
Metabolic Imaging Biomarkers of Response to Signaling Inhibition Therapy in Melanoma
 , ,
, , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Melanoma Cell Lines
2.3. Cell Cultures
2.4. Extracellular Glucose and Lactate Measurements
2.5. Oxygen Consumption and Extracellular Acidification Rates
2.6. In Vitro Measurement of Intracellular Metabolites by High-Resolution 1H MRS
2.7. Mouse Xenografts for the Noninvasive 1H and 31P MRS Studies
2.8. In Vivo 1H and 31P MRS Experiments
2.9. Tumor Volume Measurement
2.10. Statistical Analysis
3. Results
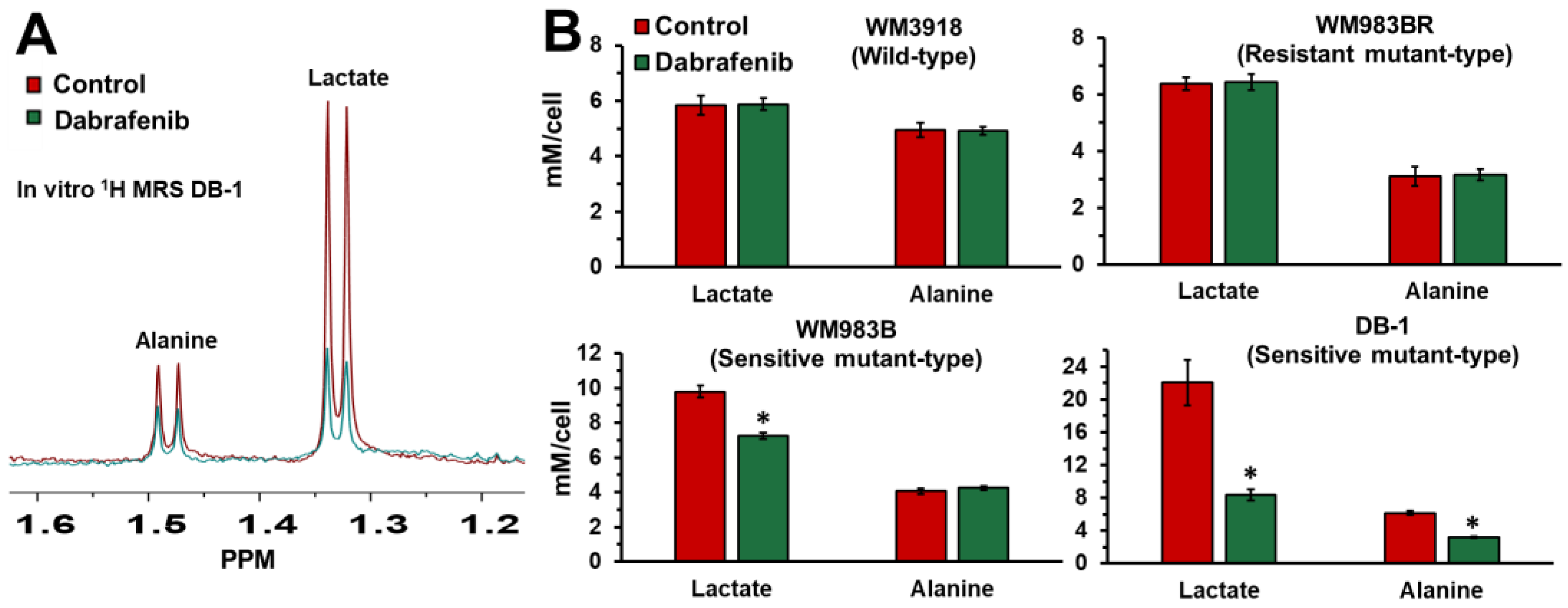
3.1. Impact of Dabrafenib on Intracellular Lactate and Alanine in Human Melanoma Cell Lines
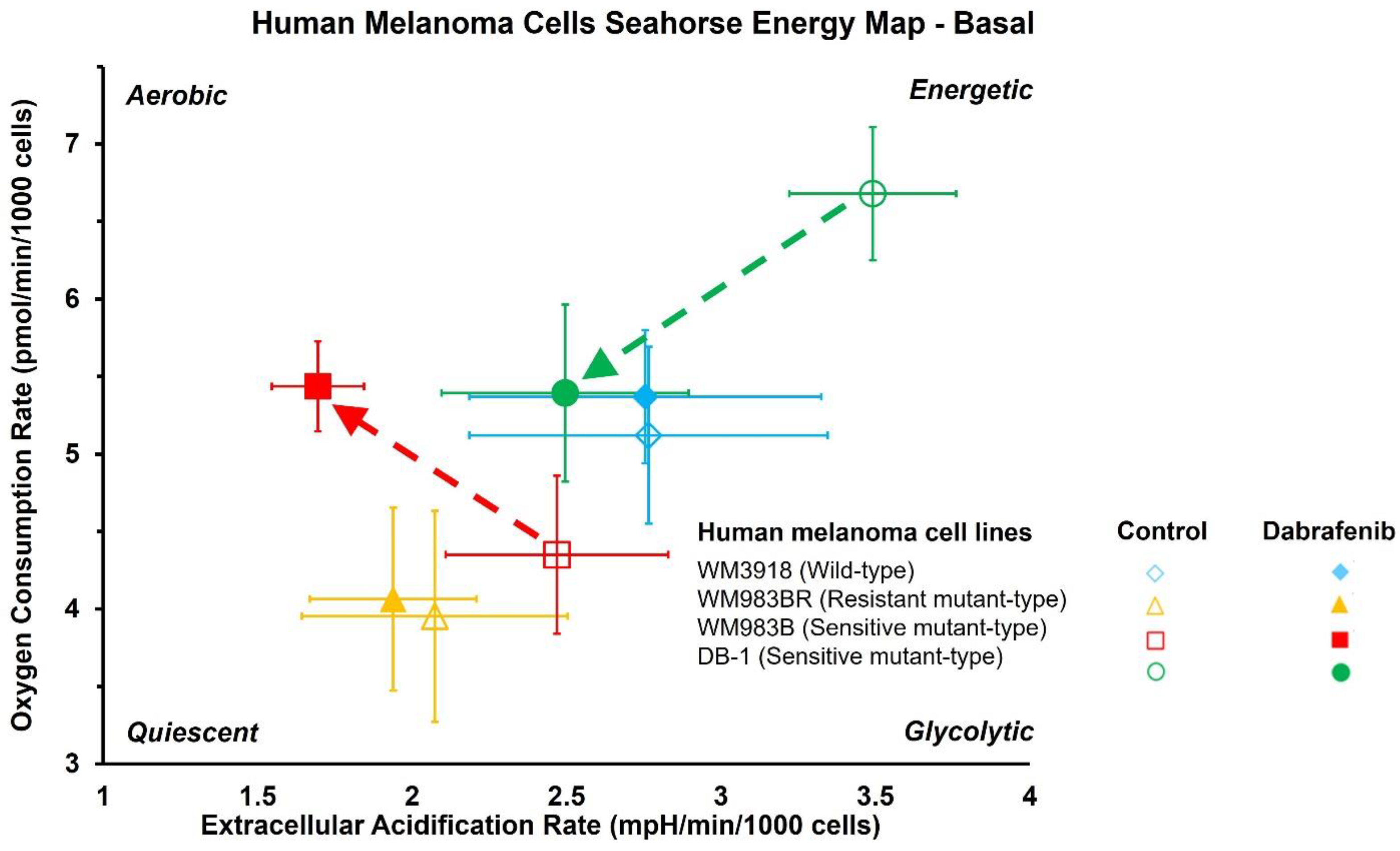
3.2. Impact of Dabrafenib on Oxygen Consumption Rate (OCR) and Extracellular Acidification Rate (ECAR) in Melanoma Cell Lines
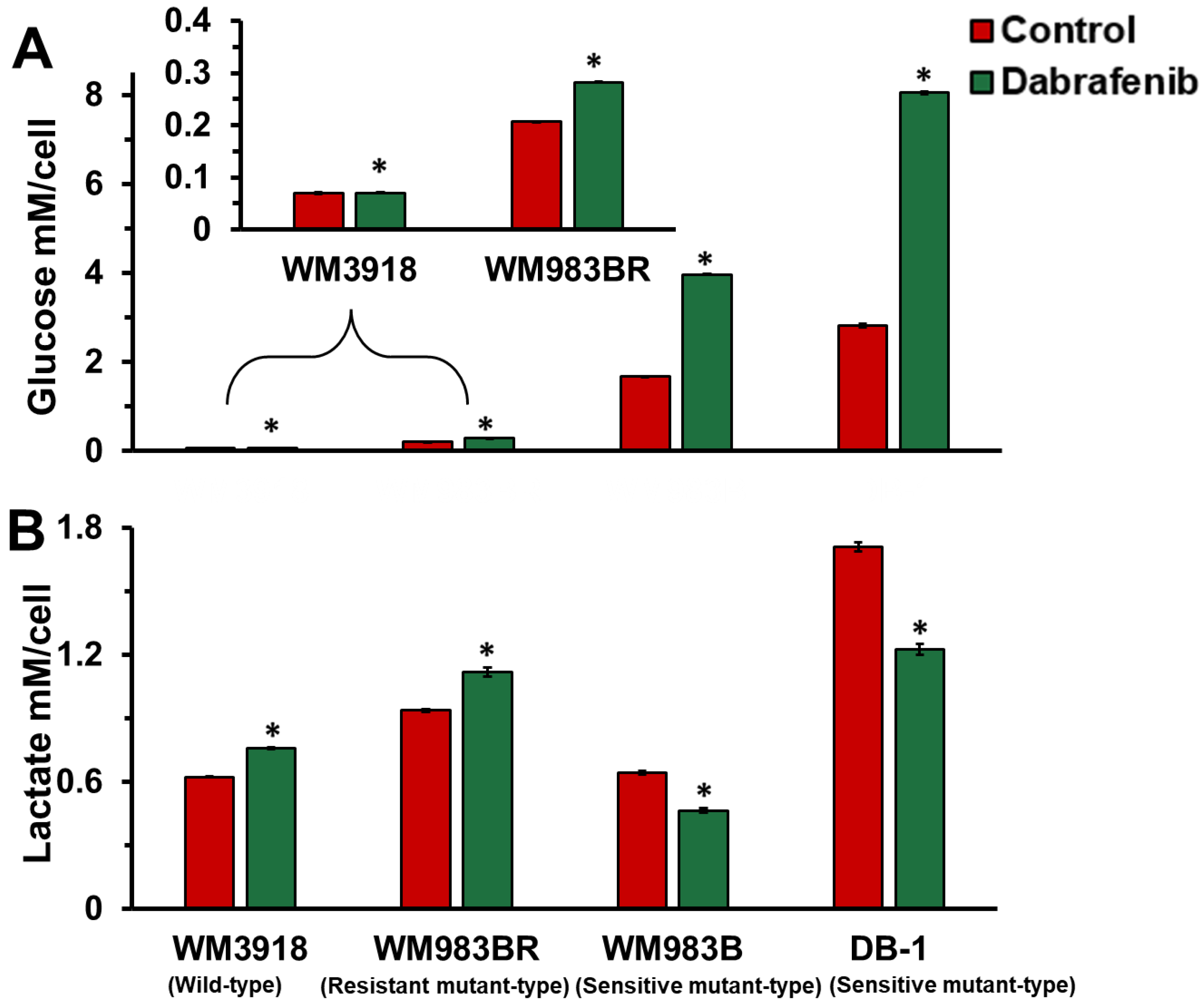
3.3. Dabrafenib Impact on Extracellular Glucose Consumption and Lactate Flux in Melanoma
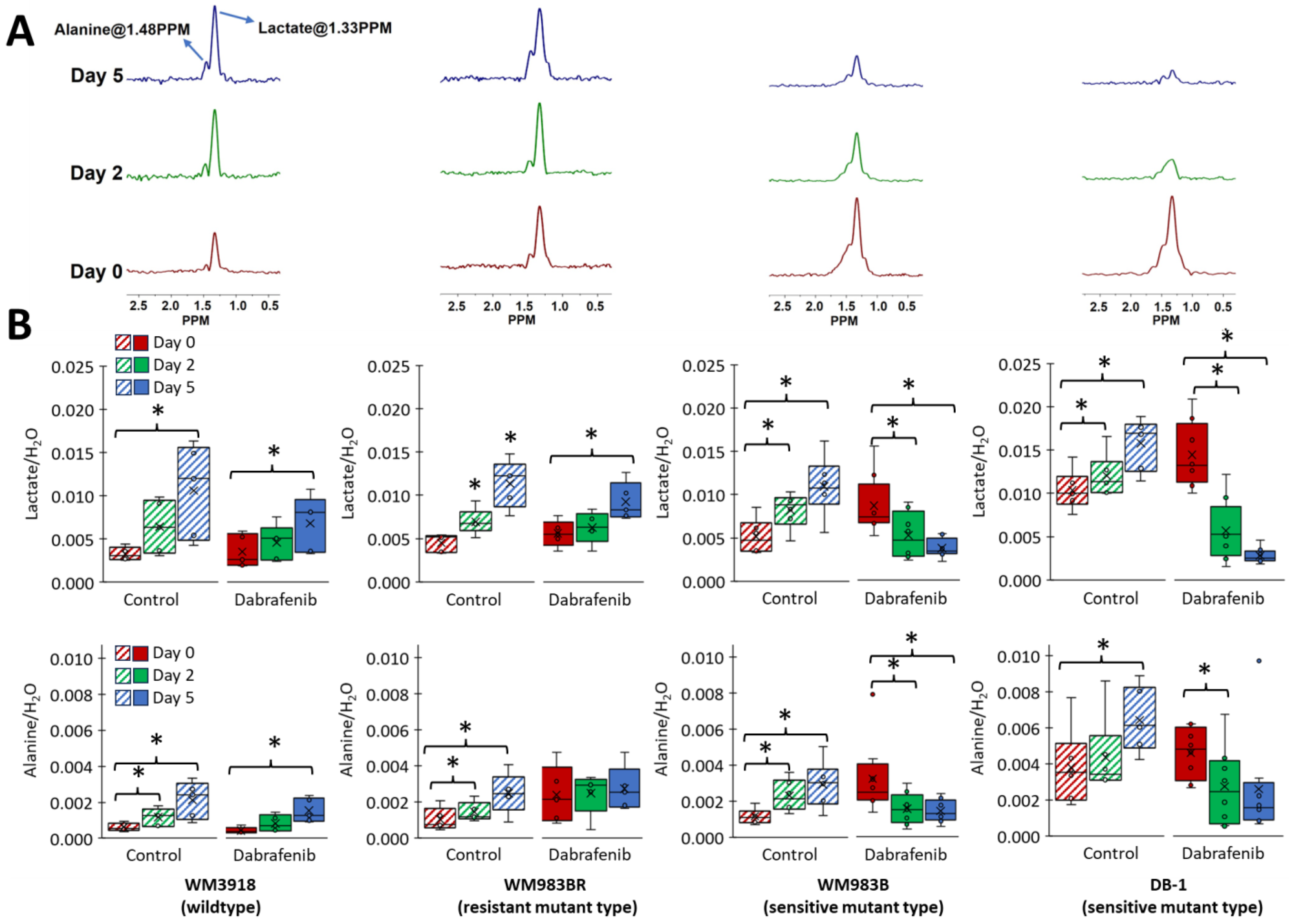
3.4. Impact of Dabrafenib on Intracellular Lactate and Alanine Values in Melanoma Xenografts
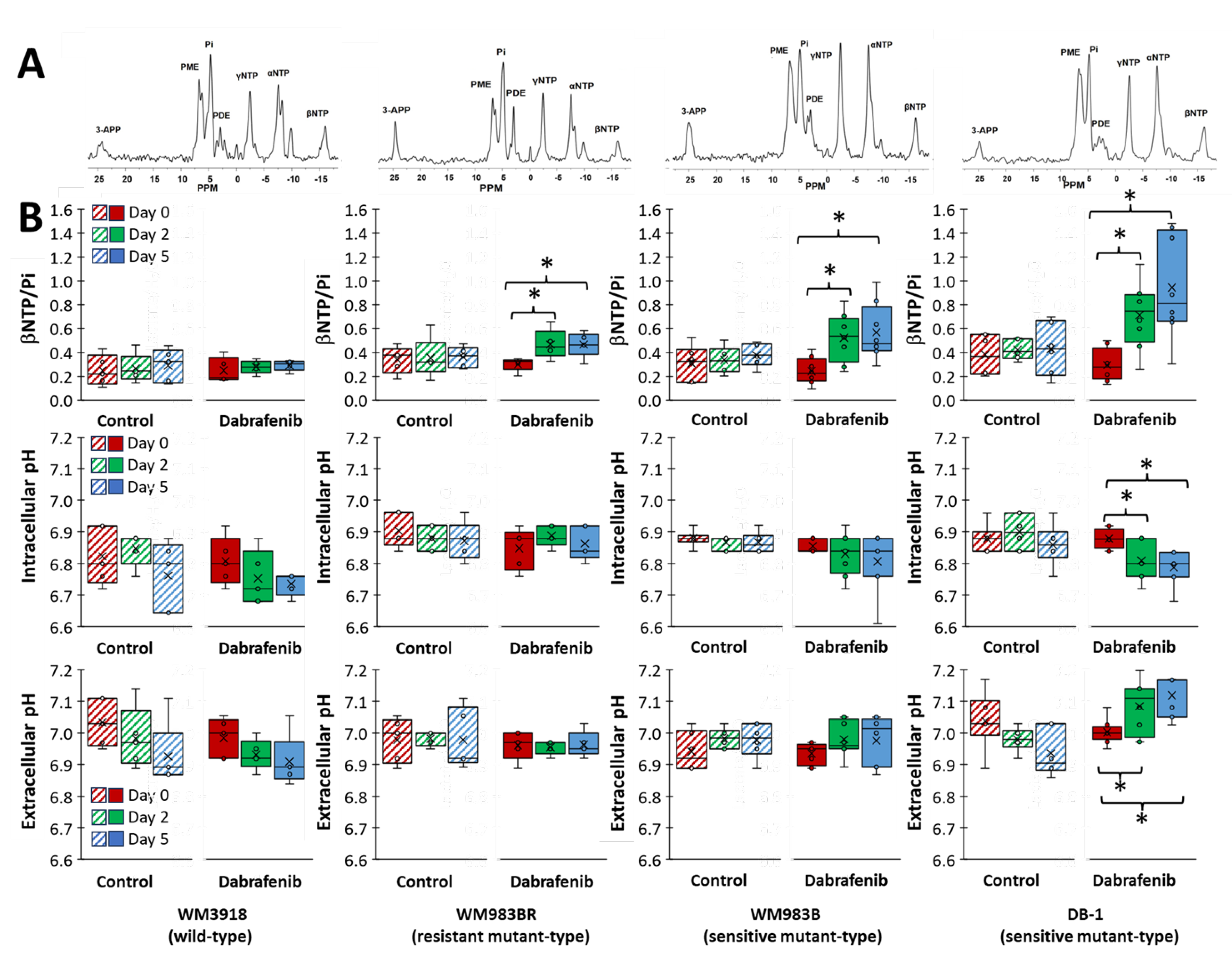
3.5. Impact of Dabrafenib on Bioenergetics and pH in Melanoma Xenografts
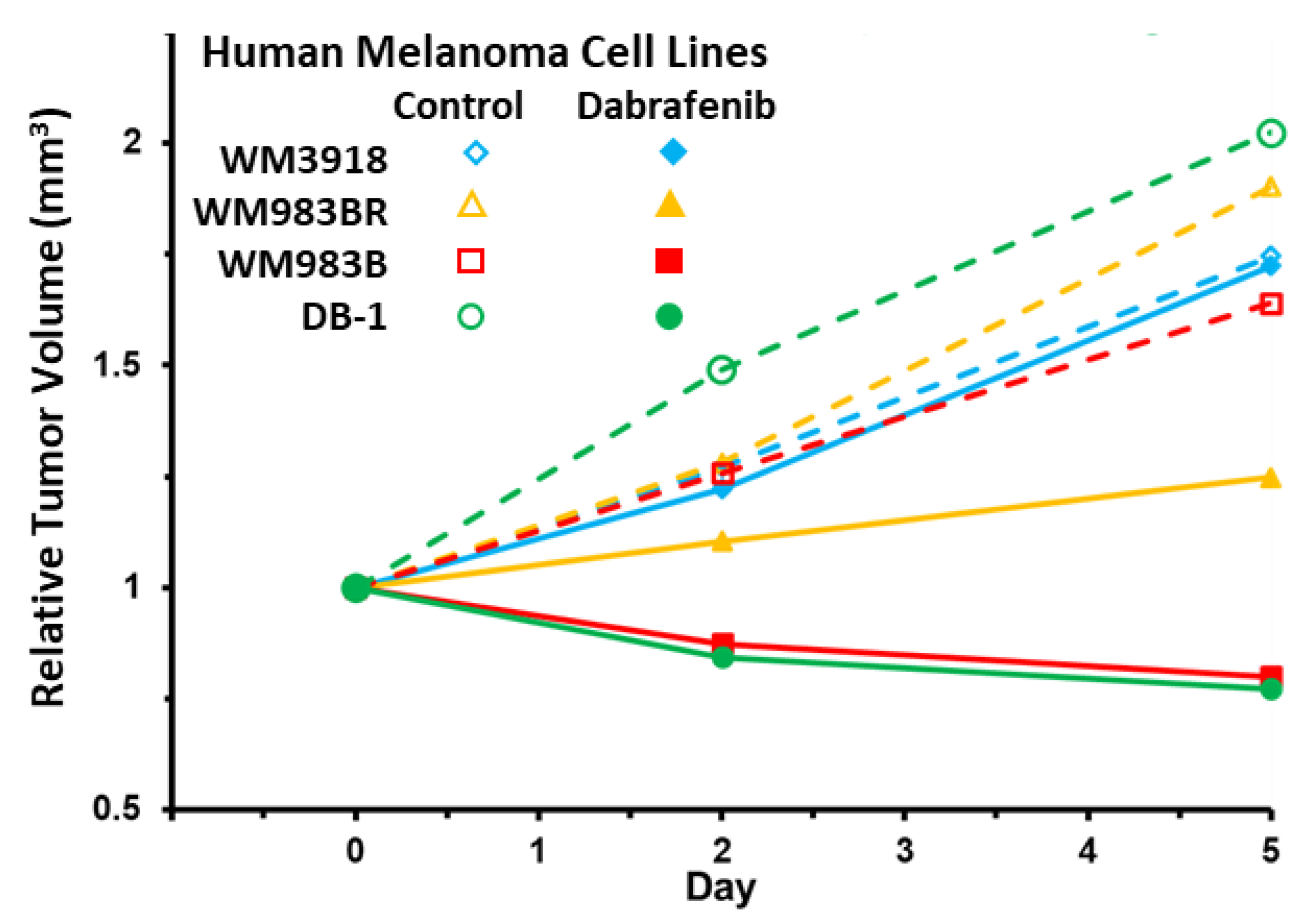
3.6. Impact of Dabrafenib on Tumor Burden in Melanoma Xenografts
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Domingues, B.; Lopes, J.M.; Soares, P.; Pópulo, H. Melanoma treatment in review. Immunotargets Ther. 2018, 7, 35–49. [Google Scholar] [CrossRef]
- Davis, L.E.; Shalin, S.C.; Tackett, A.J. Current state of melanoma diagnosis and treatment. Cancer Biol. Ther. 2019, 20, 1366–1379. [Google Scholar] [CrossRef] [PubMed]
- Polkowska, M.; Ekk-Cierniakowski, P.; Czepielewska, E.; Kozłowska-Wojciechowska, M. Efficacy and safety of BRAF inhibitors and anti-CTLA4 antibody in melanoma patients-real-world data. Eur. J. Clin. Pharmacol. 2019, 75, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Spagnolo, F.; Ghiorzo, P.; Orgiano, L.; Pastorino, L.; Picasso, V.; Tornari, E.; Ottaviano, V.; Queirolo, P. BRAF-mutant melanoma: Treatment approaches, resistance mechanisms, and diagnostic strategies. Onco Targets Ther. 2015, 8, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zhang, X.; Sun, N.; Zhao, Z.; He, J. Comprehensive Analysis of the Tumor Microenvironment in Cutaneous Melanoma associated with Immune Infiltration. J. Cancer 2020, 11, 3858–3870. [Google Scholar] [CrossRef] [PubMed]
- Pavri, S.N.; Clune, J.; Ariyan, S.; Narayan, D. Malignant Melanoma: Beyond the Basics. Plast. Reconstr. Surg. 2016, 138, 330e–340e. [Google Scholar] [CrossRef] [PubMed]
- Soumoy, L.; Kindt, N.; Ghanem, G.; Saussez, S.; Journe, F. Role of Macrophage Migration Inhibitory Factor (MIF) in Melanoma. Cancers 2019, 11, 529. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Zheng, M. Advances in targeted therapy and immunotherapy for melanoma (Review). Exp. Ther. Med. 2023, 26, 416. [Google Scholar] [CrossRef]
- Perez, M.C.; Depalo, D.K.; Zager, J.S. A safety review of recently approved and late-stage trial treatments for metastatic melanoma: Systemic and regional therapies. Expert. Opin. Drug Saf. 2023, 22, 789–797. [Google Scholar] [CrossRef]
- Tinca, A.C.; Raicea, A.; Szoke, A.R.; Cocuz, I.G.; Sincu, M.C.; Niculescu, R.; Sabau, A.H.; Popelea, M.C.; Fruntelata, R.F.; Cotoi, O.S. Morphological aspects and therapeutic options in melanoma: A narrative review of the past decade. Rom. J. Morphol. Embryol. 2023, 64, 135–141. [Google Scholar] [CrossRef]
- Konieczkowski, D.J.; Johannessen, C.M.; Abudayyeh, O.; Kim, J.W.; Cooper, Z.A.; Piris, A.; Frederick, D.T.; Barzily-Rokni, M.; Straussman, R.; Haq, R.; et al. A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov. 2014, 4, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Tengda, L.; Cheng, Q.; Yi, S. Identification of Melanoma Subsets Based on DNA Methylation Sites and Construction of a Prognosis Evaluation Model. J. Oncol. 2022, 2022, 6608650. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.; Banerjee, R.; Waly, R.; Urbano, A.; Gimenez, G.; Day, R.; Eccles, M.R.; Weeks, R.J.; Chatterjee, A. Locus-Specific DNA Methylation Editing in Melanoma Cell Lines Using a CRISPR-Based System. Cancers 2021, 13, 5433. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Kuo, C.; Nicholl, M.B.; Sim, M.S.; Turner, R.R.; Morton, D.L.; Hoon, D.S. Downregulation of microRNA-29c is associated with hypermethylation of tumor-related genes and disease outcome in cutaneous melanoma. Epigenetics 2011, 6, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Platz, A.; Egyhazi, S.; Ringborg, U.; Hansson, J. Human cutaneous melanoma; a review of NRAS and BRAF mutation frequencies in relation to histogenetic subclass and body site. Mol. Oncol. 2008, 1, 395–405. [Google Scholar] [CrossRef]
- Geiger, C.E.; Mrabet-Dahbi, S.; Berger, I. The BRAF and NRAS status among distinct metastases of malignant melanoma differ significantly independent of tissue origin and temporal occurrence. Possible effect on clinical relevance? Melanoma Res. 2024, 34, 85–87. [Google Scholar] [CrossRef] [PubMed]
- Dinter, L.; Karitzky, P.C.; Schulz, A.; Wurm, A.A.; Mehnert, M.C.; Sergon, M.; Tunger, A.; Lesche, M.; Wehner, R.; Muller, A.; et al. BRAF and MEK inhibitor combinations induce potent molecular and immunological effects in NRAS-mutant melanoma cells: Insights into mode of action and resistance mechanisms. Int. J. Cancer 2023, 1–16. [Google Scholar] [CrossRef]
- Nazarian, R.; Shi, H.; Wang, Q.; Kong, X.; Koya, R.C.; Lee, H.; Chen, Z.; Lee, M.K.; Attar, N.; Sazegar, H.; et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010, 468, 973–977. [Google Scholar] [CrossRef]
- Hall, A.; Meyle, K.D.; Lange, M.K.; Klima, M.; Sanderhoff, M.; Dahl, C.; Abildgaard, C.; Thorup, K.; Moghimi, S.M.; Jensen, P.B.; et al. Dysfunctional oxidative phosphorylation makes malignant melanoma cells addicted to glycolysis driven by the (V600E)BRAF oncogene. Oncotarget 2013, 4, 584–599. [Google Scholar] [CrossRef]
- Scott, A.J.; O’Dea, K.P.; O’Callaghan, D.; Williams, L.; Dokpesi, J.O.; Tatton, L.; Handy, J.M.; Hogg, P.J.; Takata, M. Reactive oxygen species and p38 mitogen-activated protein kinase mediate tumor necrosis factor α-converting enzyme (TACE/ADAM-17) activation in primary human monocytes. J. Biol. Chem. 2011, 286, 35466–35476. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, P.; Trinh, A.; Khamari, R.; Kluza, J. Melanoma metabolism contributes to the cellular responses to MAPK/ERK pathway inhibitors. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.; Cohen, M.S. The discovery of vemurafenib for the treatment of BRAF-mutated metastatic melanoma. Expert. Opin. Drug Discov. 2016, 11, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Kirkwood, J.M.; Grob, J.J.; Simeone, E.; Grimaldi, A.M.; Maio, M.; Palmieri, G.; Testori, A.; Marincola, F.M.; Mozzillo, N. The role of BRAF V600 mutation in melanoma. J. Transl. Med. 2012, 10, 85. [Google Scholar] [CrossRef]
- Hosseini, M.; Kasraian, Z.; Rezvani, H.R. Energy metabolism in skin cancers: A therapeutic perspective. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 712–722. [Google Scholar] [CrossRef] [PubMed]
- Abildgaard, C.; Guldberg, P. Molecular drivers of cellular metabolic reprogramming in melanoma. Trends Mol. Med. 2015, 21, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Haq, R.; Shoag, J.; Andreu-Perez, P.; Yokoyama, S.; Edelman, H.; Rowe, G.C.; Frederick, D.T.; Hurley, A.D.; Nellore, A.; Kung, A.L.; et al. Oncogenic BRAF regulates oxidative metabolism via PGC1α and MITF. Cancer Cell 2013, 23, 302–315. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, F.; Lim, J.H.; Chim, H.; Bhalla, K.; Girnun, G.; Pierce, K.; Clish, C.B.; Granter, S.R.; Widlund, H.R.; Spiegelman, B.M.; et al. PGC1α expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell 2013, 23, 287–301. [Google Scholar] [CrossRef]
- Fischer, G.M.; Vashisht Gopal, Y.N.; McQuade, J.L.; Peng, W.; DeBerardinis, R.J.; Davies, M.A. Metabolic strategies of melanoma cells: Mechanisms, interactions with the tumor microenvironment, and therapeutic implications. Pigment. Cell Melanoma Res. 2018, 31, 11–30. [Google Scholar] [CrossRef]
- Jose, C.; Bellance, N.; Rossignol, R. Choosing between glycolysis and oxidative phosphorylation: A tumor’s dilemma? Biochim. Biophys. Acta 2011, 1807, 552–561. [Google Scholar] [CrossRef]
- Roesch, A. Tumor heterogeneity and plasticity as elusive drivers for resistance to MAPK pathway inhibition in melanoma. Oncogene 2015, 34, 2951–2957. [Google Scholar] [CrossRef]
- Thomas, N.E.; Edmiston, S.N.; Alexander, A.; Groben, P.A.; Parrish, E.; Kricker, A.; Armstrong, B.K.; Anton-Culver, H.; Gruber, S.B.; From, L.; et al. Association Between NRAS and BRAF Mutational Status and Melanoma-Specific Survival Among Patients With Higher-Risk Primary Melanoma. JAMA Oncol. 2015, 1, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Proietti, I.; Skroza, N.; Michelini, S.; Mambrin, A.; Balduzzi, V.; Bernardini, N.; Marchesiello, A.; Tolino, E.; Volpe, S.; Maddalena, P.; et al. BRAF Inhibitors: Molecular Targeting and Immunomodulatory Actions. Cancers 2020, 12, 1823. [Google Scholar] [CrossRef] [PubMed]
- Śmiech, M.; Leszczyński, P.; Kono, H.; Wardell, C.; Taniguchi, H. Emerging BRAF Mutations in Cancer Progression and Their Possible Effects on Transcriptional Networks. Genes 2020, 11, 1342. [Google Scholar] [CrossRef] [PubMed]
- Botton, T.; Talevich, E.; Mishra, V.K.; Zhang, T.; Shain, A.H.; Berquet, C.; Gagnon, A.; Judson, R.L.; Ballotti, R.; Ribas, A.; et al. Genetic Heterogeneity of BRAF Fusion Kinases in Melanoma Affects Drug Responses. Cell Rep. 2019, 29, 573–588.e577. [Google Scholar] [CrossRef] [PubMed]
- Pickup, S.; Lee, S.C.; Mancuso, A.; Glickson, J.D. Lactate imaging with Hadamard-encoded slice-selective multiple quantum coherence chemical-shift imaging. Magn. Reson. Med. 2008, 60, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Nath, K.; Nelson, D.S.; Ho, A.M.; Lee, S.C.; Darpolor, M.M.; Pickup, S.; Zhou, R.; Heitjan, D.F.; Leeper, D.B.; Glickson, J.D. (31) P and (1) H MRS of DB-1 melanoma xenografts: Lonidamine selectively decreases tumor intracellular pH and energy status and sensitizes tumors to melphalan. NMR Biomed. 2013, 26, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Nath, K.; Roman, J.; Nelson, D.S.; Guo, L.; Lee, S.C.; Orlovskiy, S.; Muriuki, K.; Heitjan, D.F.; Pickup, S.; Leeper, D.B.; et al. Effect of Differences in Metabolic Activity of Melanoma Models on Response to Lonidamine plus Doxorubicin. Sci. Rep. 2018, 8, 14654. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Rai, S.K.; Gunnam, A.; Mannava, M.K.C.; Nangia, A.K. Improving the Dissolution Rate of the Anticancer Drug Dabrafenib. Cryst. Growth Des. 2020, 20, 1035–1046. [Google Scholar] [CrossRef]
- Sumimoto, H.; Imabayashi, F.; Iwata, T.; Kawakami, Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J. Exp. Med. 2006, 203, 1651–1656. [Google Scholar] [CrossRef]
- Zhang, P.; Guan, H.; Yuan, S.; Cheng, H.; Zheng, J.; Zhang, Z.; Liu, Y.; Yu, Y.; Meng, Z.; Zheng, X.; et al. Targeting myeloid derived suppressor cells reverts immune suppression and sensitizes BRAF-mutant papillary thyroid cancer to MAPK inhibitors. Nat. Commun. 2022, 13, 1588. [Google Scholar] [CrossRef]
- Sullivan, R.J.; Flaherty, K.T. BRAF in Melanoma: Pathogenesis, Diagnosis, Inhibition, and Resistance. J. Skin. Cancer 2011, 2011, 423239. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oxygen Consumption Rate (OCR; pmol/min/cell) | ||||||
|---|---|---|---|---|---|---|
| Group | Basal | p-Value | Stressed | p-Value | ||
| Control | Dabrafenib | Control | Dabrafenib | |||
| WM3918 | 5.12 ± 0.57 | 5.37 ± 0.43 | 7.47 ± 1.02 | 7.86 ± 1.41 | ||
| WM983BR | 3.95 ± 0.68 | 4.06 ± 0.59 | 5.52 ± 0.81 | 5.96 ± 0.55 | ||
| WM983B | 4.35 ± 0.51 | 5.44 ± 0.29 | 0.004 | 6.97 ± 1.13 | 12.6 ± 2.42 | 0.002 |
| DB-1 | 6.68 ± 0.43 | 5.39 ± 0.57 | 0.004 | 15.6 ± 2.34 | 7.97 ± 1.64 | <0.001 |
| Extracellular Acidification Rate (ECAR; pmol/min/cell) | ||||||
| WM3918 | 2.77 ± 0.58 | 2.76 ± 0.57 | 4.63 ± 0.86 | 4.76 ± 0.96 | ||
| WM983BR | 2.07 ± 0.43 | 1.94 ± 0.27 | 3.44 ± 0.66 | 3.25 ± 0.52 | ||
| WM983B | 2.47 ± 0.36 | 1.69 ± 0.15 | 0.002 | 4.73 ± 0.57 | 3.29 ± 0.21 | 0.002 |
| DB-1 | 3.49 ± 0.27 | 2.50 ± 0.40 | <0.01 | 5.35 ± 0.32 | 4.26 ± 0.46 | 0.009 |
| Human Melanoma Cell Lines | Relative Tumor Volume (Mean ± S.E.M.) | |||
|---|---|---|---|---|
| Day 0 | Day 2 | Day 5 | ||
| WM3918 | Control | 1.00 ± 0.12 * | 1.27 ± 0.13 ** | 1.75 ± 0.09 *** |
| Dabrafenib | 1.00 ± 0.03 * | 1.22 ± 0.09 ** | 1.72 ± 0.17 *** | |
| WM983BR | Control | 1.00 ± 0.15 * | 1.28 ± 0.18 ** | 1.90 ± 0.32 *** |
| Dabrafenib | 1.00 ± 0.08 | 1.10 ± 0.11 | 1.25 ± 0.13 *** | |
| WM983B | Control | 1.00 ± 0.13 * | 1.26 ± 0.16 ** | 1.64 ± 0.20 *** |
| Dabrafenib | 1.00 ± 0.08 # | 0.87 ± 0.07 ## | 0.80 ± 0.07 ### | |
| DB-1 | Control | 1.00 ± 0.20 * | 1.49 ± 0.16 ** | 2.02 ± 0.20 *** |
| Dabrafenib | 1.00 ± 0.17 # | 0.84 ± 0.16 ## | 0.77 ± 0.16 ### | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gupta, P.K.; Orlovskiy, S.; Arias-Mendoza, F.; Nelson, D.S.; Osborne, A.; Pickup, S.; Glickson, J.D.; Nath, K. Metabolic Imaging Biomarkers of Response to Signaling Inhibition Therapy in Melanoma. Cancers 2024, 16, 365. https://doi.org/10.3390/cancers16020365
Gupta PK, Orlovskiy S, Arias-Mendoza F, Nelson DS, Osborne A, Pickup S, Glickson JD, Nath K. Metabolic Imaging Biomarkers of Response to Signaling Inhibition Therapy in Melanoma. Cancers. 2024; 16(2):365. https://doi.org/10.3390/cancers16020365
Chicago/Turabian StyleGupta, Pradeep Kumar, Stepan Orlovskiy, Fernando Arias-Mendoza, David S. Nelson, Aria Osborne, Stephen Pickup, Jerry D. Glickson, and Kavindra Nath. 2024. "Metabolic Imaging Biomarkers of Response to Signaling Inhibition Therapy in Melanoma" Cancers 16, no. 2: 365. https://doi.org/10.3390/cancers16020365
APA StyleGupta, P. K., Orlovskiy, S., Arias-Mendoza, F., Nelson, D. S., Osborne, A., Pickup, S., Glickson, J. D., & Nath, K. (2024). Metabolic Imaging Biomarkers of Response to Signaling Inhibition Therapy in Melanoma. Cancers, 16(2), 365. https://doi.org/10.3390/cancers16020365