

Dual Checkpoint Aptamer Immunotherapy: Unveiling Tailored Cancer Treatment Targeting CTLA-4 and NKG2A

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Initial Aptamer Generation
2.2. Iterative Selection and Mutation
2.3. Secondary and Tertiary Structure Prediction
2.4. Oligonucleotides
2.5. Docking Simulation
2.6. Cell Lines
2.7. Competitive Inhibition by ELISA
2.8. Molar to Molar Competition Assay by ELISA
2.9. ELISA-Based Binding Assay of AYA22T-R2-13 with CTLA-4/CD152 Protein
2.10. Flow Cytometry-Based Binding Assay of AYA22T-Aptamers to CTLA4 Protein
2.11. PBMCs Isolation
2.12. Isolation and Stimulation of CD8+ T Cells and NK Cells
2.13. Surface Staining of Tumor Cells
2.14. Surface Staining of Immune Cells including Enriched CD8 T Cells, Total T Cells and NK Cells
2.15. Competitive Binding of AYA22T-R2-13 to CTLA4/NKG2A Surface Receptors on CD8 T Cells
2.16. Detection of Cell Surface CTLA4 Binding to AYA22T-R2-13 via Competitive Binding Inhibition Assay
2.17. Sulforhodamine B (SRB) Cell Cytotoxicity Assay
2.18. CD8+ T-Cell-Mediated Tumor Cell Killing Assay
2.19. NK Cell-Mediated Tumor Cell Killing Assay
2.20. Killing Assay in the Presence of Human Serum
2.21. Lactate Dehydrogenase (LDH) Assay
2.22. Surface and Intra-Cellular Staining
3. Statistical Analysis
4. Results
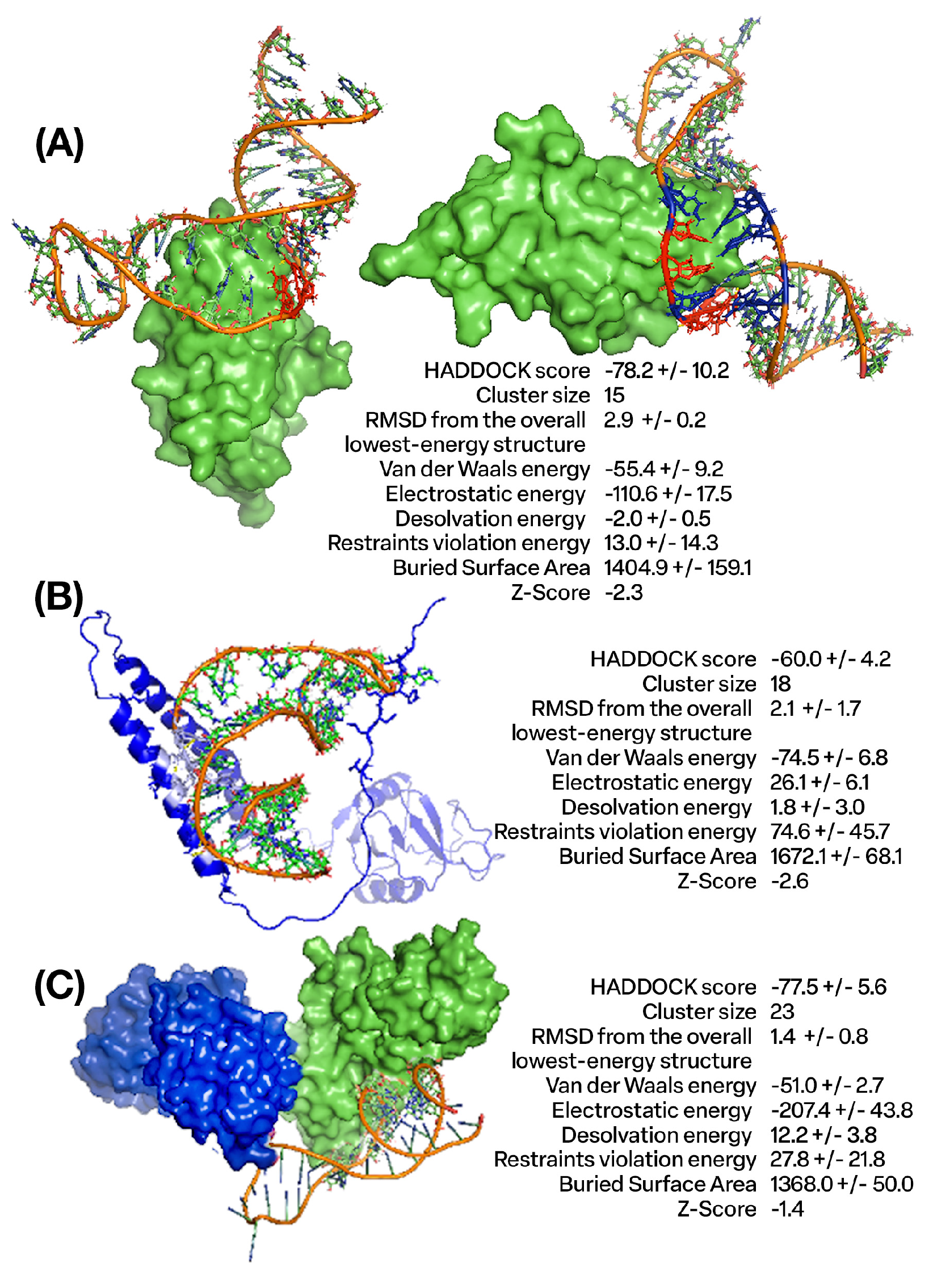
4.1. Design and Molecular Docking Analysis of Aptamers Targeting CTLA4 and NKG2A Proteins Using In Silico Approaches
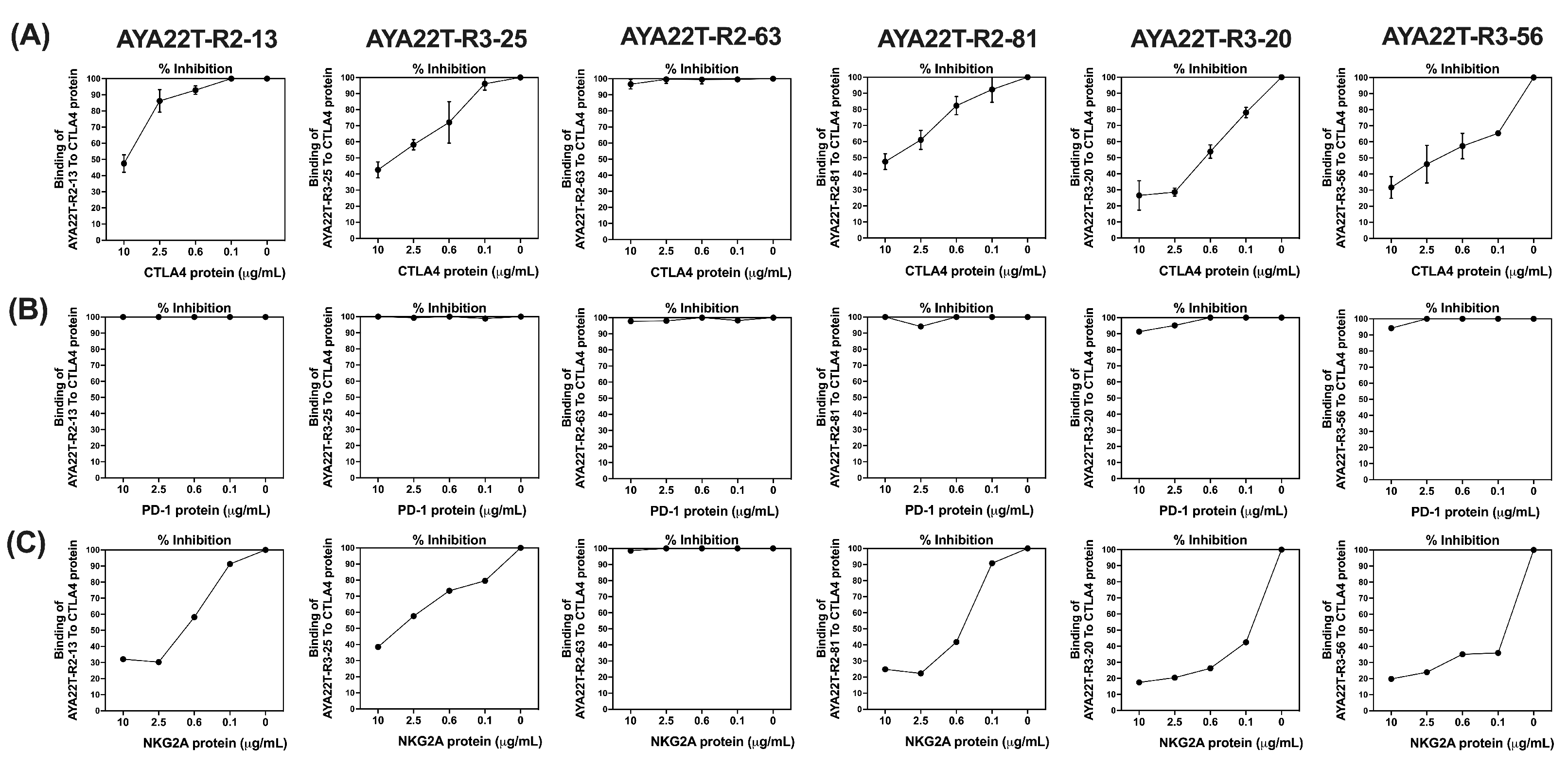
4.2. Binding Specificity Assessment of CTLA4/NKG2A Aptamers (AYA22T) to CTLA4 and NKG2A Proteins
4.3. Detection of AYA22T Aptamers’ Binding to CTLA4 Protein
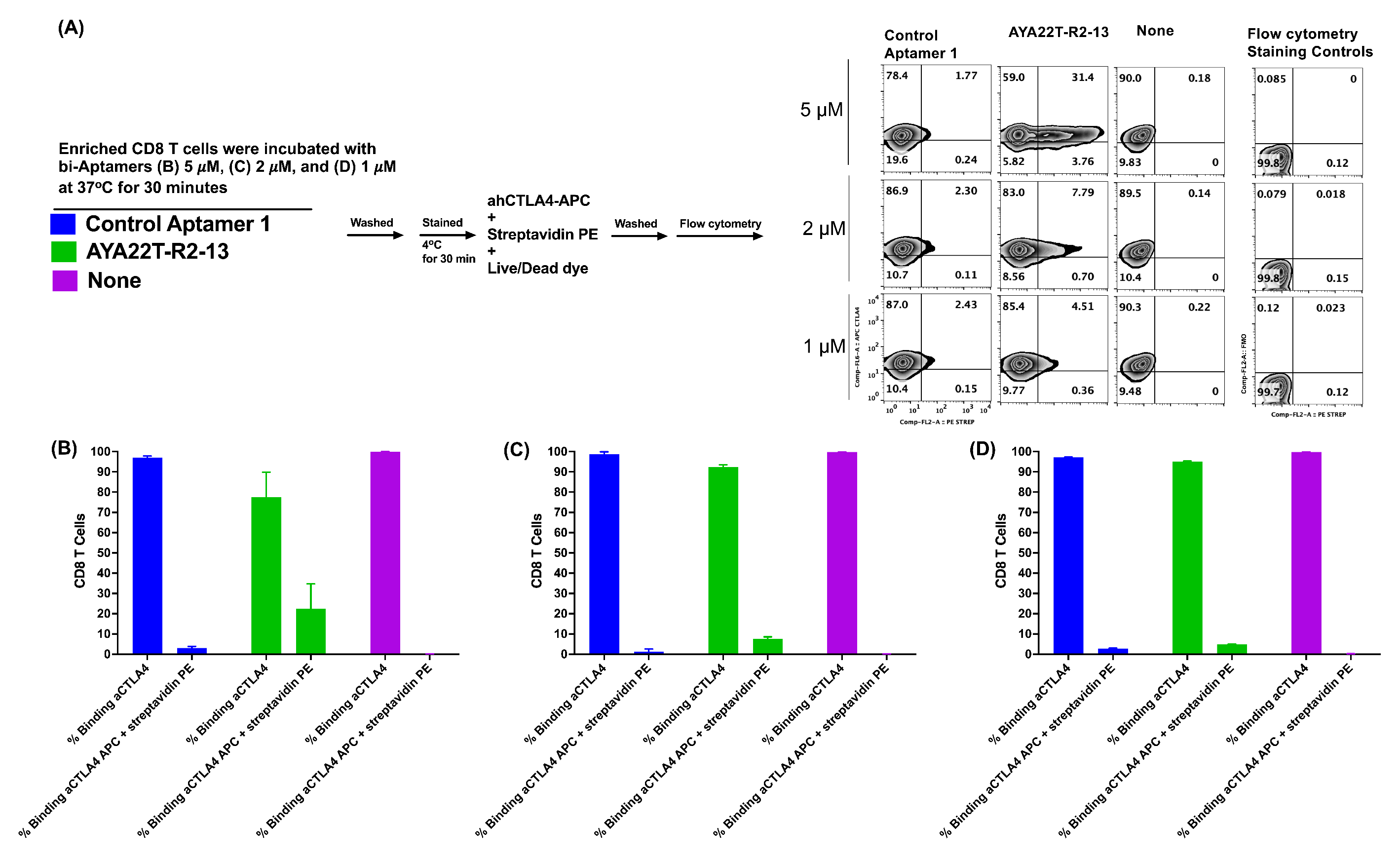
4.4. Detection of AYA22T Aptamers’ Binding to CD8 T Cells and NK Cells
4.5. Specificity of Binding of AYA22T-R2-13 to CTLA4 and NKG2A on Activated CD8 T Cells
4.6. Detection of Cell Surface Binding of AYA22T-R2-13 via Competitive Co-Staining Binding Inhibition Assay
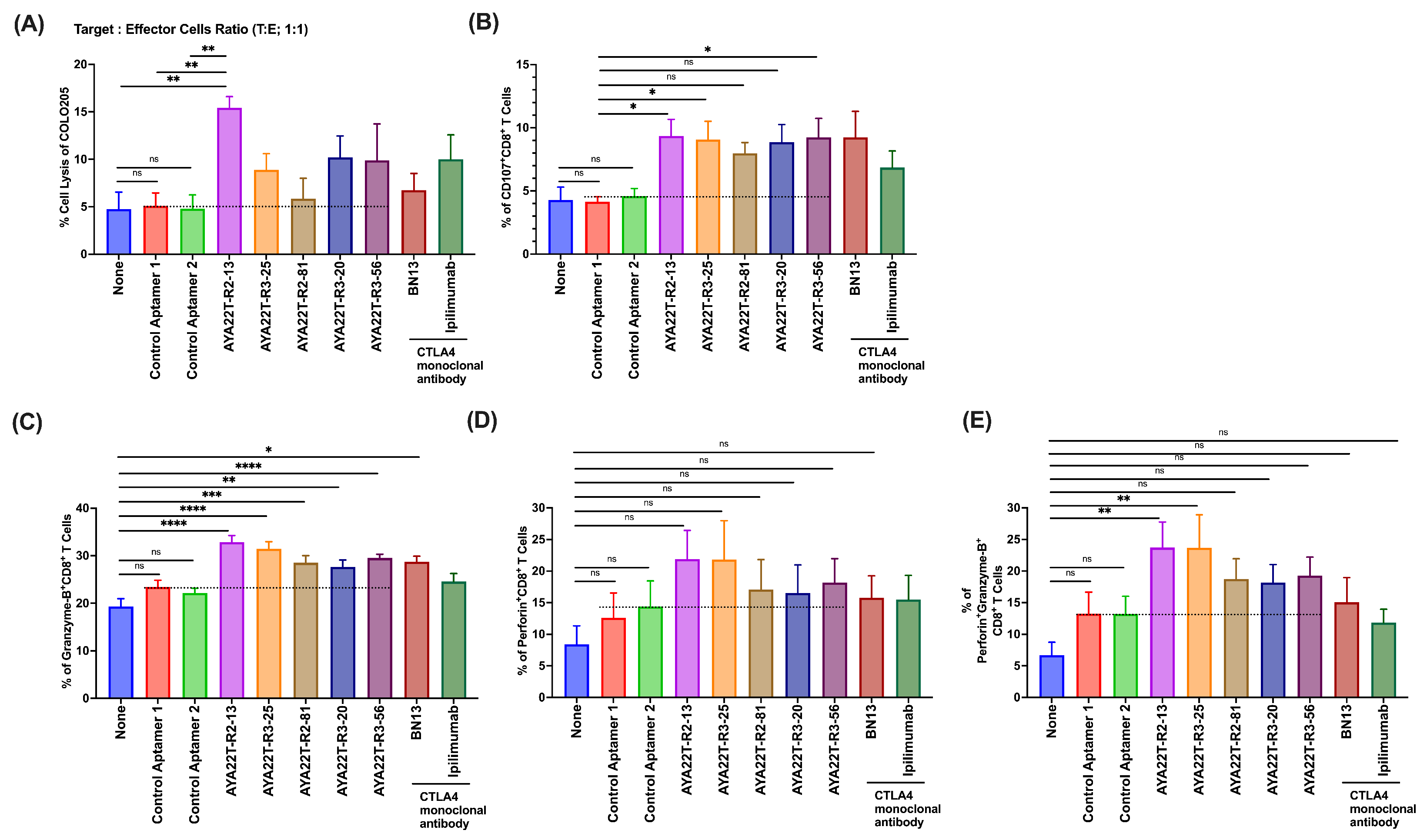
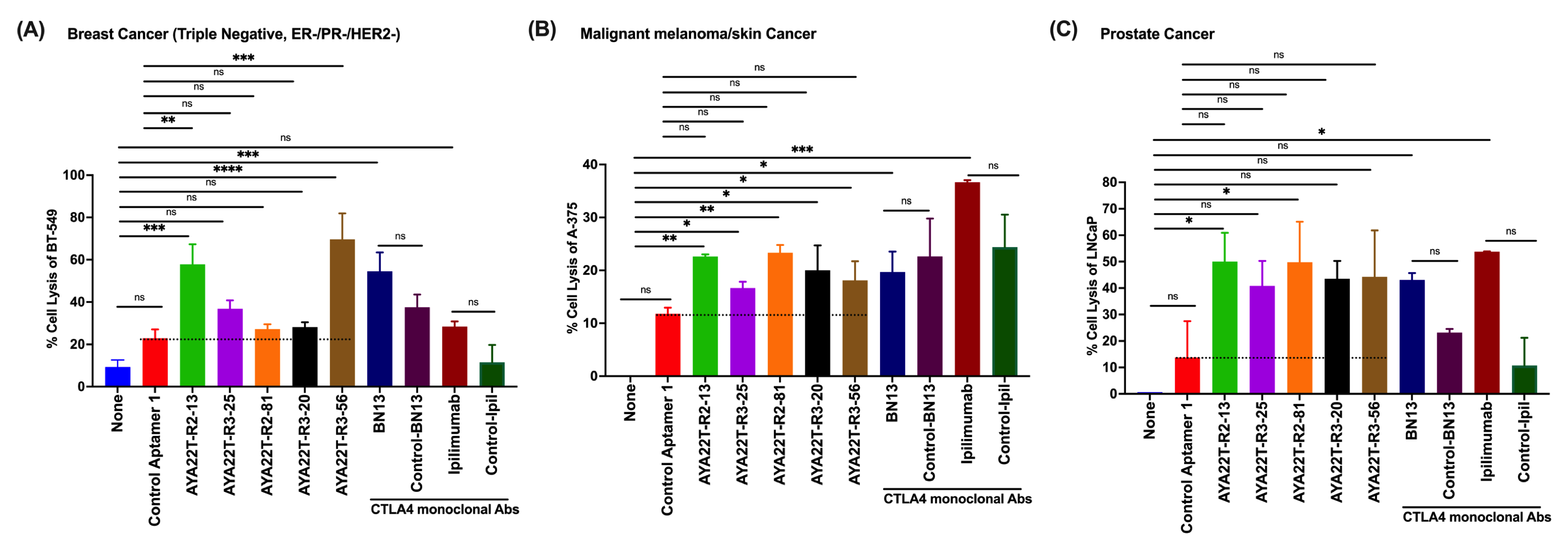
4.7. Assessment of Cytotoxicity of AYA22T-R2-13 on Tumors and Immune Cells
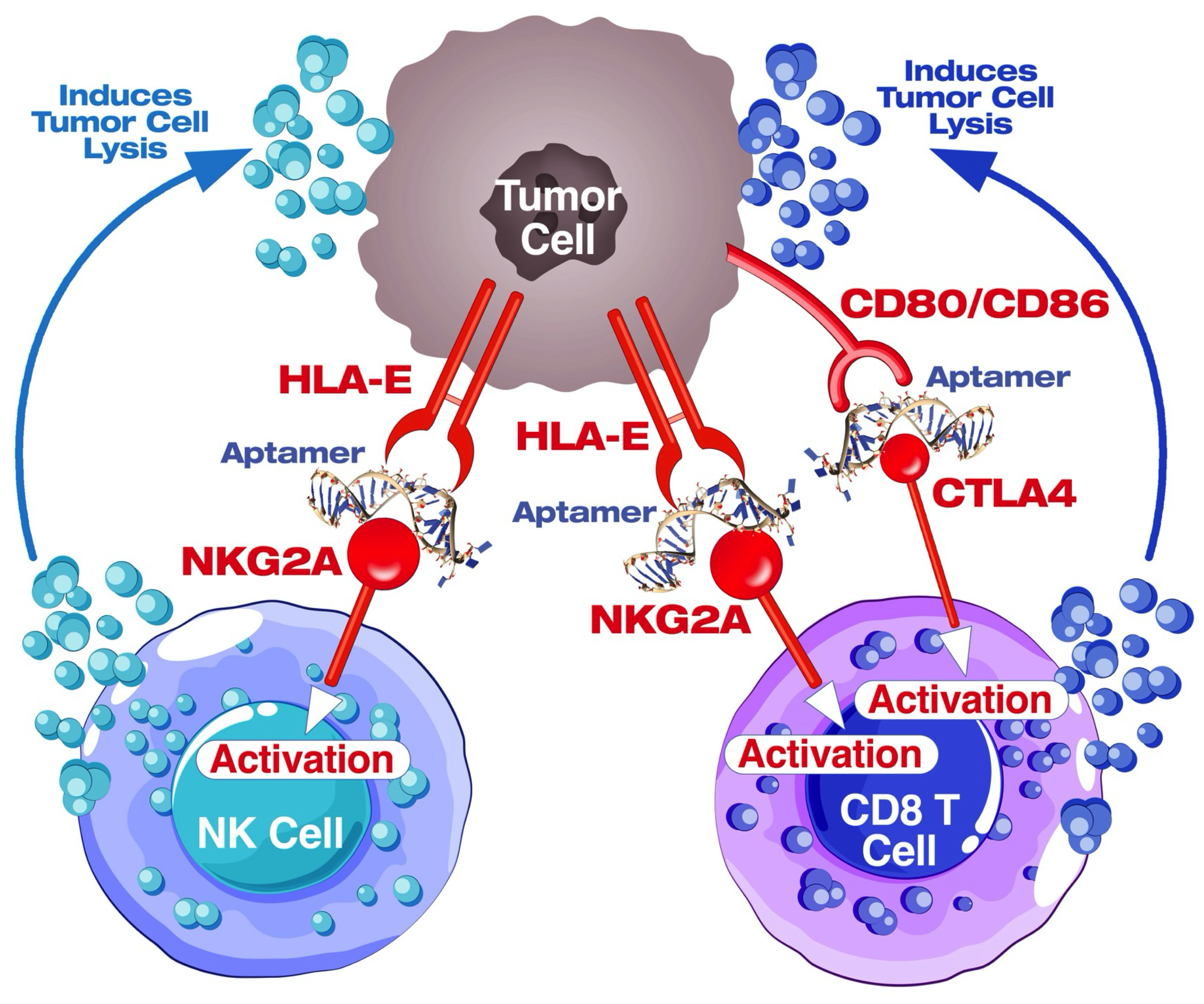
4.8. Blockade of the CTLA4-CD80/86 Axis Using AYA22T-R2-13 Unleashes CTL-Mediated Lysis of Tumor Cells In Vitro
4.9. AYA22T-R2-13 Unleashes NK Cell-Mediated Lysis of Tumor Cells In Vitro
5. Discussion
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CTLA-4 | cytotoxic T-lymphocyte–associated antigen 4 |
| PD-1 | programmed death 1 |
| PD-L1 | programmed cell death-ligand 1 |
| IDO | indoleamine 2,3-dioxygenase |
| NK | natural killer |
| irAEs | immune-related adverse events |
| A | adenine |
| U | uracil |
| G | guanine |
| C | cytosine |
| dU | deoxyuridine |
| PBMCs | peripheral blood mononuclear cells |
| LDH | lactate dehydrogenase |
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. An Update on Cancer Deaths in the United States 2022. Available online: https://www.cdc.gov/nchs/hus/topics/cancer-deaths.htm (accessed on 15 December 2023).
- American Cancer Society. Cancer Fact & Figures 2023. Available online: https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/2023-cancer-facts-figures.html (accessed on 15 December 2023).
- Negoita, S.; Chen, H.S.; Sanchez, P.V.; Sherman, R.L.; Henley, S.J.; Siegel, R.L.; Sung, H.; Scott, S.; Benard, V.B.; Kohler, B.A.; et al. Annual Report to the Nation on the Status of Cancer, part 2: Early assessment of the COVID-19 pandemic’s impact on cancer diagnosis. Cancer 2023, 130, 117–127. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. Cancer Trends Progress Reports: Financial Burden Of Cancer Care (Update as of April 2022). Available online: https://progressreport.cancer.gov/ (accessed on 15 December 2023).
- Valiullina, A.K.; Zmievskaya, E.A.; Ganeeva, I.A.; Zhuravleva, M.N.; Garanina, E.E.; Rizvanov, A.A.; Petukhov, A.V.; Bulatov, E.R. Evaluation of CAR-T Cells’ Cytotoxicity against Modified Solid Tumor Cell Lines. Biomedicines 2023, 11, 626. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Dai, Z.; Wu, W.; Wang, Z.; Zhang, N.; Zhang, L.; Zeng, W.J.; Liu, Z.; Cheng, Q. Regulatory mechanisms of immune checkpoints PD-L1 and CTLA-4 in cancer. J. Exp. Clin. Cancer Res. 2021, 40, 184. [Google Scholar] [CrossRef] [PubMed]
- Pistillo, M.P.; Tazzari, P.L.; Palmisano, G.L.; Pierri, I.; Bolognesi, A.; Ferlito, F.; Capanni, P.; Polito, L.; Ratta, M.; Pileri, S.; et al. CTLA-4 is not restricted to the lymphoid cell lineage and can function as a target molecule for apoptosis induction of leukemic cells. Blood J. Am. Soc. Hematol. 2003, 101, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Kosmaczewska, A.; Ciszak, L.; Suwalska, K.; Wolowiec, D.; Frydecka, I. CTLA-4 overexpression in CD19+/CD5+ cells correlates with the level of cell cycle regulators and disease progression in B-CLL patients. Leukemia 2005, 19, 301–304. [Google Scholar] [CrossRef]
- Joshi, A.D.; Hegde, G.V.; Dickinson, J.D.; Mittal, A.K.; Lynch, J.C.; Eudy, J.D.; Armitage, J.O.; Bierman, P.J.; Bociek, R.G.; Devetten, M.P.; et al. ATM, CTLA4, MNDA, and HEM1 in high versus low CD38–expressing B-cell chronic lymphocytic leukemia. Clin. Cancer Res. 2007, 13, 5295–5304. [Google Scholar] [CrossRef]
- Mao, H.; Zhang, L.; Yang, Y.; Zuo, W.; Bi, Y.; Gao, W.; Deng, B.; Sun, J.; Shao, Q.; Qu, X. New insights of CTLA-4 into its biological function in breast cancer. Curr. Cancer Drug Targets 2010, 10, 728–736. [Google Scholar] [CrossRef]
- Kamiya, T.; Seow, S.V.; Wong, D.; Robinson, M.; Campana, D. Blocking expression of inhibitory receptor NKG2A overcomes tumor resistance to NK cells. J. Clin. Investig. 2019, 129, 2094–2106. [Google Scholar] [CrossRef]
- André, P.; Denis, C.; Soulas, C.; Bourbon-Caillet, C.; Lopez, J.; Arnoux, T.; Bléry, M.; Bonnafous, C.; Gauthier, L.; Morel, A.; et al. Anti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell 2018, 175, 1731–1743. [Google Scholar] [CrossRef]
- Ribas, A. Overcoming immunologic tolerance to melanoma: Targeting CTLA-4 with tremelimumab (CP-675,206). Oncologist 2008, 13, 10–15. [Google Scholar] [CrossRef]
- Bertrand, A.; Kostine, M.; Barnetche, T.; Truchetet, M.E.; Schaeverbeke, T. Immune related adverse events associated with anti-CTLA-4 antibodies: Systematic review and meta-analysis. BMC Med. 2015, 13, 211. [Google Scholar] [CrossRef]
- Karimi, A.; Alilou, S.; Mirzaei, H.R. Adverse events following administration of anti-CTLA4 antibody ipilimumab. Front. Oncol. 2021, 11, 624780. [Google Scholar] [CrossRef]
- Food and Drug Administration. Imjudo® (Tremelimumab-actl) Injection, for Intravenous Use. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/761289s000lbl.pdf (accessed on 15 December 2023).
- Fecher, L.A.; Agarwala, S.S.; Hodi, F.S.; Weber, J.S. Ipilimumab and its toxicities: A multidisciplinary approach. Oncologist 2013, 18, 733–743. [Google Scholar] [CrossRef]
- Santulli-Marotto, S.; Nair, S.K.; Rusconi, C.; Sullenger, B.; Gilboa, E. Multivalent RNA aptamers that inhibit CTLA-4 and enhance tumor immunity. Cancer Res. 2003, 63, 7483–7489. [Google Scholar]
- Huang, B.T.; Lai, W.Y.; Chang, Y.C.; Wang, J.W.; Yeh, S.D.; Lin, E.P.Y.; Yang, P.C. A CTLA-4 antagonizing DNA aptamer with antitumor effect. Mol. Ther. Nucleic Acids 2017, 8, 520–528. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Zhang, D.; Wang, Y.; Wu, M.; Zhang, C.; Zheng, Y.; Zheng, A.; Liu, X. A highly stable multifunctional aptamer for enhancing antitumor immunity against hepatocellular carcinoma by blocking dual immune checkpoints. Biomater. Sci. 2021, 9, 4159–4168. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Antczak, M.; Popenda, M.; Zok, T.; Sarzynska, J.; Ratajczak, T.; Tomczyk, K.; Adamiak, R.; Szachniuk, M. New functionality of RNAComposer: An application to shape the axis of miR160 precursor structure. Acta Biochim. Pol. 2016, 63, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Popenda, M.; Szachniuk, M.; Antczak, M.; Purzycka, K.J.; Lukasiak, P.; Bartol, N.; Blazewicz, J.; Adamiak, R.W. Automated 3D structure composition for large RNAs. Nucleic Acids Res. 2012, 40, e112. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Luo, H.; Gubu, A.; Yu, S.; Zhang, H.; Dai, H.; Zhang, Y.; Zhang, B.; Ma, Y.; Lu, A.; et al. Chemically modified aptamers for improving binding affinity to the target proteins via enhanced non-covalent bonding. Front. Cell Dev. Biol. 2023, 11, 1091809. [Google Scholar] [CrossRef]
- Ayass, M.A.; Griko, N.; Pashkov, V.; Tripathi, T.; Cao, W.; Javan, N.; Dai, J.; Zhang, J.; Zhu, K.; Abi-Mosleh, L. High-Affinity Neutralizing DNA Aptamers against SARS-CoV-2 Spike Protein Variants. COVID 2023, 3, 520–542. [Google Scholar] [CrossRef]
- Honorato, R.V.; Koukos, P.I.; Jiménez-García, B.; Tsaregorodtsev, A.; Verlato, M.; Giachetti, A.; Rosato, A.; Bonvin, A.M. Structural biology in the clouds: The WeNMR-EOSC ecosystem. Front. Mol. Biosci. 2021, 8, 729513. [Google Scholar] [CrossRef]
- Van Zundert, G.; Rodrigues, J.; Trellet, M.; Schmitz, C.; Kastritis, P.; Karaca, E.; Melquiond, A.; van Dijk, M.; De Vries, S.; Bonvin, A. The HADDOCK2. 2 web server: User-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Rasheed, Z.; Ali, R. Reactive oxygen species damaged human serum albumin in patients with type 1 diabetes mellitus: Biochemical and immunological studies. Life Sci. 2006, 79, 2320–2328. [Google Scholar] [CrossRef]
- Baysal, H.; De Pauw, I.; Zaryouh, H.; De Waele, J.; Peeters, M.; Pauwels, P.; Vermorken, J.B.; Smits, E.; Lardon, F.; Jacobs, J.; et al. Cetuximab-induced natural killer cell cytotoxicity in head and neck squamous cell carcinoma cell lines: Investigation of the role of cetuximab sensitivity and HPV status. Br. J. Cancer 2020, 123, 752–761. [Google Scholar] [CrossRef]
- Limame, R.; Wouters, A.; Pauwels, B.; Fransen, E.; Peeters, M.; Lardon, F.; De Wever, O.; Pauwels, P. Comparative analysis of dynamic cell viability, migration and invasion assessments by novel real-time technology and classic endpoint assays. PLoS ONE 2012, 7, e46536. [Google Scholar] [CrossRef] [PubMed]
- Raskov, H.; Orhan, A.; Christensen, J.P.; Gögenur, I. Cytotoxic CD8+ T cells in cancer and cancer immunotherapy. Br. J. Cancer 2021, 124, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Fasano, M.; Della Corte, C.M.; Di Liello, R.; Barra, G.; Sparano, F.; Viscardi, G.; Iacovino, M.L.; Paragliola, F.; Famiglietti, V.; Ciaramella, V.; et al. Induction of natural killer antibody-dependent cell cytotoxicity and of clinical activity of cetuximab plus avelumab in non-small cell lung cancer. ESMO Open 2020, 5, e000753. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.B.; Zheng, C.Y.; Giscombe, R.; Lefvert, A. Regulation of surface and intracellular expression of CTLA-4 on human peripheral T cells. Scand. J. Immunol. 2001, 54, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Vandenborre, K.; Van Gool, S.; Kasran, A.; Ceuppens, J.; Boogaerts, M.; Vandenberghe, P. Interaction of CTLA-4 (CD152) with CD80 or CD86 inhibits human T-cell activation. Immunology 1999, 98, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Jorgovanovic, D.; Song, M.; Wang, L.; Zhang, Y. Roles of IFN-γ in tumor progression and regression: A review. Biomark. Res. 2020, 8, 49. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Yang, J.; Cai, Y.; Fu, S.; Zhang, N.; Fu, X.; Li, L. IFN-γ-mediated inhibition of lung cancer correlates with PD-L1 expression and is regulated by PI3K-AKT signaling. Int. J. Cancer 2018, 143, 931–943. [Google Scholar] [CrossRef] [PubMed]
- Zahavi, D.J.; Weiner, L.M. Targeting multiple receptors to increase checkpoint blockade efficacy. Int. J. Mol. Sci. 2019, 20, 158. [Google Scholar] [CrossRef]
- Zhu, S.; Zhang, T.; Zheng, L.; Liu, H.; Song, W.; Liu, D.; Li, Z.; Pan, C.x. Combination strategies to maximize the benefits of cancer immunotherapy. J. Hematol. Oncol. 2021, 14, 156. [Google Scholar] [CrossRef]
- Patel, S.A.; Minn, A.J. Combination cancer therapy with immune checkpoint blockade: Mechanisms and strategies. Immunity 2018, 48, 417–433. [Google Scholar] [CrossRef]
- van Montfoort, N.; Borst, L.; Korrer, M.J.; Sluijter, M.; Marijt, K.A.; Santegoets, S.J.; van Ham, V.J.; Ehsan, I.; Charoentong, P.; André, P.; et al. NKG2A blockade potentiates CD8 T cell immunity induced by cancer vaccines. Cell 2018, 175, 1744–1755. [Google Scholar] [CrossRef]
- Wei, S.C.; Anang, N.A.A.; Sharma, R.; Andrews, M.C.; Reuben, A.; Levine, J.H.; Cogdill, A.P.; Mancuso, J.J.; Wargo, J.A.; Pe’er, D.; et al. Combination anti–CTLA-4 plus anti–PD-1 checkpoint blockade utilizes cellular mechanisms partially distinct from monotherapies. Proc. Natl. Acad. Sci. USA 2019, 116, 22699–22709. [Google Scholar] [CrossRef] [PubMed]
- Hannani, D.; Vétizou, M.; Enot, D.; Rusakiewicz, S.; Chaput, N.; Klatzmann, D.; Desbois, M.; Jacquelot, N.; Vimond, N.; Chouaib, S.; et al. Anticancer immunotherapy by CTLA-4 blockade: Obligatory contribution of IL-2 receptors and negative prognostic impact of soluble CD25. Cell Res. 2015, 25, 208–224. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.M.; Wang, C.J.; Ryan, G.A.; Clough, L.E.; Qureshi, O.S.; Goodall, M.; Abbas, A.K.; Sharpe, A.H.; Sansom, D.M.; Walker, L.S. Ctla-4 controls regulatory T cell peripheral homeostasis and is required for suppression of pancreatic islet autoimmunity. J. Immunol. 2009, 182, 274–282. [Google Scholar] [CrossRef]
- Schneider, H.; Valk, E.; Leung, R.; Rudd, C.E. CTLA-4 activation of phosphatidylinositol 3-kinase (PI 3-K) and protein kinase B (PKB/AKT) sustains T-cell anergy without cell death. PLoS ONE 2008, 3, e3842. [Google Scholar] [CrossRef]
- Hegel, J.K.; Knieke, K.; Kolar, P.; Reiner, S.L.; Brunner-Weinzierl, M.C. CD152 (CTLA-4) regulates effector functions of CD8+ T lymphocytes by repressing Eomesodermin. Eur. J. Immunol. 2009, 39, 883–893. [Google Scholar] [CrossRef]
- Rudd, C.E. CTLA-4 co-receptor impacts on the function of Treg and CD8+ T-cell subsets. Eur. J. Immunol. 2009, 39, 687–690. [Google Scholar] [CrossRef] [PubMed]
- Daneshmandi, S.; Wegiel, B.; Seth, P. Blockade of lactate dehydrogenase-A (LDH-A) improves efficacy of anti-programmed cell death-1 (PD-1) therapy in melanoma. Cancers 2019, 11, 450. [Google Scholar] [CrossRef] [PubMed]
- Kumar Kulabhusan, P.; Hussain, B.; Yüce, M. Current perspectives on aptamers as diagnostic tools and therapeutic agents. Pharmaceutics 2020, 12, 646. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.S.; Sansom, D.M. The emerging role of CTLA4 as a cell-extrinsic regulator of T cell responses. Nat. Rev. Immunol. 2011, 11, 852–863. [Google Scholar] [CrossRef] [PubMed]
- Krummel, M.F.; Allison, J.P. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med. 1995, 182, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.C.; Hughes, M.; Kammula, U.; Royal, R.; Sherry, R.M.; Topalian, S.L.; Suri, K.B.; Levy, C.; Allen, T.; Mavroukakis, S.; et al. Ipilimumab (anti-CTLA4 antibody) causes regression of metastatic renal cell cancer associated with enteritis and hypophysitis. J. Immunother. 2007, 30, 825. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Neyns, B.; Linette, G.; Negrier, S.; Lutzky, J.; Thomas, L.; Waterfield, W.; Schadendorf, D.; Smylie, M.; Guthrie, T.; et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: A randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol. 2010, 11, 155–164. [Google Scholar] [CrossRef]
- Nishino, M.; Ramaiya, N.H.; Hatabu, H.; Hodi, F.S. Monitoring immune-checkpoint blockade: Response evaluation and biomarker development. Nat. Rev. Clin. Oncol. 2017, 14, 655–668. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. Immune checkpoint targeting in cancer therapy: Toward combination strategies with curative potential. Cell 2015, 161, 205–214. [Google Scholar] [CrossRef]
- Martin, A.; Chahwan, R.; Parsa, J.Y.; Scharff, M.D. Somatic hypermutation: The molecular mechanisms underlying the production of effective high-affinity antibodies. In Molecular Biology of B Cells; Elsevier: Amsterdam, The Netherlands, 2015; pp. 363–388. [Google Scholar]
- Barroso-Sousa, R.; Jain, E.; Cohen, O.; Kim, D.; Buendia-Buendia, J.; Winer, E.; Lin, N.; Tolaney, S.; Wagle, N. Prevalence and mutational determinants of high tumor mutation burden in breast cancer. Ann. Oncol. 2020, 31, 387–394. [Google Scholar] [CrossRef]
- Mouradov, D.; Sloggett, C.; Jorissen, R.N.; Love, C.G.; Li, S.; Burgess, A.W.; Arango, D.; Strausberg, R.L.; Buchanan, D.; Wormald, S.; et al. Colorectal cancer cell lines are representative models of the main molecular subtypes of primary cancer. Cancer Res. 2014, 74, 3238–3247. [Google Scholar] [CrossRef] [PubMed]
- Sabri, M.Z.; Hamid, A.A.A.; Hitam, S.M.S.; Rahim, M.Z.A. In-silico selection of aptamer: A review on the revolutionary approach to understand the aptamer design and interaction through computational chemistry. Mater. Today Proc. 2019, 19, 1572–1581. [Google Scholar] [CrossRef]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ayass, M.A.; Tripathi, T.; Griko, N.; Okyay, T.; Ramankutty Nair, R.; Zhang, J.; Zhu, K.; Melendez, K.; Pashkov, V.; Abi-Mosleh, L. Dual Checkpoint Aptamer Immunotherapy: Unveiling Tailored Cancer Treatment Targeting CTLA-4 and NKG2A. Cancers 2024, 16, 1041. https://doi.org/10.3390/cancers16051041
Ayass MA, Tripathi T, Griko N, Okyay T, Ramankutty Nair R, Zhang J, Zhu K, Melendez K, Pashkov V, Abi-Mosleh L. Dual Checkpoint Aptamer Immunotherapy: Unveiling Tailored Cancer Treatment Targeting CTLA-4 and NKG2A. Cancers. 2024; 16(5):1041. https://doi.org/10.3390/cancers16051041
Chicago/Turabian StyleAyass, Mohamad Ammar, Trivendra Tripathi, Natalya Griko, Tutku Okyay, Ramya Ramankutty Nair, Jin Zhang, Kevin Zhu, Kristen Melendez, Victor Pashkov, and Lina Abi-Mosleh. 2024. "Dual Checkpoint Aptamer Immunotherapy: Unveiling Tailored Cancer Treatment Targeting CTLA-4 and NKG2A" Cancers 16, no. 5: 1041. https://doi.org/10.3390/cancers16051041
APA StyleAyass, M. A., Tripathi, T., Griko, N., Okyay, T., Ramankutty Nair, R., Zhang, J., Zhu, K., Melendez, K., Pashkov, V., & Abi-Mosleh, L. (2024). Dual Checkpoint Aptamer Immunotherapy: Unveiling Tailored Cancer Treatment Targeting CTLA-4 and NKG2A. Cancers, 16(5), 1041. https://doi.org/10.3390/cancers16051041