
Low-Dose Naltrexone as an Adjuvant in Combined Anticancer Therapy



Abstract
:Simple Summary
Abstract
1. Introduction
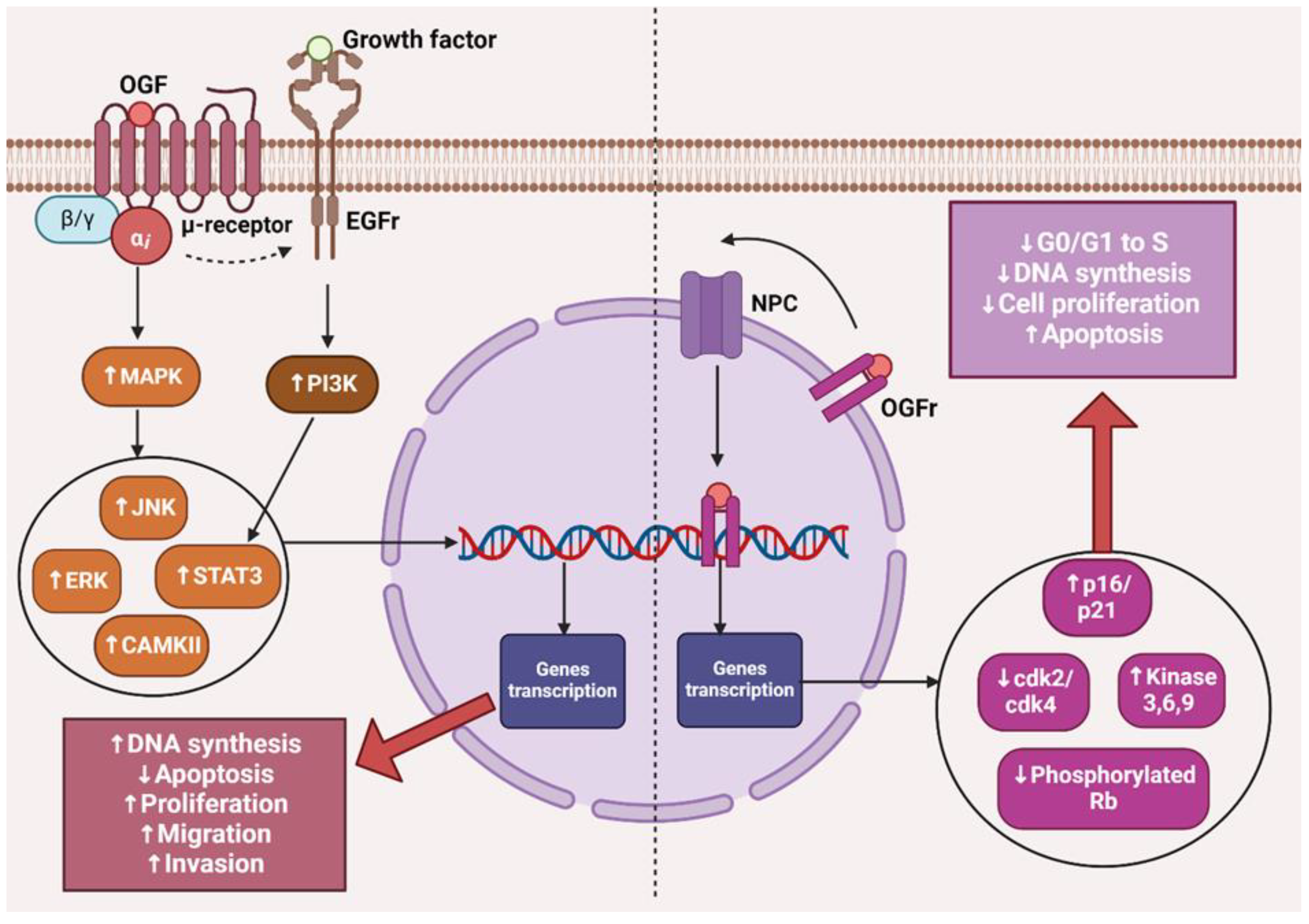
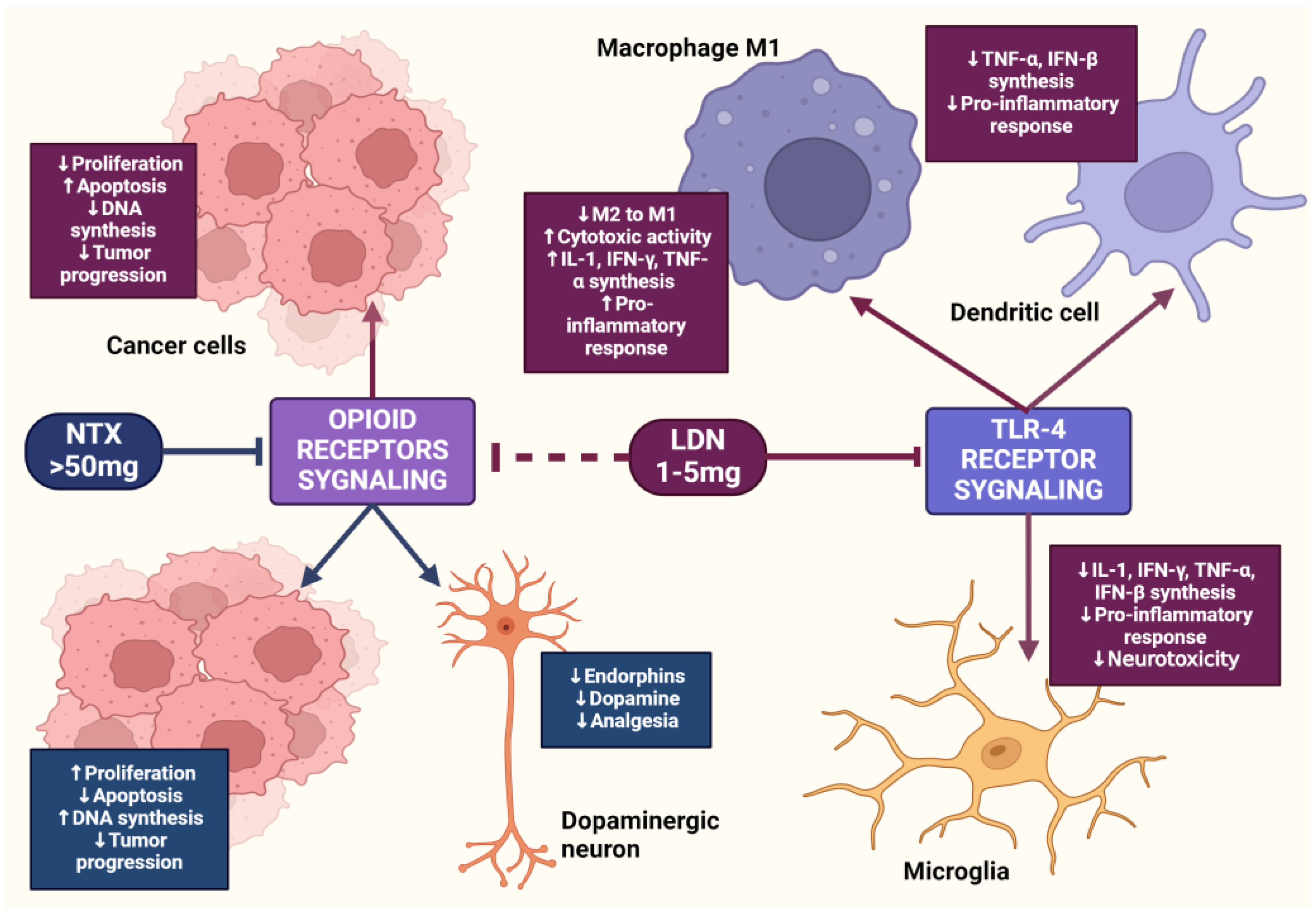
2. The µ Receptor and Cancer
3. The OGF–OGFr Axis
4. Naltrexone
4.1. Naltrexone Pharmacokinetics
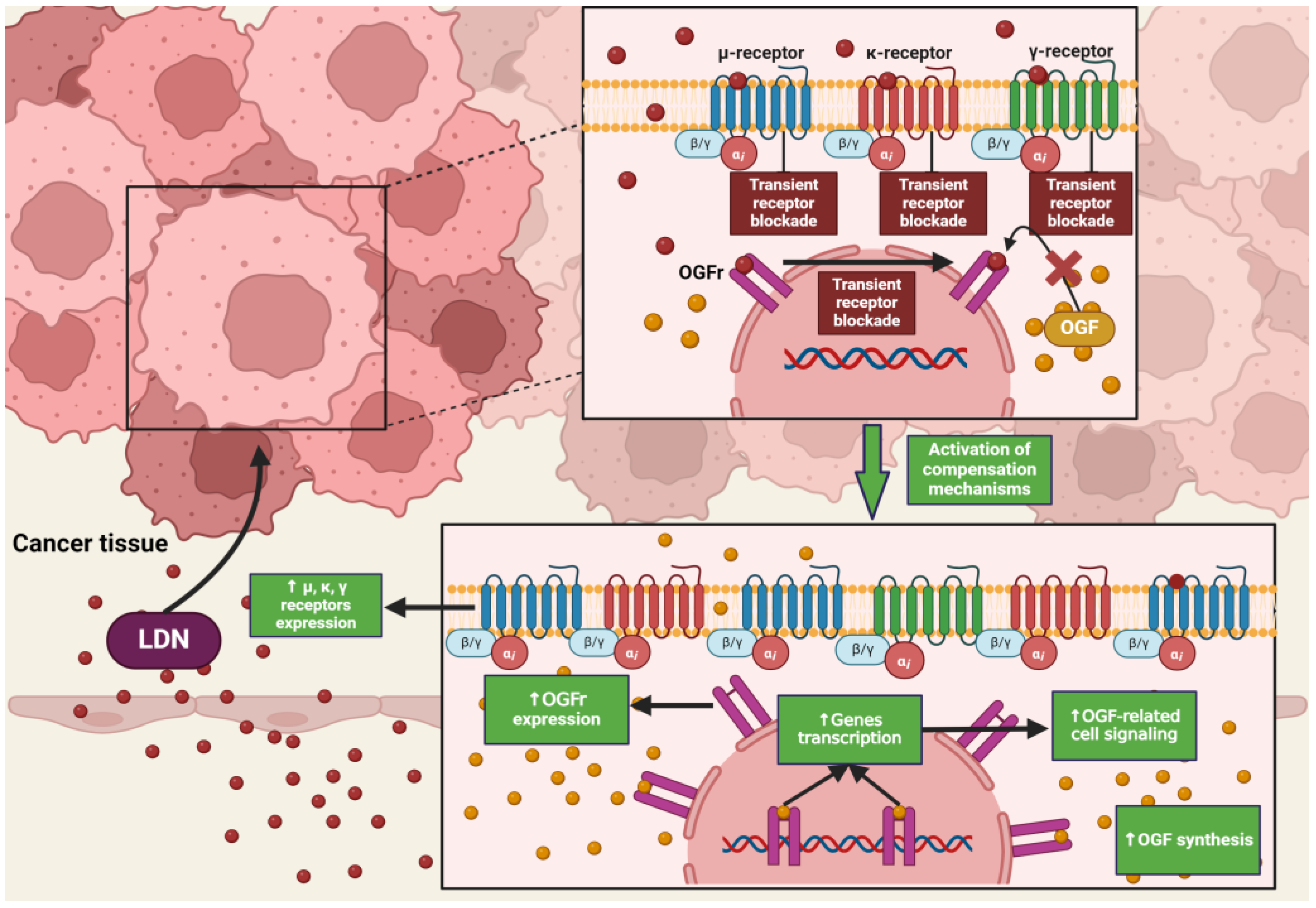
4.2. Low Doses of Naltrexone
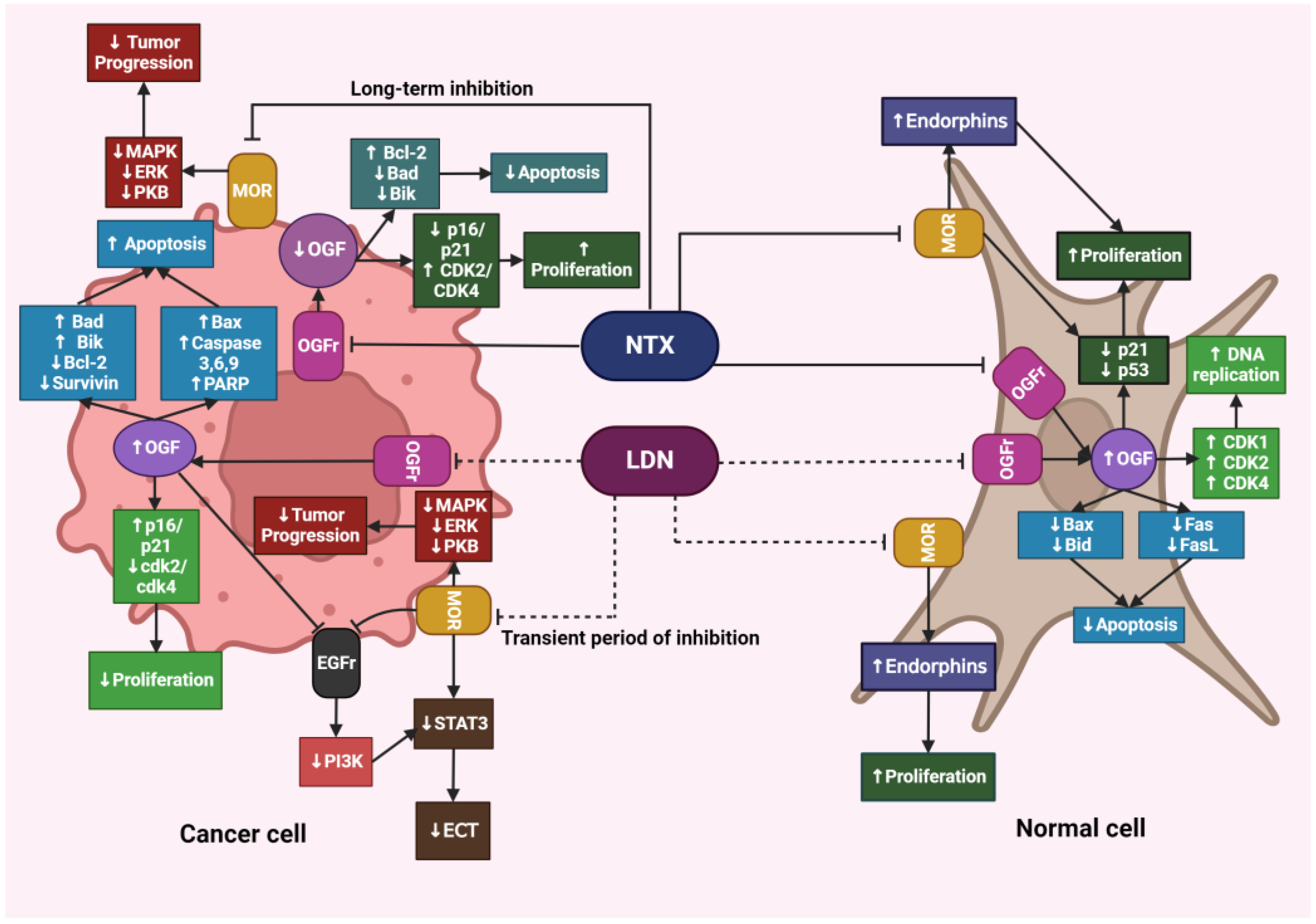
4.3. The Impact of LDN on Healthy Tissues
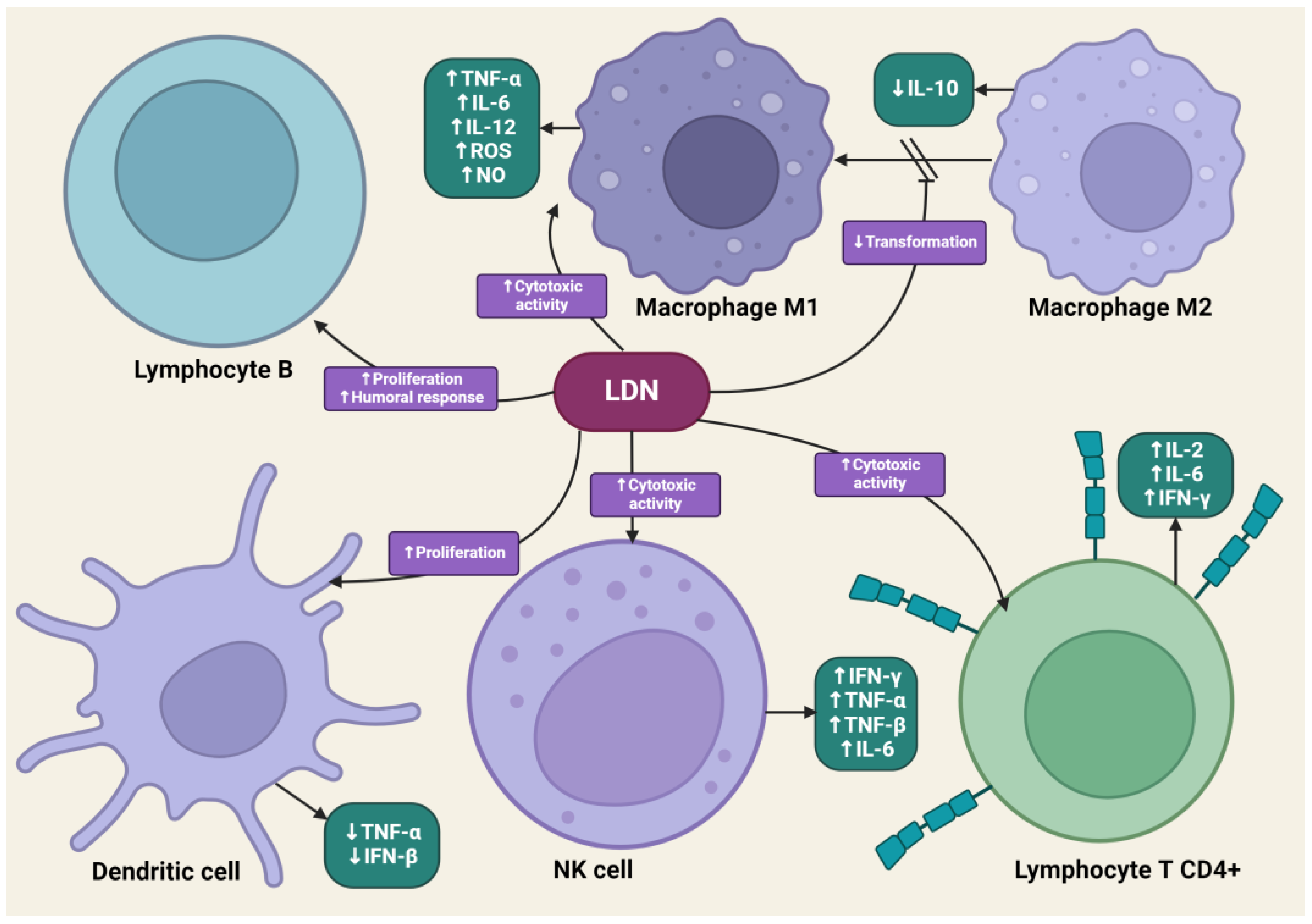
4.4. The Impact of LDN on the Immune System
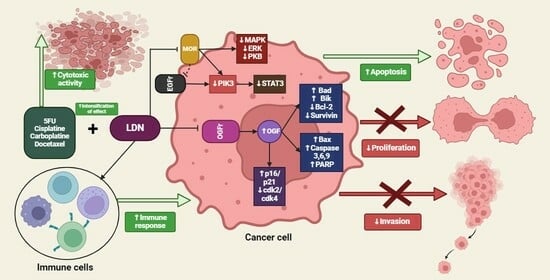
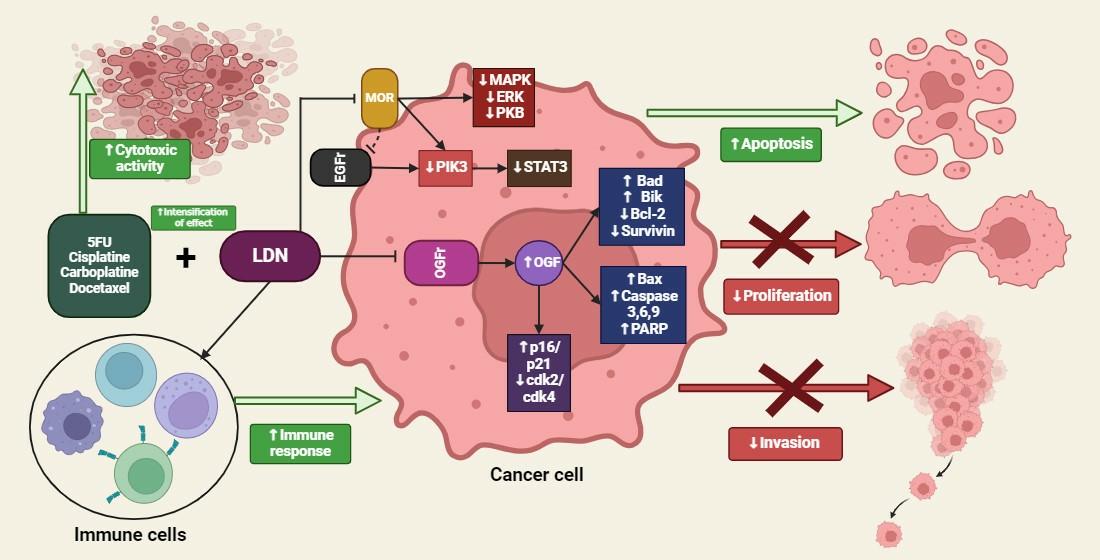
4.5. The Impact of LDN on Cancer Cells
4.6. Differences in the Action of LDN and NTX at Standard Doses
4.7. LDN in In Vitro and In Vivo Experimental Models
4.8. Synergistic Therapy
4.8.1. LDN and Immunotherapy
4.8.2. LDN and Cisplatin
4.8.3. LDN and Carboplatin
4.8.4. LDN and 5-Fluorouracil
4.8.5. Low-Dose Methylnaltrexone and 5-Fluorouracil
4.8.6. Low-Dose Methylnaltrexone and Docetaxel
4.8.7. LDN and Cannabidiol
4.8.8. LDN and Propranolol
4.8.9. LDN and Vitamin D
4.8.10. LDN and α-Lipoic Acid
4.9. LDN in Clinical Trials
5. Summary of the LDN Effects on Cancer Cells
6. Further Perspectives
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Van den Beuken-van Everdingen, M.H.J.; De Rijke, J.M.; Kessels, A.G.; Schouten, H.C.; Van Kleef, M.; Patijn, J. Prevalence of Pain in Patients with Cancer: A Systematic Review of the Past 40 Years. Ann. Oncol. 2007, 18, 1437–1449. [Google Scholar] [CrossRef]
- Montagna, G.; Gupta, H.V.; Hannum, M.; Tan, K.S.; Lee, J.; Scarpa, J.R.; Plitas, G.; Irie, T.; McCormick, P.J.; Fischer, G.W.; et al. Intraoperative Opioids Are Associated with Improved Recurrence-Free Survival in Triple-Negative Breast Cancer. Br. J. Anaesth. 2021, 126, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Patrad, E.; Khalighfard, S.; Khori, V.; Alizadeh, A.M. The other side of the coin: Positive view on the role of opioids in cancer. Eur. J. Pharmacol. 2022, 923, 174888. [Google Scholar] [CrossRef] [PubMed]
- Belltall, A.; Mazzinari, G.; Diaz-Cambronero, O.; Eroles, P.; Argente Navarro, M.P. Antagonists of the Mu-Opioid Receptor in the Cancer Patient: Fact or Fiction? Curr. Oncol. Rep. 2022, 24, 1337–1349. [Google Scholar] [CrossRef]
- Caraceni, A.; Hanks, G.; Kaasa, S.; Bennett, M.I.; Brunelli, C.; Cherny, N.; Dale, O.; De Conno, F.; Fallon, M.; Hanna, M.; et al. Use of opioid analgesics in the treatment of cancer pain: Evidence-based recommendations from the EAPC. Lancet Oncol. 2012, 13, e58–e68. [Google Scholar] [CrossRef]
- Machelska, H.; Celik, M.Ö. Advances in Achieving Opioid Analgesia Without Side Effects. Front. Pharmacol. 2018, 9, 1388. [Google Scholar] [CrossRef] [PubMed]
- Vallejo, R.; De Leon-Casasola, O.; Benyamin, R. Opioid therapy and immunosuppression: A review. Am. J. Ther. 2004, 11, 354–365. [Google Scholar] [CrossRef]
- Farooqui, M.; Li, Y.; Rogers, T.; Poonawala, T.; Griffin, R.J.; Song, C.W.; Gupta, K. COX-2 inhibitor celecoxib prevents chronic morphine-induced promotion of angiogenesis, tumour growth, metastasis and mortality, without compromising analgesia. Br. J. Cancer 2007, 97, 1523–1531. [Google Scholar] [CrossRef]
- Chen, C.; Farooqui, M.; Gupta, K. Morphine stimulates vascular endothelial growth factor-like signaling in mouse retinal endothelial cells. Curr. Neurovasc. Res. 2006, 3, 171–180. [Google Scholar] [CrossRef]
- Mathew, B.; Lennon, F.E.; Siegler, J.H.; Mirzapoiazova, T.; Mambetsariev, N.; Sammani, S.; Gerhold, L.M.; Riviere, P.J.L.; Chen, C.-T.; Garcia, J.G.N.; et al. Novel Role of the Mu Opioid Receptor in Lung Cancer Progression: A Laboratory Study. Anesth. Analg. 2011, 112, 558. [Google Scholar] [CrossRef]
- Jorand, R.; Biswas, S.; Wakefield, D.L.; Tobin, S.J.; Golfetto, O.; Hilton, K.; Ko, M.; Ramos, J.W.; Small, A.R.; Chu, P.; et al. Molecular signatures of mu opioid receptor and somatostatin receptor 2 in pancreatic cancer. Mol. Biol. Cell 2016, 27, 3659–3672. [Google Scholar] [CrossRef] [PubMed]
- Nylund, G.; Pettersson, A.; Bengtsson, C.; Khorram-Manesh, A.; Nordgren, S.; Delbro, D.S. Functional Expression of μ-Opioid Receptors in the Human Colon Cancer Cell Line, HT-29, and Their Localization in Human Colon. Dig. Dis. Sci. 2008, 53, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-F.; Xu, Q.-X.; Liao, L.-D.; Xu, X.-E.; Wu, J.-Y.; Wu, Z.-Y.; Shen, J.-H.; Li, E.-M.; Xu, L.-Y. Association of mu-opioid receptor expression with lymph node metastasis in esophageal squamous cell carcinoma. Esophageal Dis. 2015, 28, 196–203. [Google Scholar] [CrossRef]
- Zhang, H.; Sun, M.; Zhou, D.; Gorur, A.; Sun, Z.; Zeng, W.; Cata, J.P.; Chen, W.; Miao, C. Increased mu-opioid receptor expression is associated with reduced disease-free and overall survival in laryngeal squamous cell carcinoma. Br. J. Anaesth. 2020, 125, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.-S.; Yao, R.-Y.; Zhuang, L.-K.; Qi, W.-W.; Lv, J.; Zhou, F.; Qiu, W.-S.; Yue, L. MOR1 expression in gastric cancer: A biomarker associated with poor outcomes. Clin. Transl. Sci. 2015, 8, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.T.; Pan, J.H.; Chen, Y.H.; Xing, W.; Yan, Y.; Yuan, Y.F.; Zeng, W.A. The mu-opioid receptor is a molecular marker for poor prognosis in hepatocellular carcinoma and represents a potential therapeutic target. Br. J. Anaesth. 2019, 122, e157–e167. [Google Scholar] [CrossRef]
- Singleton, P.A.; Mirzapoiazova, T.; Hasina, R.; Salgia, R.; Moss, J. Increased μ-opioid receptor expression in metastatic lung cancer. Br. J. Anaesth. 2014, 113 (Suppl. 1), i103–i108. [Google Scholar] [CrossRef]
- Huang, H.; Liu, B.; Qu, N.; Zhang, S.; Bai, X.; Handley, M.; Shan, F. Research Progress of Opioid Growth Factor in Immune-Related Diseases and Cancer Diseases. Int. Immunopharmacol. 2021, 99, 107713. [Google Scholar] [CrossRef]
- Cheng, F.; McLaughlin, P.J.; Verderame, M.F.; Zagon, I.S. The OGF-OGFr axis utilizes the p16INK4a and p21WAF1/CIP1 pathways to restrict normal cell proliferation. Mol. Biol. Cell 2009, 20, 319–327. [Google Scholar] [CrossRef]
- McLaughlin, P.J.; Wylie, J.D.; Bloom, G.; Griffith, J.W.; Zagon, I.S. Chronic exposure to the opioid growth factor, [Met5]-enkephalin, during pregnancy: Maternal and preweaning effects. Pharmacol. Biochem. Behav. 2002, 71, 171–181. [Google Scholar] [CrossRef]
- Cheng, F.; McLaughlin, P.J.; Verderame, M.F.; Zagon, I.S. Dependence on Nuclear Localization Signals of the Opioid Growth Factor Receptor in the Regulation of Cell Proliferation. Exp. Biol. Med. 2009, 234, 532–541. [Google Scholar] [CrossRef]
- Zagon, I.S.; Goodman, S.R.; McLaughlin, P.J. Characterization of Opioid Binding Sites in Murine Neuroblastoma. Brain Res. 1988, 449, 80–88. [Google Scholar] [CrossRef]
- Zagon, I.S.; Gibo, D.; McLaughlin, P.J. Expression of Zeta (Zeta), a Growth-Related Opioid Receptor, in Metastatic Adenocarcinoma of the Human Cerebellum. J. Natl. Cancer Inst. 1990, 82, 325–327. [Google Scholar] [CrossRef]
- Martin, W.R. Opioid Antagonists. Pharmacol. Rev. 1967, 19, 463–521. [Google Scholar] [PubMed]
- Zagon, I.S.; Sassani, J.W.; McLaughlin, P.J. Re-epithelialization of the rat cornea is accelerated by blockade of opioid receptors. Brain Res. 1998, 798, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Rayess, H.; Wang, M.B.; Srivatsan, E.S. Cellular Senescence and Tumor Suppressor Gene P16. Int. J. Cancer 2012, 130, 1715–1725. [Google Scholar] [CrossRef] [PubMed]
- Abbas, T.; Dutta, A. P21 in Cancer: Intricate Networks and Multiple Activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef]
- Donahue, R.N.; McLaughlin, P.J.; Zagon, I.S. Low-Dose Naltrexone Targets the Opioid Growth Factor–Opioid Growth Factor Receptor Pathway to Inhibit Cell Proliferation: Mechanistic Evidence from a Tissue Culture Model. Exp. Biol. Med. 2011, 236, 1036–1050. [Google Scholar] [CrossRef]
- Zagon, I.S.; Goodman, S.R.; McLaughlin, P.J. Demonstration and characterization of zeta (zeta), a growth-related opioid receptor, in a neuroblastoma cell line. Brain Res. 1990, 511, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Qu, N.; Wang, X.; Meng, Y.; Shan, F. Prospective oncotarget for gynecological cancer: Opioid growth factor (OGF)—Opioid growth factor receptor (OGFr) axis. Int. Immunopharmacol. 2019, 75, 105723. [Google Scholar] [CrossRef] [PubMed]
- Sudakin, D. Naltrexone: Not Just for Opioids Anymore. J. Med. Toxicol. 2016, 12, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Wentland, M.P.; Lou, R.; Lu, Q.; Bu, Y.; Denhardt, C.; Jin, J.; Ganorkar, R.; VanAlstine, M.A.; Guo, C.; Cohen, D.J.; et al. Syntheses of Novel High Affinity Ligands for Opioid Receptors. Bioorg. Med. Chem. Lett. 2009, 19, 2289–2294. [Google Scholar] [CrossRef] [PubMed]
- Weerts, E.M.; Kim, Y.K.; Wand, G.S.; Dannals, R.F.; Lee, J.S.; Frost, J.J.; McCaul, M.E. Differences in Delta- and Mu-Opioid Receptor Blockade Measured by Positron Emission Tomography in Naltrexone-Treated Recently Abstinent Alcohol-Dependent Subjects. Neuropsychopharmacology 2008, 33, 653–665. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.C.; Ma, J.D.; Morello, C.M.; Atayee, R.S.; Best, B.M. Naltrexone Metabolism and Concomitant Drug Concentrations in Chronic Pain Patients. J. Anal. Toxicol. 2014, 38, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Onakpoya, I.J.; Lee, J.J.; Mahtani, K.R.; Aronson, J.K.; Heneghan, C.J. Naltrexone–Bupropion (Mysimba) in Management of Obesity: A Systematic Review and Meta-analysis of Unpublished Clinical Study Reports. Br. J. Clin. Pharmacol. 2020, 86, 646–667. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, E.J. Hormetic Mechanisms. Crit. Rev. Toxicol. 2013, 43, 580–606. [Google Scholar] [CrossRef] [PubMed]
- Center for Substance Abuse Treatment. Chapter 4—Oral Naltrexone. In Incorporating Alcohol Pharmacotherapies into Medical Practice; Treatment Improvement Protocol (TIP) Series No. 49; Substance Abuse and Mental Health Services Administration (US): Rockville, MD, USA, 2009. Available online: https://www.ncbi.nlm.nih.gov/books/NBK64042/ (accessed on 15 March 2024).
- Hospira Inc. Naloxone Prescribing Information 2024; Hospira Inc.: Lake Forest, IL, USA, 2024. [Google Scholar]
- Stancil, S.L.; Nolte, W.; Pearce, R.E.; Staggs, V.S.; Leeder, J.S. The Impact of Age and Genetics on Naltrexone Biotransformation. Drug Metab. Dispos. 2022, 50, 168–173. [Google Scholar] [CrossRef]
- Meyer, M.C.; Straughn, A.B.; Lo, M.W.; Schary, W.L.; Whitney, C.C. Bioequivalence, dose-proportionality, and pharmacokinetics of naltrexone after oral administration. J. Clin. Psychiatry 1984, 45, 15–19. [Google Scholar]
- AlRabiah, H.; Ahad, A.; Mostafa, G.A.; Al-Jenoobi, F.I. Effect of Naltrexone Hydrochloride on Cytochrome P450 1A2, 2C9, 2D6, and 3A4 Activity in Human Liver Microsomes. Eur. J. Drug Metab. Pharmacokinet. 2018, 43, 707–713. [Google Scholar] [CrossRef]
- McLaughlin, P.J.; Zagon, I.S. Duration of opioid receptor blockade determines biotherapeutic response. Biochem. Pharmacol. 2015, 97, 236–246. [Google Scholar] [CrossRef]
- Brown, N.; Panksepp, J. Low-Dose Naltrexone for Disease Prevention and Quality of Life. Med. Hypotheses 2009, 72, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Donahue, R.N.; McLaughlin, P.J.; Zagon, I.S. Low-dose naltrexone suppresses ovarian cancer and exhibits enhanced inhibition in combination with cisplatin. Exp. Biol. Med. 2011, 236, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Donahue, R.N.; McLaughlin, P.J.; Zagon, I.S. The opioid growth factor (OGF) and low dose naltrexone (LDN) suppress human ovarian cancer progression in mice. Gynecol. Oncol. 2011, 122, 382–388. [Google Scholar] [CrossRef] [PubMed]
- Tempel, A.; Gardner, E.L.; Zukin, R.S. Neurochemical and Functional Correlates of Naltrexone-Induced Opiate Receptor up-Regulation. J. Pharmacol. Exp. Ther. 1985, 232, 439–444. [Google Scholar]
- Sher, L. Combined Dexamethasone Suppression-Corticotropin-Releasing Hormone Stimulation Test in Studies of Depression, Alcoholism, and Suicidal Behavior. Sci. World J. 2006, 6, 1398–1404. [Google Scholar] [CrossRef]
- Zagon, I.S.; McLaughlin, P.J. Opioid antagonists inhibit the growth of metastatic murine neuroblastoma. Cancer Lett. 1983, 21, 89–94. [Google Scholar] [CrossRef]
- Zagon, I.S.; McLaughlin, P.J. Naltrexone modulates tumor response in mice with neuroblastoma. Science 1983, 221, 671–673. [Google Scholar] [CrossRef] [PubMed]
- San-Emeterio, E.P.; Hurlé, M.A. Modulation of brain apoptosis-related proteins by the opioid antagonist naltrexone in mice. Neurosci. Lett. 2006, 403, 276–279. [Google Scholar] [CrossRef]
- Meng, Y.; Gao, X.; Chen, W.; Plotnikoff, N.P.; Griffin, N.; Zhang, G.; Shan, F. Methionine enkephalin (MENK) mounts antitumor effect via regulating dendritic cells (DCs). Int. Immunopharmacol. 2017, 44, 61–71. [Google Scholar] [CrossRef]
- Meng, J.; Meng, Y.; Plotnikoff, N.P.; Youkilis, G.; Griffin, N.; Shan, F. Low dose naltrexone (LDN) enhances maturation of bone marrow dendritic cells (BMDCs). Int. Immunopharmacol. 2013, 17, 1084–1089. [Google Scholar] [CrossRef]
- Machado, M.C.; Da Costa-Neto, J.M.; Portela, R.D.; D’Assis, M.J.M.H.; Martins-Filho, O.A.; Barrouin-Melo, S.M.; Borges, N.F.; Silva, F.L.; Estrela-Lima, A. The effect of naltrexone as a carboplatin chemotherapy-associated drug on the immune response, quality of life and survival of dogs with mammary carcinoma. PLoS ONE 2018, 13, e0204830. [Google Scholar] [CrossRef]
- Boyadjieva, N.I.; Sarkar, D.K. Opioid-Like Activity of Naltrexone on Natural Killer Cell Cytolytic Activity and Cytokine Production in Splenocytes: Effects of Alcohol. J. Interferon Cytokine Res. 2010, 30, 15–22. [Google Scholar] [CrossRef]
- Azizi, H.; Mirzaeei, H.; Nasiri, A.A.; Bazi, A.; Mirzapour, A.; Khatami, M.; Nahavandi, K.H.; Azimi, A.; Yaghoobi, H. Naltrexone; as an efficient adjuvant in induction of Th1 immunity and protection against Fasciola hepatica infection. Exp. Parasitol. 2018, 189, 66–71. [Google Scholar] [CrossRef]
- Yi, Z.; Guo, S.; Hu, X.; Wang, X.; Zhang, X.; Griffin, N.; Shan, F. Functional modulation on macrophage by low dose naltrexone (LDN). Int. Immunopharmacol. 2016, 39, 397–402. [Google Scholar] [CrossRef]
- Rakaee, M.; Busund, L.R.; Jamaly, S.; Paulsen, E.E.; Richardsen, E.; Andersen, S.; Al-Saad, S.; Bremnes, R.M.; Donnem, T.; Kilvaer, T.K. Prognostic Value of Macrophage Phenotypes in Resectable Non-Small Cell Lung Cancer Assessed by Multiplex Immunohistochemistry. Neoplasia 2019, 21, 282–293. [Google Scholar] [CrossRef]
- Cheng, H.; Wang, Z.; Fu, L.; Xu, T. Macrophage Polarization in the Development and Progression of Ovarian Cancers: An Overview. Front. Oncol. 2019, 9, 421. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, M.M.; Cappello, P.; Nguyen, A.V.; Ralainirina, N.; Hardamon, C.R.; Foubert, P.; Schmid, M.C.; Sun, P.; Mose, E.; Bouvet, M.; et al. Macrophage PI3Kγ Drives Pancreatic Ductal Adenocarcinoma Progression. Cancer Discov. 2016, 6, 870–885. [Google Scholar] [CrossRef] [PubMed]
- Zagon, I.S.; Donahue, R.N.; Bonneau, R.H.; McLaughlin, P.J. B lymphocyte proliferation is suppressed by the opioid growth factor-opioid growth factor receptor axis: Implication for the treatment of autoimmune diseases. Immunobiology 2011, 216, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Yasaghi, M.; Mahdavi, M. Potentiation of Human Papilloma Vaccine Candidate Using Naloxone/Alum Mixture as an Adjuvant: Increasing Immunogenicity of HPV-16E7d Vaccine. Iran J. Basic Med. Sci. 2016, 19, 1003–1009. [Google Scholar]
- Ji, R.-R.; Xu, Z.-Z.; Gao, Y.-J. Emerging Targets in Neuroinflammation-Driven Chronic Pain. Nat. Rev. Drug Discov. 2014, 13, 533–548. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern Recognition Receptors and Inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Selfridge, B.R.; Wang, X.; Zhang, Y.; Yin, H.; Grace, P.M.; Watkins, L.R.; Jacobson, A.E.; Rice, K.C. Structure-Activity Relationships of (+)-Naltrexone-Inspired Toll-Like Receptor 4 (TLR4) Antagonists. J. Med. Chem. 2015, 58, 5038–5052. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, Y.; Peng, Y.; Hutchinson, M.R.; Rice, K.C.; Yin, H.; Watkins, L.R. Pharmacological Characterization of the Opioid Inactive Isomers (+)-naltrexone and (+)-naloxone as Antagonists of Toll-like Receptor 4. Br. J. Pharmacol. 2016, 173, 856–869. [Google Scholar] [CrossRef] [PubMed]
- Toljan, K.; Vrooman, B. Low-Dose Naltrexone (LDN)—Review of Therapeutic Utilization. Med. Sci. 2018, 6, 82. [Google Scholar] [CrossRef] [PubMed]
- Raknes, G.; Småbrekke, L. Low Dose Naltrexone in Multiple Sclerosis: Effects on Medication Use. A Quasi-Experimental Study. PLoS ONE 2017, 12, e0187423. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, Y.P. Low Dose Naltrexone Therapy in Multiple Sclerosis. Med. Hypotheses 2005, 64, 721–724. [Google Scholar] [CrossRef]
- Younger, J.; Mackey, S. Fibromyalgia Symptoms Are Reduced by Low-Dose Naltrexone: A Pilot Study. Pain Med. 2009, 10, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Raknes, G.; Småbrekke, L. Low Dose Naltrexone: Effects on Medication in Rheumatoid and Seropositive Arthritis. A Nationwide Register-Based Controlled Quasi-Experimental before-after Study. PLoS ONE 2019, 14, e0212460. [Google Scholar]
- Ma, M.; Wang, X.; Liu, N.; Shan, F.; Feng, Y. Low-Dose Naltrexone Inhibits Colorectal Cancer Progression and Promotes Apoptosis by Increasing M1-Type Macrophages and Activating the Bax/Bcl-2/Caspase-3/PARP Pathway. Int. Immunopharmacol. 2020, 83, 106388. [Google Scholar] [CrossRef]
- Liu, W.M.; Scott, K.A.; Dennis, J.L.; Kaminska, E.; Levett, A.J.; Dalgleish, A.G. Naltrexone at Low Doses Upregulates a Unique Gene Expression Not Seen with Normal Doses: Implications for Its Use in Cancer Therapy. Int. J. Oncol. 2016, 49, 793–802. [Google Scholar] [CrossRef]
- Liu, N.; Ma, M.; Qu, N.; Wang, R.; Chen, H.; Hu, F.; Gao, S.; Shan, F. Low-dose naltrexone inhibits the epithelial-mesenchymal transition of cervical cancer cells in vitro and effects indirectly on tumor-associated macrophages in vivo. Int. Immunopharmacol. 2020, 86, 106718. [Google Scholar] [CrossRef]
- Liu, N.; Yan, L.; Shan, F.; Wang, X.; Qu, N.; Handley, M.K.; Ma, M. Low-dose naltrexone plays antineoplastic role in cervical cancer progression through suppressing PI3K/AKT/mTOR pathway. Transl. Oncol. 2021, 14, 101028. [Google Scholar] [CrossRef]
- McLaughlin, P.J.; Stucki, J.K.; Zagon, I.S. Modulation of the Opioid Growth Factor ([Met5]-Enkephalin)–Opioid Growth Factor Receptor Axis: Novel Therapies for Squamous Cell Carcinoma of the Head and Neck. Head Neck 2012, 34, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Qu, N.; Meng, Y.; Handley, M.K.; Wang, C.; Shan, F. Preclinical and clinical studies into the bioactivity of low-dose naltrexone (LDN) for oncotherapy. Int. Immunopharmacol. 2021, 96, 107714. [Google Scholar] [CrossRef] [PubMed]
- Zagon, I.S.; McLaughlin, P.J. Opioid growth factor and the treatment of human pancreatic cancer: A review. World J. Gastroenterol. 2014, 20, 2218–2223. [Google Scholar] [CrossRef] [PubMed]
- Budka, J.; Kowalski, S.; Chylinska, M.; Dzierzbicka, K.; Inkielewicz-Stepniak, I. Opioid Growth Factor and its Derivatives as Potential Non-toxic Multifunctional Anticancer and Analgesic Compounds. Curr. Med. Chem. 2021, 28, 673–686. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, M.L.; Rodríguez, F.D.; Coveñas, R. Involvement of the Opioid Peptide Family in Cancer Progression. Biomedicines 2023, 11, 1993. [Google Scholar] [CrossRef]
- Lei, Z.N.; Tian, Q.; Teng, Q.X.; Wurpel, J.N.D.; Zeng, L.; Pan, Y.; Chen, Z.S. Understanding and targeting resistance mechanisms in cancer. MedComm 2023, 4, e265. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Zhou, C.; Ren, S. Role of IL-2 in Cancer Immunotherapy. Oncoimmunology 2016, 5, e1163462. [Google Scholar] [CrossRef]
- Yui, M.A.; Sharp, L.L.; Havran, W.L.; Rothenberg, E.V. Preferential Activation of an IL-2 Regulatory Sequence Transgene in TCR Gamma Delta and NKT Cells: Subset-Specific Differences in IL-2 Regulation. J. Immunol. 2004, 172, 4691–4699. [Google Scholar] [CrossRef]
- Paliard, X.; De Waal Malefijt, R.; Yssel, H.; Blanchard, D.; Chrétien, I.; Abrams, J.; De Vries, J.; Spits, H. Simultaneous Production of IL-2, IL-4, and IFN-Gamma by Activated Human CD4+ and CD8+ T Cell Clones. J. Immunol. 1988, 141, 849–855. [Google Scholar] [CrossRef]
- Leonard, W.J.; Krönke, M.; Peffer, N.J.; Depper, J.M.; Greene, W.C. Interleukin 2 Receptor Gene Expression in Normal Human T Lymphocytes. Proc. Natl. Acad. Sci. USA 1985, 82, 6281–6285. [Google Scholar] [CrossRef]
- Littman, D.R.; Rudensky, A.Y. Th17 and Regulatory T Cells in Mediating and Restraining Inflammation. Cell 2010, 140, 845–858. [Google Scholar] [CrossRef]
- Rosenberg, S.A. IL-2: The first effective immunotherapy for human cancer. J. Immunol. 2014, 192, 5451–5458. [Google Scholar] [CrossRef]
- Atkins, M.B.; Sparano, J.; Fisher, R.I.; Weiss, G.R.; Margolin, K.A.; Fink, K.I.; Rubinstein, L.; Louie, A.; Mier, J.W.; Gucalp, R. Randomized Phase II Trial of High-Dose Interleukin-2 Either Alone or in Combination with Interferon Alfa-2b in Advanced Renal Cell Carcinoma. J. Clin. Oncol. 1993, 11, 661–670. [Google Scholar] [CrossRef]
- McDermott, D.F.; Regan, M.M.; Clark, J.I.; Flaherty, L.E.; Weiss, G.R.; Logan, T.F.; Kirkwood, J.M.; Gordon, M.S.; Sosman, J.A.; Ernstoff, M.S.; et al. Randomized Phase III Trial of High-Dose Interleukin-2 versus Subcutaneous Interleukin-2 and Interferon in Patients with Metastatic Renal Cell Carcinoma. J. Clin. Oncol. 2005, 23, 133–141. [Google Scholar] [CrossRef]
- Negrier, S.; Escudier, B.; Lasset, C.; Douillard, J.Y.; Savary, J.; Chevreau, C.; Ravaud, A.; Mercatello, A.; Peny, J.; Mousseau, M.; et al. Recombinant Human Interleukin-2, Recombinant Human Interferon Alfa-2a, or Both in Metastatic Renal-Cell Carcinoma. Groupe Français d’Immunothérapie. N. Engl. J. Med. 1998, 338, 1272–1278. [Google Scholar] [CrossRef] [PubMed]
- Mazumder, A.; Grimm, E.A.; Zhang, H.Z.; Rosenberg, S.A. Lysis of Fresh Human Solid Tumors by Autologous Lymphocytes Activated in Vitro with Lectins. Cancer Res. 1982, 42, 913–918. [Google Scholar] [PubMed]
- Dudley, M.E.; Wunderlich, J.R.; Yang, J.C.; Sherry, R.M.; Topalian, S.L.; Restifo, N.P.; Royal, R.E.; Kammula, U.; White, D.E.; Mavroukakis, S.A.; et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J. Clin. Oncol. 2005, 23, 2346–2357. [Google Scholar] [CrossRef] [PubMed]
- Keilholz, U.; Goey, S.H.; Punt, C.J.; Proebstle, T.M.; Salzmann, R.; Scheibenbogen, C.; Schadendorf, D.; Liénard, D.; Enk, A.; Dummer, R.; et al. Interferon Alfa-2a and Interleukin-2 with or without Cisplatin in Metastatic Melanoma: A Randomized Trial of the European Organization for Research and Treatment of Cancer Melanoma Cooperative Group. J. Clin. Oncol. 1997, 15, 2579–2588. [Google Scholar] [CrossRef] [PubMed]
- Van Elsas, A.; Hurwitz, A.A.; Allison, J.P. Combination Immunotherapy of B16 Melanoma Using Anti–Cytotoxic T Lymphocyte–Associated Antigen 4 (Ctla-4) and Granulocyte/Macrophage Colony-Stimulating Factor (Gm-Csf)-Producing Vaccines Induces Rejection of Subcutaneous and Metastatic Tumors Accompanied by Autoimmune Depigmentation. J. Exp. Med. 1999, 190, 355–366. [Google Scholar]
- Maker, A.V.; Phan, G.Q.; Attia, P.; Yang, J.C.; Sherry, R.M.; Topalian, S.L.; Kammula, U.S.; Royal, R.E.; Haworth, L.R.; Levy, C.; et al. Tumor Regression and Autoimmunity in Patients Treated with Cytotoxic T Lymphocyte–Associated Antigen 4 Blockade and Interleukin 2: A Phase I/II Study. Ann. Surg. Oncol. 2005, 12, 1005–1016. [Google Scholar] [CrossRef]
- Hannani, D.; Vétizou, M.; Enot, D.; Rusakiewicz, S.; Chaput, N.; Klatzmann, D.; Desbois, M.; Jacquelot, N.; Vimond, N.; Chouaib, S.; et al. Anticancer Immunotherapy by CTLA-4 Blockade: Obligatory Contribution of IL-2 Receptors and Negative Prognostic Impact of Soluble CD25. Cell Res. 2015, 25, 208–224. [Google Scholar] [CrossRef] [PubMed]
- Parkitny, L.; Younger, J. Reduced Pro-Inflammatory Cytokines after Eight Weeks of Low-Dose Naltrexone for Fibromyalgia. Biomedicines 2017, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Lissoni, P.; Malugani, F.; Bordin, V.; Conti, A.; Maestroni, G.; Tancini, G. A New Neuroimmunotherapeutic Strategy of Subcutaneous Low-Dose Interleukin-2 plus the Long-Acting Opioid Antagonist Naltrexone in Metastatic Cancer Patients Progressing on Interleukin-2 Alone. Neuro. Endocrinol. Lett. 2002, 23, 255–258. [Google Scholar]
- Lissoni, P.; Malugani, F.; Malysheva, O.; Kozlov, V.; Laudon, M.; Conti, A.; Maestroni, G. Neuroimmunotherapy of Untreatable Metastatic Solid Tumors with Subcutaneous Low-Dose Interleukin-2, Melatonin and Naltrexone: Modulation of Interleukin-2-Induced Antitumor Immunity by Blocking the Opioid System. Neuro. Endocrinol. Lett. 2002, 23, 341–344. [Google Scholar] [PubMed]
- Aboalsoud, A.; El-Ghaiesh, S.H.; Abd Elmonem, F.F.; Salem, M.L.; Abdel Rahman, M.N. The effect of low-dose naltrexone on solid Ehrlich carcinoma in mice: The role of OGFr, BCL2, and immune response. Int. Immunopharmacol. 2020, 78, 106068. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-Z.; Li, X.-L.; Sun, S.; Xie, J.-T.; Aung, H.H.; Tong, R.; McEntee, E.; Yuan, C.-S. Methylnaltrexone, a Peripherally Acting Opioid Receptor Antagonist, Enhances Tumoricidal Effects of 5-Fu on Human Carcinoma Cells. Anticancer Res. 2009, 29, 2927–2932. [Google Scholar] [PubMed]
- Singleton, P.A.; Garcia, J.G.N.; Moss, J. Synergistic Effects of Methylnaltrexone with 5-Fluorouracil and Bevacizumab on Inhibition of Vascular Endothelial Growth Factor–Induced Angiogenesis. Mol. Cancer Ther. 2008, 7, 1669–1679. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Chiwaki, F.; Sawada, Y.; Ashikawa, M.; Aoyagi, K.; Fujita, T.; Yanagihara, K.; Komatsu, M.; Narita, M.; Suzuki, T.; et al. Peripheral Opioid Antagonist Enhances the Effect of Anti-Tumor Drug by Blocking a Cell Growth-Suppressive Pathway In Vivo. PLoS ONE 2015, 10, e0123407. [Google Scholar] [CrossRef]
- Massi, P.; Solinas, M.; Cinquina, V.; Parolaro, D. Cannabidiol as Potential Anticancer Drug. Br. J. Clin. Pharmacol. 2013, 75, 303–312. [Google Scholar] [CrossRef]
- Liu, W.M.; Hall, N.K.; Liu, H.S.Y.; Hood, F.L.; Dalgleish, A.G. Combination of Cannabidiol with Low-dose Naltrexone Increases the Anticancer Action of Chemotherapy in Vitro and in Vivo. Oncol. Rep. 2022, 47, 76. [Google Scholar] [CrossRef] [PubMed]
- Murugan, S.; Rousseau, B.; Sarkar, D.K. Beta 2 Adrenergic Receptor Antagonist Propranolol and Opioidergic Receptor Antagonist Naltrexone Produce Synergistic Effects on Breast Cancer Growth Prevention by Acting on Cancer Cells and Immune Environment in a Preclinical Model of Breast Cancer. Cancers 2021, 13, 4858. [Google Scholar] [CrossRef] [PubMed]
- Jordan, B.A. Oligomerization of Opioid Receptors with Beta 2-Adrenergic Receptors: A Role in Trafficking and Mitogen-Activated Protein Kinase Activation. Proc. Natl. Acad. Sci. USA 2001, 98, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Khan, A. Long-term remission of adenoid cystic tongue carcinoma with low dose naltrexone and vitamin D3—A case report. Oral Health Dent. Manag. 2014, 13, 721–724. [Google Scholar] [PubMed]
- Berkson, B.M.; Calvo Riera, F. The Long-Term Survival of a Patient with Stage IV Renal Cell Carcinoma Following an Integrative Treatment Approach Including the Intravenous α-Lipoic Acid/Low-Dose Naltrexone Protocol. Integr. Cancer Ther. 2017, 17, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Kadhim, M.M.; Turki Jalil, A.; Qasim Alasheqi, M.; Alsaikhan, F.; Khalimovna Mukhamedova, N.; Alexis Ramírez-Coronel, A.; Hassan Jawhar, Z.; Ramaiah, P.; Najafi, M. The interactions of docetaxel with tumor microenvironment. Int. Immunopharmacol. 2023, 119, 110214. [Google Scholar] [CrossRef]
- Peters, K. Effects of Low Dose Naltrexone on Quality of Life in High Grade Glioma Patients: A Placebo-Controlled, Double-Blind Randomized Trial; Clinical Trial Registration NCT01303835. 2015. Available online: https://www.clinicaltrials.gov (accessed on 15 March 2024).
- Lissoni, P.; Meregalli, S.; Fossati, V.; Barni, S.; Tancini, G.; Barigozzi, P.; Frigerio, F. Radioendocrine Therapy of Brain Tumors with the Long Acting Opioid Antagonist Naltrexone in Association with Radiotherapy. Tumori 1993, 79, 198–201. [Google Scholar] [CrossRef]
- Brown University. Low Dose Naltrexone for Metastatic Melanoma, Castrate Resistant Prostate Cancer and Renal Cancer: A Phase II Brown University Oncology Group Research Project; Clinical Trial Registration NCT01650350. 2022. Available online: https://www.clinicaltrials.gov (accessed on 15 March 2024).
- Stephenson, R. A Phase I Study to Evaluate the Safety of Naltrexone and Propranolol in Combination with Standard of Care Ipilimumab and Nivolumab in Patients with Advanced Melanoma; Clinical Trial Registration NCT05968690. 2023. Available online: https://www.clinicaltrials.gov (accessed on 15 March 2024).
- Janku, F.; Johnson, L.K.; Karp, D.D.; Atkins, J.T.; Singleton, P.A.; Moss, J. Treatment with Methylnaltrexone Is Associated with Increased Survival in Patients with Advanced Cancer. Ann. Oncol. 2016, 27, 2032–2038. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LDN/Low-Dose MNTX Concentration/Dose | Cancer | Results | References |
|---|---|---|---|
| LDN 0.5, 1.5, 2, 3 and 5 mg/mL | In vitro model of Hela and Siha, cervical cancer cells | LDN inhibits the proliferation of cervical cancer cells in a time- and dose-dependent manner. After 48 h of LDN treatment, the IC50 was 1.26 mg/mL. After treatment with LDN for 48 h, the inhibition rates of different concentrations (0.5 mg/mL, 1.5 mg/mL, 2 mg/mL, 3 mg/mL, 5 mg/mL) were 17.27 ± 5%, 47.44 ± 3%, 68.59 ± 4%, 84.68 ± 1%, and 95.47 ± 1%, respectively. | [73] |
| LDN 1 nM LDN 10 nM | In vitro model of human colorectal cancer cell lines HCT116 and human lung cancer cell lines A549 | Cell counting experiments revealed that the reduction in cell number was associated with a fall in cell viability, which suggests an active cytotoxic response was achieved. Flow cytometric analysis of the cell cycle showed significant increases in the sub-G1 peak following an LDN-then-recovery schedule with concomitant emptying of cells from G1 and G2. | [72] |
| Low-dose MNTX 0.10–100 nM | In vitro model of human non-small cell lung cancer cells lines NSCLC | Treatment with MNTX inhibited cell invasion and anchorage-independent growth by 50–80%. | [10] |
| LDN intraperitoneal (IP) injection 0.1 mg/kg | SCC-1 oral squamous cell carcinoma xenografts in Foxn1nu (nude) mice | LDN increased the latency from visible to measurable tumors up to 1.6-fold. OGF, low-dose naltrexone, and imiquimod treatment markedly reduced tumor volume and weight and decreased DNA synthesis in tumors. | [76] |
| LDN 0.1 mg/kg | SKOV-3 human ovarian cancer xenografts in athymic nu/nu female mice | LDN-treated mice displayed a visible reduction in tumor burden relative to the control group. Compared to the total number of nodules detected in the control group, animals treated with LDN displayed a 39% reduction. | [45] |
| LDN 0.5 mg/kg, LDN 5 mg/kg LDN 10 mg/kg | Hela and Siha human cervical cancer xenografts in BALB/C nude mice | LDN significantly decreased the expression of PI3K, PDK1, and mTOR. There was no difference in the expression of VEGF and AKT, but the expression of pVEGFR2 and pAKT was downregulated. The expression of pVEGFR, PI3K, PDK1, pAKT, and mTOR significantly reduced after LDN treatment, especially in the 10 mg/kg group. Compared to the control group, the 10 mg/kg LDN treatment group showed significant differences in tumor growth inhibition from day 22 of the treatment, while the 5 mg/kg LDN-treated group showed such differences from day 31. The time of significant difference in mice treated with 0.5 mg/kg LDN was 34 days | [74] |
| LDN 0.5 mg/kg, LDN 5 mg/kg, LDN 10 mg/kg | Human cervical cancer cell lines Hela and Siha, xeno-grafts in BALB/C nude mice | The ratio of M2 macrophage membrane markers labeled with CD206+ showed a decrease in the LDN group compared with the control group. The proportion of TAMs significantly reduced after LDN treatment, especially in the 10 mg/kg group. LDN suppressed the M2 macrophages and reduced the expression of IL-10. | [73] |
| Co-Treatments | Cancer | Mechanism/Results | References |
|---|---|---|---|
| LDN (10–5 mol/L), Taxol (10–9 or 10–10 mol/L), Cisplatin (0.01 or 0.001 µg/mL) | In vitro studies conducted on human ovarian cancer cell line SKOV-3 | The number of cells exposed short-term to LDN and taxol was 36–61% lower compared to cells exposed only to LDN, and 19–31% lower compared to cells exposed only to taxol. The number of cells exposed to the short-term effects of LDN and cisplatin was reduced by 21–42% compared to cells exposed only to cisplatin. | [44] |
| Methylnaltrexone (1 µM) 5-Fluorouracil (10 µM) | In vitro studies conducted on human colorectal cancer cell lines SW-480, human breast cancer MCF-7, and non-small cell lung cancer cells | Inhibition of growth and proliferation by 63.5% in SW-480 cells, 58.3% in MCF-7 cells, and 81.3% in non-small cell lung cancer cells compared to groups treated only with 5FU. | [100] |
| MNTX (100 nmol/L), 5-FU (5 μmol/L), Bevacizumab (25 ng/mL) | In vitro studies conducted on human pulmonary microvascular EC (HPMVEC) | Methylnaltrexone (MNTX), synergistically with 5-FU and bevacizumab, inhibited vascular endothelial growth factor (VEGF)-induced human pulmonary microvascular endothelial cell (EC) proliferation and migration. MNTX inhibited EC proliferation with an IC(50) of approximately 100 nmol/L. The addition of MNTX to EC shifted the IC(50) of 5-FU from approximately 5 micromol/L to approximately 7 nmol/L. The addition of 50 MNTX shifted the IC(50) of bevacizumab in inhibiting EC migration from approximately 25 to approximately 6 ng/mL. RPTPμ activation inhibits VEGF-induced Src activation (target of bevacizumab). MNTX-induced Src inactivation results in activation of p190 RhoGAP and inhibition of active RhoA, which prevents reorganization of the actin cytoskeleton (targeted by 5-FU) and the resulting EC proliferation (targeted by 5-FU) and migration. | [101] |
| Naltrexone (10 nM–10 µM) Cannabidiol (CBD) (1 µM) | A549 (human lung cancer) and HCT116 (human colorectal cancer) cells | LDN and CBD reduced the number of cells. There was a 35% reduction in cell numbers when using LDN before CBD compared to a 22% reduction when using CBD before LDN. | [104] |
| LDN (0.1 mg/kg daily), Taxol (3 mg/kg, days 0, 7, 14, 21, 28, 35), Cisplatin (4 mg/kg days 0 and 7) intraperitoneal injections | Human ovarian cancer xenografts in female nude mice | Administration of NTX for six hours every two days, but not continuously, reduced DNA synthesis and cell replication compared to the control group. The combination of LDN with cisplatin, but not taxol, resulted in an additive inhibitory effect on tumorigenesis with enhanced depression of DNA synthesis and angiogenesis. | [44] |
| LDN (0.1 mg/kg), 5FU (20 mg/kg) subcutaneous injection | Human ovarian cancer xenografts in nude mice | A decrease in tumor mass and volume and an increase in the number of splenocytes, with a tendency to decrease the number of MDSC cells were observed. LDN led to an increase in OGFr both alone and in combination with 5FU, increased serum IFN-γ levels, but decreased when combined with 5-FU. The use of LDN and 5FU increased the expression of p21 and decreased Bcl2. | [99] |
| Low-Dose Methylnaltrexone (0.3 mg/kg) Docetaxel (Doc) (0.5 mg/kg) | 60As6 human gastric cancer xenograft in female C.B17/Icr-scid mice | The growth of cells obtained from mice treated with a low-dose MNTX and Doc was significantly lower compared to mice treated with Doc only (Doc: 65.3 ± 6.6%, Doc/MNTX: 40.5 ± 7.1%). The use of Doc and low-dose MNTX polytherapy significantly extended life and alleviated cancer-related pain compared to mice treated with Doc only. | [102] |
| LDN (1.2 µg/mouse), CBD (35 µg/mouse), Gemcitabine (9 µg/mouse) | HCT116 colon cancer xenograft in athymic nu/nu BALB/c mice | The use of both compounds enhanced the effects of gemcitabine, without toxic effects. | [104] |
| NTX (0.001 µM to 200 µM) Propranolol (PRO) (0.001 µM to 200 µM) | Human breast cancer cells MDA-MB-231, MDA-MB-468, and T47D MDA-MB-231 xenograft in nude rat | Antitumor effects were observed due to the arrest of cell growth. NTX promoted PRO effects on expanded NK cells from the spleens and PBMCs of tumor xenografted animals. PRO and NTX increased the levels of NK cell-modulating cytokines while decreasing the levels of Th1 inflammatory cytokines. | [105] |
| LDN (0.1 mg/kg dose every 24 h for 24 weeks) orally, Carboplatin (300 mg/m2) intravenously | 60 female dogs with mammary neoplasia | The higher serum concentrations of beta-endorphin and met-enkephalin, fewer chemotherapy-related side effects, and better quality of life and survival rates in the LDN-treated groups than in LDN-untreated groups. Evaluation of clinical and pathological parameters indicated a significant association between the use of LDN and prolonged survival, as well as enhanced quality of life. | [53] |
| NTX (100 mg) orally IL-2 (6 million lU/day subcutaneously for 6 days/week for 4 weeks) | 14 consecutive untreatable metastatic solid tumor patients | The concomitant administration of NTX induced a significantly higher increase in lymphocyte mean number than that achieved with IL-2 plus MLT alone. | [44] |
| LDN (4.5 mg) α-Lipoic Acid (ALA) (300–600 mg) | 64-year-old male patient diagnosed with metastatic renal cell carcinoma (RCC) | ALA could inhibit cancer cell growth by inhibiting the pro-inflammatory transcription factor, nuclear factor κ light chain enhancer of activated B cells (NF-κB). ALA, by inhibiting pyruvate dehydrogenase kinase (PDK), increases the activity of pyruvate dehydrogenase (PDHC), i.e., enzymes in the Warburg effect, inhibiting tumor development. Short-term opioid receptor blockade caused by LDN increases the production of enkephalin peptide, which, upon binding to OGFr, inhibits the proliferation of cancer cells. | [108] |
| LDN (3 mg) Vitamin D (10,000 IU daily) | 58-year-old patient suffering from tonsillar-cystic tongue cancer without metastases | The patient has achieved nearly a four-year remission of his cancer based on his clinical status and the last MRI scan. LDN increases levels of the endogenous opioid methionine-enkephalin, which regulates cell proliferation and may inhibit the growth of cancer cells. | [107] |
| NCT Number | Status | Cancer | Treatment | Phase | Participants | Results/Comments | References |
|---|---|---|---|---|---|---|---|
| NCT05968690 | Study Start (Actual) | Advanced Melanoma | Propranolol 30 mg + Naltrexone 4.5 mg | I | 12 | Study Completion (Estimated) 30 September 2025 | [109] |
| NCT01650350 | Enrollment (Actual) | Melanoma, Prostate Cancer, Renal Cancer | LDN, 5 mg/day − (1 cycle = 28 days) | II | 7 | Results N/A | [108] |
| NCT01303835 | Enrollment (Actual) | Glioma | LDN, 4.5 mg | II | 110 | QOL and fatigue changes between baseline and post-concurrent chemotherapy and radiation therapy were not significantly different between patients receiving LDN or placebo. | [107] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciwun, M.; Tankiewicz-Kwedlo, A.; Pawlak, D. Low-Dose Naltrexone as an Adjuvant in Combined Anticancer Therapy. Cancers 2024, 16, 1240. https://doi.org/10.3390/cancers16061240
Ciwun M, Tankiewicz-Kwedlo A, Pawlak D. Low-Dose Naltrexone as an Adjuvant in Combined Anticancer Therapy. Cancers. 2024; 16(6):1240. https://doi.org/10.3390/cancers16061240
Chicago/Turabian StyleCiwun, Marianna, Anna Tankiewicz-Kwedlo, and Dariusz Pawlak. 2024. "Low-Dose Naltrexone as an Adjuvant in Combined Anticancer Therapy" Cancers 16, no. 6: 1240. https://doi.org/10.3390/cancers16061240
APA StyleCiwun, M., Tankiewicz-Kwedlo, A., & Pawlak, D. (2024). Low-Dose Naltrexone as an Adjuvant in Combined Anticancer Therapy. Cancers, 16(6), 1240. https://doi.org/10.3390/cancers16061240