
PIK Your Poison: The Effects of Combining PI3K and CDK Inhibitors against Metastatic Cutaneous Squamous Cell Carcinoma In Vitro
 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. General Cell Culture and Maintenance
2.2. Nucleic Acid Extraction, Sequencing, and Analysis
2.3. Identification of Recurrently Mutated and Relevant Genes
2.4. Drug Sourcing and Storage
2.5. Two−Dimensional Single−Agent Drug Viability Assay
2.6. Two−Dimensional Drug Combination Synergy Assay
2.7. Drug Synergy Analysis
2.8. Three−Dimensional Drug Viability Assay
2.9. AnnexinV−FITC Apoptosis Assay
2.10. Cell Migration Assay
2.11. Cell Cycle Analysis
2.12. Cell Lysate Preperation of Western Blot Analysis
3. Results
3.1. Multi−Omic Analysis of mcSCC Cell Lines
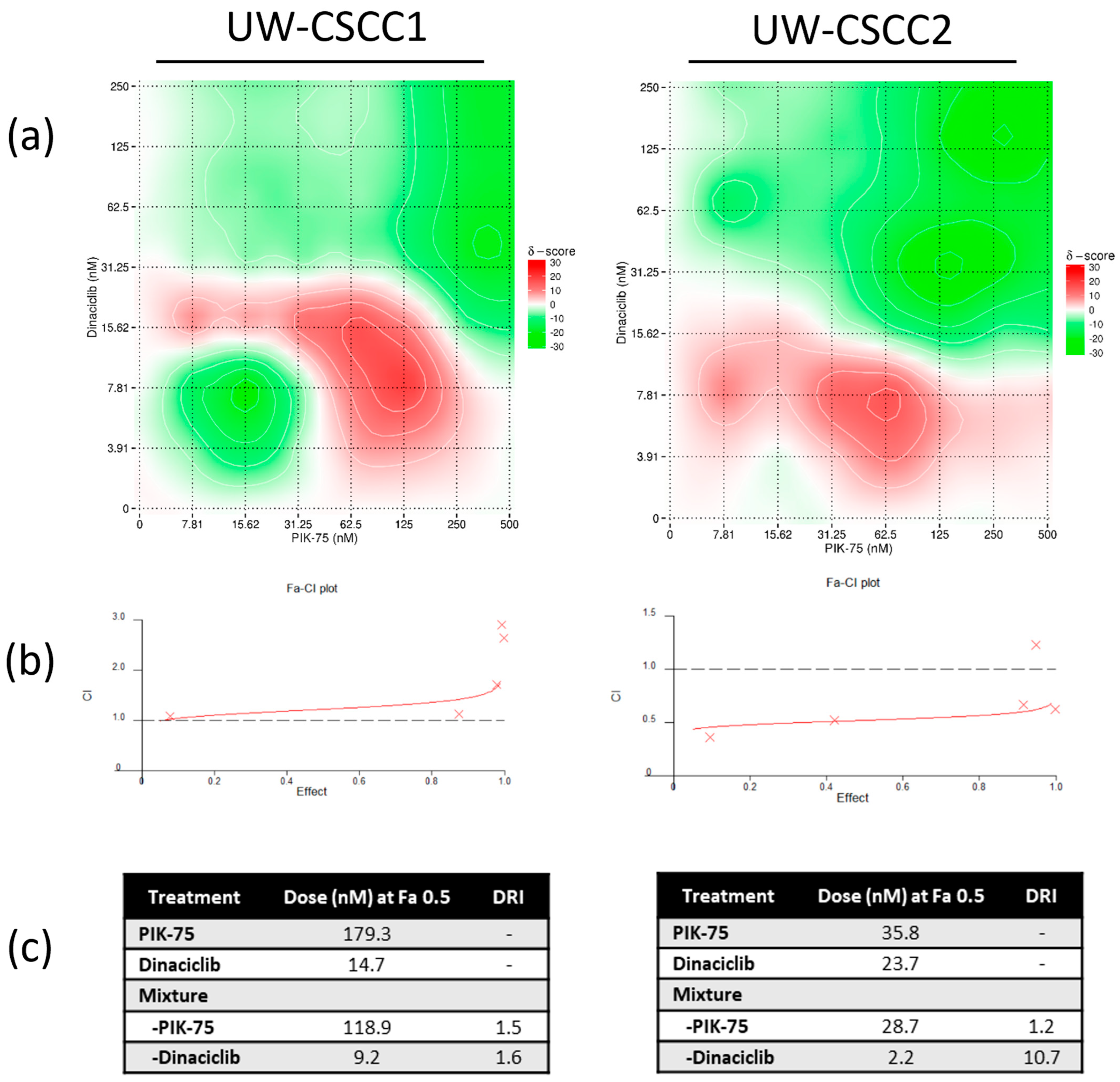
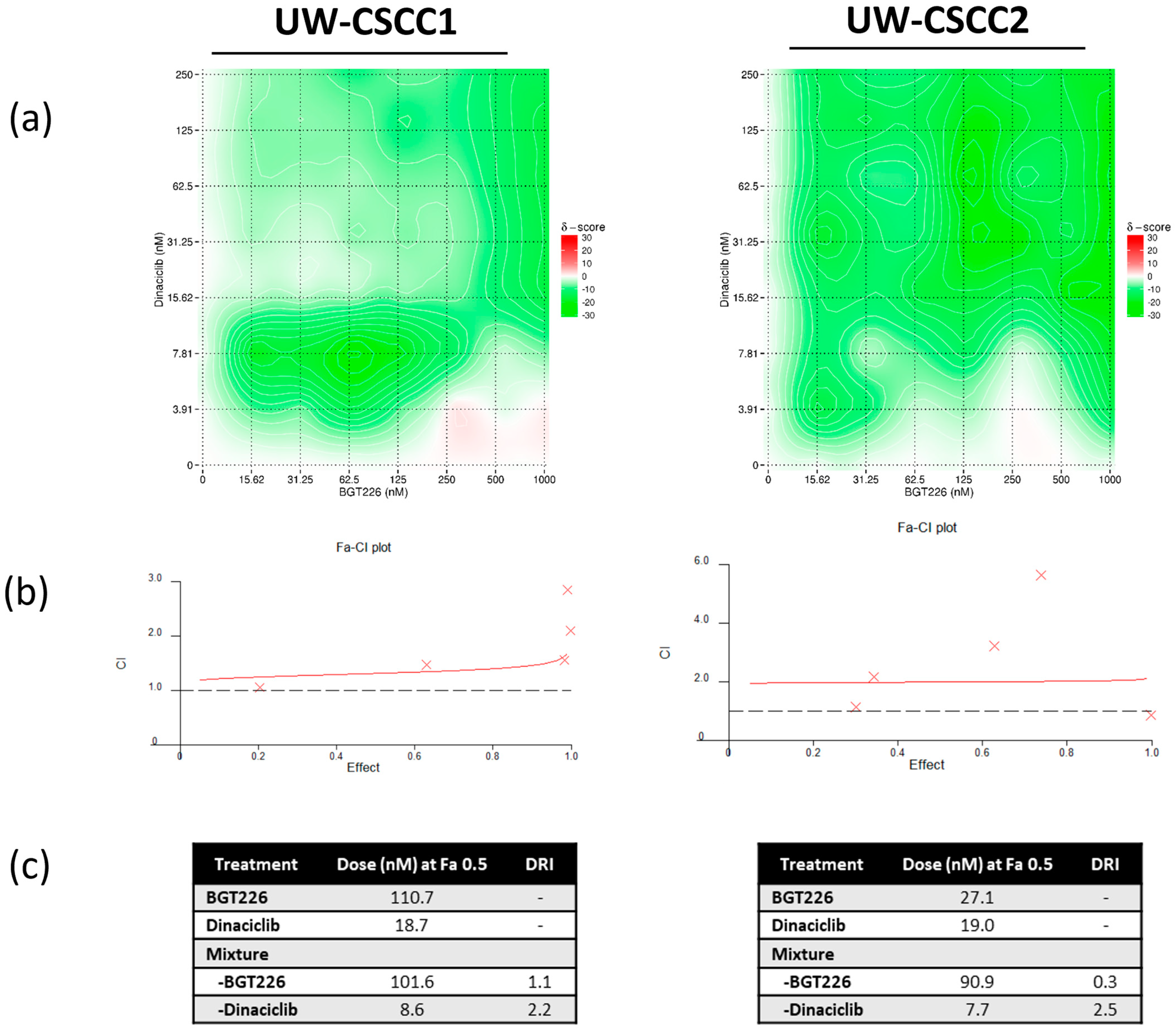
3.2. Two−Dimensional Cell Viability Assays and Drug Synergy Studies
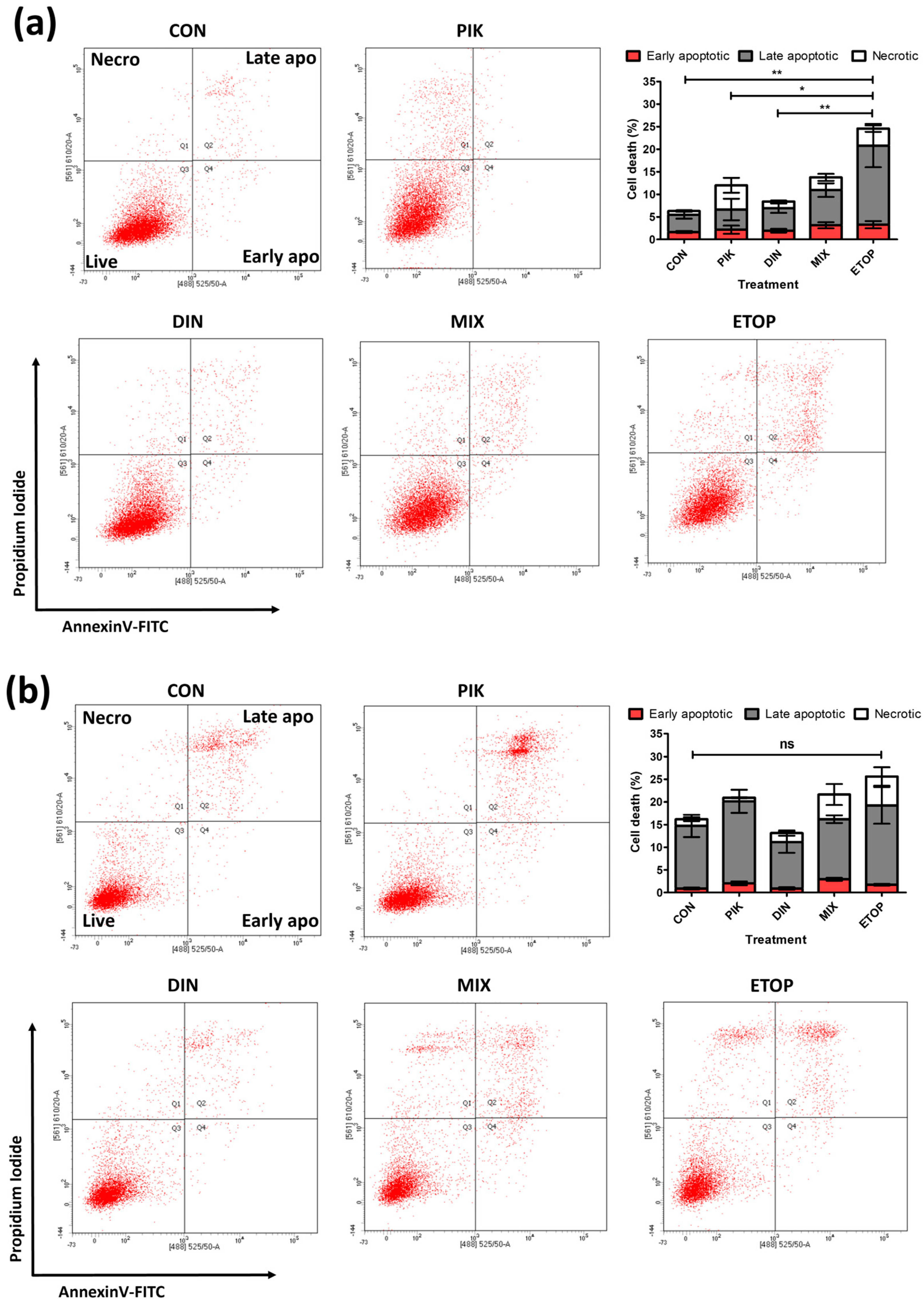
3.3. Combination PI3K−CDKi Effect on 2D Cell Apoptosis, Cell Cycle Distribution, and Motility
3.4. Cell Signalling Effects
3.5. Three−Dimensional Cell Viability Assays and Drug Synergy Studies
4. Discussion
4.1. UW-CSCC1 and UW-CSCC2 Present Distinct Molecular Profiles for Cell Cycle− and PI3K−Related Pathways
4.2. PI3K and Cell Cycle Inhibitors Potently Reduced cSCC Cell Viability In Vitro
4.3. Synergistic Effect of PI3K and CDK Dual Inhibition
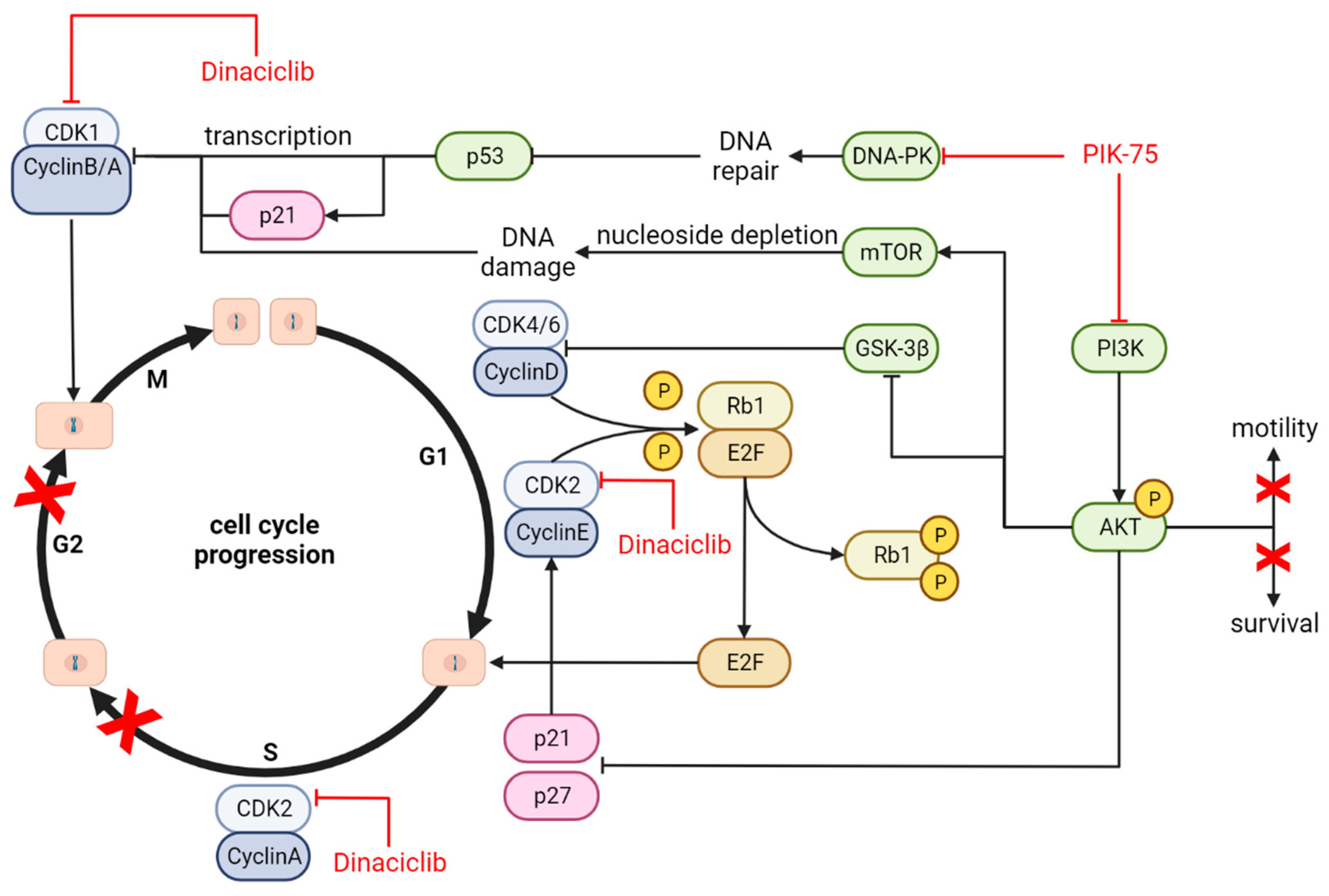
4.4. Mechanism of Action and Downstream Effects
4.5. Limitations and Future Directions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wilson, A.; Goltsman, D.; Nankervis, J.; Clark, J.; Gupta, R.; Ashford, B. Defining the incidence of cutaneous squamous cell carcinoma in coastal NSW Australia. Australas. J. Dermatol. 2022, 63, 213–216. [Google Scholar] [CrossRef]
- Mannella, V.; Boehm, K.; Celik, S.; Ali, T.; Mirza, A.N.; El Hasnaouy, M.; Kaffa, A.; Lyu, Y.; Kafaei Golahmadi, D.; Leigh, I.M.; et al. Growth and Viability of Cutaneous Squamous Cell Carcinoma Cell Lines Display Different Sensitivities to Isoform−Specific Phosphoinositide 3-Kinase Inhibitors. Int. J. Mol. Sci. 2021, 22, 3567. [Google Scholar] [CrossRef] [PubMed]
- Migden, M.R.; Khushalani, N.I.; Chang, A.L.S.; Lewis, K.D.; Schmults, C.D.; Hernandez−Aya, L.; Meier, F.; Schadendorf, D.; Guminski, A.; Hauschild, A.; et al. Cemiplimab in locally advanced cutaneous squamous cell carcinoma: Results from an open−label, phase 2, single−arm trial. Lancet. Oncol. 2020, 21, 294–305. [Google Scholar] [CrossRef]
- Migden, M.R.; Rischin, D.; Schmults, C.D.; Guminski, A.; Hauschild, A.; Lewis, K.D.; Chung, C.H.; Hernandez-Aya, L.; Lim, A.M.; Chang, A.L.S.; et al. PD−1 Blockade with Cemiplimab in Advanced Cutaneous Squamous−Cell Carcinoma. N. Engl. J. Med. 2018, 379, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Duggan, S.; Deeks, E.D. Cemiplimab: A Review in Advanced Cutaneous Squamous Cell Carcinoma. Drugs 2020, 80, 813–819. [Google Scholar] [CrossRef] [PubMed]
- Stanganelli, I.; Spagnolo, F.; Argenziano, G.; Ascierto, P.A.; Bassetto, F.; Bossi, P.; Donato, V.; Massi, D.; Massone, C.; Patuzzo, R.; et al. The Multidisciplinary Management of Cutaneous Squamous Cell Carcinoma: A Comprehensive Review and Clinical Recommendations by a Panel of Experts. Cancers 2022, 14, 377. [Google Scholar] [CrossRef] [PubMed]
- Yeo, N.; Genenger, B.; Aghmesheh, M.; Thind, A.; Napaki, S.; Perry, J.; Ashford, B.; Ranson, M.; Brungs, D. Sex as a Predictor of Response to Immunotherapy in Advanced Cutaneous Squamous Cell Carcinoma. Cancers 2023, 15, 5026. [Google Scholar] [CrossRef] [PubMed]
- Fania, L.; Didona, D.; Di Pietro, F.R.; Verkhovskaia, S.; Morese, R.; Paolino, G.; Donati, M.; Ricci, F.; Coco, V.; Ricci, F.; et al. Cutaneous Squamous Cell Carcinoma: From Pathophysiology to Novel Therapeutic Approaches. Biomedicines 2021, 9, 171. [Google Scholar] [CrossRef] [PubMed]
- Gellrich, F.F.; Hüning, S.; Beissert, S.; Eigentler, T.; Stockfleth, E.; Gutzmer, R.; Meier, F. Medical treatment of advanced cutaneous squamous−cell carcinoma. J. Eur. Acad. Dermatol. Venereol. 2019, 33, 38–43. [Google Scholar] [CrossRef]
- Bernat−Peguera, A.; Navarro−Ventura, J.; Lorenzo−Sanz, L.; da Silva−Diz, V.; Bosio, M.; Palomero, L.; Penin, R.M.; Pérez Sidelnikova, D.; Bermejo, J.O.; Taberna, M.; et al. FGFR Inhibition Overcomes Resistance to EGFR−targeted Therapy in Epithelial−like Cutaneous Carcinoma. Clin. Cancer Res. 2021, 27, 1491–1504. [Google Scholar] [CrossRef]
- Janus, J.M.; O’Shaughnessy, R.F.L.; Harwood, C.A.; Maffucci, T. Phosphoinositide 3−Kinase−Dependent Signalling Pathways in Cutaneous Squamous Cell Carcinomas. Cancers 2017, 9, 86. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.-T.; Zhao, P.; Yang, M.-L.; Lv, G.-Z.; Zhao, T.-L. GDC−0084 inhibits cutaneous squamous cell carcinoma cell growth. Biochem. Biophys. Res. Commun. 2018, 503, 1941–1948. [Google Scholar] [CrossRef] [PubMed]
- Lin, N.; Moroi, Y.; Uchi, H.; Fukiwake, N.; Dainichi, T.; Takeuchi, S.; Takahara, M.; Tu, Y.; Furue, M.; Urabe, K. Significance of the expression of phosphorylated−STAT3, −Akt, and −ERK1/2 in several tumors of the epidermis. J. Dermatol. Sci. 2007, 48, 71–73. [Google Scholar] [CrossRef]
- Pickering, C.R.; Zhou, J.H.; Lee, J.J.; Drummond, J.A.; Peng, S.A.; Saade, R.E.; Tsai, K.Y.; Curry, J.L.; Tetzlaff, M.T.; Lai, S.Y.; et al. Mutational landscape of aggressive cutaneous squamous cell carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 6582–6592. [Google Scholar] [CrossRef] [PubMed]
- Al−Rohil, R.N.; Tarasen, A.J.; Carlson, J.A.; Wang, K.; Johnson, A.; Yelensky, R.; Lipson, D.; Elvin, J.A.; Vergilio, J.A.; Ali, S.M.; et al. Evaluation of 122 advanced−stage cutaneous squamous cell carcinomas by comprehensive genomic profiling opens the door for new routes to targeted therapies. Cancer 2016, 122, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-S.; Shin, S.; Jung, S.-H.; Park, Y.M.; Park, G.S.; Lee, S.H.; Chung, Y.-J. Genomic Progression of Precancerous Actinic Keratosis to Squamous Cell Carcinoma. J. Investig. Dermatol. 2022, 142, 528–538. [Google Scholar] [CrossRef]
- Barrette, K.; Van Kelst, S.; Wouters, J.; Marasigan, V.; Fieuws, S.; Agostinis, P.; van den Oord, J.; Garmyn, M. Epithelial−mesenchymal transition during invasion of cutaneous squamous cell carcinoma is paralleled by AKT activation. Br. J. Dermatol. 2014, 171, 1014–1021. [Google Scholar] [CrossRef]
- Genenger, B.; Perry, J.R.; Ashford, B.; Ranson, M. A tEMTing target? Clinical and experimental evidence for epithelial−mesenchymal transition in the progression of cutaneous squamous cell carcinoma (a scoping systematic review). Discov. Oncol. 2022, 13, 42. [Google Scholar] [CrossRef]
- Minaei, E.; Mueller, S.A.; Ashford, B.; Thind, A.S.; Mitchell, J.; Perry, J.R.; Genenger, B.; Clark, J.R.; Gupta, R.; Ranson, M. Cancer Progression Gene Expression Profiling Identifies the Urokinase Plasminogen Activator Receptor as a Biomarker of Metastasis in Cutaneous Squamous Cell Carcinoma. Front. Oncol. 2022, 12, 1188. [Google Scholar] [CrossRef] [PubMed]
- Thind, A.S.; Ashford, B.; Strbenac, D.; Mitchell, J.; Lee, J.; Mueller, S.A.; Minaei, E.; Perry, J.R.; Ch’ng, S.; Iyer, N.G.; et al. Whole genome analysis reveals the genomic complexity in metastatic cutaneous squamous cell carcinoma. Front. Oncol. 2022, 12, 919118. [Google Scholar] [CrossRef]
- Hedberg, M.L.; Berry, C.T.; Moshiri, A.S.; Xiang, Y.; Yeh, C.J.; Attilasoy, C.; Capell, B.C.; Seykora, J.T. Molecular Mechanisms of Cutaneous Squamous Cell Carcinoma. Int. J. Mol. Sci. 2022, 23, 3478. [Google Scholar] [CrossRef] [PubMed]
- Hanker, A.B.; Kaklamani, V.; Arteaga, C.L. Challenges for the Clinical Development of PI3K Inhibitors: Strategies to Improve Their Impact in Solid Tumors. Cancer Discov. 2019, 9, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Rodon, J.; Dienstmann, R.; Serra, V.; Tabernero, J. Development of PI3K inhibitors: Lessons learned from early clinical trials. Nat. Rev. Clin. Oncol. 2013, 10, 143–153. [Google Scholar] [CrossRef]
- Wang, X.; Ding, J.; Meng, L.H. PI3K isoform−selective inhibitors: Next−generation targeted cancer therapies. Acta Pharmacol. Sin. 2015, 36, 1170–1176. [Google Scholar] [CrossRef]
- Smirnova, T.; Zhou, Z.N.; Flinn, R.J.; Wyckoff, J.; Boimel, P.J.; Pozzuto, M.; Coniglio, S.J.; Backer, J.M.; Bresnick, A.R.; Condeelis, J.S.; et al. Phosphoinositide 3−kinase signaling is critical for ErbB3−driven breast cancer cell motility and metastasis. Oncogene 2012, 31, 706–715. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.K.; Gustafson, W.C.; Charron, E.; Houseman, B.T.; Zunder, E.; Goga, A.; Gray, N.S.; Pollok, B.; Oakes, S.A.; James, C.D.; et al. Dual blockade of lipid and cyclin−dependent kinases induces synthetic lethality in malignant glioma. Proc. Natl. Acad. Sci. USA 2012, 109, 12722–12727. [Google Scholar] [CrossRef] [PubMed]
- Mohiuddin, I.S.; Kang, M.H. DNA−PK as an Emerging Therapeutic Target in Cancer. Front. Oncol. 2019, 9, 635. [Google Scholar] [CrossRef] [PubMed]
- Duong, H.Q.; Yi, Y.W.; Kang, H.J.; Hong, Y.B.; Tang, W.; Wang, A.; Seong, Y.S.; Bae, I. Inhibition of NRF2 by PIK-75 augments sensitivity of pancreatic cancer cells to gemcitabine. Int. J. Oncol. 2014, 44, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Bose, P.; Simmons, G.L.; Grant, S. Cyclin−dependent kinase inhibitor therapy for hematologic malignancies. Expert. Opin. Investig. Drugs 2013, 22, 723–738. [Google Scholar] [CrossRef]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin−dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef]
- Gupta, P.; Narayanan, S.; Yang, D.-H. Chapter 9—CDK Inhibitors as Sensitizing Agents for Cancer Chemotherapy. In Protein Kinase Inhibitors as Sensitizing Agents for Chemotherapy; Chen, Z.−S., Yang, D.−H., Eds.; Academic Press: Cambridge, MA, USA, 2019; Volume 4, pp. 125–149. [Google Scholar]
- Gerson, S.L.; Caimi, P.F.; William, B.M.; Creger, R.J. Chapter 57—Pharmacology and Molecular Mechanisms of Antineoplastic Agents for Hematologic Malignancies. In Hematology, 7th ed.; Hoffman, R., Benz, E.J., Silberstein, L.E., Heslop, H.E., Weitz, J.I., Anastasi, J., Salama, M.E., Abutalib, S.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 849–912. [Google Scholar]
- Cragg, G.M.; Newman, D.J. 1.08—Natural Product Sources of Drugs: Plants, Microbes, Marine Organisms, and Animals. In Comprehensive Medicinal Chemistry II; Taylor, J.B., Triggle, D.J., Eds.; Elsevier: Oxford, UK, 2007; pp. 355–403. [Google Scholar]
- Walker, A.J.; Wedam, S.; Amiri−Kordestani, L.; Bloomquist, E.; Tang, S.; Sridhara, R.; Chen, W.; Palmby, T.R.; Fourie Zirkelbach, J.; Fu, W.; et al. FDA Approval of Palbociclib in Combination with Fulvestrant for the Treatment of Hormone Receptor−Positive, HER2−Negative Metastatic Breast Cancer. Clin. Cancer Res. 2016, 22, 4968–4972. [Google Scholar] [CrossRef] [PubMed]
- Parry, D.; Guzi, T.; Shanahan, F.; Davis, N.; Prabhavalkar, D.; Wiswell, D.; Seghezzi, W.; Paruch, K.; Dwyer, M.P.; Doll, R.; et al. Dinaciclib (SCH 727965), a novel and potent cyclin−dependent kinase inhibitor. Mol. Cancer Ther. 2010, 9, 2344–2353. [Google Scholar] [CrossRef] [PubMed]
- Nemunaitis, J.J.; Small, K.A.; Kirschmeier, P.; Zhang, D.; Zhu, Y.; Jou, Y.-M.; Statkevich, P.; Yao, S.-L.; Bannerji, R. A first−in−human, phase 1, dose−escalation study of dinaciclib, a novel cyclin−dependent kinase inhibitor, administered weekly in subjects with advanced malignancies. J. Transl. Med. 2013, 11, 259. [Google Scholar] [CrossRef] [PubMed]
- Gojo, I.; Sadowska, M.; Walker, A.; Feldman, E.J.; Iyer, S.P.; Baer, M.R.; Sausville, E.A.; Lapidus, R.G.; Zhang, D.; Zhu, Y.; et al. Clinical and laboratory studies of the novel cyclin−dependent kinase inhibitor dinaciclib (SCH 727965) in acute leukemias. Cancer Chemother. Pharmacol. 2013, 72, 897–908. [Google Scholar] [CrossRef] [PubMed]
- Murphy, A.G.; Zahurak, M.; Shah, M.; Weekes, C.D.; Hansen, A.; Siu, L.L.; Spreafico, A.; LoConte, N.; Anders, N.M.; Miles, T.; et al. A Phase I Study of Dinaciclib in Combination with MK−2206 in Patients with Advanced Pancreatic Cancer. Clin. Transl. Sci. 2020, 13, 1178–1188. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.; Ashford, B.; Thind, A.S.; Gauthier, M.E.; Minaei, E.; Major, G.; Iyer, N.G.; Gupta, R.; Clark, J.; Ranson, M. Comprehensive Mutational and Phenotypic Characterization of New Metastatic Cutaneous Squamous Cell Carcinoma Cell Lines Reveal Novel Drug Susceptibilities. Int. J. Mol. Sci. 2020, 21, 9536. [Google Scholar] [CrossRef]
- Hu, C.; Dadon, T.; Chenna, V.; Yabuuchi, S.; Bannerji, R.; Booher, R.; Strack, P.; Azad, N.; Nelkin, B.D.; Maitra, A. Combined Inhibition of Cyclin−Dependent Kinases (Dinaciclib) and AKT (MK−2206) Blocks Pancreatic Tumor Growth and Metastases in Patient−Derived Xenograft Models. Mol. Cancer Ther. 2015, 14, 1532–1539. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.J.; Yeh, Y.Y.; Smith, L.L.; Wagner, A.J.; Hessler, J.; Gupta, S.; Flynn, J.; Jones, J.; Zhang, X.; Bannerji, R.; et al. The novel cyclin−dependent kinase inhibitor dinaciclib (SCH727965) promotes apoptosis and abrogates microenvironmental cytokine protection in chronic lymphocytic leukemia cells. Leukemia 2012, 26, 2554–2557. [Google Scholar] [CrossRef] [PubMed]
- Boukamp, P.; Petrussevska, R.T.; Breitkreutz, D.; Hornung, J.; Markham, A.; Fusenig, N.E. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 1988, 106, 761–771. [Google Scholar] [CrossRef]
- Kolde, R. pheatmap: Pretty Heatmaps (v1.0.12). Available online: https://CRAN.R−project.org/package=pheatmap (accessed on 20 October 2023).
- Brungs, D.; Minaei, E.; Piper, A.K.; Perry, J.; Splitt, A.; Carolan, M.; Ryan, S.; Wu, X.J.; Corde, S.; Tehei, M.; et al. Establishment of novel long−term cultures from EpCAM positive and negative circulating tumour cells from patients with metastatic gastroesophageal cancer. Sci. Rep. 2020, 10, 539. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou−Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Y.; Hanna, G.J.; Laga, A.C.; Haddad, R.I.; Lorch, J.H.; Hammerman, P.S. Genomic Analysis of Metastatic Cutaneous Squamous Cell Carcinoma. Clin. Cancer Res. 2015, 21, 1447–1456. [Google Scholar] [CrossRef]
- Kusaba, Y.; Kajihara, I.; Sakamoto, R.; Maeda−Otsuka, S.; Yamada−Kanazawa, S.; Sawamura, S.; Makino, K.; Aoi, J.; Masuguchi, S.; Fukushima, S. PIK3CA mutations in cutaneous squamous cell carcinoma. Intractable Rare Dis. Res. 2023, 12, 206–207. [Google Scholar] [CrossRef] [PubMed]
- Hollander, M.C.; Blumenthal, G.M.; Dennis, P.A. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat. Rev. Cancer 2011, 11, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Vogt, P.K. Helical domain and kinase domain mutations in p110α of phosphatidylinositol 3−kinase induce gain of function by different mechanisms. Natl. Acad. Sci. 2008, 105, 2652–2657. [Google Scholar] [CrossRef]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef]
- Herrera−Abreu, M.T.; Palafox, M.; Asghar, U.; Rivas, M.A.; Cutts, R.J.; Garcia−Murillas, I.; Pearson, A.; Guzman, M.; Rodriguez, O.; Grueso, J.; et al. Early Adaptation and Acquired Resistance to CDK4/6 Inhibition in Estrogen Receptor–Positive Breast Cancer. Cancer Res. 2016, 76, 2301–2313. [Google Scholar] [CrossRef]
- García−Sancha, N.; Corchado−Cobos, R.; Pérez−Losada, J.; Cañueto, J. MicroRNA Dysregulation in Cutaneous Squamous Cell Carcinoma. Int. J. Mol. Sci. 2019, 20, 2181. [Google Scholar] [CrossRef]
- Muguruma, M.; Teraoka, S.; Miyahara, K.; Ueda, A.; Asaoka, M.; Okazaki, M.; Kawate, T.; Kuroda, M.; Miyagi, Y.; Ishikawa, T. Differences in drug sensitivity between two−dimensional and three−dimensional culture systems in triple−negative breast cancer cell lines. Biochem. Biophys. Res. Commun. 2020, 533, 268–274. [Google Scholar] [CrossRef]
- Bonelli, M.; Terenziani, R.; Zoppi, S.; Fumarola, C.; La Monica, S.; Cretella, D.; Alfieri, R.; Cavazzoni, A.; Digiacomo, G.; Galetti, M.; et al. Dual Inhibition of CDK4/6 and PI3K/AKT/mTOR Signaling Impairs Energy Metabolism in MPM Cancer Cells. Int. J. Mol. Sci. 2020, 21, 5165. [Google Scholar] [CrossRef]
- Zhao, S.; Zhang, H.; Yang, N.; Yang, J. A narrative review about CDK4/6 inhibitors in the setting of drug resistance: Updates on biomarkers and therapeutic strategies in breast cancer. Transl. Cancer Res. 2023, 12, 1617–1634. [Google Scholar] [CrossRef]
- Ornelas, I.M.; Silva, T.M.; Fragel−Madeira, L.; Ventura, A.L. Inhibition of PI3K/Akt pathway impairs G2/M transition of cell cycle in late developing progenitors of the avian embryo retina. PLoS ONE 2013, 8, e53517. [Google Scholar] [CrossRef]
- Juvekar, A.; Hu, H.; Yadegarynia, S.; Lyssiotis, C.A.; Ullas, S.; Lien, E.C.; Bellinger, G.; Son, J.; Hok, R.C.; Seth, P.; et al. Phosphoinositide 3−kinase inhibitors induce DNA damage through nucleoside depletion. Proc. Natl. Acad. Sci. USA 2016, 113, E4338–E4347. [Google Scholar] [CrossRef] [PubMed]
- Strock, C.J.; Park, J.I.; Nakakura, E.K.; Bova, G.S.; Isaacs, J.T.; Ball, D.W.; Nelkin, B.D. Cyclin−dependent kinase 5 activity controls cell motility and metastatic potential of prostate cancer cells. Cancer Res. 2006, 66, 7509–7515. [Google Scholar] [CrossRef]
- Pozo, K.; Bibb, J.A. The Emerging Role of Cdk5 in Cancer. Trends Cancer 2016, 2, 606–618. [Google Scholar] [CrossRef] [PubMed]
- Kölsch, V.; Charest, P.G.; Firtel, R.A. The regulation of cell motility and chemotaxis by phospholipid signaling. J. Cell Sci. 2008, 121, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Cain, R.J.; Ridley, A.J. Phosphoinositide 3−kinases in cell migration. Biol. Cell 2009, 101, 13–29. [Google Scholar] [CrossRef]
- Arcaro, A.; Guerreiro, A.S. The phosphoinositide 3−kinase pathway in human cancer: Genetic alterations and therapeutic implications. Curr. Genom. 2007, 8, 271–306. [Google Scholar] [CrossRef]
- Qie, S.; Diehl, J.A. Cyclin D1, cancer progression, and opportunities in cancer treatment. J. Mol. Med. 2016, 94, 1313–1326. [Google Scholar] [CrossRef]
- Maret, W. Analyzing free zinc(II) ion concentrations in cell biology with fluorescent chelating molecules. Metallomics 2015, 7, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Naderali, E.; Valipour, B.; Khaki, A.A.; Soleymani Rad, J.; Alihemmati, A.; Rahmati, M.; Nozad Charoudeh, H. Positive Effects of PI3K/Akt Signaling Inhibition on PTEN and P53 in Prevention of Acute Lymphoblastic Leukemia Tumor Cells. Adv. Pharm. Bull. 2019, 9, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. Are p27 and p21 Cytoplasmic Oncoproteins? Cell Cycle 2002, 1, 391–393. [Google Scholar] [CrossRef]
- Bock, C.; Datlinger, P.; Chardon, F.; Coelho, M.A.; Dong, M.B.; Lawson, K.A.; Lu, T.; Maroc, L.; Norman, T.M.; Song, B.; et al. High−content CRISPR screening. Nat. Rev. Methods Primers 2022, 2, 8. [Google Scholar] [CrossRef]
- Alyateem, G.; Wade, H.M.; Bickert, A.A.; Lipsey, C.C.; Mondal, P.; Smith, M.D.; Labib, R.M.; Mock, B.A.; Robey, R.W.; Gottesman, M.M. Use of CRISPR−based screens to identify mechanisms of chemotherapy resistance. Cancer Gene Ther. 2023, 30, 1043–1050. [Google Scholar] [CrossRef] [PubMed]
- Nykänen, N.; Mäkelä, R.; Arjonen, A.; Härmä, V.; Lewandowski, L.; Snowden, E.; Blaesius, R.; Jantunen, I.; Kuopio, T.; Kononen, J.; et al. Ex Vivo Drug Screening Informed Targeted Therapy for Metastatic Parotid Squamous Cell Carcinoma. Front. Oncol. 2021, 11, 735820. [Google Scholar] [CrossRef]
- Kenerson, H.L.; Sullivan, K.M.; Seo, Y.D.; Stadeli, K.M.; Ussakli, C.; Yan, X.; Lausted, C.; Pillarisetty, V.G.; Park, J.O.; Riehle, K.J.; et al. Tumor slice culture as a biologic surrogate of human cancer. Ann. Transl. Med. 2020, 8, 114. [Google Scholar] [CrossRef]
- Capala, M.E.; Pachler, K.S.; Lauwers, I.; de Korte, M.A.; Verkaik, N.S.; Mast, H.; Jonker, B.P.; Sewnaik, A.; Hardillo, J.A.; Keereweer, S.; et al. Ex Vivo Functional Assay for Evaluating Treatment Response in Tumor Tissue of Head and Neck Squamous Cell Carcinoma. Cancers 2023, 15, 478. [Google Scholar] [CrossRef]
- Donnadieu, J.; Lachaier, E.; Peria, M.; Saidak, Z.; Dakpe, S.; Ikoli, J.F.; Chauffert, B.; Page, C.; Galmiche, A. Short−term culture of tumour slices reveals the heterogeneous sensitivity of human head and neck squamous cell carcinoma to targeted therapies. BMC Cancer 2016, 16, 273. [Google Scholar] [CrossRef]
- Voabil, P.; de Bruijn, M.; Roelofsen, L.M.; Hendriks, S.H.; Brokamp, S.; van den Braber, M.; Broeks, A.; Sanders, J.; Herzig, P.; Zippelius, A.; et al. An ex vivo tumor fragment platform to dissect response to PD−1 blockade in cancer. Nat. Med. 2021, 27, 1250–1261. [Google Scholar] [CrossRef]
- Rosenblat, G.; Meretski, S.; Segal, J.; Tarshis, M.; Schroeder, A.; Zanin−Zhorov, A.; Lion, G.; Ingber, A.; Hochberg, M. Polyhydroxylated fatty alcohols derived from avocado suppress inflammatory response and provide non−sunscreen protection against UV−induced damage in skin cells. Arch. Dermatol. Res. 2011, 303, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Hochberg, M.; Kunicher, N.; Gilead, L.; Maly, A.; Falk, H.; Ingber, A.; Panet, A. Tropism of herpes simplex virus type 1 to nonmelanoma skin cancers. Br. J. Dermatol. 2011, 164, 273–281. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | UW-CSCC1 | UW-CSCC2 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Mutation Type | Impact (CADD) | TPM | CNV | Mutation Type | Impact (CADD) | TPM | CNV | ||
| Cell cycle | CCNB3 | 1.4 | 1.98 | missense | 1.945 | 6.3 | 1.89 | ||
| CDK12 | synonymous | 10.79 | 51 | 3.98 | 60 | 3.80 | |||
| synonymous | 7.65 | ||||||||
| CDKN2A | 451 | 3.81 | stop-gained | n.a. | 124 | 2.83 | |||
| NOTCH1 | 14 | 4.06 | stop-gained | 48 | 7 | 5.83 | |||
| synonymous | 6.043 | ||||||||
| NOTCH2 | 67 | 3.03 | stop gained | 41 | 11 | 1.37 | |||
| PAK4 | missense | 25.6 | 99 | 4.92 | 109 | 3.03 | |||
| PAK5 | missense | 31 | 0.05 | 4.02 | 0.2 | 6.73 | |||
| RB1 | in−frame insertion | n.a. | 28 | 2.98 | 37 | 2.46 | |||
| TP53 | missense | 29.5 | 109 | 3.15 | stop-gained | 36 | 153 | 3.38 | |
| missense | 31 | missense | 28.4 | ||||||
| WEE1 * | 46 | 3.15 | stop-gained | n.a. | 61 | 3.29 | |||
| PI3K/AKT/mTOR | AKT3 | missense | 28.2 | 62 | 3.01 | synonymous | 12.69 | 22 | 2.92 |
| EGFR | missense | 26 | 20 | 2.31 | 196 | 3.47 | |||
| ERBB2 | synonymous | 10.74 | 80 | 3.98 | 70 | 3.80 | |||
| ERBB3 | missense | 25.6 | 5.7 | 3.01 | missense | 23.4 | 21 | 2.94 | |
| missense | 23.5 | ||||||||
| HRAS | 20 | 3.15 | missense | n.a. | 57 | 3.29 | |||
| PIK3C2A | 27 | 3.15 | missense | 17.23 | 36 | 3.29 | |||
| PIK3C2B | missense | 26.3 | 5.3 | 3.01 | 20 | 2.92 | |||
| PIK3CG | missense | 24.1 | 2.4 | 3.04 | missense | 20.7 | 1.4 | 2.98 | |
| PIK3R5 | missense | 22.7 | 0.11 | 3.15 | 0.01 | 3.38 | |||
| PIK3R6 | frameshift | n.a. | 0.04 | 3.15 | 0.04 | 3.38 | |||
| complex substitution | n.a. | ||||||||
| missense | 16.13 | ||||||||
| missense | n.a. | ||||||||
| SMAD3 | 103 | 2.09 | complex substitution | n.a. | 233 | 3.99 | |||
| TGFBR1 | stop gained | 38 | 73 | 4.92 | 29 | 3.03 | |||
| Cell Line | IC50 (nM) | ||
|---|---|---|---|
| PIK-75 | BGT226 | Dinaciclib | |
| UW-CSCC1 | 220 ± 32 * | 223 ± 43 | 19 ± 2.8 |
| UW-CSCC2 | 56 ± 3 * | 195 ± 42 | 20 ± 2.4 |
| HaCaT | No effect | NA | No effect |
| Variable | UW-CSCC1 | UW-CSCC2 | ||
|---|---|---|---|---|
| PIK:DIN | BGT/DIN | PIK:DIN | BGT/DIN | |
| 2D synergy assay | Strongly additive | Weakly additive | Synergistic | Antagonistic |
| 3D synergy assays | Additive | Synergistic | Weakly additive | Synergistic |
| Apoptosis | Weakly additive, PIK-75 dominates | Not Assessed | Weakly additive, PIK-75 dominates | Not Assessed |
| Cell cycle phase | No synergy, PIK-75 effect dominates | Not Assessed | No synergy, PIK-75 effect dominates | Not Assessed |
| Motility | Weakly additive, dinaciclib effect strongly dominates | Not Assessed | Weakly additive, dinaciclib effect dominates | Not Assessed |
| Cell signalling | No synergy, some effects dominated by either drug | Not Assessed | No synergy, some effects dominated by either drug | Not Assessed |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perry, J.R.; Genenger, B.; Thind, A.S.; Ashford, B.; Ranson, M. PIK Your Poison: The Effects of Combining PI3K and CDK Inhibitors against Metastatic Cutaneous Squamous Cell Carcinoma In Vitro. Cancers 2024, 16, 370. https://doi.org/10.3390/cancers16020370
Perry JR, Genenger B, Thind AS, Ashford B, Ranson M. PIK Your Poison: The Effects of Combining PI3K and CDK Inhibitors against Metastatic Cutaneous Squamous Cell Carcinoma In Vitro. Cancers. 2024; 16(2):370. https://doi.org/10.3390/cancers16020370
Chicago/Turabian StylePerry, Jay R., Benjamin Genenger, Amarinder Singh Thind, Bruce Ashford, and Marie Ranson. 2024. "PIK Your Poison: The Effects of Combining PI3K and CDK Inhibitors against Metastatic Cutaneous Squamous Cell Carcinoma In Vitro" Cancers 16, no. 2: 370. https://doi.org/10.3390/cancers16020370
APA StylePerry, J. R., Genenger, B., Thind, A. S., Ashford, B., & Ranson, M. (2024). PIK Your Poison: The Effects of Combining PI3K and CDK Inhibitors against Metastatic Cutaneous Squamous Cell Carcinoma In Vitro. Cancers, 16(2), 370. https://doi.org/10.3390/cancers16020370