07 |
| Traditional Chemotherapy Agents | ||||||||||||||
| Cisplatin | 11,300 ± 1900 | 3800 ± 1100 | 6500 ± 800 | 1000 ± 100 | 900 ± 200 | 2400 ± 300 | 1200 ± 100 | 1900 ± 200 | 400 ± 100 | 7500 ± 1300 | 5200 ± 520 | 28,000 ± 1600 | 5842 ± 610 | 4.33 |
| Oxaliplatin | 900 ± 200 | 1800 ± 200 | 500 ± 100 | 160 ± 100 | 1600 ± 100 | 4100 ± 500 | 2900 ± 400 | 900 ± 200 | 3000 ± 1200 | 900 ± 200 | nd | 800 ± 100 | 1463 ± 320 | nd |
| Carboplatin | >50,000 | >50,000 | >50,000 | 9200 ± 2900 | 14,000 ± 1000 | 24,000 ± 2200 | 15,000 ± 1200 | 19,000 ± 1200 | 5700 ± 200 | >50,000 | >50,000 | >50,000 | 32,242 ± 1450 | 3.33 |
| NSAIDs (Axial Ligands) | ||||||||||||||
| NPX | >50,000 | >50,000 | >50,000 | >50,000 | >50,000 | >50,000 | >50,000 | >50,000 | >50,000 | >50,000 | >50,000 | >50,000 | >50,000 | 1.00 |
| ACE | >50,000 | >50,000 | >50,000 | >50,000 | >50,000 | >50,000 | >50,000 | >50,000 | >50,000 | >50,000 | >50,000 | >50,000 | >50,000 | 1.00 |
| Compounds | ROS Production in Different Time Intervals (RFU) | ||
|---|---|---|---|
| 24 h | 48 h | 72 h | |
| Control | 100 ± 17 | 130 ± 11 | 131 ± 12 |
| Cisplatin | 299 ± 25 | 370 ± 22 | 392 ± 21 |
| TBHP | 658 ± 28 | 504 ± 15 | 497 ± 10 |
| PHENSS | 174 ± 2 | 172 ± 9 | 176 ± 7 |
| 5MESS | 204 ± 4 | 205 ± 3 | 188 ± 3 |
| 56MESS | 240 ± 5 | 218 ± 3 | 255 ± 4 |
| PHENSS(IV)(OH)2 | 144 ± 5 | 273 ± 4 | 303 ± 1 |
| 5MESS(IV)(OH)2 | 234 ± 1 | 323 ± 9 | 335 ± 2 |
| 56MESS(IV)(OH)2 | 259 ± 3 | 356 ± 11 | 438 ± 7 |
| NPX | 187 ± 12 | 178 ± 13 | 15 ± 5 |
| 1 | 235 ± 23 | 215 ± 19 | 63 ± 7 |
| 2 | 290 ± 17 | 261 ± 15 | 84 ± 9 |
| 3 | 297 ± 21 | 287 ± 11 | 104 ± 15 |
| ACE | 279 ± 7 | 260 ± 9 | 132 ± 13 |
| 4 | 303 ± 8 | 298 ± 17 | 168 ± 15 |
| 5 | 313 ± 11 | 305 ± 10 | 192 ± 7 |
| 6 | 344 ± 17 | 312 ± 13 | 202 ± 10 |
| Compounds | MtMP Changes in Different Time Intervals (RFU) | ||
|---|---|---|---|
| 24 h | 48 h | 72 h | |
| Control | 495 ± 7 | 412 ± 15 | 411 ± 5 |
| Cisplatin | 256 ± 9 | 236 ± 13 | 199 ± 11 |
| FCCP | 194 ± 10 | 143 ± 5 | 80 ± 5 |
| PHENSS | 289 ± 17 | 271 ± 15 | 237 ± 10 |
| 5MESS | 210 ± 15 | 204 ± 6 | 144 ± 5 |
| 56MESS | 156 ± 10 | 162 ± 13 | 103 ± 7 |
| PHENSS(IV)(OH)2 | 321 ± 17 | 241 ± 10 | 216 ± 8 |
| 5MESS(IV)(OH)2 | 301 ± 14 | 214 ± 9 | 198 ± 4 |
| 56MESS(IV)(OH)2 | 245 ± 16 | 177 ± 11 | 125 ± 12 |
| NPX | 499 ± 10 | 397 ± 13 | 304 ± 9 |
| 1 | 382 ± 11 | 211 ± 12 | 163 ± 5 |
| 2 | 368 ± 9 | 160 ± 13 | 130 ± 7 |
| 3 | 351 ± 5 | 152 ± 8 | 104 ± 6 |
| ACE | 494 ± 6 | 371 ± 10 | 295 ± 13 |
| 4 | 416 ± 8 | 343 ± 8 | 184 ± 5 |
| 5 | 412 ± 4 | 284 ± 12 | 140 ± 5 |
| 6 | 387 ± 7 | 284 ± 11 | 112 ± 8 |
| Compounds | Percentage COX-2 Inhibitory Activity (%) |
|---|---|
| 72 h | |
| Control | 84 ± 3 |
| Cisplatin | 9 ± 2 |
| PHENSS | 8 ± 4 |
| 5MESS | 6 ± 3 |
| 56MESS | 5 ± 4 |
| NPX | 83 ± 5 |
| 1 | 55 ± 3 |
| 2 | 44 ± 6 |
| 3 | 37 ± 4 |
| ACE | 76 ± 8 |
| 4 | 48 ± 3 |
| 5 | 36 ± 2 |
| 6 | 32 ± 3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aputen, A.D.; Elias, M.G.; Gilbert, J.; Sakoff, J.A.; Gordon, C.P.; Scott, K.F.; Aldrich-Wright, J.R. Versatile Platinum(IV) Prodrugs of Naproxen and Acemetacin as Chemo-Anti-Inflammatory Agents. Cancers 2023, 15, 2460. https://doi.org/10.3390/cancers15092460
Aputen AD, Elias MG, Gilbert J, Sakoff JA, Gordon CP, Scott KF, Aldrich-Wright JR. Versatile Platinum(IV) Prodrugs of Naproxen and Acemetacin as Chemo-Anti-Inflammatory Agents. Cancers. 2023; 15(9):2460. https://doi.org/10.3390/cancers15092460
Chicago/Turabian StyleAputen, Angelico D., Maria George Elias, Jayne Gilbert, Jennette A. Sakoff, Christopher P. Gordon, Kieran F. Scott, and Janice R. Aldrich-Wright. 2023. "Versatile Platinum(IV) Prodrugs of Naproxen and Acemetacin as Chemo-Anti-Inflammatory Agents" Cancers 15, no. 9: 2460. https://doi.org/10.3390/cancers15092460
APA StyleAputen, A. D., Elias, M. G., Gilbert, J., Sakoff, J. A., Gordon, C. P., Scott, K. F., & Aldrich-Wright, J. R. (2023). Versatile Platinum(IV) Prodrugs of Naproxen and Acemetacin as Chemo-Anti-Inflammatory Agents. Cancers, 15(9), 2460. https://doi.org/10.3390/cancers15092460