
Antibody–Drug Conjugates: The Dynamic Evolution from Conventional to Next-Generation Constructs


Abstract
:Simple Summary
Abstract
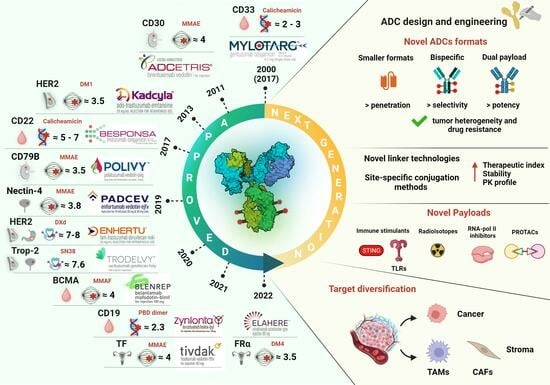
1. Introduction
2. Key Components of ADCs and Mechanism of Action
3. Target Antigen Selection: A Balance between Expression and Internalization
4. Antibody: The Precision Guide in ADC Therapy
5. From Traditional to Novel: Diversifying the ADC Payload Landscape
6. The Linker—A Balancing Bridge
7. Conjugation Technology
8. Existing Challenges and Opportunities of Next-Generation ADCs
Toxicity and Drug Resistance
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Joubert, N.; Beck, A.; Dumontet, C.; Denevault-Sabourin, C. Antibody-Drug Conjugates: The Last Decade. Pharmaceuticals 2020, 13, 245. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Li, S.; Han, S.; Shi, C.; Zhang, Y. Antibody drug conjugate: The “biological missile” for targeted cancer therapy. Signal Transduct. Target. Ther. 2022, 7, 93. [Google Scholar] [CrossRef] [PubMed]
- Dumontet, C.; Reichert, J.M.; Senter, P.D.; Lambert, J.M.; Beck, A. Antibody–drug conjugates come of age in oncology. Nat. Rev. Drug Discov. 2023, 22, 641–661. [Google Scholar] [CrossRef] [PubMed]
- Gogia, P.; Ashraf, H.; Bhasin, S.; Xu, Y. Antibody-Drug Conjugates: A Review of Approved Drugs and Their Clinical Level of Evidence. Cancers 2023, 15, 3886. [Google Scholar] [CrossRef] [PubMed]
- Esapa, B.; Jiang, J.; Cheung, A.; Chenoweth, A.; Thurston, D.E.; Karagiannis, S.N. Target Antigen Attributes and Their Contributions to Clinically Approved Antibody-Drug Conjugates (ADCs) in Haematopoietic and Solid Cancers. Cancers 2023, 15, 1845. [Google Scholar] [CrossRef] [PubMed]
- Maecker, H.; Jonnalagadda, V.; Bhakta, S.; Jammalamadaka, V.; Junutula, J.R. Exploration of the antibody–drug conjugate clinical landscape. mAbs 2023, 15, 2229101. [Google Scholar] [CrossRef] [PubMed]
- Staudacher, A.H.; Brown, M.P. Antibody drug conjugates and bystander killing: Is antigen-dependent internalisation required? Br. J. Cancer 2017, 117, 1736–1742. [Google Scholar] [CrossRef]
- Ashman, N.; Bargh, J.D.; Spring, D.R. Non-internalising antibody-drug conjugates. Chem. Soc. Rev. 2022, 51, 9182–9202. [Google Scholar] [CrossRef]
- Dal Corso, A.; Gébleux, R.; Murer, P.; Soltermann, A.; Neri, D. A non-internalizing antibody-drug conjugate based on an anthracycline payload displays potent therapeutic activity in vivo. J. Control Release 2017, 264, 211–218. [Google Scholar] [CrossRef]
- Cazzamalli, S.; Corso, A.D.; Neri, D. Linker stability influences the anti-tumor activity of acetazolamide-drug conjugates for the therapy of renal cell carcinoma. J. Control Release 2017, 246, 39–45. [Google Scholar] [CrossRef]
- Liu, R.; Oldham, R.J.; Teal, E.; Beers, S.A.; Cragg, M.S. Fc-Engineering for Modulated Effector Functions-Improving Antibodies for Cancer Treatment. Antibodies 2020, 9, 64. [Google Scholar] [CrossRef] [PubMed]
- Ramdani, Y.; Lamamy, J.; Watier, H.; Gouilleux-Gruart, V. Monoclonal Antibody Engineering and Design to Modulate FcRn Activities: A Comprehensive Review. Int. J. Mol. Sci. 2022, 23, 9604. [Google Scholar] [CrossRef] [PubMed]
- Conilh, L.; Sadilkova, L.; Viricel, W.; Dumontet, C. Payload diversification: A key step in the development of antibody–drug conjugates. J. Hematol. Oncol. 2023, 16, 3. [Google Scholar] [CrossRef] [PubMed]
- Mahalingaiah, P.K.; Ciurlionis, R.; Durbin, K.R.; Yeager, R.L.; Philip, B.K.; Bawa, B.; Mantena, S.R.; Enright, B.P.; Liguori, M.J.; Van Vleet, T.R. Potential mechanisms of target-independent uptake and toxicity of antibody-drug conjugates. Pharmacol. Ther. 2019, 200, 110–125. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Li, Z.; Bussing, D.; Shah, D.K. Evaluation of Quantitative Relationship Between Target Expression and Antibody-Drug Conjugate Exposure Inside Cancer Cells. Drug Metab. Dispos. 2020, 48, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Drago, J.Z.; Modi, S.; Chandarlapaty, S. Unlocking the potential of antibody–drug conjugates for cancer therapy. Nat. Rev. Clin. Oncol. 2021, 18, 327–344. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.; Jacot, W.; Yamashita, T.; Sohn, J.; Vidal, M.; Tokunaga, E.; Tsurutani, J.; Ueno, N.T.; Prat, A.; Chae, Y.S.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Low Advanced Breast Cancer. New Engl. J. Med. 2022, 387, 9–20. [Google Scholar] [CrossRef]
- Hammood, M.; Craig, A.W.; Leyton, J.V. Impact of Endocytosis Mechanisms for the Receptors Targeted by the Currently Approved Antibody-Drug Conjugates (ADCs)-A Necessity for Future ADC Research and Development. Pharmaceuticals 2021, 14, 674. [Google Scholar] [CrossRef]
- Leyton, J.V. Improving Receptor-Mediated Intracellular Access and Accumulation of Antibody Therapeutics-The Tale of HER2. Antibodies 2020, 9, 32. [Google Scholar] [CrossRef]
- Nami, B.; Maadi, H.; Wang, Z. Mechanisms Underlying the Action and Synergism of Trastuzumab and Pertuzumab in Targeting HER2-Positive Breast Cancer. Cancers 2018, 10, 342. [Google Scholar] [CrossRef]
- Li, J.Y.; Perry, S.R.; Muniz-Medina, V.; Wang, X.; Wetzel, L.K.; Rebelatto, M.C.; Hinrichs, M.J.; Bezabeh, B.Z.; Fleming, R.L.; Dimasi, N.; et al. A Biparatopic HER2-Targeting Antibody-Drug Conjugate Induces Tumor Regression in Primary Models Refractory to or Ineligible for HER2-Targeted Therapy. Cancer Cell 2016, 29, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Andreev, J.; Thambi, N.; Perez Bay, A.E.; Delfino, F.; Martin, J.; Kelly, M.P.; Kirshner, J.R.; Rafique, A.; Kunz, A.; Nittoli, T.; et al. Bispecific Antibodies and Antibody-Drug Conjugates (ADCs) Bridging HER2 and Prolactin Receptor Improve Efficacy of HER2 ADCs. Mol. Cancer Ther. 2017, 16, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Shim, H. Bispecific Antibodies and Antibody-Drug Conjugates for Cancer Therapy: Technological Considerations. Biomolecules 2020, 10, 360. [Google Scholar] [CrossRef] [PubMed]
- Weisser, N.E.; Sanches, M.; Escobar-Cabrera, E.; O’Toole, J.; Whalen, E.; Chan, P.W.Y.; Wickman, G.; Abraham, L.; Choi, K.; Harbourne, B.; et al. An anti-HER2 biparatopic antibody that induces unique HER2 clustering and complement-dependent cytotoxicity. Nat. Commun. 2023, 14, 1394. [Google Scholar] [CrossRef] [PubMed]
- Baah, S.; Laws, M.; Rahman, K.M. Antibody-Drug Conjugates-A Tutorial Review. Molecules 2021, 26, 2943. [Google Scholar] [CrossRef] [PubMed]
- Walter, R.B. The role of CD33 as therapeutic target in acute myeloid leukemia. Expert Opin. Ther. Targets 2014, 18, 715–718. [Google Scholar] [CrossRef]
- Du, X.; Beers, R.; FitzGerald, D.J.; Pastan, I. Differential Cellular Internalization of Anti-CD19 and -CD22 Immunotoxins Results in Different Cytotoxic Activity. Cancer Res. 2008, 68, 6300–6305. [Google Scholar] [CrossRef]
- Deonarain, M.P.; Xue, Q. Tackling solid tumour therapy with small-format drug conjugates. Antib. Ther. 2020, 3, 237–245. [Google Scholar] [CrossRef]
- Strosberg, J.; El-Haddad, G.; Wolin, E.; Hendifar, A.; Yao, J.; Chasen, B.; Mittra, E.; Kunz, P.L.; Kulke, M.H.; Jacene, H.; et al. Phase 3 Trial of (177)Lu-Dotatate for Midgut Neuroendocrine Tumors. New Engl. J. Med. 2017, 376, 125–135. [Google Scholar] [CrossRef]
- Olivier, T.; Prasad, V. The approval and withdrawal of melphalan flufenamide (melflufen): Implications for the state of the FDA. Transl. Oncol. 2022, 18, 101374. [Google Scholar] [CrossRef]
- Wang, Z.; Li, H.; Gou, L.; Li, W.; Wang, Y. Antibody–drug conjugates: Recent advances in payloads. Acta Pharm. Sin. B 2023, 13, 4025–4059. [Google Scholar] [CrossRef] [PubMed]
- Sheyi, R.; de la Torre, B.G.; Albericio, F. Linkers: An Assurance for Controlled Delivery of Antibody-Drug Conjugate. Pharmaceutics 2022, 14, 396. [Google Scholar] [CrossRef] [PubMed]
- Weng, W.; Meng, T.; Zhao, Q.; Shen, Y.; Fu, G.; Shi, J.; Zhang, Y.; Wang, Z.; Wang, M.; Pan, R.; et al. Antibody–Exatecan Conjugates with a Novel Self-immolative Moiety Overcome Resistance in Colon and Lung Cancer. Cancer Discov. 2023, 13, 950–973. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, L.; Menzel, U.; Prummer, M.; Müller, P.; Buchi, M.; Kashyap, A.; Haessler, U.; Yermanos, A.; Gébleux, R.; Briendl, M.; et al. A novel anti-HER2 anthracycline-based antibody-drug conjugate induces adaptive anti-tumor immunity and potentiates PD-1 blockade in breast cancer. J. Immunother. Cancer 2019, 7, 16. [Google Scholar] [CrossRef] [PubMed]
- Fabre, M.; Ferrer, C.; Domínguez-Hormaetxe, S.; Bockorny, B.; Murias, L.; Seifert, O.; Eisler, S.A.; Kontermann, R.E.; Pfizenmaier, K.; Lee, S.Y.; et al. OMTX705, a Novel FAP-Targeting ADC Demonstrates Activity in Chemotherapy and Pembrolizumab-Resistant Solid Tumor Models. Clin. Cancer Res. 2020, 26, 3420–3430. [Google Scholar] [CrossRef]
- Hamilton, J.Z.; Klussman, K.; Mahzereh, R.; Simmons, J.; Ulrich, M.; Gardai, S.J.; Senter, P.D.; Burke, P.J. Abstract 2013: Oxidized anthracycline payloads induce anti-tumor immunogenic cell-death and show linker-dependent tolerability when delivered as ADCs. Cancer Res. 2023, 83, 2013. [Google Scholar] [CrossRef]
- Gardai, S.; Epp, A.; Law, C.-L. Abstract 2469: Brentuximab vedotin-mediated immunogenic cell death. Cancer Res. 2015, 75, 2469. [Google Scholar] [CrossRef]
- Grawunder, U. Abstract 737: Antibody drug conjugates with anthracycline payload induce tumor-selective antitumor immunity and exhibit a favorable safety profile in cynomolgus monkey toxicology studies. Cancer Res. 2018, 78, 737. [Google Scholar] [CrossRef]
- Fuentes-Antrás, J.; Genta, S.; Vijenthira, A.; Siu, L.L. Antibody-drug conjugates: In search of partners of choice. Trends Cancer 2023, 9, 339–354. [Google Scholar] [CrossRef]
- Rios-Doria, J.; Harper, J.; Rothstein, R.; Wetzel, L.; Chesebrough, J.; Marrero, A.; Chen, C.; Strout, P.; Mulgrew, K.; McGlinchey, K.; et al. Antibody–Drug Conjugates Bearing Pyrrolobenzodiazepine or Tubulysin Payloads Are Immunomodulatory and Synergize with Multiple Immunotherapies. Cancer Res. 2017, 77, 2686–2698. [Google Scholar] [CrossRef]
- Nicolò, E.; Giugliano, F.; Ascione, L.; Tarantino, P.; Corti, C.; Tolaney, S.M.; Cristofanilli, M.; Curigliano, G. Combining antibody-drug conjugates with immunotherapy in solid tumors: Current landscape and future perspectives. Cancer Treat. Rev. 2022, 106, 102395. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, M.C.; Morais, J.A.V.; Ganassin, R.; Oliveira, G.R.T.; Costa, F.C.; Morais, A.A.C.; Silveira, A.P.; Silva, V.C.M.; Longo, J.P.F.; Muehlmann, L.A. An Overview on Immunogenic Cell Death in Cancer Biology and Therapy. Pharmaceutics 2022, 14, 1564. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Xiao, D.; Xie, F.; Liu, L.; Wang, Y.; Fan, S.; Zhou, X.; Li, S. Antibody-drug conjugates: Recent advances in linker chemistry. Acta Pharm. Sin. B 2021, 11, 3889–3907. [Google Scholar] [CrossRef] [PubMed]
- Kostova, V.; Désos, P.; Starck, J.B.; Kotschy, A. The Chemistry Behind ADCs. Pharmaceuticals 2021, 14, 442. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Dai, Y.; Wu, M.; Li, L. Glutathione-mediated nanomedicines for cancer diagnosis and therapy. Chem. Eng. J. 2021, 426, 128880. [Google Scholar] [CrossRef]
- Nguyen, T.D.; Bordeau, B.M.; Balthasar, J.P. Mechanisms of ADC Toxicity and Strategies to Increase ADC Tolerability. Cancers 2023, 15, 713. [Google Scholar] [CrossRef] [PubMed]
- Sievers, E.L.; Larson, R.A.; Stadtmauer, E.A.; Estey, E.; Löwenberg, B.; Dombret, H.; Karanes, C.; Theobald, M.; Bennett, J.M.; Sherman, M.L.; et al. Efficacy and safety of gemtuzumab ozogamicin in patients with CD33-positive acute myeloid leukemia in first relapse. J. Clin. Oncol. 2001, 19, 3244–3254. [Google Scholar] [CrossRef]
- Petersdorf, S.H.; Kopecky, K.J.; Slovak, M.; Willman, C.; Nevill, T.; Brandwein, J.; Larson, R.A.; Erba, H.P.; Stiff, P.J.; Stuart, R.K.; et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood 2013, 121, 4854–4860. [Google Scholar] [CrossRef]
- Norsworthy, K.J.; Ko, C.W.; Lee, J.E.; Liu, J.; John, C.S.; Przepiorka, D.; Farrell, A.T.; Pazdur, R. FDA Approval Summary: Mylotarg for Treatment of Patients with Relapsed or Refractory CD33-Positive Acute Myeloid Leukemia. Oncologist 2018, 23, 1103–1108. [Google Scholar] [CrossRef]
- Abelman, R.O.; Wu, B.; Spring, L.M.; Ellisen, L.W.; Bardia, A. Mechanisms of Resistance to Antibody-Drug Conjugates. Cancers 2023, 15, 1278. [Google Scholar] [CrossRef]
- Zhao, H.; Gulesserian, S.; Malinao, M.C.; Ganesan, S.K.; Song, J.; Chang, M.S.; Williams, M.M.; Zeng, Z.; Mattie, M.; Mendelsohn, B.A.; et al. A Potential Mechanism for ADC-Induced Neutropenia: Role of Neutrophils in Their Own Demise. Mol. Cancer Ther. 2017, 16, 1866–1876. [Google Scholar] [CrossRef] [PubMed]
- Dorywalska, M.; Dushin, R.; Moine, L.; Farias, S.E.; Zhou, D.; Navaratnam, T.; Lui, V.; Hasa-Moreno, A.; Casas, M.G.; Tran, T.T.; et al. Molecular Basis of Valine-Citrulline-PABC Linker Instability in Site-Specific ADCs and Its Mitigation by Linker Design. Mol. Cancer Ther. 2016, 15, 958–970. [Google Scholar] [CrossRef] [PubMed]
- Balamkundu, S.; Liu, C.-F. Lysosomal-Cleavable Peptide Linkers in Antibody–Drug Conjugates. Biomedicines 2023, 11, 3080. [Google Scholar]
- Grier, K.E.; Hansen, A.H.; Haxvig, C.S.; Li, X.; Krigslund, O.; Behrendt, N.; Engelholm, L.H.; Rossi, F.; Sousa, B.C.; Harradence, G.J.; et al. Targeted delivery of alcohol-containing payloads with antibody-drug conjugates. Chem. Commun. 2023, 59, 7240–7242. [Google Scholar] [CrossRef]
- Zhou, Q. Site-Specific Antibody Conjugation for ADC and Beyond. Biomedicines 2017, 5, 64. [Google Scholar] [CrossRef] [PubMed]
- Panowski, S.; Bhakta, S.; Raab, H.; Polakis, P.; Junutula, J.R. Site-specific antibody drug conjugates for cancer therapy. MAbs 2014, 6, 34–45. [Google Scholar] [CrossRef]
- Jackson, D.Y. Processes for Constructing Homogeneous Antibody Drug Conjugates. Org. Process Res. Dev. 2016, 20, 852–866. [Google Scholar] [CrossRef]
- Sadiki, A.; Vaidya, S.; Abdollahi, M.; Bhardwaj, G.; Dolan, M.; Turna, H.; Arora, V.; Sanjeev, A.; Robinson, T.; Koid, A.; et al. Site-specific Conjugation of Native Antibody. Antib. Ther. 2020, 3, 271–284. [Google Scholar] [CrossRef]
- Dai, Z.; Zhang, X.N.; Nasertorabi, F.; Cheng, Q.; Li, J.; Katz, B.B.; Smbatyan, G.; Pei, H.; Louie, S.G.; Lenz, H.J.; et al. Synthesis of site-specific antibody-drug conjugates by ADP-ribosyl cyclases. Sci. Adv. 2020, 6, eaba6752. [Google Scholar] [CrossRef]
- Li, W.; Zhu, Z.; Chen, W.; Feng, Y.; Dimitrov, D.S. Crystallizable Fragment Glycoengineering for Therapeutic Antibodies Development. Front. Immunol. 2017, 8, 1554. [Google Scholar] [CrossRef]
- Duivelshof, B.; Deslignière, E.; Hernandez-Alba, O.; Ehkirch, A.; Toftevall, H.; Sjögren, J.; Sanglier-Cianférani, S.; Beck, A.; Guillarme, D.; D’Atri, V. Glycan-Mediated Technology for Obtaining Homogeneous Site-Specific Conjugated Antibody-Drug Conjugates: Synthesis and Analytical Characterization by Using Complementary Middle-up LC/HRMS Analysis. Anal. Chem. 2020, 92, 8170–8177. [Google Scholar] [CrossRef] [PubMed]
- Toftevall, H.; Nyhlén, H.; Olsson, F.; Sjögren, J. Antibody Conjugations via Glycosyl Remodeling. Methods Mol. Biol. 2020, 2078, 131–145. [Google Scholar] [CrossRef] [PubMed]
- van Geel, R.; Wijdeven, M.A.; Heesbeen, R.; Verkade, J.M.; Wasiel, A.A.; van Berkel, S.S.; van Delft, F.L. Chemoenzymatic Conjugation of Toxic Payloads to the Globally Conserved N-Glycan of Native mAbs Provides Homogeneous and Highly Efficacious Antibody-Drug Conjugates. Bioconjug Chem. 2015, 26, 2233–2242. [Google Scholar] [CrossRef] [PubMed]
- Wijdeven, M.A.; van Geel, R.; Hoogenboom, J.H.; Verkade, J.M.M.; Janssen, B.M.G.; Hurkmans, I.; de Bever, L.; van Berkel, S.S.; van Delft, F.L. Enzymatic glycan remodeling-metal free click (GlycoConnect™) provides homogenous antibody-drug conjugates with improved stability and therapeutic index without sequence engineering. MAbs 2022, 14, 2078466. [Google Scholar] [CrossRef]
- D’Arienzo, A.; Verrazzo, A.; Pagliuca, M.; Napolitano, F.; Parola, S.; Viggiani, M.; Caputo, R.; Puglisi, F.; Giuliano, M.; Del Mastro, L.; et al. Toxicity profile of antibody-drug conjugates in breast cancer: Practical considerations. EClinicalMedicine 2023, 62, 102113. [Google Scholar] [CrossRef]
- Yamazaki, C.M.; Yamaguchi, A.; Anami, Y.; Xiong, W.; Otani, Y.; Lee, J.; Ueno, N.T.; Zhang, N.; An, Z.; Tsuchikama, K. Antibody-drug conjugates with dual payloads for combating breast tumor heterogeneity and drug resistance. Nat. Commun. 2021, 12, 3528. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ADC (Company) | Trade Name | Target Ag | Linker | Payload | Average DAR | Approved Date | Approved Indications/ Drug Regimen | Common AEs |
|---|---|---|---|---|---|---|---|---|
| Hematological Malignancies | ||||||||
| Gemtuzumab ozogamicin (Pfizer, New York, NY, USA) | Mylotarg® | CD33 | Cleavable (hydrazone) | calicheamicin | 2–3 | 2000/5, (USA; withdrawn from the market in 2010); 2017/9 (USA), 2018/4 (EU) | Newly diagnosed AML (including pediatric patients) | Normal tissue expression of the Ag: VOD, hemorrhage, hepatotoxicity Payload-related: hepatic dysfunction, myelosuppression Other AEs: fatigue, pyrexia, nausea, vomiting, headache, infection, stomatitis, diarrhea, abdominal pain |
| Brentuximab Vedotin (Seagen, Bothell, WA, USA) | Adcetris® | CD30 | Cleavable (mc-VC-PABC) | MMAE | 4 | 2011/8 (USA); 2012/10 (EU) | R/R CD30-positive HL and systemic ALCL, including some types of PTCL and previously untreated stage III or IV cHL; in combination with chemotherapy | Payload-related: peripheral sensory neuropathy, myelosuppression Other Aes: upper respiratory tract nausea, fatigue, diarrhea, pyrexia, vomiting, arthralgia, pruritus, myalgia, alopecia |
| Inotuzumab ozogamicin (Pfizer) | Besponsa® | CD22 | Cleavable (hydrazone) | calicheamicin | 5–7 | 2017/6 (EU); 2017/8 (USA) | R/R B-cell precursor ALL | Payload-related: hepatic dysfunction, myelosuppression Other Aes: hemorrhage, pyrexia, nausea, infection, headache |
| Moxetumomab Pasudotox (AstraZeneca, Cambridge, UK) | Lumoxiti® | CD22 | Cleavable (mc-VC-PABC) | PE38 | NA | 2018/9 (USA; withdrawn in 2023/7); 2021/2 (EU, 2021/7) | R/R HCL who have failed to receive at least two systemic therapies | Body swelling, nausea, fatigue, headache, fever, constipation, anemia, diarrhea, capillary leak syndrome, and hemolytic uremic syndrome |
| Polatuzumab vedotin (Roche, Basilea, Switzerland) | Polivy® | CD79B | Cleavable (mc-VC-PABC) | MMAE | 3.5 | 2019/6 (USA); 2020/1 (EU) | R/R DLBCL, after at least two prior therapies. In combination with bendamustine plus rituximab | Payload-related: peripheral sensory neuropathy, myelosuppression |
| Belantamab mafodotin (GSK, London, UK) | Blenrep® | BCMA | Non-cleavable (mc) | MMAF | 4 | 2020/8 (USA; terminated in 2022/11); 2020/8 (EU; terminated in 2023/9) | R/R MM, after at least four treatments, including anti-CD38 mAbs, proteasome inhibitors, and immunomodulators | Payload-related: ocular toxicity Other AEs: myelosuppression, pyrexia, nausea, increased aspartate aminotransferase, keratopathy |
| Loncastuximab tesirine (ADC Therapeutics, Épalinges, Switzerland) | Zynlonta® | CD19 | Cleavable (dipeptide) | PBD dimer (SG3199) | 2.3 | 2021/4 (USA); 2022/12 (EU) | R/R large B-cell lymphoma after two or more lines of systemic therapy (adults) | Payload-related: increased gamma-glutamyl transferase, fluid retention, myelosuppression Other AEs: hyperglycemia, transaminase increase, hypoalbuminemia, musculoskeletal pain, fatigue |
| Solid cancers | ||||||||
| Ado-trastuzumab emtansine (Roche) | Kadcyla® | HER2 | Non-cleavable (SMCC) | DM1 | 3.5 | 2013/2 (USA); 2013/11 (EU) | Adjuvant treatment of patients with HER2-positive early breast cancer presenting residual invasive disease after neoadjuvant therapy | Normal tissue expression of antigen: cardiac toxicity Payload-related: myelosuppression, increased transaminases, peripheral sensory neuropathy Off-target toxicity: interstitial pneumonitis Other AEs: ocular toxicity, fatigue, nausea |
| Enfortumab vedotin (Seagen) | Padcev® | Nectin-4 | Cleavable (mc-VC-PABC) | MMAE | 3.8 | 2019/12 (USA); 2022/4 (EU) | Advanced or metastatic urothelial cancer patients previously treated with platinum chemotherapy and a PD-L1/PD-1 inhibitor | Normal tissue expression of antigen: dysgeusia Payload-related: peripheral sensory neuropathy Other AE: rash, alopecia, dry eyes and skin, pruritus, diarrhea, fatigue, alopecia, nausea, decreased appetite |
| Fam-trastuzumab deruxtecan (Daiichi Sankyo, Tokyo, Japan) | Enhertu® | HER2 | Cleavable (tetrapept) | DXd | 7–8 | 2019/12 (USA); 2021/1 (EU) | Unresectable or metastatic HER2-positive breast cancer patients after two or more prior HER2-targeting regiments; locally advanced or metastatic HER2-positive gastric or gastroesophageal junction adenocarcinoma patients after a prior trastuzumab-based regimen | Normal tissue expression of antigen: cardiac toxicity Payload-related: gastrointestinal toxicity, myelosuppression Off-target toxicity: interstitial pneumonitis, nausea, fatigue, alopecia, vomiting, decreased appetite, diarrhea, constipation |
| Sacituzumab govitecan (Immunomedics, Morris Plains, NJ, USA) | Trodelvy® | Trop-2 | Cleavable (CL2A) | SN38 | 7.6 | 2020/4 (USA); 2021/11 (EU) | Unresectable locally advanced or metastatic TNBC patients who have received two or more systemic therapies (of which at least one is for metastatic disease) | Normal tissue expression of antigen: skin rash, hyperglycemia Payload-related: myelosuppression, diarrhea Other AEs: alopecia, vomiting, nausea, constipation |
| Cetuximab sarotalocan (Rakuten Medical, San Diego, CA, USA) | Akalux® | EGFR | NA | IRDye700DX | 1.3–3.8 | 2019/9 (China) | Unresectable locally advanced or recurrent HNSCC | Application site-pain, local edema |
| Disitamab vedotin (RemeGen, Yantai, China) | Aidixi® | HER2 | Cleavable (mc-VC-PABC) | MMAE | 4 | 2021/6 (China) | Locally advanced or metastatic gastric cancer patients (including gastroesophageal junction adenocarcinoma) previously treated with at least 2 types of systemic chemotherapy | Myelosuppression, gastrointestinal diseases, fatigue, fever |
| Tisotumab vedotin (Genmab, Copenhagen, Denmark/Seagen) | Tivdak® | TF | Cleavable (mc-VC-PABC) | MMAE | 4 | 2021/9 (USA) | Adult patients with metastatic or recurrent cervical cancer or after chemotherapy | Normal tissue expression of antigen: hemorrhagic complication and conjunctival reaction Payload-related: peripheral sensory neuropathy, myelosuppression |
| Mirvetuximab soravtansine (ImmunoGen, Waltham, MA, USA) | Elahere® | FR | Cleavable (Sulfo-SPDB) | DM4 | 3.5 | 2022/11 (USA) | Adult patients with folate receptor–alpha positive ovarian cancer, fallopian tube cancer, or primary peritoneal cancer refractory platinum-based chemotherapy or after 1 to 3 prior chemotherapies | Payload-related: peripheral neuropathy, myelosuppression Off-target toxicity: ocular toxicity Other AEs: reversible ocular toxicity (uveitis and keratopathy), pneumonitis |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Metrangolo, V.; Engelholm, L.H. Antibody–Drug Conjugates: The Dynamic Evolution from Conventional to Next-Generation Constructs. Cancers 2024, 16, 447. https://doi.org/10.3390/cancers16020447
Metrangolo V, Engelholm LH. Antibody–Drug Conjugates: The Dynamic Evolution from Conventional to Next-Generation Constructs. Cancers. 2024; 16(2):447. https://doi.org/10.3390/cancers16020447
Chicago/Turabian StyleMetrangolo, Virginia, and Lars H. Engelholm. 2024. "Antibody–Drug Conjugates: The Dynamic Evolution from Conventional to Next-Generation Constructs" Cancers 16, no. 2: 447. https://doi.org/10.3390/cancers16020447
APA StyleMetrangolo, V., & Engelholm, L. H. (2024). Antibody–Drug Conjugates: The Dynamic Evolution from Conventional to Next-Generation Constructs. Cancers, 16(2), 447. https://doi.org/10.3390/cancers16020447