

Relationship between the Expression of CHK2 and p53 in Tumor Tissue and the Course of Papillary Thyroid Cancer in Patients with CHEK2 Germline Mutations

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
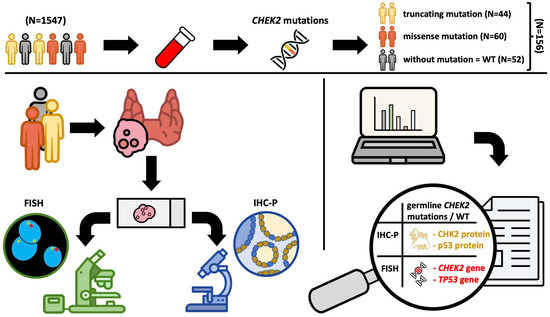
2. Materials and Methods
2.1. Patients
2.2. Description of the Test Procedure
2.3. Detection of CHEK2 Mutation
2.4. Fluorescence In Situ Hybridization (FISH)
2.5. Immunohistochemistry (IHC)
IHC Staining
2.6. Analysis of Immunohistochemical Results
2.7. Statistical Analyses
3. Results
3.1. Baseline Characteristics
3.1.1. CHK2 and p53 Expression: Impact on Clinicopathological Features, Treatment Response and Outcomes in PTC Patients with and without CHEK2 Mutations
3.1.2. Impact of CHEK2 and TP53 Gene Status, and p53 Expression on Clinicopathological Features, Treatment Response and Outcomes in PTC Patients with and without CHEK2 Mutations
3.1.3. Prognostically Favorable and Predictive Results Defined as Categories of Interest in Groups of PTC Patients with and without CHEK2 Mutations
3.1.4. Predictive Value of Clinicopathological Features, Treatment Response, and Outcome Correlations with CHK2, TP53 Status, and p53 Expression in PTC Patients with and without CHEK2 Mutations
3.1.5. Posterior Distribution of Odds Log Ratio for Clinicopathological Correlations with CHK2, TP53 Status, and p53 Expression in PTC Patients with and without CHEK2 Mutations
3.1.6. Sequential Analysis of Clinicopathological Correlations with CHK2, TP53 Status, and p53 Expression in PTC Tumors with and without CHEK2 Mutations
3.1.7. Correlation of Germline CHEK2 Mutation with p53 and CHK2 Expression, and CHEK2 and TP53 Gene Statuses in PTC Patients with and without CHEK2 Mutations
3.1.8. The Incidence of Unfavorable Prognostic Factors Linked to CHEK2 and TP53 Gene Alterations, CHK2 and p53 Expression in PTC Patients with and without CHEK2 Mutations
3.1.9. Predictive Value of CHEK2 and TP53 Gene Status, CHK2 and p53 Expression in PTC Patients with WT and/or I157T CHEK2 Mutations
3.1.10. Posterior Odds Log Ratio Distribution for CHEK2 and TP53 Gene Status, CHK2 and p53 Expression in PTC Patients with and without CHEK2 Mutations
3.1.11. Sequential Analysis of Associations Involving CHEK2 and TP53 Gene Status, CHK2 and p53 Expression in PTC Patients with and without CHEK2 Mutations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Correction Statement
References
- Davies, L.; Welch, H.G. Increasing incidence of thyroid cancer in the United States, 1973–2002. JAMA 2006, 295, 2164–2167. [Google Scholar] [CrossRef]
- Davies, L.; Welch, H.G. Current thyroid cancer trends in the United States. JAMA Otolaryngol. Head Neck Surg. 2014, 140, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Ahn, H.S.; Kim, H.J.; Welch, H.G. Korea’s thyroid-cancer “epidemic”—Screening and overdiagnosis. N. Engl. J. Med. 2014, 371, 1765–1767. [Google Scholar] [CrossRef] [PubMed]
- Carling, T.; Udelsman, R. Thyroid cancer. Annu. Rev. Med. 2014, 65, 125–137. [Google Scholar] [CrossRef]
- Cady, B.; Rossi, R. An expanded view of risk-group definition in differentiated thyroid carcinoma. Surgery 1988, 104, 947–953. [Google Scholar]
- Bartek, J.; Falck, J.; Lukas, J. CHK2 kinase—A busy messenger. Nat. Rev. Mol. Cell. Biol. 2001, 2, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Urist, M.; Prives, C. The Chk2 protein kinase. DNA Repair 2004, 3, 1039–1047. [Google Scholar] [CrossRef]
- Bahassi, E.M.; Ovesen, J.L.; Riesenberg, A.L.; Bernstein, W.Z.; E Hasty, P.; Stambrook, P.J. The checkpoint kinases Chk1 and Chk2 regulate the functional associations between hBRCA2 and Rad51 in response to DNA damage. Oncogene 2008, 27, 3977–3985. [Google Scholar] [CrossRef]
- Kauffmann, A.; Rosselli, F.; Lazar, V.; Winnepenninckx, V.; Mansuet-Lupo, A.; Dessen, P.; Oord, J.J.v.D.; Spatz, A.; Sarasin, A. High expression of DNA repair pathways is associated with metastasis in melanoma patients. Oncogene 2007, 27, 565–573. [Google Scholar] [CrossRef]
- Zhao, W.; Chen, S.; Hou, X.; Chen, G.; Zhao, Y. CHK2 Promotes Anoikis and is Associated with the Progression of Papillary Thyroid Cancer. Cell. Physiol. Biochem. 2018, 45, 1590–1602. [Google Scholar] [CrossRef]
- Dai, B.; Zhao, X.F.; Mazan-Mamczarz, K.; Hagner, P.; Corl, S.; Bahassi, E.M.; Lu, S.; Stambrook, P.J.; Shapiro, P.; Gartenhaus, R.B. Functional and molecular interactions between ERK and CHK2 in diffuse large B-cell lymphoma. Nat. Commun. 2011, 2, 402. [Google Scholar] [CrossRef]
- Bartkova, J.; Guldberg, P.; Grønbæk, K.; Koed, K.; Primdahl, H.; Møller, K.; Lukas, J.; Ørntoft, T.F.; Bartek, J. Aberrations of the Chk2 tumour suppressor in advanced urinary bladder cancer. Oncogene 2004, 23, 8545–8551. [Google Scholar] [CrossRef]
- Davidson, B.; Bjørnerem, M.; Holth, A.; Hellesylt, E.; Falkenthal, T.E.H.; Flørenes, V.A. Expression, activation and clinical relevance of CHK1 and CHK2 in metastatic high-grade serous carcinoma. Gynecol. Oncol. 2018, 150, 136–142. [Google Scholar] [CrossRef]
- Barnoud, T.; Parris, J.L.D.; Murphy, M.E. Common genetic variants in the TP53 pathway and their impact on cancer. J. Mol. Cell. Biol. 2019, 11, 578–585. [Google Scholar] [CrossRef]
- Bell, D.W.; Varley, J.M.; Szydlo, T.E.; Kang, D.H.; Wahrer, D.C.R.; Shannon, K.E.; Lubratovich, M.; Verselis, S.J.; Isselbacher, K.J.; Fraumeni, J.F.; et al. Heterozygous Germ Line hCHK2 Mutations in Li-Fraumeni Syndrome. Science 1999, 286, 2528–2531. [Google Scholar] [CrossRef]
- Soussi, T. The p53 tumor suppressor gene: From molecular biology to clinical investigation. Ann. N. Y. Acad. Sci. 2000, 910, 121–137, discussion 137–139. [Google Scholar] [CrossRef]
- Levine, A.J. p53, the cellular gatekeeper for growth and division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef]
- Levine, A.J.; Momand, J.; Finlay, C.A. The p53 tumour suppressor gene. Nature 1991, 351, 453–456. [Google Scholar] [CrossRef] [PubMed]
- el-Deiry, W.S.; Harper, J.W.; O’Connor, P.M.; Velculescu, V.E.; Canman, C.E.; Jackman, J.; Pietenpol, J.A.; Burrell, M.; Hill, D.E.; Wang, Y.; et al. WAF1/CIP1 is induced in p53-mediated G1 arrest and apoptosis. Cancer Res. 1994, 54, 1169–1174. [Google Scholar] [PubMed]
- Siolek, M.; Cybulski, C.; Gąsior-Perczak, D.; Kowalik, A.; Kozak-Klonowska, B.; Kowalska, A.; Chłopek, M.; Kluźniak, W.; Wokołorczyk, D.; Pałyga, I.; et al. CHEK2 mutations and the risk of papillary thyroid cancer. Int. J. Cancer 2015, 137, 548–552. [Google Scholar] [CrossRef] [PubMed]
- Gasior-Perczak, D.; Kowalik, A.; Gruszczyński, K.; Walczyk, A.; Siołek, M.; Pałyga, I.; Trepka, S.; Mikina, E.; Trybek, T.; Kopczyński, J.; et al. Incidence of the CHEK2 Germline Mutation and Its Impact on Clinicopathological Features, Treatment Responses, and Disease Course in Patients with Papillary Thyroid Carcinoma. Cancers 2021, 13, 470. [Google Scholar] [CrossRef]
- Kaczmarek-Ryś, M.; Ziemnicka, K.; Hryhorowicz, S.T.; Górczak, K.; Hoppe-Gołębiewska, J.; Skrzypczak-Zielińska, M.; Tomys, M.; Gołąb, M.; Szkudlarek, M.; Budny, B.; et al. The c.470 T > C CHEK2 missense variant increases the risk of differentiated thyroid carcinoma in the Great Poland population. Hered. Cancer Clin. Pract. 2015, 13, 8. [Google Scholar] [CrossRef] [PubMed]
- Wójcicka, A.; Czetwertyńska, M.; Świerniak, M.; Długosińska, J.; Maciąg, M.; Czajka, A.; Dymecka, K.; Kubiak, A.; Kot, A.; Płoski, R.; et al. Variants in the ATM-CHEK2-BRCA1 axis determine genetic predisposition and clinical presentation of papillary thyroid carcinoma. Genes Chromosom. Cancer 2014, 53, 516–523. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Integrated genomic characterization of papillary thyroid carcinoma. Cell 2014, 159, 676–690. [Google Scholar] [CrossRef]
- R Core Team. (2020). R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing. Available online: https://www.R-project.org/ (accessed on 30 January 2024).
- Gąsior-Perczak, D.; Kowalik, A.; Walczyk, A.; Siołek, M.; Gruszczyński, K.; Pałyga, I.; Mikina, E.; Trybek, T.; Kopczyński, J.; Mężyk, R.; et al. Coexisting Germline CHEK2 and Somatic BRAF(V600E) Mutations in Papillary Thyroid Cancer and Their Association with Clinicopathological Features and Disease Course. Cancers 2019, 11, 1744. [Google Scholar] [CrossRef]
- Haugen, B.R.; Alexander, E.K.; Bible, K.C.; Doherty, G.M.; Mandel, S.J.; Nikiforov, Y.E.; Pacini, F.; Randolph, G.W.; Sawka, A.M.; Schlumberger, M.; et al. 2015 American Thyroid Association Management Guidelines for Adult Patients with Thyroid Nodules and Differentiated Thyroid Cancer: The American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid 2016, 26, 1–133. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, M.L.; Morris, L.F.; Haugen, B.; Shah, J.; Sosa, J.A.; Rohren, E.; Subramaniam, R.M.; Hunt, J.L.; Perrier, N.D. AJCC Cancer Staging Manual; Springer: New York, NY, USA, 2017; Volume 8, pp. 1–19. [Google Scholar]
- Gąsior-Perczak, D.; Pałyga, I.; Szymonek, M.; Kowalik, A.; Walczyk, A.; Kopczyński, J.; Lizis-Kolus, K.; Trybek, T.; Mikina, E.; Szyska-Skrobot, D.; et al. The impact of BMI on clinical progress, response to treatment, and disease course in patients with differentiated thyroid cancer. PLOS ONE 2018, 13, e0204668. [Google Scholar] [CrossRef]
- Tuttle, R.M.; Tala, H.; Shah, J.; Leboeuf, R.; Ghossein, R.; Gonen, M.; Brokhin, M.; Omry, G.; Fagin, J.A.; Shaha, A. Estimating Risk of Recurrence in Differentiated Thyroid Cancer After Total Thyroidectomy and Radioactive Iodine Remnant Ablation: Using Response to Therapy Variables to Modify the Initial Risk Estimates Predicted by the New American Thyroid Association Staging System. Thyroid® 2010, 20, 1341–1349. [Google Scholar] [CrossRef]
- Momesso, D.P.; Vaisman, F.; Yang, S.P.; Bulzico, D.A.; Corbo, R.; Vaisman, M.; Tuttle, R.M. Dynamic Risk Stratification in Patients with Differentiated Thyroid Cancer Treated Without Radioactive Iodine. J. Clin. Endocrinol. Metab. 2016, 101, 2692–2700. [Google Scholar] [CrossRef]
- Kowalska, A.; Walczyk, A.; Pałyga, I.; Gąsior-Perczak, D.; Gadawska-Juszczyk, K.; Szymonek, M.; Trybek, T.; Lizis-Kolus, K.; Szyska-Skrobot, D.; Mikina, E.; et al. The Delayed Risk Stratification System in the Risk of Differentiated Thyroid Cancer Recurrence. PLOS ONE 2016, 11, e0153242. [Google Scholar] [CrossRef]
- Gąsior-Perczak, D.; Pałyga, I.; Szymonek, M.; Kowalik, A.; Walczyk, A.; Kopczyński, J.; Lizis-Kolus, K.; Słuszniak, A.; Słuszniak, J.; Łopatyński, T.; et al. Delayed risk stratification system in pT1aN0/Nx DTC patients treated without radioactive iodine. Endocr. Connect. 2017, 6, 522–527. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Bai, J.; Hao, X.; Zhang, S.; Hu, Y.; Zhang, X.; Yuan, W.; Hu, L.; Cheng, T.; Zetterberg, A.; et al. Multi-gene fluorescence in situ hybridization to detect cell cycle gene copy number aberrations in young breast cancer patients. Cell Cycle 2014, 13, 1299–1305. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.D.; Wagenmakers, E.J. Bayesian Cognitive Modeling: A Practical Course; Cambridge University Press: Cambridge, UK, 2013. [Google Scholar]
- Stolarova, L.; Kleiblova, P.; Janatova, M.; Soukupova, J.; Zemankova, P.; Macurek, L.; Kleibl, Z. CHEK2 Germline Variants in Cancer Predisposition: Stalemate Rather than Checkmate. Cells 2020, 9, 2675. [Google Scholar] [CrossRef] [PubMed]
- Ertych, N.; Stolz, A.; Valerius, O.; Braus, G.H.; Bastians, H. CHK2 – BRCA1 tumor-suppressor axis restrains oncogenic Aurora-A kinase to ensure proper mitotic microtubule assembly. Proc. Natl. Acad. Sci. USA 2016, 113, 1817–1822. [Google Scholar] [CrossRef] [PubMed]
- Cybulski, C.; Górski, B.; Huzarski, T.; Masojć, B.; Mierzejewski, M.; Dębniak, T.; Teodorczyk, U.; Byrski, T.; Gronwald, J.; Matyjasik, J.; et al. CHEK2 Is a Multiorgan Cancer Susceptibility Gene. Am. J. Hum. Genet. 2004, 75, 1131–1135. [Google Scholar] [CrossRef] [PubMed]
- Cybulski, C.; Masojć, B.; Oszutowska, D.; Jaworowska, E.; Grodzki, T.; Waloszczyk, P.; Serwatowski, P.; Pankowski, J.; Huzarski, T.; Byrski, T.; et al. Constitutional CHEK2 mutations are associated with a decreased risk of lung and laryngeal cancers. Carcinog. 2008, 29, 762–765. [Google Scholar] [CrossRef]
- Brennan, P.; McKay, J.; Moore, L.; Zaridze, D.; Mukeria, A.; Szeszenia-Dabrowska, N.; Lissowska, J.; Rudnai, P.; Fabianova, E.; Mates, D.; et al. Uncommon CHEK2 mis-sense variant and reduced risk of tobacco-related cancers: Case–control study. Hum. Mol. Genet. 2007, 16, 1794–1801. [Google Scholar] [CrossRef]
- Szymanska-Pasternak, J.; Szymanska, A.; Medrek, K.; Imyanitov, E.; Cybulski, C.; Gorski, B.; Magnowski, P.; Dziuba, I.; Gugala, K.; Debniak, B.; et al. CHEK2 variants predispose to benign, borderline and low-grade invasive ovarian tumors. Gynecol. Oncol. 2006, 102, 429–431. [Google Scholar] [CrossRef]
- Wang, Y.; McKay, J.D.; Rafnar, T.; Wang, Z.; Timofeeva, M.N.; Broderick, P.; Zong, X.; Laplana, M.; Wei, Y.; Han, Y.; et al. Rare variants of large effect in BRCA2 and CHEK2 affect risk of lung cancer. Nat. Genet. 2014, 46, 736–741. [Google Scholar] [CrossRef]
- Rudd, M.F.; Sellick, G.S.; Webb, E.L.; Catovsky, D.; Houlston, R.S. Variants in the ATM-BRCA2-CHEK2 axis predispose to chronic lymphocytic leukemia. Blood 2006, 108, 638–644. [Google Scholar] [CrossRef]
- Złowocka, E.; Cybulski, C.; Górski, B.; Dębniak, T.; Słojewski, M.; Wokołorczyk, D.; Serrano-Fernández, P.; Matyjasik, J.; van de Wetering, T.; Sikorski, A.; et al. Germline mutations in the CHEK2 kinase gene are associated with an increased risk of bladder cancer. Int. J. Cancer 2007, 122, 583–586. [Google Scholar] [CrossRef] [PubMed]
- Teodorczyk, U.; Cybulski, C.; Wokołorczyk, D.; Jakubowska, A.; Starzyńska, T.; Ławniczak, M.; Domagała, P.; Ferenc, K.; Marlicz, K.; Banaszkiewicz, Z.; et al. The risk of gastric cancer in carriers of CHEK2 mutations. Fam. Cancer 2013, 12, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Havranek, O.; Spacek, M.; Hubacek, P.; Mocikova, H.; Markova, J.; Trneny, M.; Kleibl, Z. Alterations of CHEK2 forkhead-associated domain increase the risk of Hodgkin lymphoma. Neoplasma 2011, 58, 392–395. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Fatah, T.M.; Arora, A.; Alsubhi, N.; Agarwal, D.; Moseley, P.M.; Perry, C.; Doherty, R.; Chan, S.Y.; Green, A.R.; Rakha, E.; et al. Clinicopathological Significance of ATM-Chk2 Expression in Sporadic Breast Cancers: A Comprehensive Analysis in Large Cohorts. Neoplasia 2014, 16, 982–991. [Google Scholar] [CrossRef] [PubMed]
- Eichenauer, T.; Federlein, F.; Möller, K.; Chirico, V.; Kind, S.; Lennartz, M.; Lutz, F.; Hube-Magg, C.; Höflmayer, D.; Fisch, M.; et al. High CHK2 protein expression is a strong and independent prognostic feature in ERG negative prostate cancer. Pathology 2020, 52, 421–430. [Google Scholar] [CrossRef]
- Feng, Z.; Hu, W.; Teresky, A.K.; Hernando, E.; Cordon-Cardo, C.; Levine, A.J. Declining p53 function in the aging process: A possible mechanism for the increased tumor incidence in older populations. Proc. Natl. Acad. Sci. USA 2007, 104, 16633–16638. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, N.A. Prognostic Parameters in Differentiated Thyroid Carcinomas. Surg. Pathol. Clin. 2019, 12, 883–900. [Google Scholar] [CrossRef]
- Nikiforova, M.N.; Wald, A.I.; Roy, S.; Durso, M.B.; Nikiforov, Y.E. Targeted Next-Generation Sequencing Panel (ThyroSeq) for Detection of Mutations in Thyroid Cancer. J. Clin. Endocrinol. Metab. 2013, 98, E1852–E1860. [Google Scholar] [CrossRef]
- Hsiao, S.J.; Nikiforov, Y.E. Molecular approaches to thyroid cancer diagnosis. Endocr. Relat. Cancer 2014, 21, T301–T313. [Google Scholar] [CrossRef]
- Zhao, Y.; Yu, T.; Chen, L.; Xie, D.; Wang, F.; Fu, L.; Cheng, C.; Li, Y.; Zhu, X.; Miao, G. A Germline CHEK2 Mutation in a Family with Papillary Thyroid Cancer. Thyroid® 2020, 30, 924–930. [Google Scholar] [CrossRef] [PubMed]
- García-Limones, C.; Lara-Chica, M.; Jiménez-Jiménez, C.; Pérez, M.; Moreno, P.; Muñoz, E.; A Calzado, M. CHK2 stability is regulated by the E3 ubiquitin ligase SIAH2. Oncogene 2016, 35, 4289–4301. [Google Scholar] [CrossRef] [PubMed]
- Bohgaki, M.; Hakem, A.; Halaby, M.J.; Bohgaki, T.; Li, Q.; A Bissey, P.; Shloush, J.; Kislinger, T.; Sanchez, O.; Sheng, Y.; et al. The E3 ligase PIRH2 polyubiquitylates CHK2 and regulates its turnover. Cell Death Differ. 2013, 20, 812–822. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Characteristic | Total n = 156 (100%) |
|---|---|
| Sex | |
| Female | 138 (88.5%) |
| Male | 18 (11.5%) |
| Age at diagnosis (years) | |
| <55 | 98 (62.8%) |
| ≥55 | 58 (37.2%) |
| Mean (SD) | 48.5 (13.6) |
| Median (Q1–Q3) | 49.5 (39.0–59.0) |
| Range | 18.0–73.0 |
| Tumor diameter (mm) | |
| Mean (SD) | 13.1 (13.0) |
| Median (Q1–Q3) | 8.0 (5.0–16.5) |
| Range | 1.0–84.0 |
| Tumor diameter (mm) | |
| ≤10 | 94 (60.3%) |
| >10–20 | 32 (20.5%) |
| >20–40 | 25 (16.0%) |
| >40 | 5 (3.2%) |
| Papillary cancer histologic variant | |
| Classic | 107 (68.6%) |
| Follicular | 35 (22.4%) |
| Oxyphilic | 3 (1.9%) |
| Diffuse sclerosing | 3 (1.9%) |
| Tall cell | 1 (0.6%) |
| Other * | 7 (4.5%) |
| Surgical treatment | |
| Total thyroidectomy | 152 (97.4%) |
| Lobectomy | 4 (2.6%) |
| Nodal dissection | |
| Central | 94 (60.3%) |
| Lateral | 17 (10.9%) |
| No dissection | 45 (28.8%) |
| Multifocality | |
| No | 106 (68.0%) |
| Yes | 50 (32.1%) |
| Lymph node metastases ** | |
| N0a | 76 (48.7%) |
| N0b | 45 (28.9%) |
| N1a | 18 (11.5%) |
| N1b | 17 (10.9%) |
| Distant metastases | |
| M0 | 155 (99.4%) |
| M1 | 1 (0.6%) |
| Extrathyroidal extension | |
| Negative | 124 (79.5%) |
| Microscopic | 30 (19.2%) |
| Gross | 2 (1.3%) |
| Vascular invasion | |
| No | 145 (93%) |
| Yes | 11 (7.1%) |
| Margin status | |
| R0 | 137 (87.8%) |
| R1 | 19 (12.2%) |
| Tumor stage | |
| pT1a | 94 (60.3%) |
| pT1b | 32 (20.5%) |
| pT2 | 23 (14.7%) |
| pT3a | 5 (3.2%) |
| pT3b | 2 (1.3%) |
| CHEK2 mutation status | |
| CHEK2 WT | 52 (33.3%) |
| CHEK2 mutation any | 104 (66.7%) |
| CHEK2 I157T missense mutation (any) | 60 (38.5%) |
| I157T heterozygous | 53 (34.0%) |
| I157T homozygous | 7 (4.5%) |
| CHEK2 truncating heterozygous mutation (any) | 38 (24.4%) |
| IVS2 + 1G > A | 16 (10.3%) |
| Del5395 | 10 (6.4%) |
| 1100delC | 12 (7.7%) |
| Coexistence of two heterozygous CHEK2 mutations | 6 (3.8%) |
| I157T and IVS2 + 1G > A | 3 (1.9%) |
| I157T and Del5395 | 1 (0.6%) |
| IVS2 + 1G > A and Del5395 | 2 (1.3%) |
| TNM (8th edition) | |
| I | 144 (92.3%) |
| II | 12 (7.7%) |
| ATA initial risk stratification system | |
| Low | 97 (62.2%) |
| Intermediate | 34 (21.8%) |
| High | 25 (16.0%) |
| Radioactive iodine (I-131) therapy | |
| No | 45 (28.9%) |
| Yes | 111 (71.2%) |
| More than one course of radioactive iodine therapy (I-131) | |
| No | 138 (88.5%) |
| Yes | 18 (11.5%) |
| Response to therapy | |
| Excellent | 127 (81.4%) |
| Indeterminate | 24 (15.4%) |
| Biochemically incomplete | 2 (1.3%) |
| Structurally incomplete | 3 (1.9%) |
| Final follow-up (disease outcome) | |
| NED | 145 (93.0%) |
| Indeterminate | 7 (4.5%) |
| Biochemically persistent | 1 (0.6%) |
| Structurally persistent | 3 (1.9%) |
| Follow-up, recurrence | |
| No | 151 (96.8%) |
| Yes | 5 (3.2%) |
| Death | |
| No | 151 (96.8%) |
| Yes | 5 (3.2%) |
| Follow-up (years) | |
| Median (range) | 7.0 (1.0–23.0) |
| Variable | p53, n = 156 (100%) | CHK2, n = 156 (100%) | CHEK2 (FISH), n = 151 (100%) | TP53 (FISH), n = 151 (100%) | Positive p53 with TP53 Deletion (FISH) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Positive n = 8 (5.1%) | High Expression n = 23 (14.7%) | Deletion n = 44 (29.1%) | Deletion n = 7 (4.6%) | n = 15 out of 151 (9.9%) | ||||||
| OR (95% CI) | p | OR (95% CI) | p | OR (95% CI) | p | OR (95% CI) | p | OR (95% CI) | p | |
| Sex | ||||||||||
| Female vs. male | NA (0 in cell) | 1.44 (0.31–6.71) | 0.645 | 1.08 (0.36–3.23) | 0.892 | 1.09 (0.13–9.24) | 0.938 | NA (0 in cell) | ||
| Age group | ||||||||||
| ≥55 years vs. <55 years | 2.99 (0.69–13.00) | 0.145 | 0.55 (0.20–1.49) | 0.238 | 0.75 (0.36–1.58) | 0.451 | 3.92 (0.94–16.35) | 0.061 | 3.03 (1.02–9.05) | 0.047 |
| Tumor stage | ||||||||||
| pT1a vs. pT1b, pT2, pT3a, pT3b | NA (0 in cell) | 0.96 (0.11–8.38) | 0.972 | 0.97 (0.18–5.21) | 0.973 | 0.81 (0.21–3.16) | 0.766 | 0.99 (0.33–2.93) | 0.982 | |
| TNM (8th edition) | ||||||||||
| TNM II vs. TNM I | NA (0 in cell) | 1.17 (0.24–5.73) | 0.845 | 0.52 (0.11–2.50) | 0.414 | 1.65 (0.19–14.54) | 0.652 | 0.90 (0.11–7.56) | 0.923 | |
| Multifocality | ||||||||||
| Yes vs. no | 0.69 (0.14–3.57) | 0.662 | 0.92 (0.35–2.39) | 0.857 | 1.00 (0.47–2.13) | 0.996 | 0.62 (0.12–3.08) | 0.555 | 0.52 (0.14–1.95) | 0.334 |
| Lymph node metastasis * | ||||||||||
| N1a, N1b vs. N0b, N0a | NA (0 in cell) | 0.95 (0.33–2.78) | 0.931 | 1.02 (0.44–2.35) | 0.968 | 0.40 (0.05–3.29) | 0.392 | 0.21 (0.03–1.69) | 0.144 | |
| Distant metastasis | ||||||||||
| M1 vs. M0 | NA (0 in cell) | NA (0 in cell) | NA (0 in cell) | NA (0 in cell) | 0.993 | NA (0 in cell) | ||||
| Tumor diameter | ||||||||||
| >10 and ≤20 mm vs. ≤10 mm | 1.84 (0.41–8.18) | 0.422 | 2.80 (1.00–7.88) | 0.051 | 0.78 (0.31–1.95) | 0.592 | 1.84 (0.41–8.21) | 0.422 | 1.75 (0.54–5.70) | 0.351 |
| >20 and ≤40 mm vs. ≤10 mm | NA (0 in cell) | 2.10 (0.65–6.83) | 0.217 | 1.31 (0.52–3.34) | 0.568 | 0.72 (0.08–6.43) | 0.766 | 0.38 (0.05–3.15) | 0.370 | |
| >40 mm vs. ≤10 mm | NA (0 in cell) | NA (0 in cell) | NA (0 in cell) | NA (0 in cell) | NA (0 in cell) | |||||
| Papillary cancer histologic variant | ||||||||||
| Follicular vs. Classic | 1.24 (0.23–6.68) | 0.805 | 1.02 (0.34–3.05) | 0.969 | 1.24 (0.54–2.86) | 0.618 | 0.44 (0.05–3.69) | 0.447 | 0.95 (0.25–3.68) | 0.941 |
| Other ** aggressive vs. Classic | NA (0 in cell) | 6.13 (0.80–46.91) | 0.081 | 0.86 (0.09–8.63) | 0.900 | 4.67 (0.43–50.91) | 0.206 | 3.17 (0.30–33.37) | 0.337 | |
| Other *** non-aggressive vs. Classic | 2.27 (0.24–21.56) | 0.476 | 0.68 (0.08–5.77) | 0.725 | 1.29 (0.30–5.52) | 0.728 | NA (0 in cell) | 1.19 (0.13–10.49) | 0.877 | |
| Extrathyroidal extension | ||||||||||
| Micro and gross vs. negative | 1.31 (0.25–6.83) | 0.748 | 1.09 (0.37–3.20) | 0.875 | 1.45 (0.63–3.35) | 0.385 | 1.11 (0.22–5.65) | 0.897 | 0.68 (0.20–2.31) | 0.537 |
| Vascular invasion | ||||||||||
| Yes vs. no | NA (0 in cell) | 1.31 (0.27–6.50) | 0.739 | 2.62 (0.72–9.53) | 0.145 | 8.38 (1.76–39.80) | 0.008 | 4.00 (0.94–17.10) | 0.061 | |
| Margin status | ||||||||||
| R1 vs. R0 | NA (0 in cell) | 0.65 (0.14–3.02) | 0.583 | 0.85 (0.29–2.53) | 0.772 | 0.92 (0.11–7.81) | 0.938 | 0.50 (0.06–4.05) | 0.516 | |
| ATA | ||||||||||
| Low vs intermediate, high | 1.01 (0.23–4.41) | 0.985 | 0.62 (0.25–1.50) | 0.287 | 1.05 (0.50–2.17) | 0.906 | 0.72 (0.19–2.81) | 0.639 | 1.55 (0.53–4.54) | 0.421 |
| Response to therapy | ||||||||||
| Excellent vs. others | NA (0 in cell) | 0.28 (0.11–0.72) | 0.009 | 0.85 (0.34–2.14) | 0.731 | 1.63 (0.19–13.61) | 0.653 | 0.33 (0.04–2.66) | 0.300 | |
| Radioactive iodine (I-131) therapy | ||||||||||
| Yes vs. no | 0.66 (0.15–2.89) | 0.581 | 2.12 (0.68–6.61) | 0.197 | 0.72 (0.34–1.53) | 0.391 | 3.48 (0.42–28.64) | 0.247 | 1.15 (0.34–3.81) | 0.824 |
| Number of I-131 courses | ||||||||||
| 2–9 courses vs. 0–1 course | 2.75 (0.51–14.79) | 0.239 | 0.70 (0.15–3.25) | 0.646 | 2.16 (0.79–5.89) | 0.134 | 0.92 (0.11–7.81) | 0.938 | 2.02 (0.51–7.97) | 0.317 |
| Final follow-up | ||||||||||
| NED vs. others | NA (0 in cell) | 0.17 (0.05–0.62) | 0.007 | 0.96 (0.24–3.88) | 0.951 | 0.54 (0.06–4.82) | 0.582 | 0.99 (0.12–8.42) | 0.994 | |
| Follow-up recurrence | ||||||||||
| Yes vs. no | NA (0 in cell) | 1.47 (0.16–13.74) | 0.738 | NA (0 in cell) | NA (0 in cell) | NA (0 in cell) | ||||
| Death | ||||||||||
| Yes vs. no | NA (0 in cell) | NA (0 in cell) | 0.60 (0.07–5.51) | 0.651 | 4.31 (0.43–43.20) | 0.214 | 2.36 (0.25–22.58) | 0.457 | ||
| Group | Category of Interest | Counts | Total | Proportion |
|---|---|---|---|---|
| Response to therapy | Excellent | |||
| Low CHK2 | 113 | 133 | 0.850 | |
| High CHK2 | 14 | 23 | 0.609 | |
| Final follow-up | NED | |||
| Low CHK2 | 127 | 133 | 0.955 | |
| High CHK2 | 18 | 23 | 0.783 | |
| Tumor diameter | ≤10 mm | |||
| Low CHK2 | 84 | 108 | 0.788 | |
| High CHK2 | 10 | 18 | 0.556 | |
| Papillary cancer histologic variant | Classic | |||
| Low CHK2 | 92 | 94 | 0.979 | |
| High CHK2 | 15 | 17 | 0.882 | |
| Age | <55 years | |||
| No loss of TP53 | 94 | 142 | 0.662 | |
| Deletion of TP53 | 3 | 9 | 0.333 | |
| Vascular invasion | No | |||
| No loss of TP53 | 134 | 142 | 0.944 | |
| Deletion of TP53 | 6 | 9 | 0.667 | |
| Age | <55 years | |||
| Negative p53 + no loss of TP53 | 91 | 136 | 0.669 | |
| Positive p53 + Deletion of TP53 | 6 | 15 | 0.400 | |
| Vascular invasion | No | |||
| Negative p53 + no loss of TP53 | 128 | 136 | 0.941 | |
| Positive p53 + Deletion of TP53 | 12 | 15 | 0.800 | |
| Variable | CHEK2 Truncating vs. WT | CHEK2 Missense vs. WT | CHEK2 Truncating vs. WT + Missense I157T | |||
|---|---|---|---|---|---|---|
| OR (95% CI) | p | OR (95% CI) | p | OR (95% CI) | p | |
| p53 expression | ||||||
| Positive vs. negative | 0.42 (0.04–4.19) | 0.459 | 1.17 (0.25–5.47) | 0.845 | 0.35 (0.04–2.92) | 0.331 |
| CHK2 expression | ||||||
| High vs. low | 3.48 (0.99–12.30) | 0.052 | 2.12 (0.61–7.33) | 0.236 | 2.24 (0.90–5.58) | 0.083 |
| CHEK2 gene status | ||||||
| Deletion vs. no loss of gene copy | 62.67 (16.37–239.93) | <0.001 | 1.31 (0.35–4.91) | 0.693 | 57.23 (19.36–169.24) | <0.001 |
| TP53 gene status | ||||||
| Deletion vs. no loss of gene copy | NA (0 in cell) | NA (0 in cell) | NA (0 in cell) | |||
| p53 expression and TP53 gene status | ||||||
| Positive vs. negative and deletion vs. no loss of gene copy | 3.47 (0.84–14.37) | 0.087 | 1.17 (0.25–5.47) | 0.845 | 3.33 (1.13–9.84) | 0.029 |
| Variable | CHEK2 Truncating and WT | CHEK2 Truncating and WT + Missense I157T | |||||
|---|---|---|---|---|---|---|---|
| Group | Counts | Total | Proportion | Counts | Total | Proportion | |
| CHEK2 deletion | No mutation | 4 | 51 | 0.078 | 10 | 111 | 0.090 |
| Mutation | 32 | 38 | 0.842 | 34 | 40 | 0.850 | |
| High expression of CHK2 | No mutation | 4 | 52 | 0.077 | 13 | 112 | 0.116 |
| Mutation | 9 | 40 | 0.225 | 10 | 44 | 0.227 | |
| Positive p53 and TP53 deletion | No mutation | 3 | 51 | 0.059 | 7 | 111 | 0.063 |
| Mutation | 7 | 38 | 0.184 | 8 | 40 | 0.200 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gąsior-Perczak, D.; Kowalik, A.; Kopczyński, J.; Macek, P.; Niemyska, K.; Walczyk, A.; Gruszczyński, K.; Siołek, M.; Dróżdż, T.; Kosowski, M.; et al. Relationship between the Expression of CHK2 and p53 in Tumor Tissue and the Course of Papillary Thyroid Cancer in Patients with CHEK2 Germline Mutations. Cancers 2024, 16, 815. https://doi.org/10.3390/cancers16040815
Gąsior-Perczak D, Kowalik A, Kopczyński J, Macek P, Niemyska K, Walczyk A, Gruszczyński K, Siołek M, Dróżdż T, Kosowski M, et al. Relationship between the Expression of CHK2 and p53 in Tumor Tissue and the Course of Papillary Thyroid Cancer in Patients with CHEK2 Germline Mutations. Cancers. 2024; 16(4):815. https://doi.org/10.3390/cancers16040815
Chicago/Turabian StyleGąsior-Perczak, Danuta, Artur Kowalik, Janusz Kopczyński, Paweł Macek, Kornelia Niemyska, Agnieszka Walczyk, Krzysztof Gruszczyński, Monika Siołek, Tomasz Dróżdż, Marcin Kosowski, and et al. 2024. "Relationship between the Expression of CHK2 and p53 in Tumor Tissue and the Course of Papillary Thyroid Cancer in Patients with CHEK2 Germline Mutations" Cancers 16, no. 4: 815. https://doi.org/10.3390/cancers16040815
APA StyleGąsior-Perczak, D., Kowalik, A., Kopczyński, J., Macek, P., Niemyska, K., Walczyk, A., Gruszczyński, K., Siołek, M., Dróżdż, T., Kosowski, M., Pałyga, I., Przybycień, P., Wabik, O., Góźdź, S., & Kowalska, A. (2024). Relationship between the Expression of CHK2 and p53 in Tumor Tissue and the Course of Papillary Thyroid Cancer in Patients with CHEK2 Germline Mutations. Cancers, 16(4), 815. https://doi.org/10.3390/cancers16040815