

Hyperthermia in Combination with Emerging Targeted and Immunotherapies as a New Approach in Cancer Treatment

 and
and
Abstract
:Simple Summary
Abstract
1. Introduction
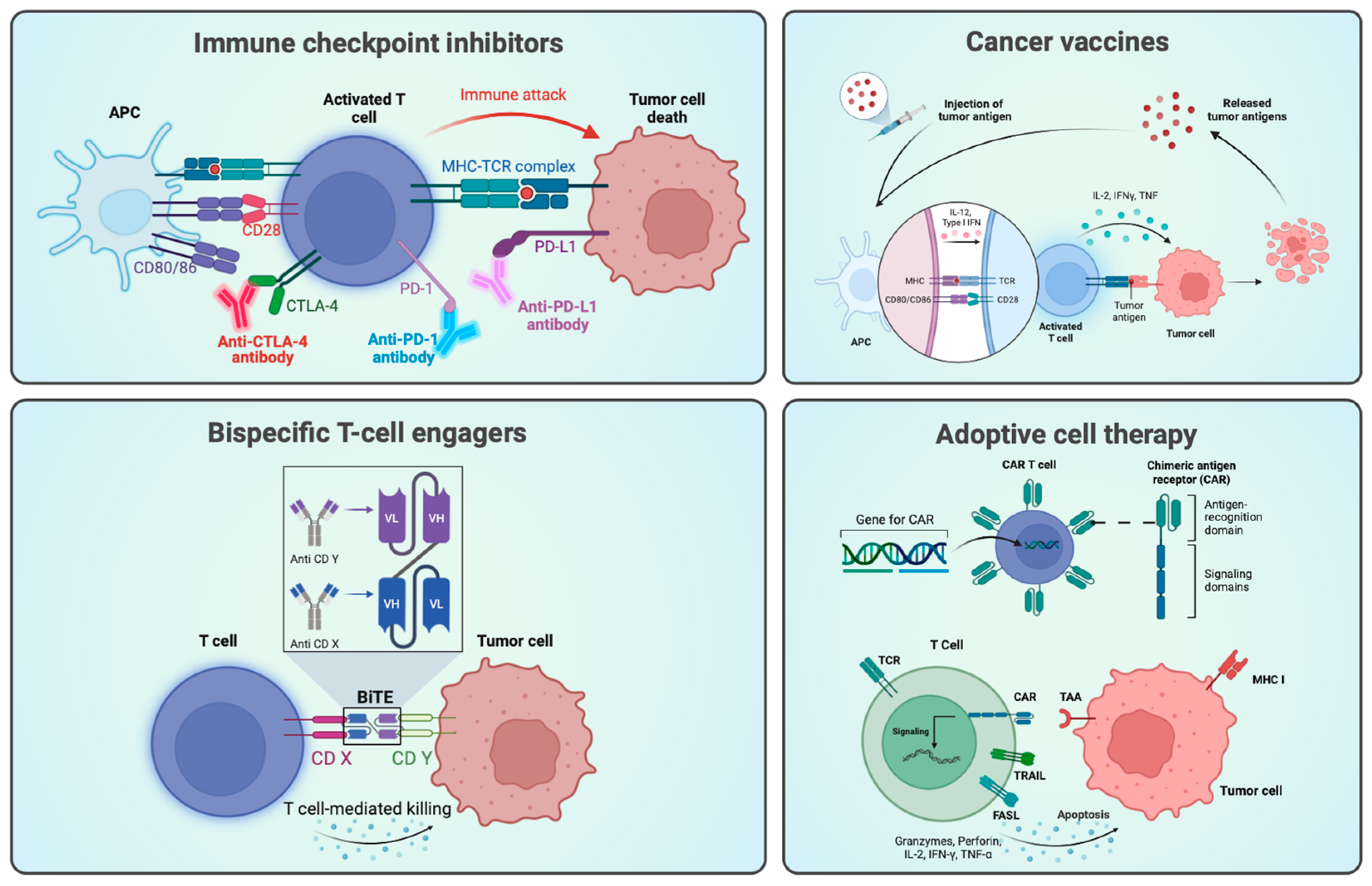
1.1. Immunotherapy and Targeted Therapy in Cancer Research
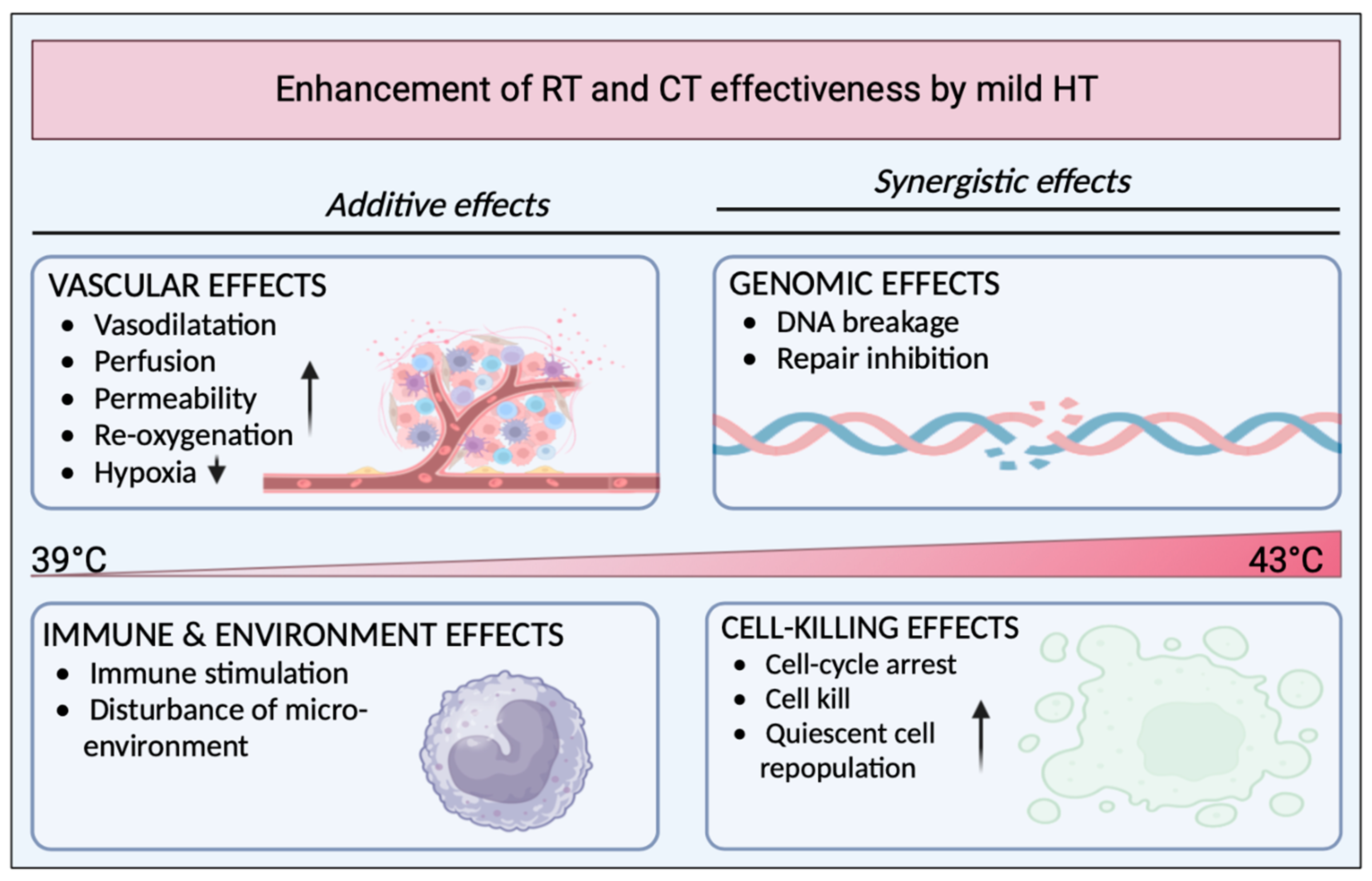
1.2. Hyperthermia as a Potential Adjuvant
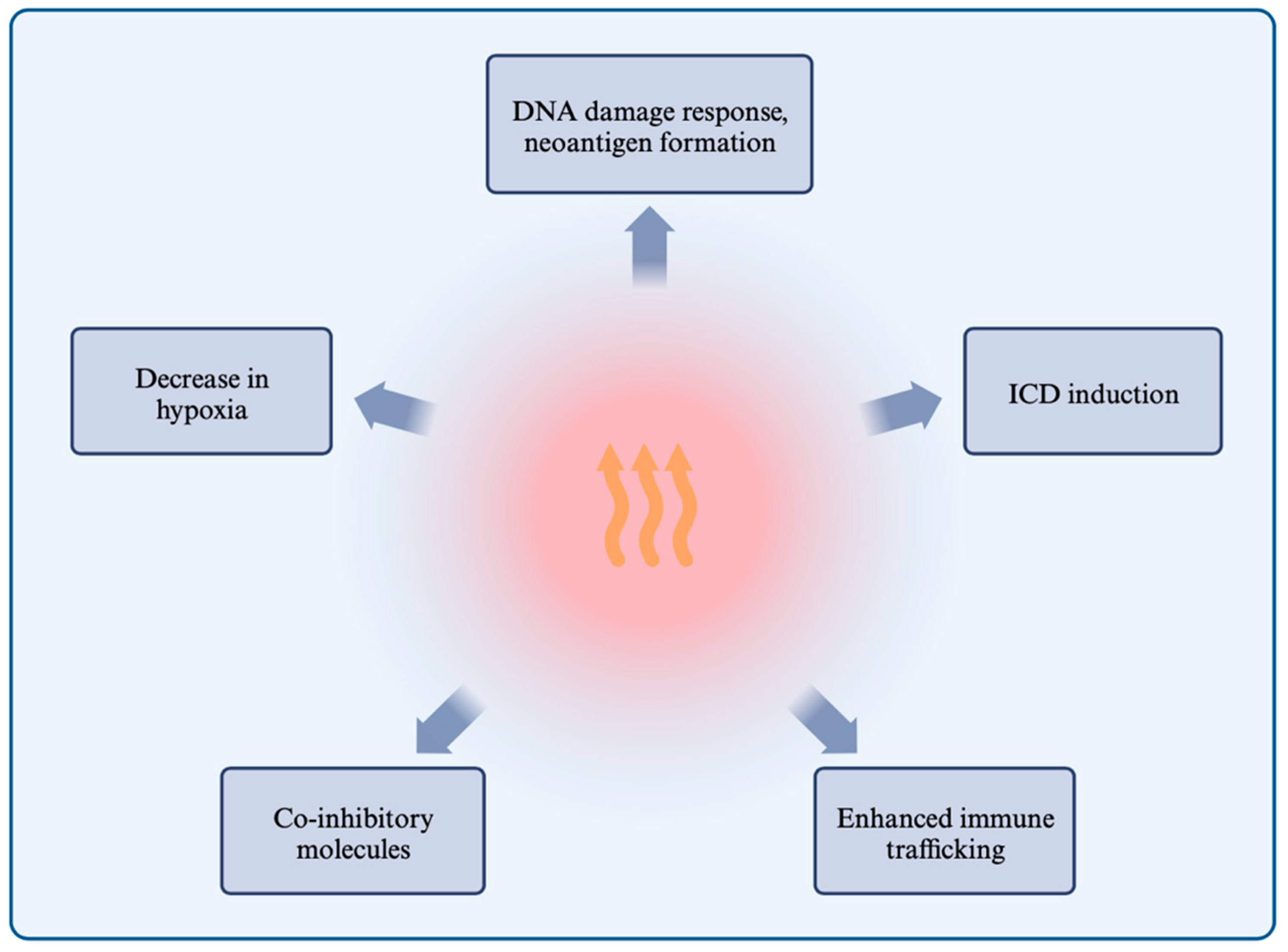
1.3. Effects of Hyperthermia on the Immune System and Responses
1.3.1. Hyperthermia Induces the DNA Damage Response and Inhibits Repair Mechanisms
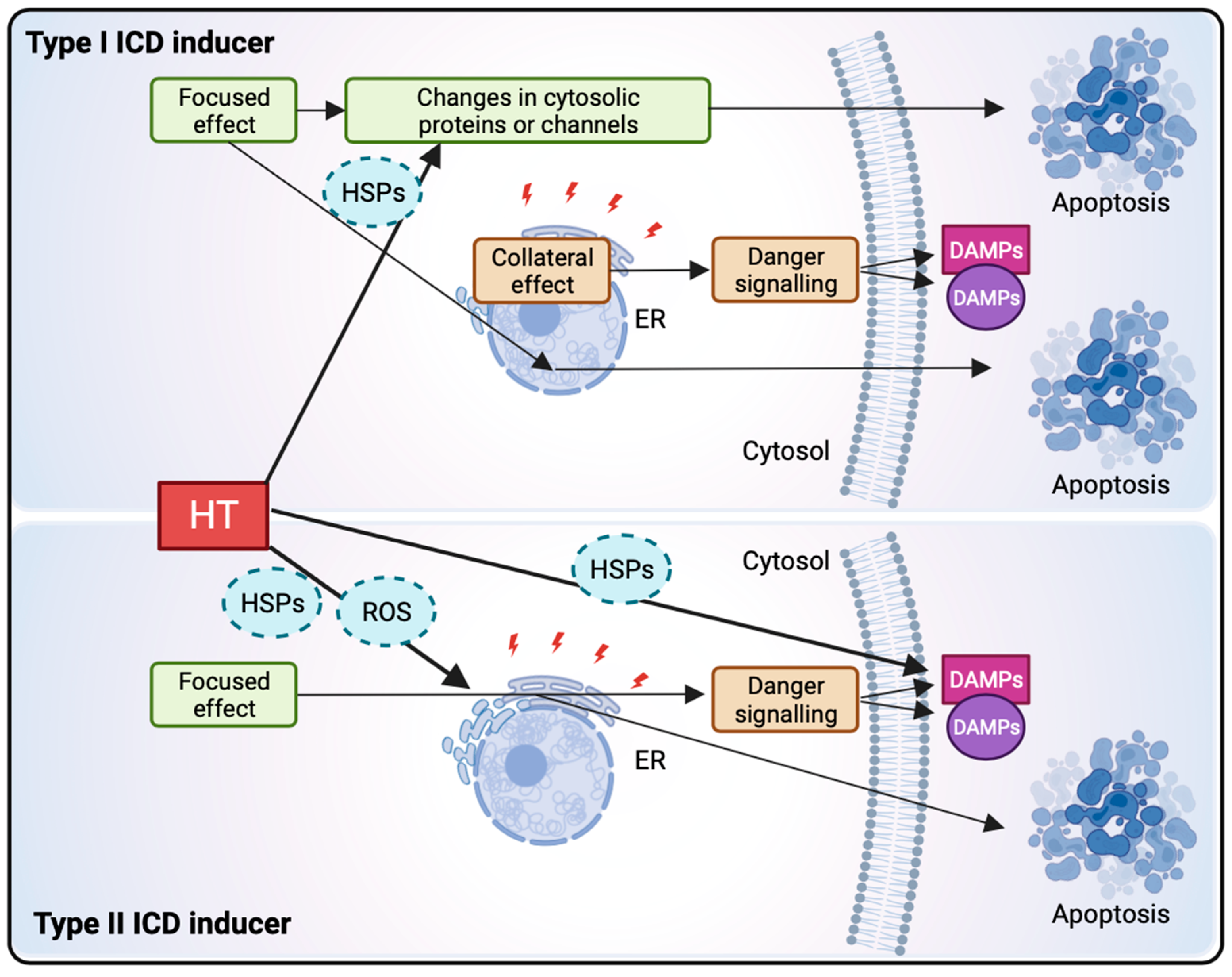
1.3.2. Hyperthermia Induces Immunogenic Cell Death through HSP Release, ER Stress, and ROS
1.3.3. Hyperthermia Enhances Immune Cell Trafficking and the Immune Response
1.3.4. Hyperthermia and the Expression of Co-Inhibitory Molecules
1.3.5. Hyperthermia Alleviates Immunosuppressive Hypoxia
2. Materials and Methods
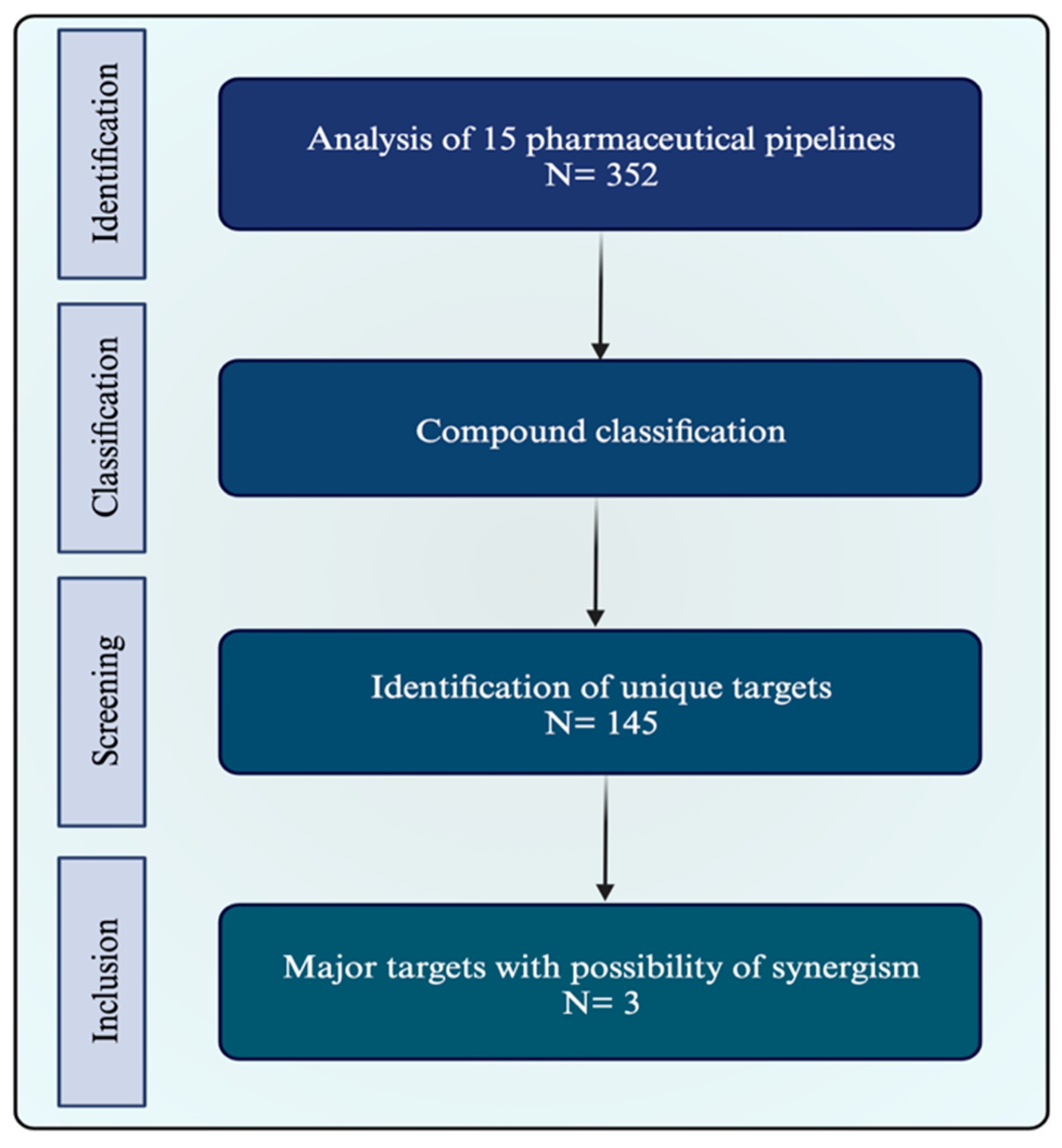
2.1. Pipeline Research
2.2. Identification of Potential Synergism
2.3. Target Characterization and Selection
3. Results
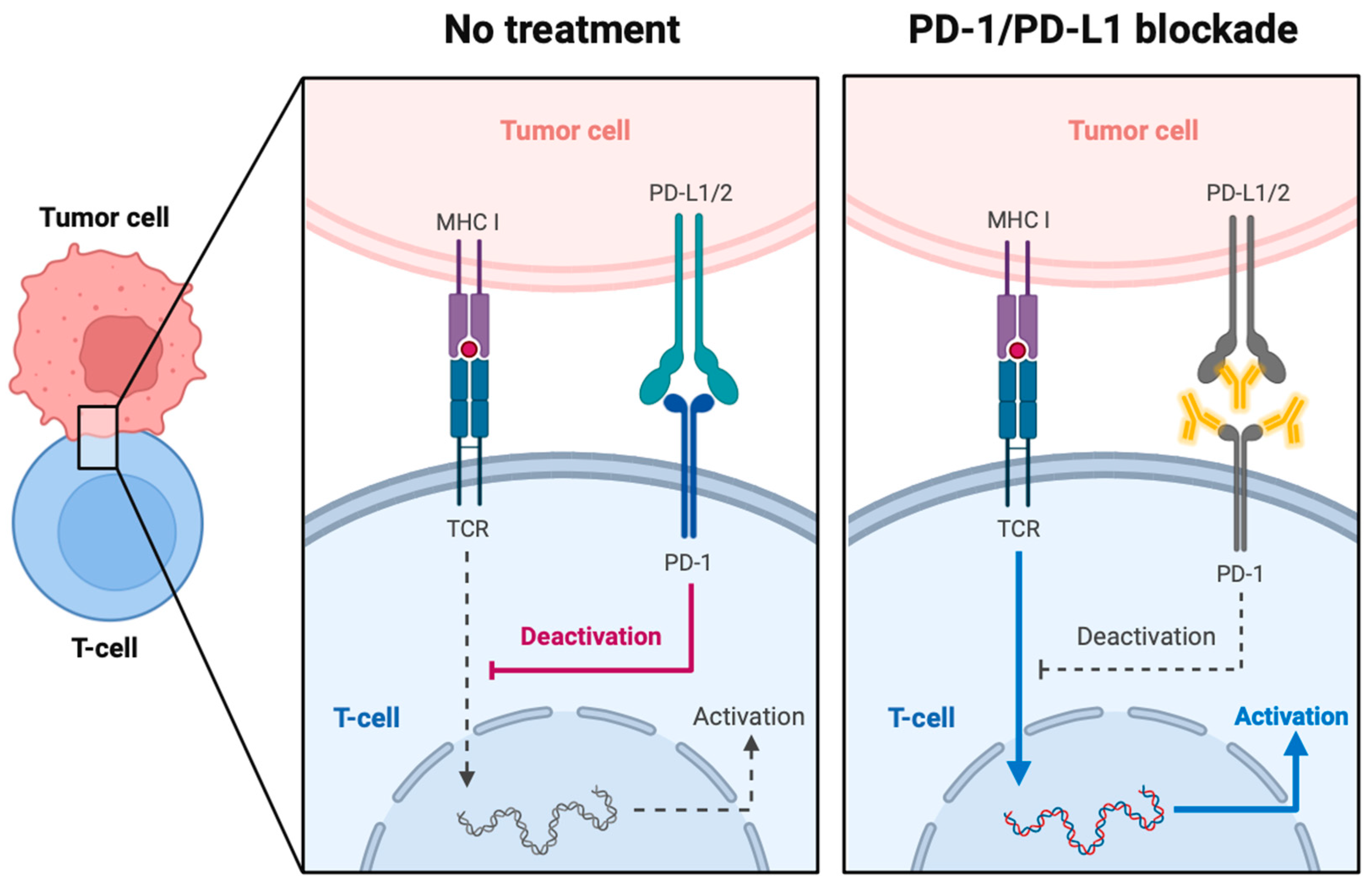
3.1. PD-1 and PD-L1
3.1.1. PD-1 and PD-L1 in Cancer and the Tumor Microenvironment
The Role of IFN-γ
Other Cellular Factors Involved in PD-L1 Induction
Hypoxia and PD-L1
3.1.2. Current Therapies
3.1.3. Benefit from Combination with Hyperthermia
3.2. Bispecific Antibodies and Immune Cell Engagers
3.2.1. CD Antigens
3.2.2. Bispecific Antibodies and Immune Cell Engagement
3.2.3. Mechanisms of Action of Bispecific Antibodies
3.2.4. T-Cell Engagement in Hematological Malignancies
3.2.5. Challenges for T-Cell Engagement in Solid Tumors
3.2.6. Potential Benefits in Combination with Hyperthermia
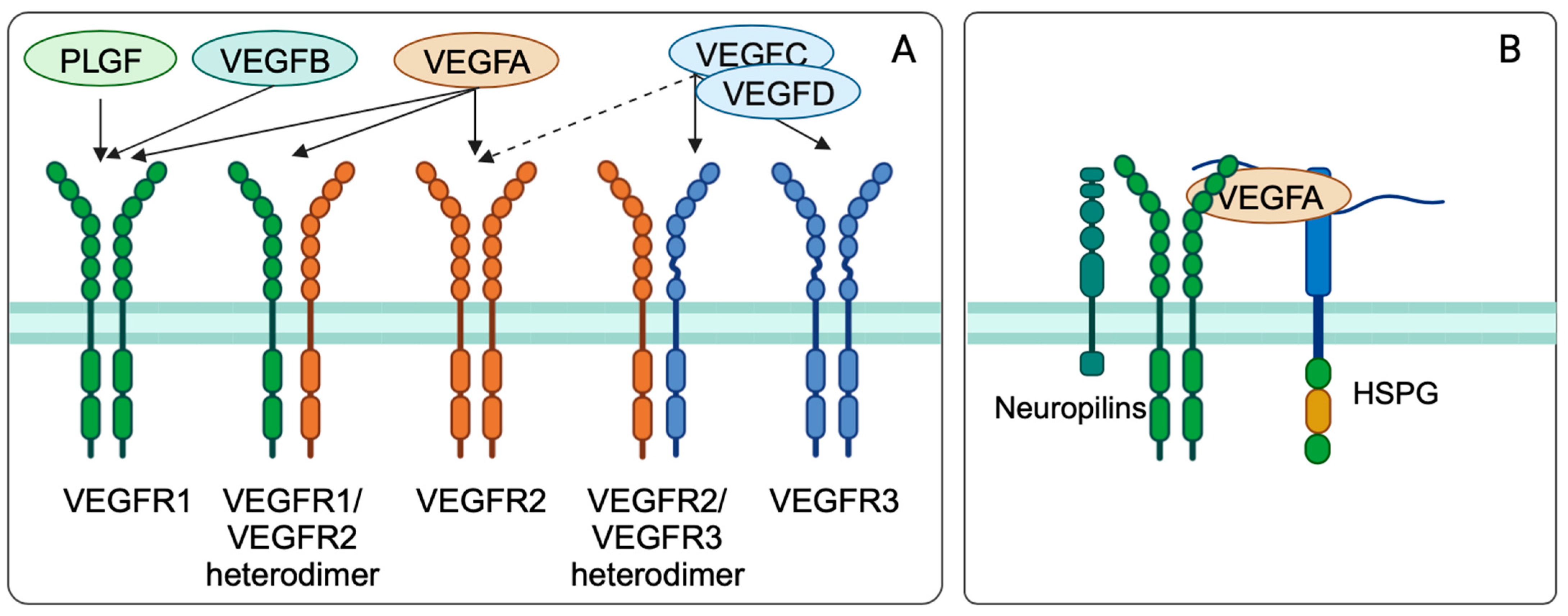
3.3. Vascular Endothelial Growth Factor (VEGF)
3.3.1. Vascular Endothelial Growth Factor Biology and Function
3.3.2. Regulation of VEGF and VEGFR Expression
3.3.3. VEGF Biology in Cancer
3.3.4. VEGF Inhibitors and Dual Targeting Therapies for Tumor Therapy
3.3.5. Challenges in the Development and Use of VEGF-A Inhibitors in Oncology
3.3.6. Hyperthermia and Anti-Angiogenic Therapy
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Barbari, C.; Fontaine, T.; Parajuli, P.; Lamichhane, N.; Jakubski, S.; Lamichhane, P.; Deshmukh, R.R. Immunotherapies and Combination Strategies for Immuno-Oncology. Int. J. Mol. Sci. 2020, 21, 5009. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Dagher, O.K.; Schwab, R.D.; Brookens, S.K.; Posey, A.D. Advances in Cancer Immunotherapies. Cell 2023, 186, 1814–1814.e1. [Google Scholar] [CrossRef] [PubMed]
- Joo, W.D.; Visintin, I.; Mor, G. Targeted Cancer Therapy—Are the Days of Systemic Chemotherapy Numbered? Maturitas 2013, 76, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Shuel, S.L. Targeted Cancer Therapies. Can. Fam. Physician 2022, 68, 515–518. [Google Scholar] [CrossRef] [PubMed]
- Targeted Therapy for Cancer—NCI. Available online: https://www.cancer.gov/about-cancer/treatment/types/targeted-therapies (accessed on 25 July 2023).
- Oei, A.L.; Kok, H.P.; Oei, S.B.; Horsman, M.R.; Stalpers, L.J.A.; Franken, N.A.P.; Crezee, J. Molecular and Biological Rationale of Hyperthermia as Radio- and Chemosensitizer. Adv. Drug Deliv. Rev. 2020, 163–164, 84–97. [Google Scholar] [CrossRef]
- Kok, H.P.; Cressman, E.N.K.; Ceelen, W.; Brace, C.L.; Ivkov, R.; Grüll, H.; ter Haar, G.; Wust, P.; Crezee, J. Heating Technology for Malignant Tumors: A Review. Int. J. Hyperth. 2020, 37, 711–741. [Google Scholar] [CrossRef]
- Levin, W.; Blair, R.M. Pettigrew Technique of Inducing Whole-Body Hyperthermia. Natl. Cancer Inst. Monogr. 1982, 61, 377–379. [Google Scholar]
- Behrouzkia, Z.; Joveini, Z.; Keshavarzi, B.; Eyvazzadeh, N.; Aghdam, R.Z. Hyperthermia: How Can It Be Used? Oman Med. J. 2016, 31, 89–97. [Google Scholar] [CrossRef]
- Maurici, C.E.; Colenbier, R.; Wylleman, B.; Brancato, L.; van Zwol, E.; Van den Bossche, J.; Timmermans, J.-P.; Giovannetti, E.; Mori da Cunha, M.G.M.C.; Bogers, J. Hyperthermia Enhances Efficacy of Chemotherapeutic Agents in Pancreatic Cancer Cell Lines. Biomolecules 2022, 12, 651. [Google Scholar] [CrossRef]
- Li, Z.; Deng, J.; Sun, J.; Ma, Y. Hyperthermia Targeting the Tumor Microenvironment Facilitates Immune Checkpoint Inhibitors. Front. Immunol. 2020, 11, 595207. [Google Scholar] [CrossRef]
- Mantso, T.; Goussetis, G.; Franco, R.; Botaitis, S.; Pappa, A.; Panayiotidis, M. Effects of Hyperthermia as a Mitigation Strategy in DNA Damage-Based Cancer Therapies. Semin. Cancer Biol. 2016, 37–38, 96–105. [Google Scholar] [CrossRef]
- Oei, A.L.; Vriend, L.E.M.; Crezee, J.; Franken, N.A.P.; Krawczyk, P.M. Effects of Hyperthermia on DNA Repair Pathways: One Treatment to Inhibit Them All. Radiat. Oncol. 2015, 10, 165. [Google Scholar] [CrossRef]
- Takahashi, A.; Mori, E.; Somakos, G.I.; Ohnishi, K.; Ohnishi, T. Heat Induces gammaH2AX Foci Formation in Mammalian Cells. Mutat. Res. 2008, 656, 88–92. [Google Scholar] [CrossRef]
- Takahashi, A.; Matsumoto, H.; Nagayama, K.; Kitano, M.; Hirose, S.; Tanaka, H.; Mori, E.; Yamakawa, N.; Yasumoto, J.-I.; Yuki, K.; et al. Evidence for the Involvement of Double-Strand Breaks in Heat-Induced Cell Killing. Cancer Res. 2004, 64, 8839–8845. [Google Scholar] [CrossRef]
- Jazayeri, A.; Falck, J.; Lukas, C.; Bartek, J.; Smith, G.C.M.; Lukas, J.; Jackson, S.P. ATM- and Cell Cycle-Dependent Regulation of ATR in Response to DNA Double-Strand Breaks. Nat. Cell Biol. 2006, 8, 37–45. [Google Scholar] [CrossRef]
- Furusawa, Y.; Iizumi, T.; Fujiwara, Y.; Zhao, Q.-L.; Tabuchi, Y.; Nomura, T.; Kondo, T. Inhibition of Checkpoint Kinase 1 Abrogates G2/M Checkpoint Activation and Promotes Apoptosis under Heat Stress. Apoptosis 2012, 17, 102–112. [Google Scholar] [CrossRef]
- Germano, G.; Lamba, S.; Rospo, G.; Barault, L.; Magrì, A.; Maione, F.; Russo, M.; Crisafulli, G.; Bartolini, A.; Lerda, G.; et al. Inactivation of DNA Repair Triggers Neoantigen Generation and Impairs Tumour Growth. Nature 2017, 552, 116–120. [Google Scholar] [CrossRef]
- Liu, Z.; Lv, J.; Dang, Q.; Liu, L.; Weng, S.; Wang, L.; Zhou, Z.; Kong, Y.; Li, H.; Han, Y.; et al. Engineering Neoantigen Vaccines to Improve Cancer Personalized Immunotherapy. Int. J. Biol. Sci. 2022, 18, 5607–5623. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Scheper, W.; Kvistborg, P. Cancer Neoantigens. Annu. Rev. Immunol. 2019, 37, 173–200. [Google Scholar] [CrossRef]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic Cell Death and DAMPs in Cancer Therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Galluzzi, L.; Apetoh, L.; Baert, T.; Birge, R.B.; Bravo-San Pedro, J.M.; Breckpot, K.; Brough, D.; Chaurio, R.; Cirone, M.; et al. Molecular and Translational Classifications of DAMPs in Immunogenic Cell Death. Front. Immunol. 2015, 6, 588. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Dudek-Peric, A.M.; Romano, E.; Agostinis, P. Immunogenic Cell Death. Int. J. Dev. Biol. 2015, 59, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Ju, X.; Wang, J.; Fan, Y.; Ren, M.; Zhang, H. Immunogenic Cell Death in Anticancer Chemotherapy and Its Impact on Clinical Studies. Cancer Lett. 2018, 438, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Adkins, I.; Fucikova, J.; Garg, A.D.; Agostinis, P.; Špíšek, R. Physical Modalities Inducing Immunogenic Tumor Cell Death for Cancer Immunotherapy. Oncoimmunology 2015, 3, e968434. [Google Scholar] [CrossRef] [PubMed]
- Kepp, O.; Zitvogel, L.; Kroemer, G. Clinical Evidence That Immunogenic Cell Death Sensitizes to PD-1/PD-L1 Blockade. Oncoimmunology 2019, 8, e1637188. [Google Scholar] [CrossRef]
- Xu, X.; Gupta, S.; Hu, W.; McGrath, B.C.; Cavener, D.R. Hyperthermia Induces the ER Stress Pathway. PLoS ONE 2011, 6, e23740. [Google Scholar] [CrossRef]
- Lee, S.; Son, B.; Park, G.; Kim, H.; Kang, H.; Jeon, J.; Youn, H.; Youn, B. Immunogenic Effect of Hyperthermia on Enhancing Radiotherapeutic Efficacy. Int. J. Mol. Sci. 2018, 19, 2795. [Google Scholar] [CrossRef]
- Katschinski, D.M.; Wiedemann, G.J.; Longo, W.; d’Oleire, F.R.; Spriggs, D.; Robins, H.I. Whole Body Hyperthermia Cytokine Induction: A Review, and Unifying Hypothesis for Myeloprotection in the Setting of Cytotoxic Therapy. Cytokine Growth Factor Rev. 1999, 10, 93–97. [Google Scholar] [CrossRef]
- Baronzio, G.F.; Seta, R.D.; D’Amico, M.; Baronzio, A.; Freitas, I.; Forzenigo, G.; Gramaglia, A.; Hager, E.D. Effects of Local and Whole Body Hyperthermia on Immunity; Landes Bioscience: Austin, TX, USA, 2013. [Google Scholar]
- Toraya-Brown, S.; Fiering, S. Local Tumour Hyperthermia as Immunotherapy for Metastatic Cancer. Int. J. Hyperth. 2014, 30, 531–539. [Google Scholar] [CrossRef]
- Redzovic, A.; Gulic, T.; Laskarin, G.; Eminovic, S.; Haller, H.; Rukavina, D. Heat-Shock Proteins 70 Induce Pro-Inflammatory Maturation Program in Decidual CD1a(+) Dendritic Cells. Am. J. Reprod. Immunol. 2015, 74, 38–53. [Google Scholar] [CrossRef]
- Wells, A.D.; Malkovsky, M. Heat Shock Proteins, Tumor Immunogenicity and Antigen Presentation: An Integrated View. Immunol. Today 2000, 21, 129–132. [Google Scholar] [CrossRef] [PubMed]
- Binder, R.J.; Han, D.K.; Srivastava, P.K. CD91: A Receptor for Heat Shock Protein Gp96. Nat. Immunol. 2000, 1, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Manjili, M.H.; Wang, X.-Y.; Park, J.; Facciponte, J.G.; Repasky, E.A.; Subjeck, J.R. Immunotherapy of Cancer Using Heat Shock Proteins. Front. Biosci. 2002, 7, d43–d52. [Google Scholar] [CrossRef] [PubMed]
- Liso, A.; Venuto, S.; Coda, A.R.D.; Giallongo, C.; Palumbo, G.A.; Tibullo, D. IGFBP-6: At the Crossroads of Immunity, Tissue Repair and Fibrosis. Int. J. Mol. Sci. 2022, 23, 4358. [Google Scholar] [CrossRef] [PubMed]
- Liso, A.; Castellani, S.; Massenzio, F.; Trotta, R.; Pucciarini, A.; Bigerna, B.; De Luca, P.; Zoppoli, P.; Castiglione, F.; Palumbo, M.C.; et al. Human Monocyte-Derived Dendritic Cells Exposed to Hyperthermia Show a Distinct Gene Expression Profile and Selective Upregulation of IGFBP6. Oncotarget 2017, 8, 60826–60840. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Benjamin, I.J.; Basu, S.; Li, Z. Heat Shock Factor 1-Independent Activation of Dendritic Cells by Heat Shock: Implication for the Uncoupling of Heat-Mediated Immunoregulation from the Heat Shock Response. Eur. J. Immunol. 2003, 33, 1754–1762. [Google Scholar] [CrossRef]
- Knippertz, I.; Stein, M.F.; Dörrie, J.; Schaft, N.; Müller, I.; Deinzer, A.; Steinkasserer, A.; Nettelbeck, D.M. Mild Hyperthermia Enhances Human Monocyte-Derived Dendritic Cell Functions and Offers Potential for Applications in Vaccination Strategies. Int. J. Hyperth. 2011, 27, 591–603. [Google Scholar] [CrossRef]
- Peer, A.J.; Grimm, M.J.; Zynda, E.R.; Repasky, E.A. Diverse Immune Mechanisms May Contribute to the Survival Benefit Seen in Cancer Patients Receiving Hyperthermia. Immunol. Res. 2010, 46, 137–154. [Google Scholar] [CrossRef]
- Moser, M.; Murphy, K.M. Dendritic Cell Regulation of TH1-TH2 Development. Nat. Immunol. 2000, 1, 199–205. [Google Scholar] [CrossRef]
- Raker, V.K.; Domogalla, M.P.; Steinbrink, K. Tolerogenic Dendritic Cells for Regulatory T Cell Induction in Man. Front. Immunol. 2015, 6, 569. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Paul, W.E. Mechanisms Underlying Lineage Commitment and Plasticity of Helper CD4+ T Cells. Science 2010, 327, 1098–1102. [Google Scholar] [CrossRef] [PubMed]
- Carlos, T.M. Leukocyte Recruitment at Sites of Tumor: Dissonant Orchestration. J. Leukoc. Biol. 2001, 70, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Evans, S.S.; Repasky, E.A.; Fisher, D.T. Fever and the Thermal Regulation of Immunity: The Immune System Feels the Heat. Nat. Rev. Immunol. 2015, 15, 335–349. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, Q.; Yuan, X.; Wang, T.; Luo, S.; Lei, H.; Xia, Y. Requirement of Heat Shock Protein 70 for Inducible Nitric Oxide Synthase Induction. Cell Signal 2013, 25, 1310–1317. [Google Scholar] [CrossRef]
- Kovalchin, J.T.; Wang, R.; Wagh, M.S.; Azoulay, J.; Sanders, M.; Chandawarkar, R.Y. In Vivo Delivery of Heat Shock Protein 70 Accelerates Wound Healing by Up-Regulating Macrophage-Mediated Phagocytosis. Wound Repair. Regen. 2006, 14, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Matsutake, T. Heat Shock Protein-Antigen Presenting Cell Interactions. Methods 2004, 32, 38–41. [Google Scholar] [CrossRef]
- Pritchard, M.T.; Li, Z.; Repasky, E.A. Nitric Oxide Production Is Regulated by Fever-Range Thermal Stimulation of Murine Macrophages. J. Leukoc. Biol. 2005, 78, 630–638. [Google Scholar] [CrossRef]
- Miller, L.; Qureshi, M.A. Induction of Heat-Shock Proteins and Phagocytic Function of Chicken Macrophage Following in Vitro Heat Exposure. Vet. Immunol. Immunopathol. 1992, 30, 179–191. [Google Scholar] [CrossRef]
- Li, S.-S.; Xu, X.-T.; Liu, W.; Xu, Z.-P.; Zhang, W.-W.; Li, Y.; Dong, X.-X.; Yang, X.-W.; Liu, F.; Wang, Y.-Z.; et al. Effects of Schistosoma japonicum heat-shock protein 40 on macrophage activation. Zhongguo Xue Xi Chong Bing Fang Zhi Za Zhi 2012, 24, 137–141+149. [Google Scholar]
- Liu, J.; Hong, S.; Feng, Z.; Xin, Y.; Wang, Q.; Fu, J.; Zhang, C.; Li, G.; Luo, L.; Yin, Z. Regulation of Lipopolysaccharide-Induced Inflammatory Response by Heat Shock Protein 27 in THP-1 Cells. Cell. Immunol. 2010, 264, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, C.; Wang, X.; Zhang, J.; Chang, Z. Heat Shock Response Inhibits IL-18 Expression through the JNK Pathway in Murine Peritoneal Macrophages. Biochem. Biophys. Res. Commun. 2002, 296, 742–748. [Google Scholar] [CrossRef] [PubMed]
- Schell, M.T.; Spitzer, A.L.; Johnson, J.A.; Lee, D.; Harris, H.W. Heat Shock Inhibits NF-kB Activation in a Dose- and Time-Dependent Manner. J. Surg. Res. 2005, 129, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Ostberg, J.R.; Dayanc, B.E.; Yuan, M.; Oflazoglu, E.; Repasky, E.A. Enhancement of Natural Killer (NK) Cell Cytotoxicity by Fever-Range Thermal Stress Is Dependent on NKG2D Function and Is Associated with Plasma Membrane NKG2D Clustering and Increased Expression of MICA on Target Cells. J. Leukoc. Biol. 2007, 82, 1322–1331. [Google Scholar] [CrossRef] [PubMed]
- Farjadian, S.; Norouzian, M.; Younesi, V.; Ebrahimpour, A.; Lotfi, R. Hyperthermia Increases Natural Killer Cell Cytotoxicity against SW-872 Liposarcoma Cell Line. Iran. J. Immunol. 2013, 10, 93–102. [Google Scholar] [PubMed]
- Repasky, E.A.; Evans, S.S.; Dewhirst, M.W. Temperature Matters! And Why It Should Matter to Tumor Immunologists. Cancer Immunol. Res. 2013, 1, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Meyer, R.E.; Braun, R.D.; Rosner, G.L.; Dewhirst, M.W. Local 42 Degrees C Hyperthermia Improves Vascular Conductance of the R3230Ac Rat Mammary Adenocarcinoma during Sodium Nitroprusside Infusion. Radiat. Res. 2000, 154, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Piazena, H.; Notter, M.; Thomsen, A.R.; Grosu, A.-L.; Scholkmann, F.; Pockley, A.G.; Multhoff, G. From Localized Mild Hyperthermia to Improved Tumor Oxygenation: Physiological Mechanisms Critically Involved in Oncologic Thermo-Radio-Immunotherapy. Cancers 2023, 15, 1394. [Google Scholar] [CrossRef]
- Fisher, D.T.; Chen, Q.; Skitzki, J.J.; Muhitch, J.B.; Zhou, L.; Appenheimer, M.M.; Vardam, T.D.; Weis, E.L.; Passanese, J.; Wang, W.-C.; et al. IL-6 Trans-Signaling Licenses Mouse and Human Tumor Microvascular Gateways for Trafficking of Cytotoxic T Cells. J. Clin. Investig. 2011, 121, 3846–3859. [Google Scholar] [CrossRef]
- Chen, Q.; Fisher, D.T.; Kucinska, S.A.; Wang, W.-C.; Evans, S.S. Dynamic Control of Lymphocyte Trafficking by Fever-Range Thermal Stress. Cancer Immunol. Immunother. 2006, 55, 299–311. [Google Scholar] [CrossRef]
- Evans, S.S.; Wang, W.-C.; Bain, M.D.; Burd, R.; Ostberg, J.R.; Repasky, E.A. Fever-Range Hyperthermia Dynamically Regulates Lymphocyte Delivery to High Endothelial Venules. Blood 2001, 97, 2727–2733. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Fuchs, S.; Böse, T.; Schmidt, H.; Hofmann, A.; Tonak, M.; Unger, R.; Kirkpatrick, C.J. Mild Heat Stress Enhances Angiogenesis in a Co-Culture System Consisting of Primary Human Osteoblasts and Outgrowth Endothelial Cells. Tissue Eng. C Methods 2014, 20, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Issels, R.D.; Noessner, E.; Lindner, L.H.; Schmidt, M.; Albertsmeier, M.; Blay, J.-Y.; Stutz, E.; Xu, Y.; Buecklein, V.; Altendorf-Hofmann, A.; et al. Immune Infiltrates in Patients with Localised High-Risk Soft Tissue Sarcoma Treated with Neoadjuvant Chemotherapy without or with Regional Hyperthermia: A Translational Research Program of the EORTC 62961-ESHO 95 Randomised Clinical Trial. Eur. J. Cancer 2021, 158, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Fezza, M.; Hilal, G.; Tahtouh, R.; Moubarak, M.; Atallah, D. HSP27 Modulates Tumoural Immune Evasion by Enhancing STAT3-Mediated Upregulation of PD-L1 and NLRC5 in Ovarian Cancer. Ecancermedicalscience 2023, 17, 1526. [Google Scholar] [CrossRef]
- Terunuma, H. Potentiating Immune System by Hyperthermia. In Hyperthermic Oncology from Bench to Bedside; Kokura, S., Yoshikawa, T., Ohnishi, T., Eds.; Springer: Singapore, 2016; pp. 127–135. ISBN 978-981-10-0719-4. [Google Scholar]
- Huang, L.; Li, Y.; Du, Y.; Zhang, Y.; Wang, X.; Ding, Y.; Yang, X.; Meng, F.; Tu, J.; Luo, L.; et al. Mild Photothermal Therapy Potentiates Anti-PD-L1 Treatment for Immunologically Cold Tumors via an All-in-One and All-in-Control Strategy. Nat. Commun. 2019, 10, 4871. [Google Scholar] [CrossRef]
- Shi, L.; Chen, L.; Wu, C.; Zhu, Y.; Xu, B.; Zheng, X.; Sun, M.; Wen, W.; Dai, X.; Yang, M.; et al. PD-1 Blockade Boosts Radiofrequency Ablation-Elicited Adaptive Immune Responses against Tumor. Clin. Cancer Res. 2016, 22, 1173–1184. [Google Scholar] [CrossRef]
- Harris, B.; Saleem, S.; Cook, N.; Searle, E. Targeting Hypoxia in Solid and Haematological Malignancies. J. Exp. Clin. Cancer Res. 2022, 41, 318. [Google Scholar] [CrossRef]
- Manalo, D.J.; Rowan, A.; Lavoie, T.; Natarajan, L.; Kelly, B.D.; Ye, S.Q.; Garcia, J.G.N.; Semenza, G.L. Transcriptional Regulation of Vascular Endothelial Cell Responses to Hypoxia by HIF-1. Blood 2005, 105, 659–669. [Google Scholar] [CrossRef]
- Abou Khouzam, R.; Brodaczewska, K.; Filipiak, A.; Zeinelabdin, N.A.; Buart, S.; Szczylik, C.; Kieda, C.; Chouaib, S. Tumor Hypoxia Regulates Immune Escape/Invasion: Influence on Angiogenesis and Potential Impact of Hypoxic Biomarkers on Cancer Therapies. Front. Immunol. 2021, 11, 613114. [Google Scholar] [CrossRef]
- Schito, L. Hypoxia-Dependent Angiogenesis and Lymphangiogenesis in Cancer. In Hypoxia and Cancer Metastasis; Advances in Experimental Medicine and Biology; Gilkes, D.M., Ed.; Springer International Publishing: Cham, Switzerland, 2019; pp. 71–85. ISBN 978-3-030-12734-3. [Google Scholar]
- Harris, A.L. Hypoxia—A Key Regulatory Factor in Tumour Growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef]
- Wilson, W.R.; Hay, M.P. Targeting Hypoxia in Cancer Therapy. Nat. Rev. Cancer 2011, 11, 393–410. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Targeting HIF-1 for Cancer Therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Hughes, V.S.; Wiggins, J.M.; Siemann, D.W. Tumor Oxygenation and Cancer Therapy—Then and Now. Br. J. Radiol. 2019, 92, 20170955. [Google Scholar] [CrossRef] [PubMed]
- Dewhirst, M.W.; Oleson, J.R.; Kirkpatrick, J.; Secomb, T.W. Accurate Three-Dimensional Thermal Dosimetry and Assessment of Physiologic Response Are Essential for Optimizing Thermoradiotherapy. Cancers 2022, 14, 1701. [Google Scholar] [CrossRef] [PubMed]
- Moon, E.J.; Sonveaux, P.; Porporato, P.E.; Danhier, P.; Gallez, B.; Batinic-Haberle, I.; Nien, Y.-C.; Schroeder, T.; Dewhirst, M.W. NADPH Oxidase-Mediated Reactive Oxygen Species Production Activates Hypoxia-Inducible Factor-1 (HIF-1) via the ERK Pathway after Hyperthermia Treatment. Proc. Natl. Acad. Sci. USA 2010, 107, 20477–20482. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Regulation of Cancer Cell Metabolism by Hypoxia-Inducible Factor 1. Semin. Cancer Biol. 2009, 19, 12–16. [Google Scholar] [CrossRef]
- Kopecka, J.; Salaroglio, I.C.; Perez-Ruiz, E.; Sarmento-Ribeiro, A.B.; Saponara, S.; De Las Rivas, J.; Riganti, C. Hypoxia as a Driver of Resistance to Immunotherapy. Drug Resist. Updates 2021, 59, 100787. [Google Scholar] [CrossRef] [PubMed]
- Yamakawa, M.; Liu, L.X.; Date, T.; Belanger, A.J.; Vincent, K.A.; Akita, G.Y.; Kuriyama, T.; Cheng, S.H.; Gregory, R.J.; Jiang, C. Hypoxia-Inducible Factor-1 Mediates Activation of Cultured Vascular Endothelial Cells by Inducing Multiple Angiogenic Factors. Circ. Res. 2003, 93, 664–673. [Google Scholar] [CrossRef]
- Schultz, K.; Fanburg, B.L.; Beasley, D. Hypoxia and Hypoxia-Inducible Factor-1α Promote Growth Factor-Induced Proliferation of Human Vascular Smooth Muscle Cells. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H2528–H2534. [Google Scholar] [CrossRef]
- Lu, H.; Forbes, R.A.; Verma, A. Hypoxia-Inducible Factor 1 Activation by Aerobic Glycolysis Implicates the Warburg Effect in Carcinogenesis*. J. Biol. Chem. 2002, 277, 23111–23115. [Google Scholar] [CrossRef]
- Courtnay, R.; Ngo, D.C.; Malik, N.; Ververis, K.; Tortorella, S.M.; Karagiannis, T.C. Cancer Metabolism and the Warburg Effect: The Role of HIF-1 and PI3K. Mol. Biol. Rep. 2015, 42, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Ai, L.; Xu, A.; Xu, J. Roles of PD-1/PD-L1 Pathway: Signaling, Cancer, and Beyond. Adv. Exp. Med. Biol. 2020, 1248, 33–59. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 Pathway: Current Researches in Cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar] [PubMed]
- Youngblood, B.; Oestreich, K.J.; Ha, S.-J.; Duraiswamy, J.; Akondy, R.S.; West, E.E.; Wei, Z.; Lu, P.; Austin, J.W.; Riley, J.L.; et al. Chronic Virus Infection Enforces Demethylation of the Locus That Encodes PD-1 in Antigen-Specific CD8+ T Cells. Immunity 2011, 35, 400–412. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.; Deng, A.; Liu, H.; Ge, G.; Liu, X. Activator Protein 1 Suppresses Antitumor T-Cell Function via the Induction of Programmed Death 1. Proc. Natl. Acad. Sci. USA 2012, 109, 15419–15424. [Google Scholar] [CrossRef] [PubMed]
- Ohaegbulam, K.C.; Assal, A.; Lazar-Molnar, E.; Yao, Y.; Zang, X. Human Cancer Immunotherapy with Antibodies to the PD-1 and PD-L1 Pathway. Trends Mol. Med. 2015, 21, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, C.; Luong, G.; Sun, Y. A Snapshot of the PD-1/PD-L1 Pathway. J. Cancer 2021, 12, 2735–2746. [Google Scholar] [CrossRef]
- Bretscher, P.A. A Two-Step, Two-Signal Model for the Primary Activation of Precursor Helper T Cells. Proc. Natl. Acad. Sci. USA 1999, 96, 185–190. [Google Scholar] [CrossRef]
- Makuku, R.; Khalili, N.; Razi, S.; Keshavarz-Fathi, M.; Rezaei, N. Current and Future Perspectives of PD-1/PDL-1 Blockade in Cancer Immunotherapy. J. Immunol. Res. 2021, 2021, e6661406. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef]
- de Visser, K.E.; Eichten, A.; Coussens, L.M. Paradoxical Roles of the Immune System during Cancer Development. Nat. Rev. Cancer 2006, 6, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Flies, D.B.; Sandler, B.J.; Sznol, M.; Chen, L. Blockade of the B7-H1/PD-1 Pathway for Cancer Immunotherapy. Yale J. Biol. Med. 2011, 84, 409–421. [Google Scholar] [PubMed]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Ge, J.; Xiang, B.; Wu, X.; Ma, J.; Zhou, M.; Li, X.; et al. Role of the Tumor Microenvironment in PD-L1/PD-1-Mediated Tumor Immune Escape. Mol. Cancer 2019, 18, 10. [Google Scholar] [CrossRef] [PubMed]
- Mimura, K.; Teh, J.L.; Okayama, H.; Shiraishi, K.; Kua, L.-F.; Koh, V.; Smoot, D.T.; Ashktorab, H.; Oike, T.; Suzuki, Y.; et al. PD-L1 Expression Is Mainly Regulated by Interferon Gamma Associated with JAK-STAT Pathway in Gastric Cancer. Cancer Sci. 2018, 109, 43–53. [Google Scholar] [CrossRef]
- Zhang, X.; Zeng, Y.; Qu, Q.; Zhu, J.; Liu, Z.; Ning, W.; Zeng, H.; Zhang, N.; Du, W.; Chen, C.; et al. PD-L1 Induced by IFN-γ from Tumor-Associated Macrophages via the JAK/STAT3 and PI3K/AKT Signaling Pathways Promoted Progression of Lung Cancer. Int. J. Clin. Oncol. 2017, 22, 1026–1033. [Google Scholar] [CrossRef]
- Moon, J.W.; Kong, S.-K.; Kim, B.S.; Kim, H.J.; Lim, H.; Noh, K.; Kim, Y.; Choi, J.-W.; Lee, J.-H.; Kim, Y.-S. IFNγ Induces PD-L1 Overexpression by JAK2/STAT1/IRF-1 Signaling in EBV-Positive Gastric Carcinoma. Sci. Rep. 2017, 7, 17810. [Google Scholar] [CrossRef]
- Liu, J.; Hamrouni, A.; Wolowiec, D.; Coiteux, V.; Kuliczkowski, K.; Hetuin, D.; Saudemont, A.; Quesnel, B. Plasma Cells from Multiple Myeloma Patients Express B7-H1 (PD-L1) and Increase Expression after Stimulation with IFN-γ and TLR Ligands via a MyD88-, TRAF6-, and MEK-Dependent Pathway. Blood 2007, 110, 296–304. [Google Scholar] [CrossRef]
- Lee, S.-K.; Seo, S.-H.; Kim, B.-S.; Kim, C.-D.; Lee, J.-H.; Kang, J.-S.; Maeng, P.J.; Lim, J.-S. IFN-Gamma Regulates the Expression of B7-H1 in Dermal Fibroblast Cells. J. Dermatol. Sci. 2005, 40, 95–103. [Google Scholar] [CrossRef]
- Parsa, A.T.; Waldron, J.S.; Panner, A.; Crane, C.A.; Parney, I.F.; Barry, J.J.; Cachola, K.E.; Murray, J.C.; Tihan, T.; Jensen, M.C.; et al. Loss of Tumor Suppressor PTEN Function Increases B7-H1 Expression and Immunoresistance in Glioma. Nat. Med. 2007, 13, 84–88. [Google Scholar] [CrossRef]
- Akbay, E.A.; Koyama, S.; Carretero, J.; Altabef, A.; Tchaicha, J.H.; Christensen, C.L.; Mikse, O.R.; Cherniack, A.D.; Beauchamp, E.M.; Pugh, T.J.; et al. Activation of the PD-1 Pathway Contributes to Immune Escape in EGFR-Driven Lung Tumors. Cancer Discov. 2013, 3, 1355–1363. [Google Scholar] [CrossRef]
- Wang, X.; Yang, L.; Huang, F.; Zhang, Q.; Liu, S.; Ma, L.; You, Z. Inflammatory Cytokines IL-17 and TNF-α up-Regulate PD-L1 Expression in Human Prostate and Colon Cancer Cells. Immunol. Lett. 2017, 184, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Yee, D.; Shah, K.M.; Coles, M.C.; Sharp, T.V.; Lagos, D. MicroRNA-155 Induction via TNF-α and IFN-γ Suppresses Expression of Programmed Death Ligand-1 (PD-L1) in Human Primary Cells. J. Biol. Chem. 2017, 292, 20683–20693. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, Y.; Yan, Z.; Yang, H.; Sun, W.; Yao, Y.; Chen, Y.; Jiang, R. IL-6 Promotes PD-L1 Expression in Monocytes and Macrophages by Decreasing Protein Tyrosine Phosphatase Receptor Type O Expression in Human Hepatocellular Carcinoma. J. Immunother. Cancer 2020, 8, e000285. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.J.; Xu, L.J.; Yang, L.; Tsai, Y.; Keng, P.C.; Chen, Y.; Lee, S.O.; Chen, Y. Radiation Alters PD-L1/NKG2D Ligand Levels in Lung Cancer Cells and Leads to Immune Escape from NK Cell Cytotoxicity via IL-6-MEK/Erk Signaling Pathway. Oncotarget 2017, 8, 80506–80520. [Google Scholar] [CrossRef] [PubMed]
- Lamano, J.B.; Lamano, J.B.; Li, Y.D.; DiDomenico, J.D.; Choy, W.; Veliceasa, D.; Oyon, D.E.; Fakurnejad, S.; Ampie, L.; Kesavabhotla, K.; et al. Glioblastoma-Derived IL-6 Induces Immunosuppressive Peripheral Myeloid Cell PD-L1 and Promotes Tumor Growth. Clin. Cancer Res. 2019, 25, 3643–3657. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Chen, X.; Shen, M.; Yang, D.; Fang, L.; Weng, G.; Tsai, Y.; Keng, P.C.; Chen, Y.; Lee, S.O. Inhibition of IL-6-JAK/Stat3 Signaling in Castration-resistant Prostate Cancer Cells Enhances the NK Cell-mediated Cytotoxicity via Alteration of PD-L1/NKG2D Ligand Levels. Mol. Oncol. 2018, 12, 269–286. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Niu, M.; Xu, L.; Luo, S.; Wu, K. Regulation of PD-L1 Expression in the Tumor Microenvironment. J. Hematol. Oncol. 2021, 14, 10. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Yuan, F.; Wang, J.; Wu, L. Oral Squamous Cell Carcinoma Suppressed Antitumor Immunity through Induction of PD-L1 Expression on Tumor-Associated Macrophages. Immunobiology 2017, 222, 651–657. [Google Scholar] [CrossRef]
- Wang, X.; Ni, S.; Chen, Q.; Ma, L.; Jiao, Z.; Wang, C.; Jia, G. Bladder Cancer Cells Induce Immunosuppression of T Cells by Supporting PD-L1 Expression in Tumour Macrophages Partially through Interleukin 10. Cell Biol. Int. 2017, 41, 177–186. [Google Scholar] [CrossRef]
- Xiong, H.-Y.; Ma, T.-T.; Wu, B.-T.; Lin, Y.; Tu, Z.-G. IL-12 Regulates B7-H1 Expression in Ovarian Cancer-Associated Macrophages by Effects on NF-κB Signalling. Asian Pac. J. Cancer Prev. 2014, 15, 5767–5772. [Google Scholar] [CrossRef]
- Hirahara, K.; Ghoreschi, K.; Yang, X.-P.; Takahashi, H.; Laurence, A.; Vahedi, G.; Sciumè, G.; Hall, A.O.; Dupont, C.D.; Francisco, L.M.; et al. Interleukin-27 Priming of T Cells Controls IL-17 Production In Trans via Induction of the Ligand PD-L1. Immunity 2012, 36, 1017–1030. [Google Scholar] [CrossRef] [PubMed]
- Gainor, J.F.; Shaw, A.T.; Sequist, L.V.; Fu, X.; Azzoli, C.G.; Piotrowska, Z.; Huynh, T.; Zhao, L.; Fulton, L.; Schultz, K.R.; et al. EGFR Mutations and ALK Rearrangements Are Associated with Low Response Rates to PD-1 Pathway Blockade in Non-Small Cell Lung Cancer (NSCLC): A Retrospective Analysis. Clin. Cancer Res. 2016, 22, 4585–4593. [Google Scholar] [CrossRef] [PubMed]
- Barsoum, I.B.; Smallwood, C.A.; Siemens, D.R.; Graham, C.H. A Mechanism of Hypoxia-Mediated Escape from Adaptive Immunity in Cancer Cells. Cancer Res. 2014, 74, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 Is a Novel Direct Target of HIF-1α, and Its Blockade under Hypoxia Enhanced MDSC-Mediated T Cell Activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Alkholifi, F.K.; Alsaffar, R.M. Dostarlimab an Inhibitor of PD-1/PD-L1: A New Paradigm for the Treatment of Cancer. Medicina 2022, 58, 1572. [Google Scholar] [CrossRef]
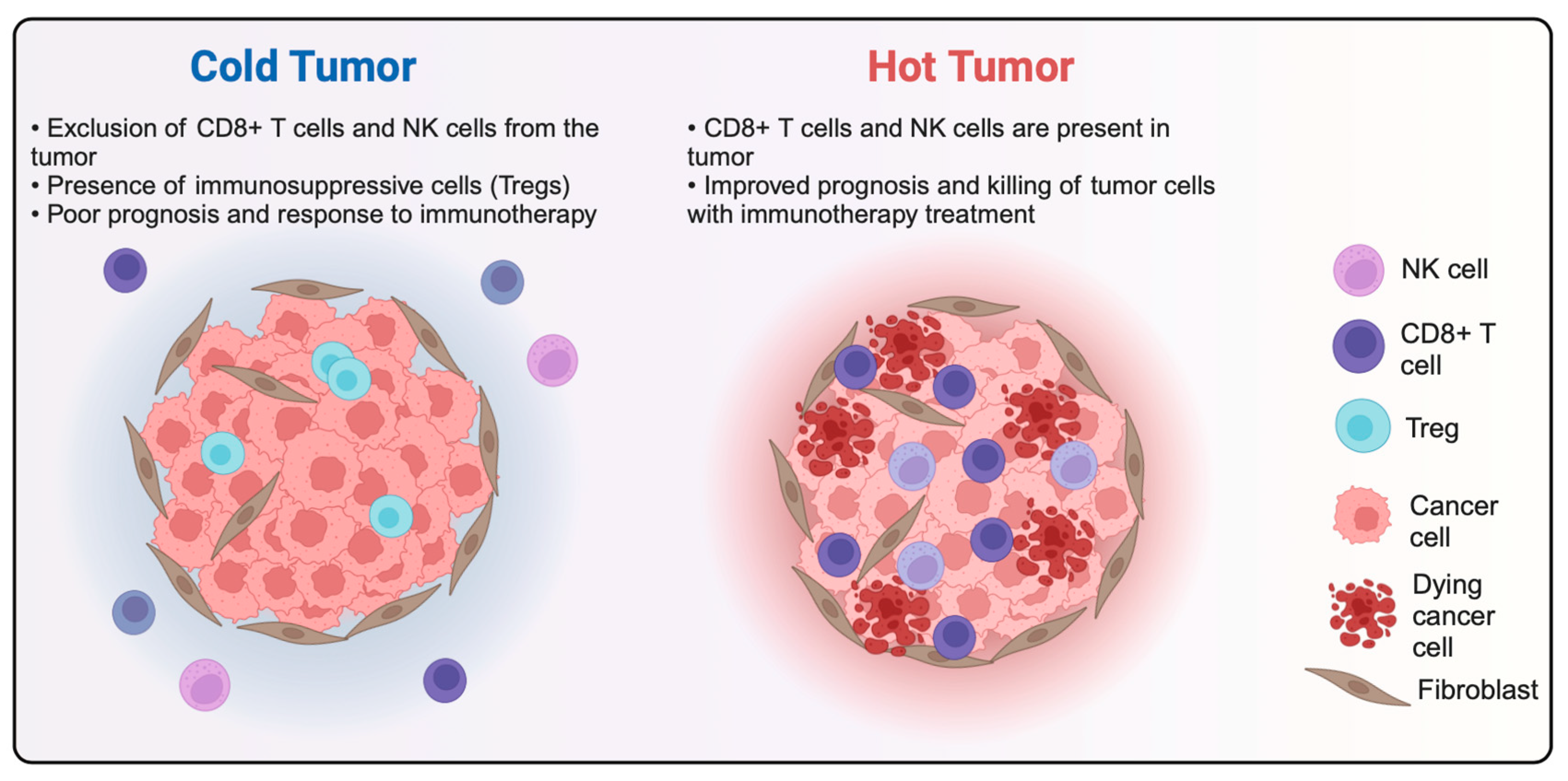
- Liu, Y.-T.; Sun, Z.-J. Turning Cold Tumors into Hot Tumors by Improving T-Cell Infiltration. Theranostics 2021, 11, 5365–5386. [Google Scholar] [CrossRef]
- Galon, J.; Bruni, D. Approaches to Treat Immune Hot, Altered and Cold Tumours with Combination Immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef]
- Noman, M.Z.; Parpal, S.; Van Moer, K.; Xiao, M.; Yu, Y.; Viklund, J.; De Milito, A.; Hasmim, M.; Andersson, M.; Amaravadi, R.K.; et al. Inhibition of Vps34 Reprograms Cold into Hot Inflamed Tumors and Improves Anti-PD-1/PD-L1 Immunotherapy. Sci. Adv. 2020, 6, eaax7881. [Google Scholar] [CrossRef]
- Knox, T.; Sahakian, E.; Banik, D.; Hadley, M.; Palmer, E.; Noonepalle, S.; Kim, J.; Powers, J.; Gracia-Hernandez, M.; Oliveira, V.; et al. Selective HDAC6 Inhibitors Improve Anti-PD-1 Immune Checkpoint Blockade Therapy by Decreasing the Anti-Inflammatory Phenotype of Macrophages and down-Regulation of Immunosuppressive Proteins in Tumor Cells. Sci. Rep. 2019, 9, 6136. [Google Scholar] [CrossRef]
- Clark, G.; Stockinger, H.; Balderas, R.; van Zelm, M.C.; Zola, H.; Hart, D.; Engel, P. Nomenclature of CD Molecules from the Tenth Human Leucocyte Differentiation Antigen Workshop. Clin. Trans. Immunol. 2016, 5, e57. [Google Scholar] [CrossRef]
- Esfandiari, A.; Cassidy, S.; Webster, R.M. Bispecific Antibodies in Oncology. Nat. Rev. Drug Discov. 2022, 21, 411–412. [Google Scholar] [CrossRef] [PubMed]
- Kontermann, R.E.; Brinkmann, U. Bispecific Antibodies. Drug Discov. Today 2015, 20, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Kontermann, R.E. Strategies to Extend Plasma Half-Lives of Recombinant Antibodies. BioDrugs 2009, 23, 93–109. [Google Scholar] [CrossRef]
- Schmidt, M.M.; Wittrup, K.D. A Modeling Analysis of the Effects of Molecular Size and Binding Affinity on Tumor Targeting. Mol. Cancer Ther. 2009, 8, 2861–2871. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Hoseini, S.S.; Xu, H.; Ponomarev, V.; Cheung, N.-K. Silencing Fc Domains in T Cell–Engaging Bispecific Antibodies Improves T-Cell Trafficking and Antitumor Potency. Cancer Immunol. Res. 2019, 7, 2013–2024. [Google Scholar] [CrossRef] [PubMed]
- Kontermann, R.E. Half-Life Extended Biotherapeutics. Expert. Opin. Biol. Ther. 2016, 16, 903–915. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, M.; Jeddi-Tehrani, M.; Golsaz-Shirazi, F.; Arjmand, M.; Bahadori, T.; Judaki, M.A.; Shiravi, F.; Zare, H.A.; Haghighat, F.N.; Mobini, M.; et al. A Novel Anti-HER2 Bispecific Antibody With Potent Tumor Inhibitory Effects In Vitro and In Vivo. Front. Immunol. 2021, 11, 600883. [Google Scholar] [CrossRef] [PubMed]
- de Goeij, B.E.C.G.; Vink, T.; ten Napel, H.; Breij, E.C.W.; Satijn, D.; Wubbolts, R.; Miao, D.; Parren, P.W.H.I. Efficient Payload Delivery by a Bispecific Antibody–Drug Conjugate Targeting HER2 and CD63. Mol. Cancer Ther. 2016, 15, 2688–2697. [Google Scholar] [CrossRef]
- Feldmann, A.; Arndt, C.; Töpfer, K.; Stamova, S.; Krone, F.; Cartellieri, M.; Koristka, S.; Michalk, I.; Lindemann, D.; Schmitz, M.; et al. Novel Humanized and Highly Efficient Bispecific Antibodies Mediate Killing of Prostate Stem Cell Antigen-Expressing Tumor Cells by CD8+ and CD4+ T Cells. J. Immunol. 2012, 189, 3249–3259. [Google Scholar] [CrossRef]
- Iizuka, A.; Nonomura, C.; Ashizawa, T.; Kondou, R.; Ohshima, K.; Sugino, T.; Mitsuya, K.; Hayashi, N.; Nakasu, Y.; Maruyama, K.; et al. A T-Cell–Engaging B7-H4/CD3-Bispecific Fab-scFv Antibody Targets Human Breast Cancer. Clin. Cancer Res. 2019, 25, 2925–2934. [Google Scholar] [CrossRef]
- Bright, R.K.; Bright, J.D.; Byrne, J.A. Overexpressed Oncogenic Tumor-Self Antigens: New Vaccine Targets. Hum. Vaccines Immunother. 2014, 10, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Akinrinmade, O.A.; Chetty, S.; Daramola, A.K.; Islam, M.; Thepen, T.; Barth, S. CD64: An Attractive Immunotherapeutic Target for M1-Type Macrophage Mediated Chronic Inflammatory Diseases. Biomedicines 2017, 5, 56. [Google Scholar] [CrossRef] [PubMed]
- Yeap, W.H.; Wong, K.L.; Shimasaki, N.; Teo, E.C.Y.; Quek, J.K.S.; Yong, H.X.; Diong, C.P.; Bertoletti, A.; Linn, Y.C.; Wong, S.C. CD16 Is Indispensable for Antibody-Dependent Cellular Cytotoxicity by Human Monocytes. Sci. Rep. 2016, 6, 34310. [Google Scholar] [CrossRef] [PubMed]
- Nagorsen, D.; Bargou, R.; Rüttinger, D.; Kufer, P.; Baeuerle, P.A.; Zugmaier, G. Immunotherapy of Lymphoma and Leukemia with T-Cell Engaging BiTE Antibody Blinatumomab. Leuk. Lymphoma 2009, 50, 886–891. [Google Scholar] [CrossRef] [PubMed]
- Kuhns, M.S.; Davis, M.M.; Garcia, K.C. Deconstructing the Form and Function of the TCR/CD3 Complex. Immunity 2006, 24, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Cromme, F.; van Bommel, P.; Walboomers, J.; Gallee, M.; Stern, P.; Kenemans, P.; Helmerhorst, T.; Stukart, M.; Meijer, C. Differences in MHC and TAP-1 Expression in Cervical Cancer Lymph Node Metastases as Compared with the Primary Tumours. Br. J. Cancer 1994, 69, 1176–1181. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, T.; Fernandez, M.A.; Sierra, A.; Garrido, A.; Herruzo, A.; Escobedo, A.; Fabra, A.; Garrido, F. High Frequency of Altered HLA Class I Phenotypes in Invasive Breast Carcinomas. Hum. Immunol. 1996, 50, 127–134. [Google Scholar] [CrossRef]
- Mehta, A.M.; Jordanova, E.S.; Kenter, G.G.; Ferrone, S.; Fleuren, G.-J. Association of Antigen Processing Machinery and HLA Class I Defects with Clinicopathological Outcome in Cervical Carcinoma. Cancer Immunol. Immunother. 2008, 57, 197–206. [Google Scholar] [CrossRef]
- Pardo, J.; Wallich, R.; Martin, P.; Urban, C.; Rongvaux, A.; Flavell, R.A.; Müllbacher, A.; Borner, C.; Simon, M.M. Granzyme B-Induced Cell Death Exerted by Ex Vivo CTL: Discriminating Requirements for Cell Death and Some of Its Signs. Cell Death Differ. 2008, 15, 567–579. [Google Scholar] [CrossRef]
- Nagorsen, D.; Baeuerle, P.A. Immunomodulatory Therapy of Cancer with T Cell-Engaging BiTE Antibody Blinatumomab. Exp. Cell Res. 2011, 317, 1255–1260. [Google Scholar] [CrossRef]
- Klein, S.C.; Boer, L.H.; de Weger, R.A.; de Gast, G.C.; Bast, E.J. Release of Cytokines and Soluble Cell Surface Molecules by PBMC after Activation with the Bispecific Antibody CD3 × CD19. Scand. J. Immunol. 1997, 46, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Piskol, R.; Ybarra, R.; Chen, Y.-J.J.; Li, J.; Slaga, D.; Hristopoulos, M.; Clark, R.; Modrusan, Z.; Totpal, K.; et al. CD3 Bispecific Antibody-Induced Cytokine Release Is Dispensable for Cytotoxic T Cell Activity. Sci. Transl. Med. 2019, 11, eaax8861. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.L.; Sherman, M.; McElroy, P.L.; Lofgren, J.A.; Moody, G.; Baeuerle, P.A.; Coxon, A.; Arvedson, T. Bispecific T Cell Engager (BiTE®) Antibody Constructs Can Mediate Bystander Tumor Cell Killing. PLoS ONE 2017, 12, e0183390. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Li, X.; Chintala, N.K.; Tano, Z.E.; Adusumilli, P.S. Driving CARs on the Uneven Road of Antigen Heterogeneity in Solid Tumors. Curr. Opin. Immunol. 2018, 51, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Koristka, S.; Cartellieri, M.; Theil, A.; Feldmann, A.; Arndt, C.; Stamova, S.; Michalk, I.; Töpfer, K.; Temme, A.; Kretschmer, K.; et al. Retargeting of Human Regulatory T Cells by Single-Chain Bispecific Antibodies. J. Immunol. 2012, 188, 1551–1558. [Google Scholar] [CrossRef]
- Duell, J.; Dittrich, M.; Bedke, T.; Mueller, T.; Eisele, F.; Rosenwald, A.; Rasche, L.; Hartmann, E.; Dandekar, T.; Einsele, H.; et al. Frequency of Regulatory T Cells Determines the Outcome of the T-Cell-Engaging Antibody Blinatumomab in Patients with B-Precursor ALL. Leukemia 2017, 31, 2181–2190. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Yang, Y.; Wang, G.; Liu, M. Current Landscape and Future Directions of Bispecific Antibodies in Cancer Immunotherapy. Front. Immunol. 2022, 13, 1035276. [Google Scholar] [CrossRef]
- Przepiorka, D.; Ko, C.-W.; Deisseroth, A.; Yancey, C.L.; Candau-Chacon, R.; Chiu, H.-J.; Gehrke, B.J.; Gomez-Broughton, C.; Kane, R.C.; Kirshner, S.; et al. FDA Approval: Blinatumomab. Clin. Cancer Res. 2015, 21, 4035–4039. [Google Scholar] [CrossRef]
- Kantarjian, H.; Stein, A.; Gökbuget, N.; Fielding, A.K.; Schuh, A.C.; Ribera, J.-M.; Wei, A.; Dombret, H.; Foà, R.; Bassan, R.; et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2017, 376, 836–847. [Google Scholar] [CrossRef]
- DiLillo, D.J.; Olson, K.; Mohrs, K.; Meagher, T.C.; Bray, K.; Sineshchekova, O.; Startz, T.; Kuhnert, J.; Retter, M.W.; Godin, S.; et al. A BCMAxCD3 Bispecific T Cell–Engaging Antibody Demonstrates Robust Antitumor Efficacy Similar to That of Anti-BCMA CAR T Cells. Blood Adv. 2021, 5, 1291–1304. [Google Scholar] [CrossRef]
- Tedder, T.F.; Klejman, G.; Schlossman, S.F.; Saito, H. Structure of the Gene Encoding the Human B Lymphocyte Differentiation Antigen CD20 (B1). J. Immunol. 1989, 142, 2560–2568. [Google Scholar] [CrossRef] [PubMed]
- Kuijpers, T.W.; Bende, R.J.; Baars, P.A.; Grummels, A.; Derks, I.A.M.; Dolman, K.M.; Beaumont, T.; Tedder, T.F.; van Noesel, C.J.M.; Eldering, E.; et al. CD20 Deficiency in Humans Results in Impaired T Cell–Independent Antibody Responses. J. Clin. Investig. 2010, 120, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Pavlasova, G.; Mraz, M. The Regulation and Function of CD20: An “Enigma” of B-Cell Biology and Targeted Therapy. Haematologica 2020, 105, 1494–1506. [Google Scholar] [CrossRef] [PubMed]
- Schultz, L.; Gardner, R. Mechanisms of and Approaches to Overcoming Resistance to Immunotherapy. Hematol. Am. Soc. Hematol. Educ. Program. 2019, 2019, 226–232. [Google Scholar] [CrossRef]
- Gardner, R.; Wu, D.; Cherian, S.; Fang, M.; Hanafi, L.-A.; Finney, O.; Smithers, H.; Jensen, M.C.; Riddell, S.R.; Maloney, D.G.; et al. Acquisition of a CD19-Negative Myeloid Phenotype Allows Immune Escape of MLL-Rearranged B-ALL from CD19 CAR-T-Cell Therapy. Blood 2016, 127, 2406–2410. [Google Scholar] [CrossRef]
- Rayes, A.; McMasters, R.L.; O’Brien, M.M. Lineage Switch in MLL-Rearranged Infant Leukemia Following CD19-Directed Therapy: Lineage Switch in MLL Rearranged Infant Leukemia. Pediatr. Blood Cancer 2016, 63, 1113–1115. [Google Scholar] [CrossRef] [PubMed]
- Fry, T.J.; Shah, N.N.; Orentas, R.J.; Stetler-Stevenson, M.; Yuan, C.M.; Ramakrishna, S.; Wolters, P.; Martin, S.; Delbrook, C.; Yates, B.; et al. CD22-CAR T Cells Induce Remissions in CD19-CAR Naïve and Resistant B-ALL. Nat. Med. 2018, 24, 20–28. [Google Scholar] [CrossRef]
- Zheng, S.; Gillespie, E.; Naqvi, A.S.; Hayer, K.E.; Ang, Z.; Torres-Diz, M.; Quesnel-Vallières, M.; Hottman, D.A.; Bagashev, A.; Chukinas, J.; et al. Modulation of CD22 Protein Expression in Childhood Leukemia by Pervasive Splicing Aberrations: Implications for CD22-Directed Immunotherapies. Blood Cancer Discov. 2022, 3, 103–115. [Google Scholar] [CrossRef]
- Ramakrishna, S.; Highfill, S.L.; Walsh, Z.; Nguyen, S.M.; Lei, H.; Shern, J.F.; Qin, H.; Kraft, I.; Stetler-Stevenson, M.; Yuan, C.M.; et al. Modulation of Target Antigen Density Improves CAR T Cell Functionality and Persistence. Clin. Cancer Res. 2019, 25, 5329–5341. [Google Scholar] [CrossRef]
- Yamashita, T.; Ji, J.; Budhu, A.; Forgues, M.; Yang, W.; Wang, H.-Y.; Jia, H.; Ye, Q.; Qin, L.-X.; Wauthier, E.; et al. EpCAM-Positive Hepatocellular Carcinoma Cells Are Tumor-Initiating Cells with Stem/Progenitor Cell Features. Gastroenterology 2009, 136, 1012–1024. [Google Scholar] [CrossRef]
- Ralhan, R.; Cao, J.; Lim, T.; MacMillan, C.; Freeman, J.L.; Walfish, P.G. EpCAM Nuclear Localization Identifies Aggressive Thyroid Cancer and Is a Marker for Poor Prognosis. BMC Cancer 2010, 10, 331. [Google Scholar] [CrossRef] [PubMed]
- Borlak, J.; Länger, F.; Spanel, R.; Schöndorfer, G.; Dittrich, C. Immune-Mediated Liver Injury of the Cancer Therapeutic Antibody Catumaxomab Targeting EpCAM, CD3 and Fcγ Receptors. Oncotarget 2016, 7, 28059–28074. [Google Scholar] [CrossRef] [PubMed]
- Akhter, M.Z.; Sharawat, S.K.; Kumar, V.; Kochat, V.; Equbal, Z.; Ramakrishnan, M.; Kumar, U.; Mathur, S.; Kumar, L.; Mukhopadhyay, A. Aggressive Serous Epithelial Ovarian Cancer Is Potentially Propagated by EpCAM+CD45+ Phenotype. Oncogene 2018, 37, 2089–2103. [Google Scholar] [CrossRef] [PubMed]
- Mohtar, M.A.; Syafruddin, S.E.; Nasir, S.N.; Low, T.Y. Revisiting the Roles of Pro-Metastatic EpCAM in Cancer. Biomolecules 2020, 10, 255. [Google Scholar] [CrossRef] [PubMed]
- Kalantari, E.; Taheri, T.; Fata, S.; Abolhasani, M.; Mehrazma, M.; Madjd, Z.; Asgari, M. Significant Co-Expression of Putative Cancer Stem Cell Markers, EpCAM and CD166, Correlates with Tumor Stage and Invasive Behavior in Colorectal Cancer. World J. Surg. Oncol. 2022, 20, 15. [Google Scholar] [CrossRef]
- Amann, M.; Lorenczewski, G.; Brischwein, K.; Kischel, R.; Lutterbuese, R.; Mangold, S.; Rau, D.; Volkland, J.; Pflanz, S.; Raum, T.; et al. Antitumor Activity of an EpCAM/CD3-Bispecific BiTE Antibody During Long-Term Treatment of Mice in the Absence of T-Cell Anergy and Sustained Cytokine Release. J. Immunother. 2009, 32, 452. [Google Scholar] [CrossRef] [PubMed]
- Salnikov, A.V.; Groth, A.; Apel, A.; Kallifatidis, G.; Beckermann, B.M.; Khamidjanov, A.; Ryschich, E.; Büchler, M.W.; Herr, I.; Moldenhauer, G. Targeting of Cancer Stem Cell Marker EpCAM by Bispecific Antibody EpCAMxCD3 Inhibits Pancreatic Carcinoma. J. Cell. Mol. Med. 2009, 13, 4023–4033. [Google Scholar] [CrossRef]
- Zhang, P.; Shi, B.; Gao, H.; Jiang, H.; Kong, J.; Yan, J.; Pan, X.; Li, K.; Zhang, P.; Yao, M.; et al. An EpCAM/CD3 Bispecific Antibody Efficiently Eliminates Hepatocellular Carcinoma Cells with Limited Galectin-1 Expression. Cancer Immunol. Immunother. 2014, 63, 121–132. [Google Scholar] [CrossRef]
- Ferrari, F.; Bellone, S.; Black, J.; Schwab, C.L.; Lopez, S.; Cocco, E.; Bonazzoli, E.; Predolini, F.; Menderes, G.; Litkouhi, B.; et al. Solitomab, an EpCAM/CD3 Bispecific Antibody Construct (BiTE®), Is Highly Active against Primary Uterine and Ovarian Carcinosarcoma Cell Lines in Vitro. J. Exp. Clin. Cancer Res. 2015, 34, 123. [Google Scholar] [CrossRef]
- Wang, C.; Chen, S.; Wu, Y.; Wu, D.; Wang, J.; Li, F. The Combination Therapy with EpCAM/CD3 BsAb and MUC-1/CD3 BsAb Elicited Antitumor Immunity by T-Cell Adoptive Immunotherapy in Lung Cancer. Int. J. Med. Sci. 2021, 18, 3380–3388. [Google Scholar] [CrossRef]
- Wang, L.; Qiao, Y.; Zong, H.; Han, L.; Ke, Y.; Pan, Z.; Chen, J.; Lu, J.; Li, J.; Ying, T.; et al. IgG-like Bispecific Antibody CD3×EpCAM Generated by Split Intein Against Colorectal Cancer. Front. Pharmacol. 2022, 13, 803059. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Suzuki, H.; Asano, T.; Tanaka, T.; Suzuki, H.; Kaneko, M.K.; Kato, Y. Development of a Novel Anti-EpCAM Monoclonal Antibody for Various Applications. Antibodies 2022, 11, 41. [Google Scholar] [CrossRef] [PubMed]
- Fiedler, W.M.; Ritter, B.; Seggewiss, R.; Bokemeyer, C.; Fettes, P.; Klinger, M.; Vieser, E.; Ruettinger, D.; Kaubitzsch, S.; Wolf, M. Phase I Safety and Pharmacology Study of the EpCAM/CD3-Bispecific BiTE Antibody MT110 in Patients with Metastatic Colorectal, Gastric, or Lung Cancer. JCO 2010, 28, 2573. [Google Scholar] [CrossRef]
- Kebenko, M.; Goebeler, M.-E.; Wolf, M.; Hasenburg, A.; Seggewiss-Bernhardt, R.; Ritter, B.; Rautenberg, B.; Atanackovic, D.; Kratzer, A.; Rottman, J.B.; et al. A Multicenter Phase 1 Study of Solitomab (MT110, AMG 110), a Bispecific EpCAM/CD3 T-Cell Engager (BiTE®) Antibody Construct, in Patients with Refractory Solid Tumors. Oncoimmunology 2018, 7, e1450710. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhao, C.; Jia, Y.; Wang, S.; Ma, X.; Wang, T.; Huang, S.; Pei, M.; Wang, X.; Zhou, P. 539P Interim Results of a Phase I Study of M701, a Recombinant Anti-EpCAM and Anti-CD3 Bispecific Antibody in EpCAM-Positive Cancer Patients with Malignant Ascites. Ann. Oncol. 2021, 32, S603. [Google Scholar] [CrossRef]
- Oberst, M.D.; Fuhrmann, S.; Mulgrew, K.; Amann, M.; Cheng, L.; Lutterbuese, P.; Richman, L.; Coats, S.; Baeuerle, P.A.; Hammond, S.A. CEA/CD3 Bispecific Antibody MEDI-565/AMG 211 Activation of T Cells and Subsequent Killing of Human Tumors Is Independent of Mutations Commonly Found in Colorectal Adenocarcinomas. MAbs 2014, 6, 1571–1584. [Google Scholar] [CrossRef] [PubMed]
- Bacac, M.; Klein, C.; Umana, P. CEA TCB: A Novel Head-to-Tail 2:1 T Cell Bispecific Antibody for Treatment of CEA-Positive Solid Tumors. OncoImmunology 2016, 5, e1203498. [Google Scholar] [CrossRef] [PubMed]
- Bacac, M.; Fauti, T.; Sam, J.; Colombetti, S.; Weinzierl, T.; Ouaret, D.; Bodmer, W.; Lehmann, S.; Hofer, T.; Hosse, R.J.; et al. A Novel Carcinoembryonic Antigen T-Cell Bispecific Antibody (CEA TCB) for the Treatment of Solid Tumors. Clin. Cancer Res. 2016, 22, 3286–3297. [Google Scholar] [CrossRef]
- Segal, N.H.; Saro, J.; Melero, I.; Ros, W.; Argiles, G.; Marabelle, A.; Ruiz, M.E.R.; Albanell, J.; Calvo, E.; Moreno, V.; et al. Phase I Studies of the Novel Carcinoembryonic Antigen T-Cell Bispecific (CEA-CD3 TCB) Antibody as a Single Agent and in Combination with Atezolizumab: Preliminary Efficacy and Safety in Patients (Pts) with Metastatic Colorectal Cancer (mCRC). Ann. Oncol. 2017, 28, v134. [Google Scholar] [CrossRef]
- Tabernero, J.; Melero, I.; Ros, W.; Argiles, G.; Marabelle, A.; Rodriguez-Ruiz, M.E.; Albanell, J.; Calvo, E.; Moreno, V.; Cleary, J.M.; et al. Phase Ia and Ib Studies of the Novel Carcinoembryonic Antigen (CEA) T-Cell Bispecific (CEA CD3 TCB) Antibody as a Single Agent and in Combination with Atezolizumab: Preliminary Efficacy and Safety in Patients with Metastatic Colorectal Cancer (mCRC). JCO 2017, 35, 3002. [Google Scholar] [CrossRef]
- Wang, N.; Patel, H.; Schneider, I.C.; Kai, X.; Varshney, A.K.; Zhou, L. An Optimal Antitumor Response by a Novel CEA/CD3 Bispecific Antibody for Colorectal Cancers. Antib. Ther. 2021, 4, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Fortmüller, K.; Alt, K.; Gierschner, D.; Wolf, P.; Baum, V.; Freudenberg, N.; Wetterauer, U.; Elsässer-Beile, U.; Bühler, P. Effective Targeting of Prostate Cancer by Lymphocytes Redirected by a PSMA × CD3 Bispecific Single-Chain Diabody. Prostate 2011, 71, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, M.; Raum, T.; Lutterbuese, R.; Voelkel, M.; Deegen, P.; Rau, D.; Kischel, R.; Hoffmann, P.; Brandl, C.; Schuhmacher, J.; et al. Regression of Human Prostate Cancer Xenografts in Mice by AMG 212/BAY2010112, a Novel PSMA/CD3-Bispecific BiTE Antibody Cross-Reactive with Non-Human Primate Antigens. Mol. Cancer Ther. 2012, 11, 2664–2673. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Hoyos, G.; Sewell, T.; Bader, R.; Bannink, J.; Chenault, R.A.; Daugherty, M.; Dasovich, M.; Fang, H.; Gottschalk, R.; Kumer, J.; et al. MOR209/ES414, a Novel Bispecific Antibody Targeting PSMA for the Treatment of Metastatic Castration-Resistant Prostate Cancer. Mol. Cancer Ther. 2016, 15, 2155–2165. [Google Scholar] [CrossRef] [PubMed]
- Leconet, W.; Liu, H.; Guo, M.; Le Lamer-Déchamps, S.; Molinier, C.; Kim, S.; Vrlinic, T.; Oster, M.; Liu, F.; Navarro, V.; et al. Anti-PSMA/CD3 Bispecific Antibody Delivery and Antitumor Activity Using a Polymeric Depot Formulation. Mol. Cancer Ther. 2018, 17, 1927–1940. [Google Scholar] [CrossRef] [PubMed]
- Chiu, D.; Tavaré, R.; Haber, L.; Aina, O.H.; Vazzana, K.; Ram, P.; Danton, M.; Finney, J.; Jalal, S.; Krueger, P.; et al. A PSMA-Targeting CD3 Bispecific Antibody Induces Antitumor Responses That Are Enhanced by 4-1BB Costimulation. Cancer Immunol. Res. 2020, 8, 596–608. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; Ma, J.S.Y.; Kim, M.S.; Laborda, E.; Choi, S.-H.; Hampton, E.N.; Yun, H.; Nunez, V.; Muldong, M.T.; Wu, C.N.; et al. A PSMA-Targeted Bispecific Antibody for Prostate Cancer Driven by a Small-Molecule Targeting Ligand. Sci. Adv. 2021, 7, eabi8193. [Google Scholar] [CrossRef]
- Markowski, M.C.; Kilari, D.; Eisenberger, M.A.; McKay, R.R.; Dreicer, R.; Trikha, M.; Heath, E.I.; Li, J.; Garzone, P.D.; Young, T.S. Phase I Study of CCW702, a Bispecific Small Molecule-Antibody Conjugate Targeting PSMA and CD3 in Patients with Metastatic Castration-Resistant Prostate Cancer (mCRPC). JCO 2021, 39, TPS5094. [Google Scholar] [CrossRef]
- Lim, E.A.; Schweizer, M.T.; Chi, K.N.; Aggarwal, R.R.; Agarwal, N.; Gulley, J.L.; Attiyeh, E.F.; Greger, J.; Wu, S.; Jaiprasart, P.; et al. Safety and Preliminary Clinical Activity of JNJ-63898081 (JNJ-081), a PSMA and CD3 Bispecific Antibody, for the Treatment of Metastatic Castrate-Resistant Prostate Cancer (mCRPC). JCO 2022, 40, 279. [Google Scholar] [CrossRef]
- Lim, E.A.; Schweizer, M.T.; Chi, K.N.; Aggarwal, R.; Agarwal, N.; Gulley, J.; Attiyeh, E.; Greger, J.; Wu, S.; Jaiprasart, P.; et al. Phase 1 Study of Safety and Preliminary Clinical Activity of JNJ-63898081, a PSMA and CD3 Bispecific Antibody, for Metastatic Castration-Resistant Prostate Cancer. Clin. Genitourin. Cancer 2023, 21, 366–375. [Google Scholar] [CrossRef]
- Junttila, T.T.; Li, J.; Johnston, J.; Hristopoulos, M.; Clark, R.; Ellerman, D.; Wang, B.-E.; Li, Y.; Mathieu, M.; Li, G.; et al. Antitumor Efficacy of a Bispecific Antibody That Targets HER2 and Activates T Cells. Cancer Res. 2014, 74, 5561–5571. [Google Scholar] [CrossRef] [PubMed]
- Vaishampayan, U.; Thakur, A.; Rathore, R.; Kouttab, N.; Lum, L.G. Phase I Study of Anti-CD3 x Anti-Her2 Bispecific Antibody in Metastatic Castrate Resistant Prostate Cancer Patients. Prostate Cancer 2015, 2015, e285193. [Google Scholar] [CrossRef]
- Lopez-Albaitero, A.; Xu, H.; Guo, H.; Wang, L.; Wu, Z.; Tran, H.; Chandarlapaty, S.; Scaltriti, M.; Janjigian, Y.; de Stanchina, E.; et al. Overcoming Resistance to HER2-Targeted Therapy with a Novel HER2/CD3 Bispecific Antibody. OncoImmunology 2017, 6, e1267891. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Zhang, J.; Yan, Y.; Yao, X.; Fang, L.; Xiong, H.; Liu, Y.; Chu, Q.; Zhou, P.; Wu, K. A Novel Asymmetrical Anti-HER2/CD3 Bispecific Antibody Exhibits Potent Cytotoxicity for HER2-Positive Tumor Cells. J. Exp. Clin. Cancer Res. 2019, 38, 355. [Google Scholar] [CrossRef] [PubMed]
- Vaishampayan, U.N.; Thakur, A.; Chen, W.; Deol, A.; Patel, M.; Dobson, K.; Dickow, B.; Schalk, D.; Schienschang, A.; Whitaker, S.; et al. Phase II Trial of Pembrolizumab and Anti-CD3 x Anti-HER2 Bispecific Antibody-Armed Activated T Cells in Metastatic Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2023, 29, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Asano, R.; Arai, K.; Shimomura, I.; Ogata, H.; Kawaguchi, H.; Hayashi, H.; Ohtsuka, H.; Yoshida, H.; Katayose, Y.; et al. In Vitro and in Vivo Antitumor Effects of Recombinant Bispecific Antibodies Based on Humanized Anti-EGFR Antibody. Oncol. Rep. 2011, 26, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.; He, Q.; Li, W.; Li, X.; Han, H.; Jin, M.; Liu, C.; Tao, H.; Ma, J.; Gao, B. Anti-CD3 x EGFR Bispecific Antibody Redirects Cytokine-Induced Killer Cells to Glioblastoma In Vitro and In Vivo. Oncol. Rep. 2015, 34, 2567–2575. [Google Scholar] [CrossRef]
- Lum, L.G.; Choi, M.; Thakur, A.; Deol, A.; Fields, K.; Tomaszewski, E.; Schalk, D.; Kondadasule, V.; Dyson, G.; Mahaseth, H.; et al. Five Advanced Pancreatic Cancer Patients in a Phase I Study of Anti-CD3 x Anti-EGFR Bispecific Antibody Armed Activated T Cells (BATS). J. ImmunoTherapy Cancer 2015, 3, P55. [Google Scholar] [CrossRef]
- Lum, L.G.; Choi, M.; Le, T.M.; Thakur, A.; Deol, A.; Ballen, K.K.; Volodin, L.; Kindwall-Keller, T.L.; Liu, Q.; Dyson, G.; et al. Targeting Advanced Pancreatic Cancer with Activated t Cells Armed with Anti-CD3 x Anti-EGFR Bispecific Antibody. JCO 2018, 36, 4108. [Google Scholar] [CrossRef]
- Harwood, S.L.; Alvarez-Cienfuegos, A.; Nuñez-Prado, N.; Compte, M.; Hernández-Pérez, S.; Merino, N.; Bonet, J.; Navarro, R.; Van Bergen en Henegouwen, P.M.P.; Lykkemark, S.; et al. ATTACK, a Novel Bispecific T Cell-Recruiting Antibody with Trivalent EGFR Binding and Monovalent CD3 Binding for Cancer Immunotherapy. OncoImmunology 2018, 7, e1377874. [Google Scholar] [CrossRef]
- Lum, L.G.; Le, T.M.; Choi, M.; Thakur, A.; Reilley, M.; Kunk, P.R.; Deol, A.; Ballen, K.K.; Kindwall-Keller, T.L.; Schalk, D.; et al. Clinical and Immune Responses Using Anti-CD3 x Anti-EGFR Bispecific Antibody Armed T Cells (BATs) for Locally Advanced or Metastatic Pancreatic Cancer. JCO 2019, 37, 4135. [Google Scholar] [CrossRef]
- Lum, L.G.; Thakur, A.; Choi, M.; Deol, A.; Kondadasula, V.; Schalk, D.; Fields, K.; Dufrense, M.; Philip, P.; Dyson, G.; et al. Clinical and Immune Responses to Anti-CD3 x Anti-EGFR Bispecific Antibody Armed Activated T Cells (EGFR BATs) in Pancreatic Cancer Patients. OncoImmunology 2020, 9, 1773201. [Google Scholar] [CrossRef] [PubMed]
- Boustany, L.M.; LaPorte, S.L.; Wong, L.; White, C.; Vinod, V.; Shen, J.; Yu, W.; Koditek, D.; Winter, M.B.; Moore, S.J.; et al. A Probody T Cell–Engaging Bispecific Antibody Targeting EGFR and CD3 Inhibits Colon Cancer Growth with Limited Toxicity. Cancer Res. 2022, 82, 4288–4298. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.T.F.; Sharma, V.; Mendelsohn, A.; Wei, Q.; Li, J.; Yu, B.; Larrick, J.W.; Lum, L.G. Broad Reactivity and Enhanced Potency of Recombinant Anti-EGFR × Anti-CD3 Bispecific Antibody-Armed Activated T Cells against Solid Tumours. Ann. Med. 2022, 54, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C.; Selitsky, S.R.; Chai, S.; Armistead, P.M.; Vincent, B.G.; Serody, J.S. Alternative Tumour-Specific Antigens. Nat. Rev. Cancer 2019, 19, 465–478. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Tebentafusp: First Approval. Drugs 2022, 82, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Davies, C.d.L.; Berk, D.A.; Pluen, A.; Jain, R.K. Comparison of IgG Diffusion and Extracellular Matrix Composition in Rhabdomyosarcomas Grown in Mice versus in Vitro as Spheroids Reveals the Role of Host Stromal Cells. Br. J. Cancer 2002, 86, 1639–1644. [Google Scholar] [CrossRef] [PubMed]
- Kuczek, D.E.; Larsen, A.M.H.; Thorseth, M.-L.; Carretta, M.; Kalvisa, A.; Siersbæk, M.S.; Simões, A.M.C.; Roslind, A.; Engelholm, L.H.; Noessner, E.; et al. Collagen Density Regulates the Activity of Tumor-Infiltrating T Cells. J. Immunother. Cancer 2019, 7, 68. [Google Scholar] [CrossRef]
- Chen, W.; Liang, X.; Peterson, A.J.; Munn, D.H.; Blazar, B.R. The Indoleamine 2,3-Dioxygenase Pathway Is Essential for Human Plasmacytoid Dendritic Cell-Induced Adaptive T Regulatory Cell Generation. J. Immunol. 2008, 181, 5396–5404. [Google Scholar] [CrossRef]
- Mezzapelle, R.; Leo, M.; Caprioglio, F.; Colley, L.S.; Lamarca, A.; Sabatino, L.; Colantuoni, V.; Crippa, M.P.; Bianchi, M.E. CXCR4/CXCL12 Activities in the Tumor Microenvironment and Implications for Tumor Immunotherapy. Cancers 2022, 14, 2314. [Google Scholar] [CrossRef]
- Ma, H.; Wang, H.; Sove, R.J.; Jafarnejad, M.; Tsai, C.-H.; Wang, J.; Giragossian, C.; Popel, A.S. A Quantitative Systems Pharmacology Model of T Cell Engager Applied to Solid Tumor. AAPS J. 2020, 22, 85. [Google Scholar] [CrossRef] [PubMed]
- Wright, K.; de Silva, K.; Purdie, A.C.; Plain, K.M. Comparison of Methods for miRNA Isolation and Quantification from Ovine Plasma. Sci. Rep. 2020, 10, 825. [Google Scholar] [CrossRef] [PubMed]
- Weddell, J. Mechanistically Modeling Peripheral Cytokine Dynamics Following Bispecific Dosing in Solid Tumors. CPT Pharmacomet. Syst. Pharmacology 2023, 12, 1726–1737. [Google Scholar] [CrossRef]
- Chen, X.; Oppenheim, J.J. Resolving the Identity Myth: Key Markers of Functional CD4+FoxP3+ Regulatory T Cells. Int. Immunopharmacol. 2011, 11, 1489–1496. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.Q.; Bull, J.M. Hyperthermia Enhances Th1 Differentiation and Downregulates FOX3 Expression in Tregs. J. Allergy Clin. Immunol. 2012, 129, AB10. [Google Scholar] [CrossRef]
- Atanackovic, D.; Nierhaus, A.; Neumeier, M.; Hossfeld, D.; Hegewisch-Becker, S. 41.8 °C Whole Body Hyperthermia as an Adjunct to Chemotherapy Induces Prolonged T Cell Activation in Patients with Various Malignant Diseases. Cancer Immunol. Immunother. 2002, 51, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Mace, T.A.; Zhong, L.; Kokolus, K.M.; Repasky, E.A. Effector CD8+ T Cell IFN-γ Production and Cytotoxicity Are Enhanced by Mild Hyperthermia. Int. J. Hyperth. 2012, 28, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Song, C.W.; Park, H.J.; Lee, C.K.; Griffin, R. Implications of Increased Tumor Blood Flow and Oxygenation Caused by Mild Temperature Hyperthermia in Tumor Treatment. Int. J. Hyperth. 2005, 21, 761–767. [Google Scholar] [CrossRef]
- Gopanenko, A.V.; Kosobokova, E.N.; Kosorukov, V.S. Main Strategies for the Identification of Neoantigens. Cancers 2020, 12, 2879. [Google Scholar] [CrossRef]
- Melincovici, C.S.; Bo, A.B.; Mihu, C.; Istrate, M.; Moldovan, I.-M.; Roman, A.L.; Mihu, C.M. Vascular Endothelial Growth Factor (VEGF)—Key Factor in Normal and Pathological Angiogenesis. Rom. J. Morphol. Embryol. 2018, 59, 455–467. [Google Scholar]
- Pandey, A.K.; Singhi, E.K.; Arroyo, J.P.; Ikizler, T.A.; Gould, E.R.; Brown, J.; Beckman, J.A.; Harrison, D.G.; Moslehi, J. Mechanisms of VEGF (Vascular Endothelial Growth Factor) Inhibitor–Associated Hypertension and Vascular Disease. Hypertension 2018, 71, e1–e8. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Shibuya, M. The Vascular Endothelial Growth Factor (VEGF)/VEGF Receptor System and Its Role under Physiological and Pathological Conditions. Clin. Sci. 2005, 109, 227–241. [Google Scholar] [CrossRef] [PubMed]
- Thomas, K.A. Vascular Endothelial Growth Factor Receptors. Encycl. Biol. Chem. 2013, 509–514. [Google Scholar] [CrossRef]
- Olsson, A.-K.; Dimberg, A.; Kreuger, J.; Claesson-Welsh, L. VEGF Receptor Signalling? In Control of Vascular Function. Nat. Rev. Mol. Cell Biol. 2006, 7, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Nieves, B.J.; D’Amore, P.A.; Bryan, B.A. The Function of Vascular Endothelial Growth Factor. Biofactors 2009, 35, 332–337. [Google Scholar] [CrossRef]
- Shibuya, M.; Claesson-Welsh, L. Signal Transduction by VEGF Receptors in Regulation of Angiogenesis and Lymphangiogenesis. Exp. Cell Res. 2006, 312, 549–560. [Google Scholar] [CrossRef]
- Gabrilovich, D.; Ishida, T.; Oyama, T.; Ran, S.; Kravtsov, V.; Nadaf, S.; Carbone, D.P. Vascular Endothelial Growth Factor Inhibits the Development of Dendritic Cells and Dramatically Affects the Differentiation of Multiple Hematopoietic Lineages In Vivo: Presented in Part at the Keystone Symposium “Cellular and Molecular Biology of Dendritic Cells,” Santa Fe, NM, March 3-9, 1998, and at the Annual Meeting of the American Association for Cancer Research, March 28–April 1, 1998. Blood 1998, 92, 4150–4166. [Google Scholar] [CrossRef]
- Carmeliet, P. VEGF as a Key Mediator of Angiogenesis in Cancer. Oncology 2005, 69, 4–10. [Google Scholar] [CrossRef]
- Kerbel, R.S. Tumor Angiogenesis. N. Engl. J. Med. 2008, 358, 2039–2049. [Google Scholar] [CrossRef]
- Carmeliet, P.; Ferreira, V.; Breier, G.; Pollefeyt, S.; Kieckens, L.; Gertsenstein, M.; Fahrig, M.; Vandenhoeck, A.; Harpal, K.; Eberhardt, C.; et al. Abnormal Blood Vessel Development and Lethality in Embryos Lacking a Single VEGF Allele. Nature 1996, 380, 435–439. [Google Scholar] [CrossRef]
- Ferrara, N.; Carver-Moore, K.; Chen, H.; Dowd, M.; Lu, L.; O’Shea, K.S.; Powell-Braxton, L.; Hillan, K.J.; Moore, M.W. Heterozygous Embryonic Lethality Induced by Targeted Inactivation of the VEGF Gene. Nature 1996, 380, 439–442. [Google Scholar] [CrossRef] [PubMed]
- Shalaby, F.; Rossant, J.; Yamaguchi, T.P.; Gertsenstein, M.; Wu, X.-F.; Breitman, M.L.; Schuh, A.C. Failure of Blood-Island Formation and Vasculogenesis in Flk-1-Deficient Mice. Nature 1995, 376, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Molecular Regulation of Vessel Maturation. Nat. Med. 2003, 9, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Nagy, J.A.; Chang, S.-H.; Dvorak, A.M.; Dvorak, H.F. Why Are Tumour Blood Vessels Abnormal and Why Is It Important to Know? Br. J. Cancer 2009, 100, 865–869. [Google Scholar] [CrossRef] [PubMed]
- Shweiki, D.; Itin, A.; Soffer, D.; Keshet, E. Vascular Endothelial Growth Factor Induced by Hypoxia May Mediate Hypoxia-Initiated Angiogenesis. Nature 1992, 359, 843–845. [Google Scholar] [CrossRef] [PubMed]
- Tanimoto, K.; Makino, Y.; Pereira, T.; Poellinger, L. Mechanism of Regulation of the Hypoxia-Inducible Factor-1α by the von Hippel-Lindau Tumor Suppressor Protein. EMBO J. 2000, 19, 4298–4309. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Davis-Smyth, T. The Biology of Vascular Endothelial Growth Factor. Endocr. Rev. 1997, 18, 4–25. [Google Scholar] [CrossRef]
- Joo, Y.E.; Sohn, Y.H.; Lee, W.S.; Park, C.H.; Choi, S.K.; Rew, J.S.; Park, C.S.; Kim, S.J. Expression of Vascular Endothelial Growth Factor and P53 in Pancreatic Carcinomas. Korean J. Intern. Med. 2002, 17, 153–159. [Google Scholar] [CrossRef]
- Masood, R.; Cai, J.; Zheng, T.; Smith, D.L.; Hinton, D.R.; Gill, P.S. Vascular Endothelial Growth Factor (VEGF) Is an Autocrine Growth Factor for VEGF Receptor–Positive Human Tumors. Blood 2001, 98, 1904–1913. [Google Scholar] [CrossRef]
- Meng, L.; Fuhao, Z.; Xiaoming, Z.; Yuxiu, Z.; Zhaojun, D.; Bingcheng, L.; Meiling, X.; Shuyan, S. Survivin Is Critically Involved in VEGFR2 Signaling-Mediated Esophageal Cancer Cell Survival. Biomed. Pharmacother. 2018, 107, 139–145. [Google Scholar] [CrossRef]
- Pepper, M.S. Role of the Matrix Metalloproteinase and Plasminogen Activator–Plasmin Systems in Angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1104–1117. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, Y.; Wang, Y.; Lin, C.; Zhang, D.; Chen, J.; Ouyang, L.; Wu, F.; Zhang, J.; Chen, L. Recent Progress on Vascular Endothelial Growth Factor Receptor Inhibitors with Dual Targeting Capabilities for Tumor Therapy. J. Hematol. Oncol. 2022, 15, 89. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Hu, C.; Hui, K.; Jiang, X. The Role and Significance of VEGFR2+ Regulatory T Cells in Tumor Immunity. OTT 2017, 10, 4315–4319. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.J.; Li, B.; Winer, J.; Armanini, M.; Gillett, N.; Phillips, H.S.; Ferrara, N. Inhibition of Vascular Endothelial Growth Factor-Induced Angiogenesis Suppresses Tumour Growth in Vivo. Nature 1993, 362, 841–844. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus Irinotecan, Fluorouracil, and Leucovorin for Metastatic Colorectal Cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef]
- Margolin, K.; Gordon, M.S.; Holmgren, E.; Gaudreault, J.; Novotny, W.; Fyfe, G.; Adelman, D.; Stalter, S.; Breed, J. Phase Ib Trial of Intravenous Recombinant Humanized Monoclonal Antibody to Vascular Endothelial Growth Factor in Combination With Chemotherapy in Patients With Advanced Cancer: Pharmacologic and Long-Term Safety Data. J. Clin. Oncol. 2016, 19, 3. [Google Scholar] [CrossRef]
- Sweeney, C.J.; Miller, K.D.; Sissons, S.E.; Nozaki, S.; Heilman, D.K.; Shen, J.; Sledge, G.W. The Antiangiogenic Property of Docetaxel Is Synergistic with a Recombinant Humanized Monoclonal Antibody against Vascular Endothelial Growth Factor or 2-Methoxyestradiol but Antagonized by Endothelial Growth Factors. Cancer Res. 2001, 61, 3369–3372. [Google Scholar]
- Gerber, H.-P.; Ferrara, N. Pharmacology and Pharmacodynamics of Bevacizumab as Monotherapy or in Combination with Cytotoxic Therapy in Preclinical Studies. Cancer Res. 2005, 65, 671–680. [Google Scholar] [CrossRef]
- Jain, R.K. Normalization of Tumor Vasculature: An Emerging Concept in Antiangiogenic Therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef]
- Willett, C.G.; Boucher, Y.; di Tomaso, E.; Duda, D.G.; Munn, L.L.; Tong, R.T.; Chung, D.C.; Sahani, D.V.; Kalva, S.P.; Kozin, S.V.; et al. Direct Evidence That the VEGF-Specific Antibody Bevacizumab Has Antivascular Effects in Human Rectal Cancer. Nat. Med. 2004, 10, 145–147. [Google Scholar] [CrossRef]
- Ferrara, N.; Adamis, A.P. Ten Years of Anti-Vascular Endothelial Growth Factor Therapy. Nat. Rev. Drug Discov. 2016, 15, 385–403. [Google Scholar] [CrossRef] [PubMed]
- Strawn, L.M.; McMahon, G.; App, H.; Schreck, R.; Kuchler, W.R.; Longhi, M.P.; Hui, T.H.; Tang, C.; Levitzki, A.; Gazit, A.; et al. Flk-1 as a Target for Tumor Growth Inhibition. Cancer Res. 1996, 56, 3540–3545. [Google Scholar] [PubMed]
- Levitzki, A.; Mishani, E. Tyrphostins and Other Tyrosine Kinase Inhibitors. Annu. Rev. Biochem. 2006, 75, 93–109. [Google Scholar] [CrossRef] [PubMed]
- McTigue, M.A.; Wickersham, J.A.; Pinko, C.; Showalter, R.E.; Parast, C.V.; Tempczyk-Russell, A.; Gehring, M.R.; Mroczkowski, B.; Kan, C.-C.; Villafranca, J.E.; et al. Crystal Structure of the Kinase Domain of Human Vascular Endothelial Growth Factor Receptor 2: A Key Enzyme in Angiogenesis. Structure 1999, 7, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Krupitskaya, Y.; Wakelee, H.A. Ramucirumab, a Fully Human mAb to the Transmembrane Signaling Tyrosine Kinase VEGFR-2 for the Potential Treatment of Cancer. Curr. Opin. Investig. Drugs 2009, 10, 597–605. [Google Scholar] [PubMed]
- Dellian, M.; Witwer, B.P.; Salehi, H.A.; Yuan, F.; Jain, R.K. Quantitation and Physiological Characterization of Angiogenic Vessels in Mice: Effect of Basic Fibroblast Growth Factor, Vascular Endothelial Growth Factor/Vascular Permeability Factor, and Host Microenvironment. Am. J. Pathol. 1996, 149, 59–71. [Google Scholar]
- Itatani, Y.; Kawada, K.; Yamamoto, T.; Sakai, Y. Resistance to Anti-Angiogenic Therapy in Cancer—Alterations to Anti-VEGF Pathway. Int. J. Mol. Sci. 2018, 19, 1232. [Google Scholar] [CrossRef]
- Haibe, Y.; Kreidieh, M.; El Hajj, H.; Khalifeh, I.; Mukherji, D.; Temraz, S.; Shamseddine, A. Resistance Mechanisms to Anti-Angiogenic Therapies in Cancer. Front. Oncol. 2020, 10, 221. [Google Scholar] [CrossRef]
- Nie, W.; Ma, X.; Sang, Y.; Li, Y.; Gao, X.; Xu, G.; Shen, G.; Shi, H.; Liu, X.; Wang, F.; et al. Synergic Antitumor Effect of SKLB1002 and Local Hyperthermia in 4T1 and CT26. Clin. Exp. Med. 2014, 14, 203–213. [Google Scholar] [CrossRef]
- Kanamori, S.; Nishimura, Y.; Okuno, Y. Induction of Vascular Endothelial Growth Factor (VEGF) by Hyperthermia and/or an Angiogenesis Inhibitor. Int. J. Hyperth. 1999, 15, 267–278. [Google Scholar] [CrossRef]
- Nishimura, Y.; Murata, R.; Hiraoka, M. Combined Effects of an Angiogenesis Inhibitor (TNP-470) and Hyperthermia. Br. J. Cancer 1996, 73, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Roca, C.; Primo, L.; Valdembri, D.; Cividalli, A.; Declerck, P.; Carmeliet, P.; Gabriele, P.; Bussolino, F. Hyperthermia Inhibits Angiogenesis by a Plasminogen Activator Inhibitor 1-Dependent Mechanism. Cancer Res. 2003, 63, 1500–1507. [Google Scholar] [PubMed]
- Moon, E.J.; Brizel, D.M.; Chi, J.-T.A.; Dewhirst, M.W. The Potential Role of Intrinsic Hypoxia Markers as Prognostic Variables in Cancer. Antioxid. Redox Signal 2007, 9, 1237–1294. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Company | Number of Compounds Related to Solid Tumors | Trial Stage per Compound | |||
|---|---|---|---|---|---|
| I | II | III | IV | ||
| AbbVie (North Chicago, IL, USA) | 16 | 13 | 1 | 2 | |
| Amgen (Thousand Oaks, CA, USA) | 19 | 13 | 3 | 3 | |
| AstraZeneca (Cambridge, UK) | 34 | 23 | 10 | 9 | 2 |
| Bayer (Leverkusen, Germany) | 10 | 8 | 2 | ||
| Bristol Myers Squibb (BMS) (New Brunswick, NJ, USA) | 41 | 16 | 9 | 16 | |
| GlaxoSmithKline (GSK) (London, UK) | 17 | 8 | 4 | 5 | |
| Ipsen (Paris, France) | 3 | 1 | 2 | ||
| Johnson & Johnson (New Brunswick, NJ, USA) | 12 | 6 | 4 | 2 | |
| Merck (Branchburg, NJ, USA) | 13 | 13 | |||
| Merck Sharp & Dohme (MSD) (Rahway, NJ, USA) | 27 | 16 | 8 | 3 | |
| Novartis (Basel, Switzerland) | 28 | 13 | 6 | 9 | |
| Pfizer (New York City, NY, USA) | 31 | 18 | 5 | 8 | |
| Roche (Basel, Switzerland) | 75 | 32 | 5 | 26 | 12 |
| Sanofi (Paris, France) | 18 | 5 | 7 | 3 | 2 |
| Servier (Suresnes, France) | 8 | 6 | 2 | ||
| Drug | FDA * Approval Year | EMA * Approval Year | Cancer Type |
|---|---|---|---|
| Axitinib | 2012 | 2012 | Renal cell carcinoma |
| Bevacizumab (Avastin) | 2004 | 2005 | Colorectal cancer, lung cancer, breast cancer, renal cancers, brain cancers, ovarian cancer, cervical cancer |
| Cabozantinib | 2012 | 2016 | Medullary and differentiated thyroid carcinoma, renal cell carcinoma, hepatocellular carcinoma |
| Everolimus | 2009 | 2009 | Advanced kidney cancer, progressive or metastatic pancreatic neuroendocrine tumors, breast cancer |
| Lenalidomide | 2005 | 2008 | Multiple myeloma, mantle cell lymphoma |
| Lenvatinib mesylate | 2015 | 2015 | Thyroid cancer, renal cell carcinoma, hepatocellular carcinoma |
| Pazopanib | 2009 | 2009 | Clear cell renal carcinoma |
| Ramucirumab | 2014 | 2014 | Gastric cancer, metastatic non-small lung carcinoma, metastatic colorectal cancer, hepatocellular carcinoma |
| Regorafenib | 2012 | 2013 | Metastatic colorectal cancer, advanced gastrointestinal stromal tumors, advanced hepatocellular carcinoma |
| Sorafenib | 2005 | 2006 | Kidney cancer, liver cancer, thyroid cancer |
| Sunitinib | 2006 | 2007 | Renal cell carcinoma, pancreatic neuroendocrine tumors, gastrointestinal stromal tumors |
| Thalidomide | 2006 | 2009 | Multiple myeloma |
| Vandetanib | 2011 | 2012 | Medullary thyroid cancer |
| Ziv-aflibercept | 2012 | 2013 | Metastatic colorectal cancer |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Logghe, T.; van Zwol, E.; Immordino, B.; Van den Cruys, K.; Peeters, M.; Giovannetti, E.; Bogers, J. Hyperthermia in Combination with Emerging Targeted and Immunotherapies as a New Approach in Cancer Treatment. Cancers 2024, 16, 505. https://doi.org/10.3390/cancers16030505
Logghe T, van Zwol E, Immordino B, Van den Cruys K, Peeters M, Giovannetti E, Bogers J. Hyperthermia in Combination with Emerging Targeted and Immunotherapies as a New Approach in Cancer Treatment. Cancers. 2024; 16(3):505. https://doi.org/10.3390/cancers16030505
Chicago/Turabian StyleLogghe, Tine, Eke van Zwol, Benoît Immordino, Kris Van den Cruys, Marc Peeters, Elisa Giovannetti, and Johannes Bogers. 2024. "Hyperthermia in Combination with Emerging Targeted and Immunotherapies as a New Approach in Cancer Treatment" Cancers 16, no. 3: 505. https://doi.org/10.3390/cancers16030505
APA StyleLogghe, T., van Zwol, E., Immordino, B., Van den Cruys, K., Peeters, M., Giovannetti, E., & Bogers, J. (2024). Hyperthermia in Combination with Emerging Targeted and Immunotherapies as a New Approach in Cancer Treatment. Cancers, 16(3), 505. https://doi.org/10.3390/cancers16030505