
Immune Therapies in AL Amyloidosis—A Glimpse to the Future


Abstract
:Simple Summary
Abstract
1. Introduction
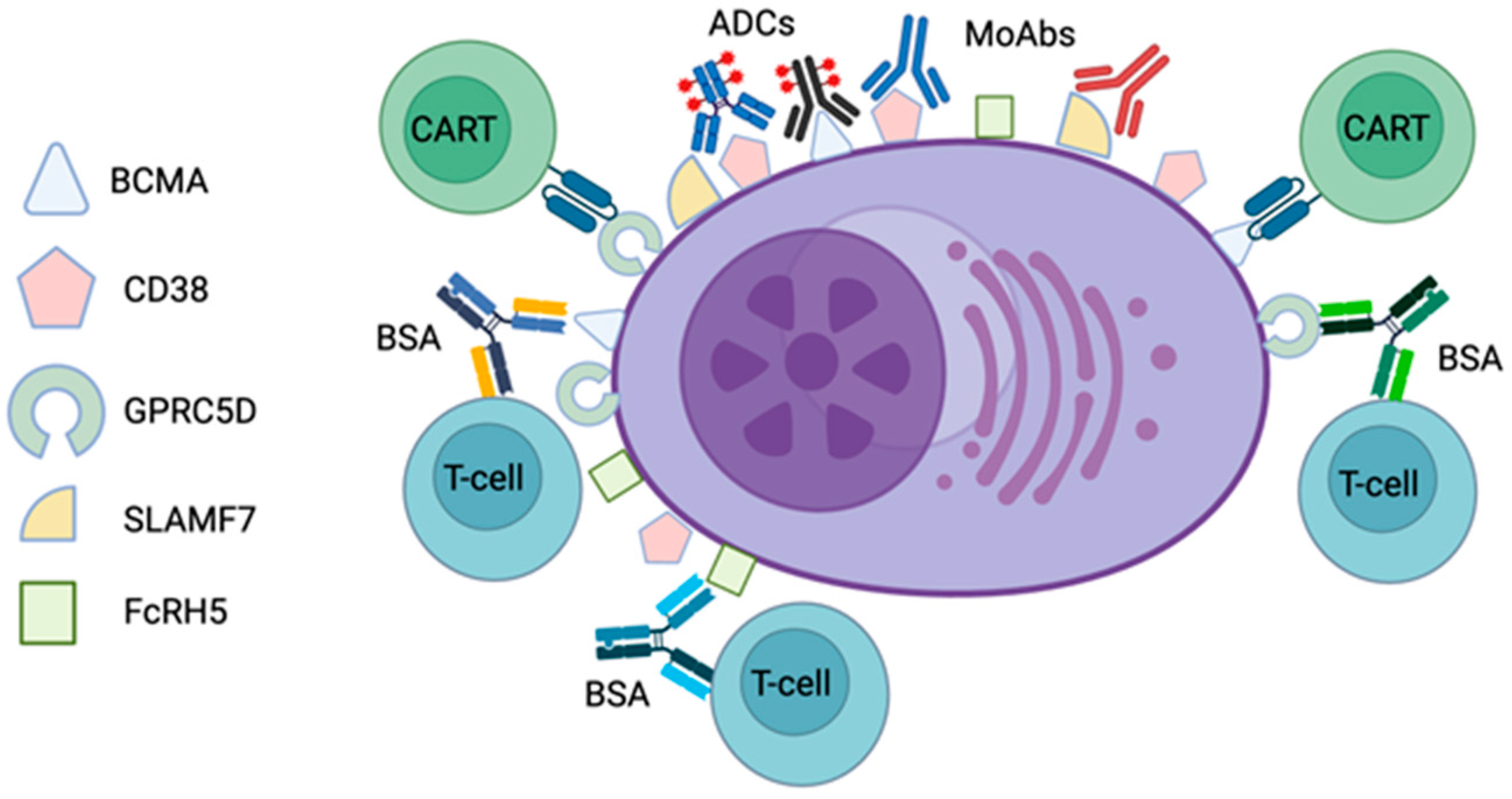
2. Monoclonal Antibodies
3. Antibody–Drug Conjugates
4. Bispecific Antibodies
5. Chimeric Antigen Receptor T-Cell Therapy
6. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Gertz, M.A.; Dispenzieri, A. Systemic Amyloidosis Recognition, Prognosis, and Therapy: A Systematic Review. JAMA 2020, 324, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Palladini, G.; Milani, P.; Merlini, G. Management of AL Amyloidosis in 2020. Blood 2020, 136, 2620–2627. [Google Scholar] [CrossRef]
- Palladini, G.; Merlini, G. How I Treat AL Amyloidosis. Blood 2022, 139, 2918–2930. [Google Scholar] [CrossRef]
- Bou Zerdan, M.; Nasr, L.; Khalid, F.; Allam, S.; Bouferraa, Y.; Batool, S.; Tayyeb, M.; Adroja, S.; Mammadii, M.; Anwer, F.; et al. Systemic AL Amyloidosis: Current Approach and Future Direction. Oncotarget 2023, 14, 384–394. [Google Scholar] [CrossRef]
- Alameda, D.; Goicoechea, I.; Vicari, M.; Arriazu, E.; Nevone, A.; Rodriguez, S.; Lasa, M.; Puig, N.; Cedena, M.T.; Alignani, D.; et al. Tumor Cells in Light-Chain Amyloidosis and Myeloma Show Distinct Transcriptional Rewiring of Normal Plasma Cell Development. Blood 2021, 138, 1583–1589. [Google Scholar] [CrossRef] [PubMed]
- Corre, J. Why Is Amyloidosis Not Multiple Myeloma? Blood 2021, 138, 1514–1515. [Google Scholar] [CrossRef]
- Costa, L.J.; Brill, I.K.; Omel, J.; Godby, K.; Kumar, S.K.; Brown, E.E. Recent Trends in Multiple Myeloma Incidence and Survival by Age, Race, and Ethnicity in the United States. Blood Adv. 2017, 1, 282–287. [Google Scholar] [CrossRef]
- Eisfeld, C.; Kajüter, H.; Möller, L.; Wellmann, I.; Shumilov, E.; Stang, A. Time Trends in Survival and Causes of Death in Multiple Myeloma: A Population-Based Study from Germany. BMC Cancer 2023, 23, 317. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.L.F.; Turesson, I.; Genell, A.; Klausen, T.W.; Knut-Bojanowska, D.; Redder, L.; Sverrisdottir, I.; Thorsen, J.; Vangsted, A.J.; Blimark, C.H. Improved Survival in Myeloma Patients–A Nationwide Registry Study of 4647 Patients ≥75 Years Treated in Denmark and Sweden. Haematologica 2022, 108, 1640–1651. [Google Scholar] [CrossRef]
- Puertas, B.; González-Calle, V.; Sobejano-Fuertes, E.; Escalante, F.; Queizán, J.A.; Bárez, A.; Labrador, J.; Alonso-Alonso, J.M.; García de Coca, A.; Cantalapiedra, A.; et al. Novel Agents as Main Drivers for Continued Improvement in Survival in Multiple Myeloma. Cancers 2023, 15, 1558. [Google Scholar] [CrossRef]
- Thorsteinsdottir, S.; Dickman, P.W.; Landgren, O.; Blimark, C.; Hultcrantz, M.; Turesson, I.; Björkholm, M.; Kristinsson, S.Y. Dramatically Improved Survival in Multiple Myeloma Patients in the Recent Decade: Results from a Swedish Population-Based Study. Haematologica 2018, 103, e412–e415. [Google Scholar] [CrossRef] [PubMed]
- Muchtar, E.; Gertz, M.A.; Kumar, S.K.; Lacy, M.Q.; Dingli, D.; Buadi, F.K.; Grogan, M.; Hayman, S.R.; Kapoor, P.; Leung, N.; et al. Improved Outcomes for Newly Diagnosed AL Amyloidosis between 2000 and 2014: Cracking the Glass Ceiling of Early Death. Blood 2017, 129, 2111–2119. [Google Scholar] [CrossRef] [PubMed]
- Sachchithanantham, S.; Offer, M.; Venner, C.; Mahmood, S.A.; Foard, D.; Rannigan, L.; Lane, T.; Gillmore, J.D.; Lachmann, H.J.; Hawkins, P.N.; et al. Clinical Profile and Treatment Outcome of Older (>75 Years) Patients with Systemic AL Amyloidosis. Haematologica 2015, 100, 1469–1476. [Google Scholar] [CrossRef] [PubMed]
- Staron, A.; Zheng, L.; Doros, G.; Connors, L.H.; Mendelson, L.M.; Joshi, T.; Sanchorawala, V. Marked Progress in AL Amyloidosis Survival: A 40-Year Longitudinal Natural History Study. Blood Cancer J. 2021, 11, 139. [Google Scholar] [CrossRef] [PubMed]
- Al Hamed, R.; Bazarbachi, A.H.; Bazarbachi, A.; Malard, F.; Harousseau, J.-L.; Mohty, M. Comprehensive Review of AL Amyloidosis: Some Practical Recommendations. Blood Cancer J. 2021, 11, 97. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, G.P.; Dispenzieri, A.; Gertz, M.A.; Lacy, M.Q.; Buadi, F.K.; Hayman, S.R.; Leung, N.; Dingli, D.; Lust, J.A.; Lin, Y.; et al. Kinetics of Organ Response and Survival Following Normalization of the Serum Free Light Chain Ratio in AL Amyloidosis. Am. J. Hematol. 2015, 90, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Chee, C.E.; Rajkumar, V.; Gertz, M.; Lacy, M.; Zeldenrust, S.; Kumar, S.; Hayman, S.R.; Buadi, F.; Russell, S.; Kyle, R.; et al. Unraveling Early Mortality in Patients with AL Amyloidosis. Blood 2010, 116, 4990. [Google Scholar] [CrossRef]
- Kumar, S.K.; Gertz, M.A.; Lacy, M.Q.; Dingli, D.; Hayman, S.R.; Buadi, F.K.; Short-Detweiler, K.; Zeldenrust, S.R.; Leung, N.; Greipp, P.R.; et al. Recent Improvements in Survival in Primary Systemic Amyloidosis and the Importance of an Early Mortality Risk Score. Mayo Clin. Proc. 2011, 86, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Wen, C.; Xia, J.; Zhang, H.; Liang, Y.; Xu, X. Targeted Immunotherapy: Harnessing the Immune System to Battle Multiple Myeloma. Cell Death Discov. 2024, 10, 1–17. [Google Scholar] [CrossRef]
- Jadoon, Y.; Siddiqui, M.A. Immunotherapy in Multiple Myeloma. Cancer Treat. Res. Commun. 2021, 29, 100468. [Google Scholar] [CrossRef]
- Palladini, G.; Milani, P.; Malavasi, F.; Merlini, G. Daratumumab in the Treatment of Light-Chain (AL) Amyloidosis. Cells 2021, 10, 545. [Google Scholar] [CrossRef] [PubMed]
- Seckinger, A.; Hillengass, J.; Emde, M.; Beck, S.; Kimmich, C.; Dittrich, T.; Hundemer, M.; Jauch, A.; Hegenbart, U.; Raab, M.-S.; et al. CD38 as Immunotherapeutic Target in Light Chain Amyloidosis and Multiple Myeloma—Association with Molecular Entities, Risk, Survival, and Mechanisms of Upfront Resistance. Front. Immunol. 2018, 9, 1676. [Google Scholar] [CrossRef] [PubMed]
- Bal, S.; Sigler, A.; Chan, A.; Chung, D.; Dogan, A.; Giralt, S.; Hassoun, H.; Landau, H. BCMA Expression in AL Amyloidosis. Clin. Lymphoma Myeloma Leuk. 2019, 19, e306. [Google Scholar] [CrossRef]
- Bal, S.; Dogan, A.; Hassoun, H.; Mailankody, S.; Giralt, S.A.; Landau, H.J. Rational Targets in Systemic AL Amyloidosis. Blood 2021, 138, 4706. [Google Scholar] [CrossRef]
- Kumar, S.K.; Callander, N.S.; Adekola, K.; Anderson, L.D.; Baljevic, M.; Campagnaro, E.; Castillo, J.J.; Costello, C.; D’Angelo, C.; Devarakonda, S.; et al. Systemic Light Chain Amyloidosis, Version 2.2023, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2023, 21, 67–81. [Google Scholar] [CrossRef]
- Morris, E.C.; Neelapu, S.S.; Giavridis, T.; Sadelain, M. Cytokine Release Syndrome and Associated Neurotoxicity in Cancer Immunotherapy. Nat. Rev. Immunol. 2022, 22, 85–96. [Google Scholar] [CrossRef]
- Swan, D.; Murphy, P.; Glavey, S.; Quinn, J. Bispecific Antibodies in Multiple Myeloma: Opportunities to Enhance Efficacy and Improve Safety. Cancers 2023, 15, 1819. [Google Scholar] [CrossRef] [PubMed]
- Alvi, R.M.; Frigault, M.J.; Fradley, M.G.; Jain, M.D.; Mahmood, S.S.; Awadalla, M.; Lee, D.H.; Zlotoff, D.A.; Zhang, L.; Drobni, Z.D.; et al. Cardiovascular Events Among Adults Treated with Chimeric Antigen Receptor T-Cells (CAR-T). J. Am. Coll. Cardiol. 2019, 74, 3099–3108. [Google Scholar] [CrossRef]
- Bonderman, D.; Pölzl, G.; Ablasser, K.; Agis, H.; Aschauer, S.; Auer-Grumbach, M.; Binder, C.; Dörler, J.; Duca, F.; Ebner, C.; et al. Diagnosis and Treatment of Cardiac Amyloidosis: An Interdisciplinary Consensus Statement. Wien. Klin. Wochenschr. 2020, 132, 742–761. [Google Scholar] [CrossRef]
- Clemmensen, T.S.; Mølgaard, H.; Sörensen, J.; Eiskjaer, H.; Andersen, N.F.; Mellemkjaer, S.; Andersen, M.J.; Tolbod, L.P.; Harms, H.J.; Poulsen, S.H. Inotropic Myocardial Reserve Deficiency Is the Predominant Feature of Exercise Haemodynamics in Cardiac Amyloidosis. Eur. J. Heart Fail. 2017, 19, 1457–1465. [Google Scholar] [CrossRef]
- Totzeck, M.; Michel, L.; Lin, Y.; Herrmann, J.; Rassaf, T. Cardiotoxicity from Chimeric Antigen Receptor-T Cell Therapy for Advanced Malignancies. Eur. Heart J. 2022, 43, 1928–1940. [Google Scholar] [CrossRef] [PubMed]
- Abramson, H.N. Immunotherapy of Multiple Myeloma: Current Status as Prologue to the Future. Int. J. Mol. Sci. 2023, 24, 15674. [Google Scholar] [CrossRef] [PubMed]
- Palladini, G.; Paiva, B.; Wechalekar, A.; Massa, M.; Milani, P.; Lasa, M.; Ravichandran, S.; Krsnik, I.; Basset, M.; Burgos, L.; et al. Minimal Residual Disease Negativity by Next-Generation Flow Cytometry Is Associated with Improved Organ Response in AL Amyloidosis. Blood Cancer J. 2021, 11, 34. [Google Scholar] [CrossRef]
- Ravichandran, S.; Cohen, O.C.; Law, S.; Foard, D.; Fontana, M.; Martinez-Naharro, A.; Whelan, C.; Gillmore, J.D.; Lachmann, H.J.; Sachchithanantham, S.; et al. Impact of Early Response on Outcomes in AL Amyloidosis Following Treatment with Frontline Bortezomib. Blood Cancer J. 2021, 11, 118. [Google Scholar] [CrossRef] [PubMed]
- Munshi, N.C.; Avet-Loiseau, H.; Anderson, K.C.; Neri, P.; Paiva, B.; Samur, M.; Dimopoulos, M.; Kulakova, M.; Lam, A.; Hashim, M.; et al. A Large Meta-Analysis Establishes the Role of MRD Negativity in Long-Term Survival Outcomes in Patients with Multiple Myeloma. Blood Adv. 2020, 4, 5988–5999. [Google Scholar] [CrossRef] [PubMed]
- Paiva, B.; San-Miguel, J.; Avet-Loiseau, H. MRD in Multiple Myeloma: Does CR Really Matter? Blood 2022, 140, 2423–2428. [Google Scholar] [CrossRef] [PubMed]
- Garderet, L.; Laubach, J.P.; Stoppa, A.-M.; Hari, P.; Cavo, M.; Ludwig, H.; Mateos, M.-V.; Luptakova, K.; Lin, J.; Yung, G.; et al. Association between Response Kinetics and Outcomes in Relapsed/Refractory Multiple Myeloma: Analysis from TOURMALINE-MM1. Leukemia 2018, 32, 2032–2036. [Google Scholar] [CrossRef] [PubMed]
- Tamariz-Amador, L.-E.; Rodríguez-Otero, P.; Jiménez-Ubieto, A.; Rosiñol, L.; Oriol, A.; Ríos, R.; Sureda, A.; Blanchard, M.J.; Hernández, M.T.; Cabañas Perianes, V.; et al. Prognostic Value of Serum Paraprotein Response Kinetics in Patients with Newly Diagnosed Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2022, 22, e844–e852. [Google Scholar] [CrossRef] [PubMed]
- Wechalekar, A.D.; Palladini, G.; Merlini, G.; Comenzo, R.L.; Jaccard, A.; Tran, N.; Pei, H.; Vasey, S.Y.; Tromp, B.; Weiss, B.M.; et al. Rapid and Deep Hematologic Responses Are Associated with Improved Major Organ Deterioration Progression-Free Survival in Newly Diagnosed AL Amyloidosis: Results from Andromeda. Blood 2020, 136, 6–7. [Google Scholar] [CrossRef]
- Shen, F.; Shen, W. Isatuximab in the Treatment of Multiple Myeloma: A Review and Comparison with Daratumumab. Technol. Cancer Res. Treat. 2022, 21, 15330338221106563. [Google Scholar] [CrossRef]
- Kastritis, E.; Palladini, G.; Minnema, M.C.; Wechalekar, A.D.; Jaccard, A.; Lee, H.C.; Sanchorawala, V.; Gibbs, S.; Mollee, P.; Venner, C.P.; et al. Daratumumab-Based Treatment for Immunoglobulin Light-Chain Amyloidosis. N. Engl. J. Med. 2021, 385, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Wechalekar, A.D.; Schonland, S.O.; Kastritis, E.; Gillmore, J.D.; Dimopoulos, M.A.; Lane, T.; Foli, A.; Foard, D.; Milani, P.; Rannigan, L.; et al. A European Collaborative Study of Treatment Outcomes in 346 Patients with Cardiac Stage III AL Amyloidosis. Blood 2013, 121, 3420–3427. [Google Scholar] [CrossRef] [PubMed]
- Wechalekar, A.D.; Sanchorawala, V. Daratumumab in AL Amyloidosis. Blood 2022, 140, 2317–2322. [Google Scholar] [CrossRef] [PubMed]
- Kastritis, E.; Minnema, M.C.; Dimopoulos, M.A.; Merlini, G.; Theodorakakou, F.; Fotiou, D.; Huart, A.; Belhadj Merzoug, K.; Papachristou, E.; Manousou, K.; et al. Efficacy and Safety of Daratumumab Monotherapy in Newly Diagnosed Patients with Stage 3B Light-Chain Amyloidosis: A Phase 2 Study By the European Myeloma Network. Blood 2023, 142, 539. [Google Scholar] [CrossRef]
- Gao, Y.-J.; Shen, K.; Chang, L.; Feng, J.; Mao, Y.; Zhang, L.; Cao, X.; Zhou, D.; Li, J. Efficacy and Safety of Daratumumab Plus Bortezomib and Dexamethasone in Newly Diagnosed Patients with Mayo 2004 Stage 3 Light-Chain Amyloidosis: A Prospective Phase 2 Study. Blood 2022, 140, 4391–4392. [Google Scholar] [CrossRef]
- Shragai, T.; Gatt, M.; Lavie, N.; Vaxman, I.; Tadmor, T.; Rouvio, O.; Zektser, M.; Horowitz, N.; Magen, H.; Ballan, M.; et al. Daratumumab for Relapsed AL Amyloidosis-When Cumulative Real-World Data Precedes Clinical Trials: A Multisite Study and Systematic Literature Review. Eur. J. Haematol. 2021, 106, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Roussel, M.; Merlini, G.; Chevret, S.; Arnulf, B.; Stoppa, A.M.; Perrot, A.; Palladini, G.; Karlin, L.; Royer, B.; Huart, A.; et al. A Prospective Phase 2 Trial of Daratumumab in Patients with Previously Treated Systemic Light-Chain Amyloidosis. Blood 2020, 135, 1531–1540. [Google Scholar] [CrossRef]
- Sanchorawala, V.; Sarosiek, S.; Schulman, A.; Mistark, M.; Migre, M.E.; Cruz, R.; Sloan, J.M.; Brauneis, D.; Shelton, A.C. Safety, Tolerability, and Response Rates of Daratumumab in Relapsed AL Amyloidosis: Results of a Phase 2 Study. Blood 2020, 135, 1541–1547. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, S.M.; Stecklein, K.; Sarow, J.; Krabak, M.; Hillengass, J.; McCarthy, P. Elotuzumab in Combination with Lenalidomide and Dexamethasone for Treatment-Resistant Immunoglobulin Light Chain Amyloidosis with Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2019, 19, e33–e36. [Google Scholar] [CrossRef]
- Lonial, S.; Lee, H.C.; Badros, A.; Trudel, S.; Nooka, A.K.; Chari, A.; Abdallah, A.-O.; Callander, N.; Lendvai, N.; Sborov, D.; et al. Belantamab Mafodotin for Relapsed or Refractory Multiple Myeloma (DREAMM-2): A Two-Arm, Randomised, Open-Label, Phase 2 Study. Lancet Oncol. 2020, 21, 207–221. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Hungria, V.T.M.; Radinoff, A.; Delimpasi, S.; Mikala, G.; Masszi, T.; Li, J.; Capra, M.; Maiolino, A.; Pappa, V.; et al. Efficacy and Safety of Single-Agent Belantamab Mafodotin versus Pomalidomide plus Low-Dose Dexamethasone in Patients with Relapsed or Refractory Multiple Myeloma (DREAMM-3): A Phase 3, Open-Label, Randomised Study. Lancet Haematol. 2023, 10, e801–e812. [Google Scholar] [CrossRef] [PubMed]
- Kastritis, E.; Palladini, G.; Dimopoulos, M.A.; Jaccard, A.; Merlini, G.; Theodorakakou, F.; Fotiou, D.; Minnema Minnema, M.C.; Wechalekar, A.; Papachristou, E.; et al. S198: Efficacy and safety of belantamab mafodotin monotherapy in patients with relapsed or refractory light chain amyloidosis: A phase 2 study by the european myeloma network. HemaSphere 2023, 7, e2301416. [Google Scholar] [CrossRef]
- Zhang, Y.; Godara, A.; Pan, S.; Toskic, D.; Mann, H.; Sborov, D.; Comenzo, R.; Kansagra, A. Belantamab Mafodotin in Patients with Relapsed/Refractory AL Amyloidosis with Myeloma. Ann. Hematol. 2022, 101, 2119–2121. [Google Scholar] [CrossRef] [PubMed]
- Khwaja, J.; Bomsztyk, J.; Mahmood, S.; Wisniowski, B.; Shah, R.; Tailor, A.; Yong, K.; Popat, R.; Rabin, N.; Kyriakou, C.; et al. High Response Rates with Single-Agent Belantamab Mafodotin in Relapsed Systemic AL Amyloidosis. Blood Cancer J. 2022, 12, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Shragai, T.; Magen, H.; Lavi, N.; Gatt, M.; Trestman, S.; Zektser, M.; Ganzel, C.; Jarchowsky, O.; Berger, T.; Tadmor, T.; et al. Real-World Experience with Belantamab Mafodotin Therapy for Relapsed/Refractory Multiple Myeloma: A Multicentre Retrospective Study. Br. J. Haematol. 2023, 200, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P.W.H.I. Bispecific Antibodies: A Mechanistic Review of the Pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef] [PubMed]
- Moreau, P.; Garfall, A.L.; van de Donk, N.W.C.J.; Nahi, H.; San-Miguel, J.F.; Oriol, A.; Nooka, A.K.; Martin, T.; Rosinol, L.; Chari, A.; et al. Teclistamab in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2022, 387, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Leung, N.; Chapman, J.A.; Bhatia, S. First Report of Teclistamab in a Patient with Relapsed AL Amyloidosis and Multiple Myeloma. eJHaem 2023, 4, 1157–1159. [Google Scholar] [CrossRef]
- Forgeard, N.; Elessa, D.; Carpinteiro, A.; Belhadj, K.; Minnema, M.C.; Roussel, M.; Huart, A.; Javaugue, V.; Pascal, L.; Royer, B.; et al. Teclistamab in Relapsed or Refractory AL Amyloidosis, a Multinational Retrospective Case Series. Blood 2024, 143, 734–737. [Google Scholar] [CrossRef]
- Chakraborty, R.; Bhutani, D.; Maurer, M.S.; Mohan, M.; Lentzsch, S.; D’Souza, A. Safety and Efficacy of Teclistamab in Systemic Immunoglobulin Light Chain Amyloidosis. Blood Cancer J. 2023, 13, 172. [Google Scholar] [CrossRef]
- Martin, T.; Jackson, C.C.; Pacaud, L.; Madduri, D.; Jagannath, S. Recent Advances in the Use of Chimeric Antigen Receptor-Expressing T-Cell Therapies for Treatment of Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2023, 23, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Xu, J.; Luo, W.; Liao, D.; Xie, W.; Wei, Q.; Zhang, Y.; Wang, X.; Wu, Z.; Kang, Y.; et al. Bispecific CS1-BCMA CAR-T Cells Are Clinically Active in Relapsed or Refractory Multiple Myeloma. Leukemia 2024, 38, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wei, G.; Zhou, L.; Zhou, J.; Chen, S.; Zhang, W.; Wang, D.; Luo, X.; Cui, J.; Huang, S.; et al. GPRC5D CAR T Cells (OriCAR-017) in Patients with Relapsed or Refractory Multiple Myeloma (POLARIS): A First-in-Human, Single-Centre, Single-Arm, Phase 1 Trial. Lancet Haematol. 2023, 10, e107–e116. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.D.; Dhakal, B.; Jain, T.; Oluwole, O.O.; Shah, G.L.; Sidana, S.; Perales, M.-A.; Pasquini, M.C. Chimeric Antigen Receptor T Cell Therapy for Myeloma: Where Are We Now and What Is Needed to Move Chimeric Antigen Receptor T Cells Forward to Earlier Lines of Therapy? Expert Panel Opinion from the American Society for Transplantation and Cellular Therapy. Transplant. Cell. Ther. Off. Publ. Am. Soc. Transplant. Cell. Ther. 2024, 30, 17–37. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Otero, P.; Ailawadhi, S.; Arnulf, B.; Patel, K.; Cavo, M.; Nooka, A.K.; Manier, S.; Callander, N.; Costa, L.J.; Vij, R.; et al. Ide-Cel or Standard Regimens in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2023, 388, 1002–1014. [Google Scholar] [CrossRef] [PubMed]
- San-Miguel, J.; Dhakal, B.; Yong, K.; Spencer, A.; Anguille, S.; Mateos, M.-V.; Fernández de Larrea, C.; Martínez-López, J.; Moreau, P.; Touzeau, C.; et al. Cilta-Cel or Standard Care in Lenalidomide-Refractory Multiple Myeloma. N. Engl. J. Med. 2023, 389, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Martino, M.; Canale, F.A.; Alati, C.; Vincelli, I.D.; Moscato, T.; Porto, G.; Loteta, B.; Naso, V.; Mazza, M.; Nicolini, F.; et al. CART-Cell Therapy: Recent Advances and New Evidence in Multiple Myeloma. Cancers 2021, 13, 2639. [Google Scholar] [CrossRef]
- Cappell, K.M.; Kochenderfer, J.N. Long-Term Outcomes Following CAR T Cell Therapy: What We Know So Far. Nat. Rev. Clin. Oncol. 2023, 20, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Verdun, N.; Marks, P. Secondary Cancers after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2024, 390, 584–586. [Google Scholar] [CrossRef]
- Martin, T.; Usmani, S.Z.; Berdeja, J.G.; Agha, M.; Cohen, A.D.; Hari, P.; Avigan, D.; Deol, A.; Htut, M.; Lesokhin, A.; et al. Ciltacabtagene Autoleucel, an Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor T-Cell Therapy, for Relapsed/Refractory Multiple Myeloma: CARTITUDE-1 2-Year Follow-Up. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2023, 41, 1265–1274. [Google Scholar] [CrossRef]
- Vainstein, V.; Avni, B.; Grisariu, S.; Kfir-Erenfeld, S.; Asherie, N.; Nachmias, B.; Auman, S.; Saban, R.; Zimran, E.; Assayag, M.; et al. Clonal Myeloid Dysplasia Following CAR T-Cell Therapy: Chicken or the Egg? Cancers 2023, 15, 3471. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Nguyen, J.; Yen, Y. Complete Spectrum of Adverse Events Associated with Chimeric Antigen Receptor (CAR)-T Cell Therapies. J. Biomed. Sci. 2023, 30, 89. [Google Scholar] [CrossRef] [PubMed]
- Oliver-Caldes, A.; Jiménez, R.; Español-Rego, M.; Cibeira, M.T.; Ortiz-Maldonado, V.; Quintana, L.F.; Castillo, P.; Guijarro, F.; Tovar, N.; Montoro, M.; et al. First Report of CART Treatment in AL Amyloidosis and Relapsed/Refractory Multiple Myeloma. J. Immunother. Cancer 2021, 9, e003783. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Ailawadhi, S.; Sher, T.; Roy, V.; Fernandez, A.; Parrondo, R.D. Anti-B Cell Maturation Antigen Chimeric Antigen Receptor T Cell Therapy for the Treatment of AL Amyloidosis and Concurrent Relapsed/Refractory Multiple Myeloma: Preliminary Efficacy and Safety. Curr. Oncol. 2023, 30, 9627–9633. [Google Scholar] [CrossRef] [PubMed]
- Sidana, S.; Hosoya, H.; Jensen, A.; Liu, L.; Goyal, A.; Hovanky, V.; Sahaf, B.; Bharadwaj, S.; Latchford, T.; Arai, S.; et al. Bendamustine vs. Fludarabine/Cyclophosphamide Lymphodepletion Prior to BCMA CAR-T Cell Therapy in Multiple Myeloma. Blood Cancer J. 2023, 13, 158. [Google Scholar] [CrossRef] [PubMed]
- Asherie, N.; Kfir-Erenfeld, S.; Avni, B.; Assayag, M.; Dubnikov, T.; Zalcman, N.; Lebel, E.; Zimran, E.; Shaulov, A.; Pick, M.; et al. Development and Manufacture of Novel Locally Produced Anti-BCMA CAR T Cells for the Treatment of Relapsed/Refractory Multiple Myeloma: Results from a Phase I Clinical Trial. Haematologica 2023, 108, 1827–1839. [Google Scholar] [CrossRef] [PubMed]
- Kfir-Erenfeld, S.; Asherie, N.; Grisariu, S.; Avni, B.; Zimran, E.; Assayag, M.; Sharon, T.D.; Pick, M.; Lebel, E.; Shaulov, A.; et al. Feasibility of a Novel Academic BCMA-CART (HBI0101) for the Treatment of Relapsed and Refractory AL Amyloidosis. Clin. Cancer Res. 2022, 28, 5156–5166. [Google Scholar] [CrossRef]
- Lebel, E.; Kfir-Erenfeld, S.; Asherie, N.; Grisariu, S.; Avni, B.; Elias, S.; Assayag, M.; Dubnikov, T.; Zalcman, N.; Pick, M.; et al. Feasibility of a Novel Academic Anti-BCMA Chimeric Antigen Receptor T-Cell (CART) (HBI0101) for the Treatment of Relapsed and Refractory AL Amyloidosis. Blood 2023, 142, 538. [Google Scholar] [CrossRef]


{kind=link}
| Treatment Type | Intervention | Phase | Disease Setting | Clinicaltrials.gov Identifier | Start Year | Completion Year | Target Patient Number |
|---|---|---|---|---|---|---|---|
| Monoclonal antibodies | Daratumumab monotherapy | 2 | Newly diagnosed, stage 3b | NCT04131309 | 2023 | 2025 | 40 |
| Daratumumab, pomalidomide, dexamethasone | 2 | R/R | NCT04270175 | 2021 | 2024 | 21 | |
| Daratumumab, bortezomib, dexamethasone | 2 | Newly diagnosed, stage 3 | NCT04474938 | 2021 | 2023 | 40 | |
| Venetoclax, daratumumab, dexamethasone | 1–2 | R/R, t(11;14) | NCT05486481 | 2023 | 2027 | 78 | |
| Daratumumab, ixazomib, dexamethasone | 1 | Newly diagnosed and R/R | NCT03283917 | 2018 | 2025 | 21 | |
| Daratumumab monotherapy | 2 | Maintenance | NCT05898646 | 2023 | 2024 | 96 | |
| Daratumumab, pomalidomide | 2 | R/R | NCT04895917 | 2021 | 2024 | 40 | |
| Isatuximab monotherapy | 2 | R/R | NCT03499808 | 2018 | 2023 | 43 | |
| Isatuximab, pomalidomide, dexamethasone | 2 | R/R | NCT05066607 | 2022 | 2025 | 46 | |
| Istauximab, bortezomib, dexamethasone, cyclophosphamide | 1 | Newly diagnosed | NCT04754945 | 2021 | 2025 | 25 | |
| Elotuzumab, lenalidomide, dexamethasone, cyclophosphamide | 2 | R/R | NCT03252600 | 2017 | 2023 | 53 | |
| Antibody-drug conjugates | STI-6129 (antibody-drug conjugate) | 1b/2a | R/R | NCT04316442 | 2021 | 2024 | 60 |
| Belantamab mafodotin | 1/2a | R/R | NCT05145816 | 2023 | 2026 | 37 | |
| Belantamab mafodotin | 2 | R/R | NCT04617925 | 2021 | 2025 | 35 | |
| CAR-T cells | FKC288 | 1 | R/R | NCT05978661 | 2023 | 2025 | 12 |
| HBI0101/NXC-201 | 1b | R/R | NCT06097832 | 2024 | 2025 | 40 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haran, A.; Vaxman, I.; Gatt, M.E.; Lebel, E. Immune Therapies in AL Amyloidosis—A Glimpse to the Future. Cancers 2024, 16, 1605. https://doi.org/10.3390/cancers16081605
Haran A, Vaxman I, Gatt ME, Lebel E. Immune Therapies in AL Amyloidosis—A Glimpse to the Future. Cancers. 2024; 16(8):1605. https://doi.org/10.3390/cancers16081605
Chicago/Turabian StyleHaran, Arnon, Iuliana Vaxman, Moshe E. Gatt, and Eyal Lebel. 2024. "Immune Therapies in AL Amyloidosis—A Glimpse to the Future" Cancers 16, no. 8: 1605. https://doi.org/10.3390/cancers16081605
APA StyleHaran, A., Vaxman, I., Gatt, M. E., & Lebel, E. (2024). Immune Therapies in AL Amyloidosis—A Glimpse to the Future. Cancers, 16(8), 1605. https://doi.org/10.3390/cancers16081605