
Current and Future Therapeutic Targets for Directed Molecular Therapies in Cholangiocarcinoma
Abstract
:Simple Summary
Abstract
1. Introduction
2. Risk Factors and Diagnostic Work-Up
3. Stage-Dependent Therapeutic Regimes
4. Current and Future Molecular-Directed Therapeutic Agents in CCA
4.1. Prevalence of Druggable Molecular Targets in Different Subtypes of CCA
4.2. Target Fibroblast Growth Factor Receptor (FGFR)
4.3. Target Isocitrate Dehydrogenase (IDH) 1/2
4.4. Target Human Epidermal Growth Factor Receptor (HER) 2
4.5. Target Epidermal Growth Factor Receptor (EGFR)/Human Epidermal Growth Factor Receptor (HER) 1
4.6. Target Neurotrophic Tyrosine Receptor Kinase (NTRK)
4.7. Targeting BRAF Alterations
4.8. Targeting Alterations in Deoxyribonucleic Acid (DNA) Damage Repair Genes (DDR Genes)
4.9. Targeting ROS1/ALK/MET Alterations
4.10. Targeting CASEIN KINASE (CK2)
4.11. Targeting Receptor Tyrosine Kinase RET
4.12. Targeting the PI3K/AKT/mTOR Signaling Pathway
4.13. Targeting the Wnt/β-Catenin Signaling Pathway
4.14. Targeting Cyclin Dependent Kinase 4/6 (CDK4/6)
4.15. Targeting the RAS/RAF/MEK/ERK Signaling Pathway
4.16. Targeting Vascular Endothelial Growth Factor (VEGF) Signaling Pathway
4.17. Immune Checkpoint Inhibitor (CPI)
4.18. Therapy with Chimeric Antigen Receptor (CAR)-Engineered T Cells and Tumor Vaccination
4.19. Targeting Non-Coding RNA in Treatment of CCA
5. Mechanisms of Therapy Resistance in CCA
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- McGlynn, K.A.; Petrick, J.L.; El-Serag, H.B. Epidemiology of Hepatocellular Carcinoma. Hepatology 2021, 73 (Suppl. S1), 4–13. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Razumilava, N.; Gores, G.J. Cholangiocarcinoma. Lancet 2014, 383, 2168–2179. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.A.; Tavolari, S.; Brandi, G. Cholangiocarcinoma: Epidemiology and risk factors. Liver Int. 2019, 39, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.; Khan, S.A.; Hallemeier, C.L.; Kelley, R.K.; Gores, G.J. Cholangiocarcinoma—Evolving concepts and therapeutic strategies. Nat. Rev. Clin. Oncol. 2018, 15, 95–111. [Google Scholar] [CrossRef] [PubMed]
- Banales, J.M.; Marin, J.J.G.; Lamarca, A.; Rodrigues, P.M.; Khan, S.A.; Roberts, L.R.; Cardinale, V.; Carpino, G.; Andersen, J.B.; Braconi, C.; et al. Cholangiocarcinoma 2020: The next horizon in mechanisms and management. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 557–588. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.; Gores, G.J. Pathogenesis, diagnosis, and management of cholangiocarcinoma. Gastroenterology 2013, 145, 1215–1229. [Google Scholar] [CrossRef] [PubMed]
- Kefas, J.; Bridgewater, J.; Vogel, A.; Stein, A.; Primrose, J. Adjuvant therapy of biliary tract cancers. Ther. Adv. Med. Oncol. 2023, 15, 17588359231163785. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.; Bridgewater, J.; Edeline, J.; Kelley, R.K.; Klümpen, H.J.; Malka, D.; Primrose, J.N.; Rimassa, L.; Stenzinger, A.; Valle, J.W.; et al. Biliary tract cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann. Oncol. 2023, 34, 127–140. [Google Scholar] [CrossRef]
- Voesch, S.; Bitzer, M.; Blödt, S.; Follmann, M.; Freudenberger, P.; Langer, T.; Lorenz, P.; Jansen, P.L.; Steubesand, N.; Galle, P.; et al. S3-Leitlinie: Diagnostik und Therapie des hepatozellulären Karzinoms und biliärer Karzinome—Version 2.0—Juni 2021, AWMF-Registernummer: 032-053OL. Z. Gastroenterol. 2022, 60, e131–e185. [Google Scholar] [CrossRef]
- Palmer, W.C.; Patel, T. Are common factors involved in the pathogenesis of primary liver cancers? A meta-analysis of risk factors for intrahepatic cholangiocarcinoma. J. Hepatol. 2012, 57, 69–76. [Google Scholar] [CrossRef] [PubMed]
- McGee, E.E.; Jackson, S.S.; Petrick, J.L.; Van Dyke, A.L.; Adami, H.O.; Albanes, D.; Andreotti, G.; Beane-Freeman, L.E.; Berrington de Gonzalez, A.; Buring, J.E.; et al. Smoking, Alcohol, and Biliary Tract Cancer Risk: A Pooling Project of 26 Prospective Studies. J. Natl. Cancer Inst. 2019, 111, 1263–1278. [Google Scholar] [CrossRef] [PubMed]
- Tyson, G.L.; El-Serag, H.B. Risk factors for cholangiocarcinoma. Hepatology 2011, 54, 173–184. [Google Scholar] [CrossRef]
- Petrick, J.L.; Yang, B.; Altekruse, S.F.; Van Dyke, A.L.; Koshiol, J.; Graubard, B.I.; McGlynn, K.A. Risk factors for intrahepatic and extrahepatic cholangiocarcinoma in the United States: A population-based study in SEER-Medicare. PLoS ONE 2017, 12, e0186643. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.T.; Lin, Y.J.; Li, C.Y.; Tsai, P.J.; Yang, C.Y.; Liou, S.H.; Wu, T.N. Cancer Attributable to Asbestos Exposure in Shipbreaking Workers: A Matched-Cohort Study. PLoS ONE 2015, 10, e0133128. [Google Scholar] [CrossRef] [PubMed]
- Söreide, K.; Körner, H.; Havnen, J.; Söreide, J.A. Bile duct cysts in adults. Br. J. Surg. 2004, 91, 1538–1548. [Google Scholar] [CrossRef] [PubMed]
- Clements, O.; Eliahoo, J.; Kim, J.U.; Taylor-Robinson, S.D.; Khan, S.A. Risk factors for intrahepatic and extrahepatic cholangiocarcinoma: A systematic review and meta-analysis. J. Hepatol. 2020, 72, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Welzel, T.M.; Graubard, B.I.; El-Serag, H.B.; Shaib, Y.H.; Hsing, A.W.; Davila, J.A.; McGlynn, K.A. Risk factors for intrahepatic and extrahepatic cholangiocarcinoma in the United States: A population-based case-control study. Clin. Gastroenterol. Hepatol. 2007, 5, 1221–1228. [Google Scholar] [CrossRef] [PubMed]
- Fahrner, R.; Dennler, S.G.; Inderbitzin, D. Risk of malignancy in Caroli disease and syndrome: A systematic review. World J. Gastroenterol. 2020, 26, 4718–4728. [Google Scholar] [CrossRef]
- Shaib, Y.H.; El-Serag, H.B.; Davila, J.A.; Morgan, R.; McGlynn, K.A. Risk factors of intrahepatic cholangiocarcinoma in the United States: A case-control study. Gastroenterology 2005, 128, 620–626. [Google Scholar] [CrossRef]
- Welzel, T.M.; Mellemkjaer, L.; Gloria, G.; Sakoda, L.C.; Hsing, A.W.; El Ghormli, L.; Olsen, J.H.; McGlynn, K.A. Risk factors for intrahepatic cholangiocarcinoma in a low-risk population: A nationwide case-control study. Int. J. Cancer 2007, 120, 638–641. [Google Scholar] [CrossRef] [PubMed]
- Huai, J.P.; Ding, J.; Ye, X.H.; Chen, Y.P. Inflammatory bowel disease and risk of cholangiocarcinoma: Evidence from a meta-analysis of population-based studies. Asian Pac. J. Cancer Prev. 2014, 15, 3477–3482. [Google Scholar] [CrossRef] [PubMed]
- Shaib, Y.H.; El-Serag, H.B.; Nooka, A.K.; Thomas, M.; Brown, T.D.; Patt, Y.Z.; Hassan, M.M. Risk factors for intrahepatic and extrahepatic cholangiocarcinoma: A hospital-based case-control study. Am. J. Gastroenterol. 2007, 102, 1016–1021. [Google Scholar] [CrossRef] [PubMed]
- Donato, F.; Gelatti, U.; Tagger, A.; Favret, M.; Ribero, M.L.; Callea, F.; Martelli, C.; Savio, A.; Trevisi, P.; Nardi, G. Intrahepatic cholangiocarcinoma and hepatitis C and B virus infection, alcohol intake, and hepatolithiasis: A case-control study in Italy. Cancer Causes Control 2001, 12, 959–964. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.Y.; Lee, S.S.; Jung, S.W.; Jeon, S.H.; Yun, S.C.; Oh, H.C.; Kwon, S.; Lee, S.K.; Seo, D.W.; Kim, M.H.; et al. Hepatitis B virus infection and intrahepatic cholangiocarcinoma in Korea: A case-control study. Am. J. Gastroenterol. 2008, 103, 1716–1720. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.M.; Yin, Z.F.; Yang, J.M.; Li, B.; Shao, W.Y.; Xu, F.; Wang, Y.L.; Li, D.Q. Risk factors for intrahepatic cholangiocarcinoma: A case-control study in China. World J. Gastroenterol. 2008, 14, 632–635. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B.; Engels, E.A.; Landgren, O.; Chiao, E.; Henderson, L.; Amaratunge, H.C.; Giordano, T.P. Risk of hepatobiliary and pancreatic cancers after hepatitis C virus infection: A population-based study of U.S. veterans. Hepatology 2009, 49, 116–123. [Google Scholar] [CrossRef]
- Yamamoto, S.; Kubo, S.; Hai, S.; Uenishi, T.; Yamamoto, T.; Shuto, T.; Takemura, S.; Tanaka, H.; Yamazaki, O.; Hirohashi, K.; et al. Hepatitis C virus infection as a likely etiology of intrahepatic cholangiocarcinoma. Cancer Sci. 2004, 95, 592–595. [Google Scholar] [CrossRef] [PubMed]
- Vijungco, J.D.; Prinz, R.A. Management of biliary and duodenal complications of chronic pancreatitis. World J. Surg. 2003, 27, 1258–1270. [Google Scholar] [CrossRef]
- Sørensen, H.T.; Mellemkjaer, L.; Jepsen, P.; Thulstrup, A.M.; Baron, J.; Olsen, J.H.; Vilstrup, H. Risk of cancer in patients hospitalized with fatty liver: A Danish cohort study. J. Clin. Gastroenterol. 2003, 36, 356–359. [Google Scholar] [CrossRef]
- Erichsen, R.; Jepsen, P.; Vilstrup, H.; Ekbom, A.; Sørensen, H.T. Incidence and prognosis of cholangiocarcinoma in Danish patients with and without inflammatory bowel disease: A national cohort study, 1978–2003. Eur. J. Epidemiol. 2009, 24, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Petrick, J.L.; Thistle, J.E.; Zeleniuch-Jacquotte, A.; Zhang, X.; Wactawski-Wende, J.; Van Dyke, A.L.; Stampfer, M.J.; Sinha, R.; Sesso, H.D.; Schairer, C.; et al. Body Mass Index, Diabetes and Intrahepatic Cholangiocarcinoma Risk: The Liver Cancer Pooling Project and Meta-analysis. Am. J. Gastroenterol. 2018, 113, 1494–1505. [Google Scholar] [CrossRef] [PubMed]
- Jing, W.; Jin, G.; Zhou, X.; Zhou, Y.; Zhang, Y.; Shao, C.; Liu, R.; Hu, X. Diabetes mellitus and increased risk of cholangiocarcinoma: A meta-analysis. Eur. J. Cancer Prev. 2012, 21, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Fernández Peláez, J.M.; Sánchez Martín, E.; Tirado Miranda, R.; Navarro Martínez, A.; Alamillo Sanz, A. Hemochromatosis and hilar cholangiocarcinoma: Report of a case. Rev. Esp. Enferm. Dig. 2000, 92, 474–475. [Google Scholar] [PubMed]
- Sulpice, L.; Rayar, M.; Boucher, E.; Pele, F.; Pracht, M.; Meunier, B.; Boudjema, K. Intrahepatic cholangiocarcinoma: Impact of genetic hemochromatosis on outcome and overall survival after surgical resection. J. Surg. Res. 2013, 180, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Morcos, M.; Dubois, S.; Bralet, M.P.; Belghiti, J.; Degott, C.; Terris, B. Primary liver carcinoma in genetic hemochromatosis reveals a broad histologic spectrum. Am. J. Clin. Pathol. 2001, 116, 738–743. [Google Scholar] [CrossRef]
- Qian, M.B.; Utzinger, J.; Keiser, J.; Zhou, X.N. Clonorchiasis. Lancet 2016, 387, 800–810. [Google Scholar] [CrossRef] [PubMed]
- Kamsa-ard, S.; Kamsa-ard, S.; Luvira, V.; Suwanrungruang, K.; Vatanasapt, P.; Wiangnon, S. Risk Factors for Cholangiocarcinoma in Thailand: A Systematic Review and Meta-Analysis. Asian Pac. J. Cancer Prev. 2018, 19, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Qian, M.B.; Zhou, X.N. Global burden of cancers attributable to liver flukes. Lancet Glob. Health 2017, 5, e139. [Google Scholar] [CrossRef]
- Wongjarupong, N.; Assavapongpaiboon, B.; Susantitaphong, P.; Cheungpasitporn, W.; Treeprasertsuk, S.; Rerknimitr, R.; Chaiteerakij, R. Non-alcoholic fatty liver disease as a risk factor for cholangiocarcinoma: A systematic review and meta-analysis. BMC Gastroenterol. 2017, 17, 149. [Google Scholar] [CrossRef]
- Claessen, M.M.; Vleggaar, F.P.; Tytgat, K.M.; Siersema, P.D.; van Buuren, H.R. High lifetime risk of cancer in primary sclerosing cholangitis. J. Hepatol. 2009, 50, 158–164. [Google Scholar] [CrossRef] [PubMed]
- de Valle, M.B.; Björnsson, E.; Lindkvist, B. Mortality and cancer risk related to primary sclerosing cholangitis in a Swedish population-based cohort. Liver Int. 2012, 32, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Boonstra, K.; Weersma, R.K.; van Erpecum, K.J.; Rauws, E.A.; Spanier, B.W.; Poen, A.C.; van Nieuwkerk, K.M.; Drenth, J.P.; Witteman, B.J.; Tuynman, H.A.; et al. Population-based epidemiology, malignancy risk, and outcome of primary sclerosing cholangitis. Hepatology 2013, 58, 2045–2055. [Google Scholar] [CrossRef]
- Catanzaro, E.; Gringeri, E.; Burra, P.; Gambato, M. Primary Sclerosing Cholangitis-Associated Cholangiocarcinoma: From Pathogenesis to Diagnostic and Surveillance Strategies. Cancers 2023, 15, 4947. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Ghoz, H.M.; Peeraphatdit, T.; Baichoo, E.; Addissie, B.D.; Harmsen, W.S.; Therneau, T.M.; Olson, J.E.; Chaiteerakij, R.; Roberts, L.R. Aspirin use and the risk of cholangiocarcinoma. Hepatology 2016, 64, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; You, L.; Xie, W.; Ning, L.; Lang, J. Smoking and risk of cholangiocarcinoma: A systematic review and meta-analysis. Oncotarget 2017, 8, 100570–100581. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, Y.; Wada, I.; Fukumoto, M. Alpha-particle carcinogenesis in Thorotrast patients: Epidemiology, dosimetry, pathology, and molecular analysis. J. Environ. Pathol. Toxicol. Oncol. 2001, 20, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.A.; Marcano-Bonilla, L.; Roberts, L.R. Gallbladder cancer: Epidemiology and genetic risk associations. Chin. Clin. Oncol. 2019, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Koshiol, J.; Zhu, B.; Wang, R.; Hildesheim, A.; Gao, Y.-T.; Egner, P.A.; Yuan, J.-M.; Groopman, J.D. Association of aflatoxin with gallbladder cancer in a case-control study nested within a Chinese cohort. Int. J. Cancer 2024, 154, 801–806. [Google Scholar] [CrossRef]
- Madhawi, R.; Pandey, A.; Raj, S.; Mandal, M.; Devi, S.; Sinha, P.K.; Singh, R.K. Geographical pattern of carcinoma gallbladder in Bihar and its association with river Ganges and arsenic levels: Retrospective individual consecutive patient data from Regional Cancer Centre. South Asian J. Cancer 2018, 7, 167–170. [Google Scholar] [CrossRef]
- Rawla, P.; Sunkara, T.; Thandra, K.C.; Barsouk, A. Epidemiology of gallbladder cancer. Clin. Exp. Hepatol. 2019, 5, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Jain, K.; Sreenivas, V.; Velpandian, T.; Kapil, U.; Garg, P.K. Risk factors for gallbladder cancer: A case-control study. Int. J. Cancer 2013, 132, 1660–1666. [Google Scholar] [CrossRef] [PubMed]
- Inzunza, M.; Irarrazaval, M.J.; Pozo, P.; Pimentel, F.; Crovari, F.; Ibañez, L. Gallbladder polyps: Correlation and agreement between ultrasonographic and histopathological findings in a population with high incidence of gallbladder cancer. Arq. Bras. Cir. Dig. 2023, 36, e1732. [Google Scholar] [CrossRef] [PubMed]
- Stinton, L.M.; Shaffer, E.A. Epidemiology of gallbladder disease: Cholelithiasis and cancer. Gut Liver 2012, 6, 172–187. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Roa, J.C.; Tapia, O.; Dursun, N.; Bagci, P.; Basturk, O.; Cakir, A.; Losada, H.; Sarmiento, J.; Adsay, V. Hyalinizing cholecystitis and associated carcinomas: Clinicopathologic analysis of a distinctive variant of cholecystitis with porcelain-like features and accompanying diagnostically challenging carcinomas. Am. J. Surg. Pathol. 2011, 35, 1104–1113. [Google Scholar] [CrossRef] [PubMed]
- Schnelldorfer, T. Porcelain gallbladder: A benign process or concern for malignancy? J. Gastrointest. Surg. 2013, 17, 1161–1168. [Google Scholar] [CrossRef]
- Alvaro, D.; Bragazzi, M.C.; Benedetti, A.; Fabris, L.; Fava, G.; Invernizzi, P.; Marzioni, M.; Nuzzo, G.; Strazzabosco, M.; Stroffolini, T. Cholangiocarcinoma in Italy: A national survey on clinical characteristics, diagnostic modalities and treatment. Results from the “Cholangiocarcinoma” committee of the Italian Association for the Study of Liver disease. Dig. Liver Dis. 2011, 43, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Khuntikeo, N.; Chamadol, N.; Yongvanit, P.; Loilome, W.; Namwat, N.; Sithithaworn, P.; Andrews, R.H.; Petney, T.N.; Promthet, S.; Thinkhamrop, K.; et al. Cohort profile: Cholangiocarcinoma screening and care program (CASCAP). BMC Cancer 2015, 15, 459. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.X.; Chen, L.D.; Liu, L.N.; Zhang, Y.F.; Guo, L.H.; Liu, C. Contrast-enhanced ultrasound of intrahepatic cholangiocarcinoma: Correlation with pathological examination. Br. J. Radiol. 2012, 85, 1029–1037. [Google Scholar] [CrossRef]
- Wildner, D.; Bernatik, T.; Greis, C.; Seitz, K.; Neurath, M.F.; Strobel, D. CEUS in hepatocellular carcinoma and intrahepatic cholangiocellular carcinoma in 320 patients—Early or late washout matters: A subanalysis of the DEGUM multicenter trial. Ultraschall. Med. 2015, 36, 132–139. [Google Scholar] [CrossRef]
- Fábrega-Foster, K.; Ghasabeh, M.A.; Pawlik, T.M.; Kamel, I.R. Multimodality imaging of intrahepatic cholangiocarcinoma. Hepatobiliary Surg. Nutr. 2017, 6, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Choi, J.Y.; Chung, Y.E. Evaluation of biliary malignancies using multidetector-row computed tomography. J. Comput. Assist. Tomogr. 2010, 34, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Valls, C.; Gumà, A.; Puig, I.; Sanchez, A.; Andía, E.; Serrano, T.; Figueras, J. Intrahepatic peripheral cholangiocarcinoma: CT evaluation. Abdom Imaging 2000, 25, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Iavarone, M.; Piscaglia, F.; Vavassori, S.; Galassi, M.; Sangiovanni, A.; Venerandi, L.; Forzenigo, L.V.; Golfieri, R.; Bolondi, L.; Colombo, M. Contrast enhanced CT-scan to diagnose intrahepatic cholangiocarcinoma in patients with cirrhosis. J. Hepatol. 2013, 58, 1188–1193. [Google Scholar] [CrossRef] [PubMed]
- Szklaruk, J.; Bhosale, P. Hepatocellular carcinoma: MRI and CT examination. Isr. Med. Assoc. J. 2007, 9, 153–155. [Google Scholar] [PubMed]
- Navaneethan, U.; Njei, B.; Venkatesh, P.G.; Lourdusamy, V.; Sanaka, M.R. Endoscopic ultrasound in the diagnosis of cholangiocarcinoma as the etiology of biliary strictures: A systematic review and meta-analysis. Gastroenterol. Rep. 2015, 3, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Navaneethan, U.; Njei, B.; Lourdusamy, V.; Konjeti, R.; Vargo, J.J.; Parsi, M.A. Comparative effectiveness of biliary brush cytology and intraductal biopsy for detection of malignant biliary strictures: A systematic review and meta-analysis. Gastrointest. Endosc. 2015, 81, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Navaneethan, U.; Hasan, M.K.; Lourdusamy, V.; Njei, B.; Varadarajulu, S.; Hawes, R.H. Single-operator cholangioscopy and targeted biopsies in the diagnosis of indeterminate biliary strictures: A systematic review. Gastrointest. Endosc. 2015, 82, 608–614.e2. [Google Scholar] [CrossRef] [PubMed]
- Jhaveri, K.S.; Hosseini-Nik, H. MRI of cholangiocarcinoma. J. Magn. Reson. Imaging 2015, 42, 1165–1179. [Google Scholar] [CrossRef]
- Lamarca, A.; Barriuso, J.; Chander, A.; McNamara, M.G.; Hubner, R.A.; ÓReilly, D.; Manoharan, P.; Valle, J.W. (18)F-fluorodeoxyglucose positron emission tomography ((18)FDG-PET) for patients with biliary tract cancer: Systematic review and meta-analysis. J. Hepatol. 2019, 71, 115–129. [Google Scholar] [CrossRef]
- Patel, A.H.; Harnois, D.M.; Klee, G.G.; LaRusso, N.F.; Gores, G.J. The utility of CA 19-9 in the diagnoses of cholangiocarcinoma in patients without primary sclerosing cholangitis. Am. J. Gastroenterol. 2000, 95, 204–207. [Google Scholar] [CrossRef] [PubMed]
- Bitzer, M.; Voesch, S.; Albert, J.; Bartenstein, P.; Bechstein, W.; Blödt, S.; Brunner, T.; Dombrowski, F.; Evert, M.; Follmann, M.; et al. S3-Leitlinie: Diagnostik und Therapie biliärer Karzinome. Z. Gastroenterol. 2022, 60, 219–238. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Bao, H.; Huang, Z.; Liang, Z.; Lin, N.; Ni, C.; Xu, Y. Non-Coding RNA in Cholangiocarcinoma: An Update. FBL 2023, 28, 173. [Google Scholar] [CrossRef]
- Ge, X.; Wang, Y.; Nie, J.; Li, Q.; Tang, L.; Deng, X.; Wang, F.; Xu, B.; Wu, X.; Zhang, X.; et al. The diagnostic/prognostic potential and molecular functions of long non-coding RNAs in the exosomes derived from the bile of human cholangiocarcinoma. Oncotarget 2017, 8, 69995–70005. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Zhu, B.; Meng, D.; Shen, X.; Li, X.; Wang, Z.; Li, L. Down-regulation of lncRNA-NEF indicates poor prognosis in intrahepatic cholangiocarcinoma. Biosci. Rep. 2019, 39, BSR20181573. [Google Scholar] [CrossRef]
- Li, J.; Jiang, X.; Xu, Y.; Kang, P.; Huang, P.; Meng, N.; Wang, H.; Zheng, W.; Wang, H.; Wang, Z.; et al. YY1-induced DLEU1/miR-149-5p Promotes Malignant Biological Behavior of Cholangiocarcinoma through Upregulating YAP1/TEAD2/SOX2. Int. J. Biol. Sci. 2022, 18, 4301–4315. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Leng, K.; Yao, Y.; Kang, P.; Liao, G.; Han, Y.; Shi, G.; Ji, D.; Huang, P.; Zheng, W.; et al. A Circular RNA, Cholangiocarcinoma-Associated Circular RNA 1, Contributes to Cholangiocarcinoma Progression, Induces Angiogenesis, and Disrupts Vascular Endothelial Barriers. Hepatology 2021, 73, 1419–1435. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.M.; Li, Z.L.; Li, J.L.; Xu, Y.; Leng, K.M.; Cui, Y.F.; Sun, D.J. A novel prognostic biomarker for cholangiocarcinoma: circRNA Cdr1as. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 365–371. [Google Scholar] [CrossRef]
- Sun, D.; Li, F.; Liu, L.; Yu, S.; Wang, H.; Gao, X.; Liu, G.; Zhao, Y.; Qiu, G.; Jiang, X. PSMA3-AS1 induced by transcription factor PAX5 promotes cholangiocarcinoma proliferation, migration and invasion by sponging miR-376a-3p to up-regulate LAMC1. Aging 2022, 14, 509–525. [Google Scholar] [CrossRef]
- Jiang, X.; Li, J.; Wang, W.; Hu, Z.; Guan, C.; Zhao, Y.; Li, W.; Cui, Y. AR-induced ZEB1-AS1 represents poor prognosis in cholangiocarcinoma and facilitates tumor stemness, proliferation and invasion through mediating miR-133b/HOXB8. Aging 2020, 12, 1237–1255. [Google Scholar] [CrossRef]
- Li, J.; Jiang, X.; Li, Z.; Huang, L.; Ji, D.; Yu, L.; Zhou, Y.; Cui, Y. SP1-induced HOXD-AS1 promotes malignant progression of cholangiocarcinoma by regulating miR-520c-3p/MYCN. Aging 2020, 12, 16304–16325. [Google Scholar] [CrossRef]
- Guan, C.; Zhao, Y.; Wang, W.; Hu, Z.; Liu, L.; Li, W.; Jiang, X. Knockdown of lncRNA SNHG20 Suppressed the Proliferation of Cholangiocarcinoma by Sponging miR-520f-3p. Cancer Biother. Radiopharm. 2024, 39, 178–187. [Google Scholar] [CrossRef]
- Li, J.; Guan, C.; Hu, Z.; Liu, L.; Su, Z.; Kang, P.; Jiang, X.; Cui, Y. Yin Yang 1-induced LINC00667 up-regulates pyruvate dehydrogenase kinase 1 to promote proliferation, migration and invasion of cholangiocarcinoma cells by sponging miR-200c-3p. Hum. Cell 2021, 34, 187–200. [Google Scholar] [CrossRef]
- Hu, Z.; Huang, L.; Wang, W.; Guan, C.; Zhao, Y.; Liu, L.; Jiang, X. Long Non-coding RNA FOXD2-AS1 Promotes Proliferation, Migration, and Invasion in Cholangiocarcinoma Through Regulating miR-760/E2F3 Axis. Dig. Dis. Sci. 2022, 67, 546–558. [Google Scholar] [CrossRef]
- Wu, Y.; Hayat, K.; Hu, Y.; Yang, J. Long Non-Coding RNAs as Molecular Biomarkers in Cholangiocarcinoma. Front. Cell Dev. Biol. 2022, 10, 890605. [Google Scholar] [CrossRef] [PubMed]
- Bagante, F.; Spolverato, G.; Weiss, M.; Alexandrescu, S.; Marques, H.P.; Aldrighetti, L.; Maithel, S.K.; Pulitano, C.; Bauer, T.W.; Shen, F.; et al. Surgical Management of Intrahepatic Cholangiocarcinoma in Patients with Cirrhosis: Impact of Lymphadenectomy on Peri-Operative Outcomes. World J. Surg. 2018, 42, 2551–2560. [Google Scholar] [CrossRef]
- Ebata, T.; Mizuno, T.; Yokoyama, Y.; Igami, T.; Sugawara, G.; Nagino, M. Surgical resection for Bismuth type IV perihilar cholangiocarcinoma. Br. J. Surg. 2018, 105, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Schnitzbauer, A.A.; Eberhard, J.; Bartsch, F.; Brunner, S.M.; Ceyhan, G.O.; Walter, D.; Fries, H.; Hannes, S.; Hecker, A.; Li, J.; et al. The MEGNA Score and Preoperative Anemia are Major Prognostic Factors After Resection in the German Intrahepatic Cholangiocarcinoma Cohort. Ann. Surg. Oncol. 2020, 27, 1147–1155. [Google Scholar] [CrossRef] [PubMed]
- Shroff, R.T.; Kennedy, E.B.; Bachini, M.; Bekaii-Saab, T.; Crane, C.; Edeline, J.; El-Khoueiry, A.; Feng, M.; Katz, M.H.G.; Primrose, J.; et al. Adjuvant Therapy for Resected Biliary Tract Cancer: ASCO Clinical Practice Guideline. J. Clin. Oncol. 2019, 37, 1015–1027. [Google Scholar] [CrossRef]
- Primrose, J.N.; Fox, R.P.; Palmer, D.H.; Malik, H.Z.; Prasad, R.; Mirza, D.; Anthony, A.; Corrie, P.; Falk, S.; Finch-Jones, M.; et al. Capecitabine compared with observation in resected biliary tract cancer (BILCAP): A randomised, controlled, multicentre, phase 3 study. Lancet Oncol. 2019, 20, 663–673. [Google Scholar] [CrossRef]
- Oh, D.Y.; Lee, K.H.; Lee, D.W.; Yoon, J.; Kim, T.Y.; Bang, J.H.; Nam, A.R.; Oh, K.S.; Kim, J.M.; Lee, Y.; et al. Gemcitabine and cisplatin plus durvalumab with or without tremelimumab in chemotherapy-naive patients with advanced biliary tract cancer: An open-label, single-centre, phase 2 study. Lancet Gastroenterol. Hepatol. 2022, 7, 522–532. [Google Scholar] [CrossRef] [PubMed]
- EASL-ILCA Clinical Practice Guidelines on the management of intrahepatic cholangiocarcinoma. J. Hepatol. 2023, 79, 181–208. [CrossRef] [PubMed]
- Kelley, R.K.; Ueno, M.; Yoo, C.; Finn, R.S.; Furuse, J.; Ren, Z.; Yau, T.; Klümpen, H.-J.; Chan, S.L.; Ozaka, M.; et al. Pembrolizumab in combination with gemcitabine and cisplatin compared with gemcitabine and cisplatin alone for patients with advanced biliary tract cancer (KEYNOTE-966): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2023, 401, 1853–1865. [Google Scholar] [CrossRef]
- Lamarca, A.; Palmer, D.H.; Wasan, H.S.; Ross, P.J.; Ma, Y.T.; Arora, A.; Falk, S.; Gillmore, R.; Wadsley, J.; Patel, K.; et al. Second-line FOLFOX chemotherapy versus active symptom control for advanced biliary tract cancer (ABC-06): A phase 3, open-label, randomised, controlled trial. Lancet Oncol. 2021, 22, 690–701. [Google Scholar] [CrossRef] [PubMed]
- Yoo, C.; Kim, K.-p.; Jeong, J.H.; Kim, I.; Kang, M.J.; Cheon, J.; Kang, B.W.; Ryu, H.; Lee, J.S.; Kim, K.W.; et al. Liposomal irinotecan plus fluorouracil and leucovorin versus fluorouracil and leucovorin for metastatic biliary tract cancer after progression on gemcitabine plus cisplatin (NIFTY): A multicentre, open-label, randomised, phase 2b study. Lancet Oncol. 2021, 22, 1560–1572. [Google Scholar] [CrossRef] [PubMed]
- Hyung, J.; Kim, I.; Kim, K.P.; Ryoo, B.Y.; Jeong, J.H.; Kang, M.J.; Cheon, J.; Kang, B.W.; Ryu, H.; Lee, J.S.; et al. Treatment with Liposomal Irinotecan Plus Fluorouracil and Leucovorin for Patients with Previously Treated Metastatic Biliary Tract Cancer: The Phase 2b NIFTY Randomized Clinical Trial. JAMA Oncol. 2023, 9, 692–699. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.; Wenzel, P.; Folprecht, G.; Schütt, P.; Wege, H.; Kretzschmar, A.; Jacobasch, L.; Ziegenhagen, N.; Boeck, S.; Kanzler, S.; et al. 53MO Nal-IRI and 5-FU/LV compared to 5-FU/LV in patients with cholangio- and gallbladder carcinoma previously treated with gemcitabine-based therapies (NALIRICC–AIO-HEP-0116). Ann. Oncol. 2022, 33, S563–S564. [Google Scholar] [CrossRef]
- Becker, N.S.; Rodriguez, J.A.; Barshes, N.R.; O’Mahony, C.A.; Goss, J.A.; Aloia, T.A. Outcomes analysis for 280 patients with cholangiocarcinoma treated with liver transplantation over an 18-year period. J. Gastrointest. Surg. 2008, 12, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Darwish Murad, S.; Kim, W.R.; Harnois, D.M.; Douglas, D.D.; Burton, J.; Kulik, L.M.; Botha, J.F.; Mezrich, J.D.; Chapman, W.C.; Schwartz, J.J.; et al. Efficacy of neoadjuvant chemoradiation, followed by liver transplantation, for perihilar cholangiocarcinoma at 12 US centers. Gastroenterology 2012, 143, 88–98.e3, quiz e14. [Google Scholar] [CrossRef]
- Vugts, J.J.A.; Gaspersz, M.P.; Roos, E.; Franken, L.C.; Olthof, P.B.; Coelen, R.J.S.; van Vugt, J.L.A.; Labeur, T.A.; Brouwer, L.; Besselink, M.G.H.; et al. Eligibility for Liver Transplantation in Patients with Perihilar Cholangiocarcinoma. Ann. Surg. Oncol. 2021, 28, 1483–1492. [Google Scholar] [CrossRef]
- Matsuo, K.; Rocha, F.G.; Ito, K.; D’Angelica, M.I.; Allen, P.J.; Fong, Y.; Dematteo, R.P.; Gonen, M.; Endo, I.; Jarnagin, W.R. The Blumgart preoperative staging system for hilar cholangiocarcinoma: Analysis of resectability and outcomes in 380 patients. J. Am. Coll. Surg. 2012, 215, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Verlingue, L.; Malka, D.; Allorant, A.; Massard, C.; Ferté, C.; Lacroix, L.; Rouleau, E.; Auger, N.; Ngo, M.; Nicotra, C.; et al. Precision medicine for patients with advanced biliary tract cancers: An effective strategy within the prospective MOSCATO-01 trial. Eur. J. Cancer 2017, 87, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Farha, N.; Dima, D.; Ullah, F.; Kamath, S. Precision Oncology Targets in Biliary Tract Cancer. Cancers 2023, 15, 2105. [Google Scholar] [CrossRef] [PubMed]
- Valery, M.; Vasseur, D.; Fachinetti, F.; Boilève, A.; Smolenschi, C.; Tarabay, A.; Antoun, L.; Perret, A.; Fuerea, A.; Pudlarz, T.; et al. Targetable Molecular Alterations in the Treatment of Biliary Tract Cancers: An Overview of the Available Treatments. Cancers 2023, 15, 4446. [Google Scholar] [CrossRef] [PubMed]
- Chong, D.Q.; Zhu, A.X. The landscape of targeted therapies for cholangiocarcinoma: Current status and emerging targets. Oncotarget 2016, 7, 46750–46767. [Google Scholar] [CrossRef] [PubMed]
- Goyal, L.; Kongpetch, S.; Crolley, V.E.; Bridgewater, J. Targeting FGFR inhibition in cholangiocarcinoma. Cancer Treat. Rev. 2021, 95, 102170. [Google Scholar] [CrossRef] [PubMed]
- Moasser, M.M. The oncogene HER2: Its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene 2007, 26, 6469–6487. [Google Scholar] [CrossRef] [PubMed]
- Amatu, A.; Sartore-Bianchi, A.; Siena, S. NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. ESMO Open 2016, 1, e000023. [Google Scholar] [CrossRef] [PubMed]
- Gönül Geyik, Ö.; Anichini, G.; Ulukaya, E.; Marra, F.; Raggi, C. DNA Damage Response Inhibitors in Cholangiocarcinoma: Current Progress and Perspectives. Cells 2022, 11, 1463. [Google Scholar] [CrossRef] [PubMed]
- Kabbara, K.W.; Cannon, T.; Winer, A.; Wadlow, R.C. Molecular Pathogenesis of Cholangiocarcinoma: Implications for Disease Classification and Therapy. Oncology 2022, 36, 492–498. [Google Scholar] [CrossRef]
- Dai, S.; Zhou, Z.; Chen, Z.; Xu, G.; Chen, Y. Fibroblast Growth Factor Receptors (FGFRs): Structures and Small Molecule Inhibitors. Cells 2019, 8, 614. [Google Scholar] [CrossRef]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.; Segatto, O.; Stenzinger, A.; Saborowski, A. FGFR2 Inhibition in Cholangiocarcinoma. Annu. Rev. Med. 2023, 74, 293–306. [Google Scholar] [CrossRef] [PubMed]
- Neumann, O.; Burn, T.C.; Allgäuer, M.; Ball, M.; Kirchner, M.; Albrecht, T.; Volckmar, A.-L.; Beck, S.; Endris, V.; Goldschmid, H.; et al. Genomic architecture of FGFR2 fusions in cholangiocarcinoma and its implication for molecular testing. Br. J. Cancer 2022, 127, 1540–1549. [Google Scholar] [CrossRef] [PubMed]
- Babina, I.S.; Turner, N.C. Advances and challenges in targeting FGFR signalling in cancer. Nat. Rev. Cancer 2017, 17, 318–332. [Google Scholar] [CrossRef] [PubMed]
- Arai, Y.; Totoki, Y.; Hosoda, F.; Shirota, T.; Hama, N.; Nakamura, H.; Ojima, H.; Furuta, K.; Shimada, K.; Okusaka, T.; et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology 2014, 59, 1427–1434. [Google Scholar] [CrossRef] [PubMed]
- Farshidfar, F.; Zheng, S.; Gingras, M.C.; Newton, Y.; Shih, J.; Robertson, A.G.; Hinoue, T.; Hoadley, K.A.; Gibb, E.A.; Roszik, J.; et al. Integrative Genomic Analysis of Cholangiocarcinoma Identifies Distinct IDH-Mutant Molecular Profiles. Cell Rep. 2017, 18, 2780–2794. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Borad, M.J.; Kelley, R.K.; Wang, Y.; Abdel-Wahab, R.; Meric-Bernstam, F.; Baggerly, K.A.; Kaseb, A.O.; Al-Shamsi, H.O.; Ahn, D.H.; et al. Cholangiocarcinoma with FGFR Genetic Aberrations: A Unique Clinical Phenotype. JCO Precis. Oncol. 2018, 2, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lowery, M.A.; Ptashkin, R.; Jordan, E.; Berger, M.F.; Zehir, A.; Capanu, M.; Kemeny, N.E.; O’Reilly, E.M.; El-Dika, I.; Jarnagin, W.R.; et al. Comprehensive Molecular Profiling of Intrahepatic and Extrahepatic Cholangiocarcinomas: Potential Targets for Intervention. Clin. Cancer Res. 2018, 24, 4154–4161. [Google Scholar] [CrossRef]
- Liu, P.C.C.; Koblish, H.; Wu, L.; Bowman, K.; Diamond, S.; DiMatteo, D.; Zhang, Y.; Hansbury, M.; Rupar, M.; Wen, X.; et al. INCB054828 (pemigatinib), a potent and selective inhibitor of fibroblast growth factor receptors 1, 2, and 3, displays activity against genetically defined tumor models. PLoS ONE 2020, 15, e0231877. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Sahai, V.; Hollebecque, A.; Vaccaro, G.; Melisi, D.; Al-Rajabi, R.; Paulson, A.S.; Borad, M.J.; Gallinson, D.; Murphy, A.G.; et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: A multicentre, open-label, phase 2 study. Lancet Oncol. 2020, 21, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Bibeau, K.; Féliz, L.; Lihou, C.F.; Ren, H.; Abou-Alfa, G.K. Progression-Free Survival in Patients with Cholangiocarcinoma with or without FGF/FGFR Alterations: A FIGHT-202 Post Hoc Analysis of Prior Systemic Therapy Response. JCO Precis. Oncol. 2022, 6, e2100414. [Google Scholar] [CrossRef] [PubMed]
- Sootome, H.; Fujita, H.; Ito, K.; Ochiiwa, H.; Fujioka, Y.; Ito, K.; Miura, A.; Sagara, T.; Ito, S.; Ohsawa, H.; et al. Futibatinib Is a Novel Irreversible FGFR 1-4 Inhibitor That Shows Selective Antitumor Activity against FGFR-Deregulated Tumors. Cancer Res. 2020, 80, 4986–4997. [Google Scholar] [CrossRef]
- Goyal, L.; Meric-Bernstam, F.; Hollebecque, A.; Valle, J.W.; Morizane, C.; Karasic, T.B.; Abrams, T.A.; Furuse, J.; Kelley, R.K.; Cassier, P.A.; et al. Futibatinib for FGFR2-Rearranged Intrahepatic Cholangiocarcinoma. N. Engl. J. Med. 2023, 388, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Goyal, L.; Meric-Bernstam, F.; Hollebecque, A.; Morizane, C.; Valle, J.W.; Karasic, T.B.; Abrams, T.A.; Kelley, R.K.; Cassier, P.A.; Furuse, J.; et al. Updated results of the FOENIX-CCA2 trial: Efficacy and safety of futibatinib in intrahepatic cholangiocarcinoma (iCCA) harboring FGFR2 fusions/rearrangements. J. Clin. Oncol. 2022, 40, 4009. [Google Scholar] [CrossRef]
- Guagnano, V.; Kauffmann, A.; Wöhrle, S.; Stamm, C.; Ito, M.; Barys, L.; Pornon, A.; Yao, Y.; Li, F.; Zhang, Y.; et al. FGFR genetic alterations predict for sensitivity to NVP-BGJ398, a selective pan-FGFR inhibitor. Cancer Discov. 2012, 2, 1118–1133. [Google Scholar] [CrossRef] [PubMed]
- Javle, M.; Roychowdhury, S.; Kelley, R.K.; Sadeghi, S.; Macarulla, T.; Weiss, K.H.; Waldschmidt, D.T.; Goyal, L.; Borbath, I.; El-Khoueiry, A.; et al. Infigratinib (BGJ398) in previously treated patients with advanced or metastatic cholangiocarcinoma with FGFR2 fusions or rearrangements: Mature results from a multicentre, open-label, single-arm, phase 2 study. Lancet Gastroenterol. Hepatol. 2021, 6, 803–815. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.G.; Yu, Y.; Eathiraj, S.; Wang, Y.; Savage, R.E.; Lapierre, J.M.; Schwartz, B.; Abbadessa, G. Preclinical Activity of ARQ 087, a Novel Inhibitor Targeting FGFR Dysregulation. PLoS ONE 2016, 11, e0162594. [Google Scholar] [CrossRef] [PubMed]
- Mazzaferro, V.; El-Rayes, B.F.; Droz Dit Busset, M.; Cotsoglou, C.; Harris, W.P.; Damjanov, N.; Masi, G.; Rimassa, L.; Personeni, N.; Braiteh, F.; et al. Derazantinib (ARQ 087) in advanced or inoperable FGFR2 gene fusion-positive intrahepatic cholangiocarcinoma. Br. J. Cancer 2019, 120, 165–171. [Google Scholar] [CrossRef]
- Borad, M.; Javle, M.; Shaib, W.L.; Mody, K.; Bergamo, F.; Harris, W.P.; Damjanov, N.; Macarulla, T.; Brandi, G.; Masi, G.; et al. Efficacy of derazantinib in intrahepatic cholangiocarcinoma (iCCA) patients with FGFR2 fusions, mutations or amplifications. Ann. Oncol. 2022, 33, S567–S568. [Google Scholar] [CrossRef]
- Perera, T.P.S.; Jovcheva, E.; Mevellec, L.; Vialard, J.; De Lange, D.; Verhulst, T.; Paulussen, C.; Van De Ven, K.; King, P.; Freyne, E.; et al. Discovery and Pharmacological Characterization of JNJ-42756493 (Erdafitinib), a Functionally Selective Small-Molecule FGFR Family Inhibitor. Mol. Cancer Ther. 2017, 16, 1010–1020. [Google Scholar] [CrossRef] [PubMed]
- Park, J.O.; Feng, Y.-H.; Chen, Y.-Y.; Su, W.-C.; Oh, D.-Y.; Shen, L.; Kim, K.-P.; Liu, X.; Bai, Y.; Liao, H.; et al. Updated results of a phase IIa study to evaluate the clinical efficacy and safety of erdafitinib in Asian advanced cholangiocarcinoma (CCA) patients with FGFR alterations. J. Clin. Oncol. 2019, 37, 4117. [Google Scholar] [CrossRef]
- Ahn, D.H.; Uson Junior, P.L.S.; Masci, P.; Kosiorek, H.; Halfdanarson, T.R.; Mody, K.; Babiker, H.; DeLeon, T.; Sonbol, M.B.; Gores, G.; et al. A pilot study of Pan-FGFR inhibitor ponatinib in patients with FGFR-altered advanced cholangiocarcinoma. Investig. New Drugs 2022, 40, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Voss, M.H.; Hierro, C.; Heist, R.S.; Cleary, J.M.; Meric-Bernstam, F.; Tabernero, J.; Janku, F.; Gandhi, L.; Iafrate, A.J.; Borger, D.R.; et al. A Phase I, Open-Label, Multicenter, Dose-escalation Study of the Oral Selective FGFR Inhibitor Debio 1347 in Patients with Advanced Solid Tumors Harboring FGFR Gene Alterations. Clin. Cancer Res. 2019, 25, 2699–2707. [Google Scholar] [CrossRef] [PubMed]
- Cleary, J.M.; Iyer, G.; Oh, D.-Y.; Mellinghoff, I.K.; Goyal, L.; Ng, M.C.H.; Meric-Bernstam, F.; Matos, I.; Chao, T.-Y.; Sarkouh, R.A.; et al. Final results from the phase I study expansion cohort of the selective FGFR inhibitor Debio 1,347 in patients with solid tumors harboring an FGFR gene fusion. J. Clin. Oncol. 2020, 38, 3603. [Google Scholar] [CrossRef]
- Subbiah, V.; Sahai, V.; Maglic, D.; Bruderek, K.; Touré, B.B.; Zhao, S.; Valverde, R.; O’Hearn, P.J.; Moustakas, D.T.; Schönherr, H.; et al. RLY-4008, the First Highly Selective FGFR2 Inhibitor with Activity across FGFR2 Alterations and Resistance Mutations. Cancer Discov. 2023, 13, 2012–2031. [Google Scholar] [CrossRef] [PubMed]
- Javle, M.M.; Fountzilas, C.; Li, D.; Fonkoua, L.A.K.; Fan, J.; Peng, P.; Wang, H.; Ngo, B.; Sun, C.; Ru, Q.C.; et al. Phase II study of FGFR1-3 inhibitor tinengotinib as monotherapy in patients with advanced or metastatic cholangiocarcinoma: Interim analysis. J. Clin. Oncol. 2023, 41, 539. [Google Scholar] [CrossRef]
- Grassian, A.R.; Pagliarini, R.; Chiang, D.Y. Mutations of isocitrate dehydrogenase 1 and 2 in intrahepatic cholangiocarcinoma. Curr. Opin. Gastroenterol. 2014, 30, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Kipp, B.R.; Voss, J.S.; Kerr, S.E.; Barr Fritcher, E.G.; Graham, R.P.; Zhang, L.; Highsmith, W.E.; Zhang, J.; Roberts, L.R.; Gores, G.J.; et al. Isocitrate dehydrogenase 1 and 2 mutations in cholangiocarcinoma. Hum. Pathol. 2012, 43, 1552–1558. [Google Scholar] [CrossRef]
- Borger, D.R.; Tanabe, K.K.; Fan, K.C.; Lopez, H.U.; Fantin, V.R.; Straley, K.S.; Schenkein, D.P.; Hezel, A.F.; Ancukiewicz, M.; Liebman, H.M.; et al. Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist 2012, 17, 72–79. [Google Scholar] [CrossRef]
- Rizzo, A.; Ricci, A.D.; Brandi, G. IDH inhibitors in advanced cholangiocarcinoma: Another arrow in the quiver? Cancer Treat. Res. Commun. 2021, 27, 100356. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.; Gores, G.J. Emerging molecular therapeutic targets for cholangiocarcinoma. J. Hepatol. 2017, 67, 632–644. [Google Scholar] [CrossRef]
- Zhu, A.X.; Macarulla, T.; Javle, M.M.; Kelley, R.K.; Lubner, S.J.; Adeva, J.; Cleary, J.M.; Catenacci, D.V.T.; Borad, M.J.; Bridgewater, J.A.; et al. Final Overall Survival Efficacy Results of Ivosidenib for Patients with Advanced Cholangiocarcinoma with IDH1 Mutation: The Phase 3 Randomized Clinical ClarIDHy Trial. JAMA Oncol. 2021, 7, 1669–1677. [Google Scholar] [CrossRef] [PubMed]
- Pauff, J.; Papadopoulos, K.; Janku, F.; Turk, A.; Goyal, L.; Shroff, R.; Shimizu, T.; Ikeda, M.; Azad, N.; Cleary, J.; et al. A phase I study of LY3410738, a first-in-class covalent inhibitor of mutant IDH1 in cholangiocarcinoma and other advanced solid tumors. J. Clin. Oncol. 2021, 39, TPS350. [Google Scholar] [CrossRef]
- Cleary, J.M.; Rouaisnel, B.; Daina, A.; Raghavan, S.; Roller, L.A.; Huffman, B.M.; Singh, H.; Wen, P.Y.; Bardeesy, N.; Zoete, V.; et al. Secondary IDH1 resistance mutations and oncogenic IDH2 mutations cause acquired resistance to ivosidenib in cholangiocarcinoma. NPJ Precis. Oncol. 2022, 6, 61. [Google Scholar] [CrossRef]
- Simile, M.M.; Bagella, P.; Vidili, G.; Spanu, A.; Manetti, R.; Seddaiu, M.A.; Babudieri, S.; Madeddu, G.; Serra, P.A.; Altana, M.; et al. Targeted Therapies in Cholangiocarcinoma: Emerging Evidence from Clinical Trials. Medicina 2019, 55, 42. [Google Scholar] [CrossRef] [PubMed]
- Pellino, A.; Loupakis, F.; Cadamuro, M.; Dadduzio, V.; Fassan, M.; Guido, M.; Cillo, U.; Indraccolo, S.; Fabris, L. Precision medicine in cholangiocarcinoma. Transl. Gastroenterol. Hepatol. 2018, 3, 40. [Google Scholar] [CrossRef]
- Galdy, S.; Lamarca, A.; McNamara, M.G.; Hubner, R.A.; Cella, C.A.; Fazio, N.; Valle, J.W. HER2/HER3 pathway in biliary tract malignancies; systematic review and meta-analysis: A potential therapeutic target? Cancer Metastasis Rev. 2017, 36, 141–157. [Google Scholar] [CrossRef]
- Harding, J.J.; Piha-Paul, S.A.; Shah, R.H.; Murphy, J.J.; Cleary, J.M.; Shapiro, G.I.; Quinn, D.I.; Braña, I.; Moreno, V.; Borad, M.; et al. Antitumour activity of neratinib in patients with HER2-mutant advanced biliary tract cancers. Nat. Commun. 2023, 14, 630. [Google Scholar] [CrossRef]
- Harding, J.J.; Fan, J.; Oh, D.Y.; Choi, H.J.; Kim, J.W.; Chang, H.M.; Bao, L.; Sun, H.C.; Macarulla, T.; Xie, F.; et al. Zanidatamab for HER2-amplified, unresectable, locally advanced or metastatic biliary tract cancer (HERIZON-BTC-01): A multicentre, single-arm, phase 2b study. Lancet Oncol. 2023, 24, 772–782. [Google Scholar] [CrossRef]
- Oh, D.-Y.; Yong, W.-P.; Chen, L.-T.; Kim, J.-W.; Park, J.H.; Hsu, K.; Lindmark, B.; McIntyre, N.; Collins, B.; Firth, C. Varlitinib in combination with gemcitabine and cisplatin for treatment-naïve advanced biliary tract cancer. J. Clin. Oncol. 2022, 40, 439. [Google Scholar] [CrossRef]
- Javle, M.M.; Oh, D.Y.; Ikeda, M.; Yong, W.P.; Hsu, K.; Lindmark, B.; McIntyre, N.; Firth, C. Varlitinib plus capecitabine in second-line advanced biliary tract cancer: A randomized, phase II study (TreeTopp). ESMO Open 2022, 7, 100314. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Sun, Y. Promising Molecular Targets for the Targeted Therapy of Biliary Tract Cancers: An Overview. OncoTargets Ther. 2021, 14, 1341–1366. [Google Scholar] [CrossRef] [PubMed]
- Koeberle, D.; Fritsch, R. Targeting HER2 in Biliary Tract Carcinomas: Challenges and Opportunities. Oncol. Res. Treat. 2021, 44, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.K.; Chon, H.J.; Cheon, J.; Lee, M.A.; Im, H.S.; Jang, J.S.; Kim, M.H.; Park, S.; Kang, B.; Hong, M.; et al. Trastuzumab plus FOLFOX for HER2-positive biliary tract cancer refractory to gemcitabine and cisplatin: A multi-institutional phase 2 trial of the Korean Cancer Study Group (KCSG-HB19-14). Lancet Gastroenterol. Hepatol. 2023, 8, 56–65. [Google Scholar] [CrossRef] [PubMed]
- de Vries, E.G.E.; Rüschoff, J.; Lolkema, M.; Tabernero, J.; Gianni, L.; Voest, E.; de Groot, D.J.A.; Castellano, D.; Erb, G.; Naab, J.; et al. Phase II study (KAMELEON) of single-agent T-DM1 in patients with HER2-positive advanced urothelial bladder cancer or pancreatic cancer/cholangiocarcinoma. Cancer Med. 2023, 12, 12071–12083. [Google Scholar] [CrossRef] [PubMed]
- Javle, M.; Borad, M.J.; Azad, N.S.; Kurzrock, R.; Abou-Alfa, G.K.; George, B.; Hainsworth, J.; Meric-Bernstam, F.; Swanton, C.; Sweeney, C.J.; et al. Pertuzumab and trastuzumab for HER2-positive, metastatic biliary tract cancer (MyPathway): A multicentre, open-label, phase 2a, multiple basket study. Lancet Oncol. 2021, 22, 1290–1300. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Mizuno, N.; Sunakawa, Y.; Canon, J.L.; Galsky, M.D.; Hamilton, E.; Hayashi, H.; Jerusalem, G.; Kim, S.T.; Lee, K.W.; et al. Tucatinib and Trastuzumab for Previously Treated Human Epidermal Growth Factor Receptor 2-Positive Metastatic Biliary Tract Cancer (SGNTUC-019): A Phase II Basket Study. J. Clin. Oncol. 2023, 41, 5569–5578. [Google Scholar] [CrossRef] [PubMed]
- Ostwal, V.; Mandavkar, S.; Bhargava, P.; Srinivas, S.; Kapoor, A.; Shetty, O.; Kannan, S.; Chaugule, D.; Patil, R.; Parulekar, M.; et al. Trastuzumab Plus Gemcitabine-Cisplatin for Treatment-Naïve Human Epidermal Growth Factor Receptor 2–Positive Biliary Tract Adenocarcinoma: A Multicenter, Open-Label, Phase II Study (TAB). J. Clin. Oncol. 2024, 42, 800–807. [Google Scholar] [CrossRef] [PubMed]
- Meric-Bernstam, F.; Makker, V.; Oaknin, A.; Oh, D.Y.; Banerjee, S.; González-Martín, A.; Jung, K.H.; Ługowska, I.; Manso, L.; Manzano, A.; et al. Efficacy and Safety of Trastuzumab Deruxtecan in Patients with HER2-Expressing Solid Tumors: Primary Results From the DESTINY-PanTumor02 Phase II Trial. J. Clin. Oncol. 2024, 42, 47–58. [Google Scholar] [CrossRef]
- Yoshikawa, D.; Ojima, H.; Iwasaki, M.; Hiraoka, N.; Kosuge, T.; Kasai, S.; Hirohashi, S.; Shibata, T. Clinicopathological and prognostic significance of EGFR, VEGF, and HER2 expression in cholangiocarcinoma. Br. J. Cancer 2008, 98, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Philip, P.A.; Mahoney, M.R.; Allmer, C.; Thomas, J.; Pitot, H.C.; Kim, G.; Donehower, R.C.; Fitch, T.; Picus, J.; Erlichman, C. Phase II Study of Erlotinib in Patients with Advanced Biliary Cancer. J. Clin. Oncol. 2006, 24, 3069–3074. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Park, S.H.; Chang, H.M.; Kim, J.S.; Choi, H.J.; Lee, M.A.; Jang, J.S.; Jeung, H.C.; Kang, J.H.; Lee, H.W.; et al. Gemcitabine and oxaliplatin with or without erlotinib in advanced biliary-tract cancer: A multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2012, 13, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Leone, F.; Marino, D.; Cereda, S.; Filippi, R.; Belli, C.; Spadi, R.; Nasti, G.; Montano, M.; Amatu, A.; Aprile, G.; et al. Panitumumab in combination with gemcitabine and oxaliplatin does not prolong survival in wild-type KRAS advanced biliary tract cancer: A randomized phase 2 trial (Vecti-BIL study). Cancer 2016, 122, 574–581. [Google Scholar] [CrossRef]
- Malka, D.; Cervera, P.; Foulon, S.; Trarbach, T.; de la Fouchardière, C.; Boucher, E.; Fartoux, L.; Faivre, S.; Blanc, J.F.; Viret, F.; et al. Gemcitabine and oxaliplatin with or without cetuximab in advanced biliary-tract cancer (BINGO): A randomised, open-label, non-comparative phase 2 trial. Lancet Oncol. 2014, 15, 819–828. [Google Scholar] [CrossRef] [PubMed]
- El-Khoueiry, A.B.; Rankin, C.; Siegel, A.B.; Iqbal, S.; Gong, I.Y.; Micetich, K.C.; Kayaleh, O.R.; Lenz, H.J.; Blanke, C.D. S0941: A phase 2 SWOG study of sorafenib and erlotinib in patients with advanced gallbladder carcinoma or cholangiocarcinoma. Br. J. Cancer 2014, 110, 882–887. [Google Scholar] [CrossRef] [PubMed]
- Cocco, E.; Scaltriti, M.; Drilon, A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar] [CrossRef]
- Kummar, S.; Lassen, U.N. TRK Inhibition: A New Tumor-Agnostic Treatment Strategy. Target Oncol 2018, 13, 545–556. [Google Scholar] [CrossRef]
- Marchiò, C.; Scaltriti, M.; Ladanyi, M.; Iafrate, A.J.; Bibeau, F.; Dietel, M.; Hechtman, J.F.; Troiani, T.; López-Rios, F.; Douillard, J.Y.; et al. ESMO recommendations on the standard methods to detect NTRK fusions in daily practice and clinical research. Ann. Oncol. 2019, 30, 1417–1427. [Google Scholar] [CrossRef]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; DuBois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: Integrated analysis of three phase 1-2 trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef]
- Demols, A.; Rocq, L.; Perez-Casanova, L.; Charry, M.; De Nève, N.; Ramadhan, A.; Van Campenhout, C.; De Clercq, S.; Maris, C.; Closset, J.; et al. A Two-Step Diagnostic Approach for NTRK Gene Fusion Detection in Biliary Tract and Pancreatic Adenocarcinomas. Oncologist 2023, 28, e520–e525. [Google Scholar] [CrossRef] [PubMed]
- Salama, A.K.S.; Li, S.; Macrae, E.R.; Park, J.I.; Mitchell, E.P.; Zwiebel, J.A.; Chen, H.X.; Gray, R.J.; McShane, L.M.; Rubinstein, L.V.; et al. Dabrafenib and Trametinib in Patients with Tumors with BRAF(V600E) Mutations: Results of the NCI-MATCH Trial Subprotocol H. J. Clin. Oncol. 2020, 38, 3895–3904. [Google Scholar] [CrossRef]
- Subbiah, V.; Lassen, U.; Élez, E.; Italiano, A.; Curigliano, G.; Javle, M.; de Braud, F.; Prager, G.W.; Greil, R.; Stein, A.; et al. Dabrafenib plus trametinib in patients with BRAF(V600E)-mutated biliary tract cancer (ROAR): A phase 2, open-label, single-arm, multicentre basket trial. Lancet Oncol. 2020, 21, 1234–1243. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Kreitman, R.J.; Wainberg, Z.A.; Gazzah, A.; Lassen, U.; Stein, A.; Wen, P.Y.; Dietrich, S.; de Jonge, M.J.A.; Blay, J.Y.; et al. Dabrafenib plus trametinib in BRAFV600E-mutated rare cancers: The phase 2 ROAR trial. Nat. Med. 2023, 29, 1103–1112. [Google Scholar] [CrossRef] [PubMed]
- Rose, A.A.N.; Ayodele, O.; Genta, S.; Pimentel Muniz, T.; Kelly, D.; Hodgson, K.; King, I.; Stockley, T.; Pugh, T.J.; Saeed Kamil, Z.; et al. Preliminary results of BEAVER: An investigator-initiated phase II study of binimetinib and encorafenib for the treatment of advanced solid tumors with non-V600E BRAF mutations (mts). J. Clin. Oncol. 2021, 39, e15038. [Google Scholar] [CrossRef]
- Shimoi, T.; Sunami, K.; Tahara, M.; Nishiwaki, S.; Tanaka, S.; Baba, E.; Kanai, M.; Kinoshita, I.; Shirota, H.; Hayashi, H.; et al. Dabrafenib and trametinib administration in patients with BRAF V600E/R or non-V600 BRAF mutated advanced solid tumours (BELIEVE, NCCH1901): A multicentre, open-label, and single-arm phase II trial. EClinicalMedicine 2024, 69, 102447. [Google Scholar] [CrossRef] [PubMed]
- Lamarca, A.; Barriuso, J.; McNamara, M.G.; Valle, J.W. Biliary Tract Cancer: State of the Art and potential role of DNA Damage Repair. Cancer Treat. Rev. 2018, 70, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Chae, H.; Kim, D.; Yoo, C.; Kim, K.P.; Jeong, J.H.; Chang, H.M.; Lee, S.S.; Park, D.H.; Song, T.J.; Hwang, S.; et al. Therapeutic relevance of targeted sequencing in management of patients with advanced biliary tract cancer: DNA damage repair gene mutations as a predictive biomarker. Eur. J. Cancer 2019, 120, 31–39. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef]
- Boerner, T.; Drill, E.; Pak, L.M.; Nguyen, B.; Sigel, C.S.; Doussot, A.; Shin, P.; Goldman, D.A.; Gonen, M.; Allen, P.J.; et al. Genetic Determinants of Outcome in Intrahepatic Cholangiocarcinoma. Hepatology 2021, 74, 1429–1444. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, N.J.; Bailey, M.L.; Hieter, P. Synthetic lethality and cancer. Nat. Rev. Genet. 2017, 18, 613–623. [Google Scholar] [CrossRef]
- Ahn, D.H.; Bekaii-Saab, T. Biliary tract cancer and genomic alterations in homologous recombinant deficiency: Exploiting synthetic lethality with PARP inhibitors. Chin. Clin. Oncol. 2020, 9, 6. [Google Scholar] [CrossRef] [PubMed]
- Ricci, A.D.; Rizzo, A.; Bonucci, C.; Tober, N.; Palloni, A.; Mollica, V.; Maggio, I.; Deserti, M.; Tavolari, S.; Brandi, G. PARP Inhibitors in Biliary Tract Cancer: A New Kid on the Block? Medicines 2020, 7, 54. [Google Scholar] [CrossRef] [PubMed]
- Bezrookove, V.; Patino, J.M.; Nosrati, M.; Desprez, P.Y.; McAllister, S.; Soroceanu, L.; Baron, A.; Osorio, R.; Kashani-Sabet, M.; Dar, A.A. Niraparib Suppresses Cholangiocarcinoma Tumor Growth by Inducing Oxidative and Replication Stress. Cancers 2021, 13, 4405. [Google Scholar] [CrossRef] [PubMed]
- George, T.J.; Lee, J.-H.; Hosein, P.J.; DeRemer, D.L.; Chatzkel, J.A.; Ramnaraign, B.H.; Rogers, S.C.; Markham, M.J.; Daily, K.C.; Ezenwajiaku, N.; et al. Results of a phase II trial of the PARP inhibitor, niraparib, in BAP1 and other DNA damage response pathway deficient neoplasms. J. Clin. Oncol. 2022, 40, 3122. [Google Scholar] [CrossRef]
- Golan, T.; Raitses-Gurevich, M.; Kelley, R.K.; Bocobo, A.G.; Borgida, A.; Shroff, R.T.; Holter, S.; Gallinger, S.; Ahn, D.H.; Aderka, D.; et al. Overall Survival and Clinical Characteristics of BRCA-Associated Cholangiocarcinoma: A Multicenter Retrospective Study. Oncologist 2017, 22, 804–810. [Google Scholar] [CrossRef]
- Leslie, M. IDH-Mutant Tumors Vulnerable to PARP Inhibition. Cancer Discov. 2017, 7, Of4. [Google Scholar] [CrossRef] [PubMed]
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. Cell Dev. Biol. 2020, 8, 564601. [Google Scholar] [CrossRef]
- Ghelli Luserna di Rorà, A.; Cerchione, C.; Martinelli, G.; Simonetti, G. A WEE1 family business: Regulation of mitosis, cancer progression, and therapeutic target. J. Hematol. Oncol. 2020, 13, 126. [Google Scholar] [CrossRef]
- Nam, A.R.; Jin, M.H.; Bang, J.H.; Oh, K.S.; Seo, H.R.; Oh, D.Y.; Bang, Y.J. Inhibition of ATR Increases the Sensitivity to WEE1 Inhibitor in Biliary Tract Cancer. Cancer Res. Treat. 2020, 52, 945–956. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.R.; Nam, A.R.; Bang, J.H.; Oh, K.S.; Kim, J.M.; Yoon, J.; Kim, T.Y.; Oh, D.Y. Inhibition of WEE1 Potentiates Sensitivity to PARP Inhibitor in Biliary Tract Cancer. Cancer Res. Treat. 2022, 54, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Takebe, N.; Naqash, A.R.; O’Sullivan Coyne, G.; Kummar, S.; Do, K.; Bruns, A.; Juwara, L.; Zlott, J.; Rubinstein, L.; Piekarz, R.; et al. Safety, Antitumor Activity, and Biomarker Analysis in a Phase I Trial of the Once-daily Wee1 Inhibitor Adavosertib (AZD1775) in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2021, 27, 3834–3844. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.R.; Hong, D.S.; Bendell, J.C.; Jones, S.F.; Hamilton, E.P.; Subbiah, V.; Karp, D.D.; Wang, J.S.-Z.; Aljumaily, R.; Hynes, S.; et al. A phase 1b dose-escalation study of prexasertib, a checkpoint kinase 1 (CHK1) inhibitor, in combination with cisplatin in patients with advanced cancer. J. Clin. Oncol. 2018, 36, 2579. [Google Scholar] [CrossRef]
- Li, X.Y.; Chen, J.Q.; Aisa, A.; Ding, Y.W.; Zhang, D.; Yuan, Y. Targeting BRCA-mutant biliary tract cancer: Current evidence and future perspectives. J. Dig. Dis. 2023, 24, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Gonzalez, R.; Jacobson, M.K. Characterization of polymers of adenosine diphosphate ribose generated in vitro and in vivo. Biochemistry 1987, 26, 3218–3224. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Shi, J.; Guo, H.; Yang, X.; Jiang, Y.; Long, J.; Bai, Y.; Wang, D.; Yang, X.; Wan, X.; et al. Alterations in DNA Damage Repair Genes in Primary Liver Cancer. Clin. Cancer Res. 2019, 25, 4701–4711. [Google Scholar] [CrossRef] [PubMed]
- Costa, B.A.; Tallón de Lara, P.; Park, W.; Keane, F.; Harding, J.J.; Khalil, D.N. Durable Response after Olaparib Treatment for Perihilar Cholangiocarcinoma with Germline BRCA2 Mutation. Oncol. Res. Treat. 2023, 46, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Ma, Z.; Fu, X.; Hao, Z.; Shang, H.; Shi, J.; Lei, M.; Xu, M.; Ning, S.; Hua, X. Olaparib effectively treats local recurrence of extrahepatic cholangiocarcinoma in a patient harboring a BRCA2-inactivating mutation: A case report. Ann. Transl. Med. 2021, 9, 1487. [Google Scholar] [CrossRef]
- Cheng, Y.; Zhang, J.; Qin, S.K.; Hua, H.Q. Treatment with olaparib monotherapy for BRCA2-mutated refractory intrahepatic cholangiocarcinoma: A case report. OncoTargets Ther. 2018, 11, 5957–5962. [Google Scholar] [CrossRef]
- Su, Y.L.; Ng, C.T.; Jan, Y.H.; Hsieh, Y.L.; Wu, C.L.; Tan, K.T. Remarkable Response to Olaparib in a Patient with Combined Hepatocellular-Cholangiocarcinoma Harboring a Biallelic BRCA2 Mutation. OncoTargets Ther. 2021, 14, 3895–3901. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Mahn, R.; Möhring, C.; Sadeghlar, F.; Meyer, C.; Toma, M.; Kreppel, B.; Essler, M.; Glowka, T.; Matthaei, H.; et al. Case Report: Sustained complete remission on combination therapy with olaparib and pembrolizumab in BRCA2-mutated and PD-L1-positive metastatic cholangiocarcinoma after platinum derivate. Front. Oncol. 2022, 12, 933943. [Google Scholar] [CrossRef] [PubMed]
- Xiong, F.; Gong, J.; Wang, Q. Olaparib and Pembrolizumab Treatment for BRCA1-Mutated and PD-L1-Positive Intrahepatic Cholangiocarcinoma Recurrence and Metastasis: A Case Report. OncoTargets Ther. 2020, 13, 6385–6391. [Google Scholar] [CrossRef] [PubMed]
- Trombetta, D.; Parente, P.; Latiano, T.P.; Fabrizio, F.P.; Muscarella, L.A. Identification of EML4-ALK fusion in a sporadic case of cholangiocarcinoma. Eur. J. Intern. Med. 2020, 71, 92–94. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Lizaso, A.; Mao, X.; Yang, N.; Zhang, Y. Novel AMBRA1-ALK fusion identified by next-generation sequencing in advanced gallbladder cancer responds to crizotinib: A case report. Ann. Transl. Med. 2020, 8, 1099. [Google Scholar] [CrossRef] [PubMed]
- Goyal, L.; Zheng, H.; Yurgelun, M.; Abrams, T.A.; Kwak, E.L.; Cleary, J.M.; Knowles, M.; Regan, E.; Gisondi, A.; Sheehan, S.; et al. A phase II and biomarker study of cabozantinib (XL-184) in patients (pts) with advanced cholangiocarcinoma (CCA). J. Clin. Oncol. 2015, 33, e15124. [Google Scholar] [CrossRef]
- Pant, S.; Saleh, M.; Bendell, J.; Infante, J.R.; Jones, S.; Kurkjian, C.D.; Moore, K.M.; Kazakin, J.; Abbadessa, G.; Wang, Y.; et al. A phase I dose escalation study of oral c-MET inhibitor tivantinib (ARQ 197) in combination with gemcitabine in patients with solid tumors. Ann. Oncol. 2014, 25, 1416–1421. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Liu, Q.; Li, W.; Qu, Y.; Zhang, Y.; Liu, T. Identification of a Novel EHBP1-MET Fusion in an Intrahepatic Cholangiocarcinoma Responding to Crizotinib. Oncologist 2020, 25, 1005–1008. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yadav, A.K.; Han, J.Y.; Ahn, K.S.; Jang, B.C. Anti-Growth, Anti-Angiogenic, and Pro-Apoptotic Effects by CX-4945, an Inhibitor of Casein Kinase 2, on HuCCT-1 Human Cholangiocarcinoma Cells via Control of Caspase-9/3, DR-4, STAT-3/STAT-5, Mcl-1, eIF-2α, and HIF-1α. Int. J. Mol. Sci. 2022, 23, 6353. [Google Scholar] [CrossRef]
- Jayaraman, P.S.; Gaston, K. Targeting protein kinase CK2 in the treatment of cholangiocarcinoma. Explor. Target. Antitumor. Ther. 2021, 2, 434–447. [Google Scholar] [CrossRef]
- Borad, M.J.; Bai, L.-Y.; Chen, M.-H.; Hubbard, J.M.; Mody, K.; Rha, S.Y.; Richards, D.A.; Davis, S.L.; Soong, J.; Huang, C.-E.C.E.; et al. Silmitasertib (CX-4945) in combination with gemcitabine and cisplatin as first-line treatment for patients with locally advanced or metastatic cholangiocarcinoma: A phase Ib/II study. J. Clin. Oncol. 2021, 39, 312. [Google Scholar] [CrossRef]
- Paratala, B.S.; Chung, J.H.; Williams, C.B.; Yilmazel, B.; Petrosky, W.; Williams, K.; Schrock, A.B.; Gay, L.M.; Lee, E.; Dolfi, S.C.; et al. RET rearrangements are actionable alterations in breast cancer. Nat. Commun. 2018, 9, 4821. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Cassier, P.A.; Siena, S.; Garralda, E.; Paz-Ares, L.; Garrido, P.; Nadal, E.; Vuky, J.; Lopes, G.; Kalemkerian, G.P.; et al. Pan-cancer efficacy of pralsetinib in patients with RET fusion-positive solid tumors from the phase 1/2 ARROW trial. Nat. Med. 2022, 28, 1640–1645. [Google Scholar] [CrossRef]
- Subbiah, V.; Wolf, J.; Konda, B.; Kang, H.; Spira, A.; Weiss, J.; Takeda, M.; Ohe, Y.; Khan, S.; Ohashi, K.; et al. Tumour-agnostic efficacy and safety of selpercatinib in patients with RET fusion-positive solid tumours other than lung or thyroid tumours (LIBRETTO-001): A phase 1/2, open-label, basket trial. Lancet Oncol. 2022, 23, 1261–1273. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.; Borad, M.J.; Patel, T.; Gores, G.J. Cholangiocarcinoma: Molecular pathways and therapeutic opportunities. Semin. Liver Dis. 2014, 34, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.S.; Cao, B.; Kim, J.; Al-Toubah, T.E.; Mehta, R.; Centeno, B.A.; Kim, R.D. Phase 2 study of copanlisib in combination with gemcitabine and cisplatin in advanced biliary tract cancers. Cancer 2021, 127, 1293–1300. [Google Scholar] [CrossRef]
- Damodaran, S.; Zhao, F.; Deming, D.A.; Mitchell, E.P.; Wright, J.J.; Gray, R.J.; Wang, V.; McShane, L.M.; Rubinstein, L.V.; Patton, D.R.; et al. Phase II Study of Copanlisib in Patients with Tumors with PIK3CA Mutations: Results From the NCI-MATCH ECOG-ACRIN Trial (EAY131) Subprotocol Z1F. J. Clin. Oncol. 2022, 40, 1552–1561. [Google Scholar] [CrossRef] [PubMed]
- Piha-Paul, S.A.; Taylor, M.H.; Spitz, D.; Schwartzberg, L.; Beck, J.T.; Bauer, T.M.; Meric-Bernstam, F.; Purkayastha, D.; Karpiak, L.; Szpakowski, S.; et al. Efficacy and safety of buparlisib, a PI3K inhibitor, in patients with malignancies harboring a PI3K pathway activation: A phase 2, open-label, single-arm study. Oncotarget 2019, 10, 6526–6535. [Google Scholar] [CrossRef] [PubMed]
- Ahn, D.H.; Li, J.; Wei, L.; Doyle, A.; Marshall, J.L.; Schaaf, L.J.; Phelps, M.A.; Villalona-Calero, M.A.; Bekaii-Saab, T. Results of an abbreviated phase-II study with the Akt Inhibitor MK-2206 in Patients with Advanced Biliary Cancer. Sci. Rep. 2015, 5, 12122. [Google Scholar] [CrossRef]
- Buzzoni, R.; Pusceddu, S.; Bajetta, E.; De Braud, F.; Platania, M.; Iannacone, C.; Cantore, M.; Mambrini, A.; Bertolini, A.; Alabiso, O.; et al. Activity and safety of RAD001 (everolimus) in patients affected by biliary tract cancer progressing after prior chemotherapy: A phase II ITMO study. Ann. Oncol. 2014, 25, 1597–1603. [Google Scholar] [CrossRef]
- Kim, S.T.; Lee, J.; Park, S.H.; Park, J.O.; Park, Y.S.; Kang, W.K.; Lim, H.Y. Prospective phase II trial of everolimus in PIK3CA amplification/mutation and/or PTEN loss patients with advanced solid tumors refractory to standard therapy. BMC Cancer 2017, 17, 211. [Google Scholar] [CrossRef] [PubMed]
- Bian, J.L.; Wang, M.M.; Tong, E.J.; Sun, J.; Li, M.; Miao, Z.B.; Li, Y.L.; Zhu, B.H.; Xu, J.J. Benefit of everolimus in treatment of an intrahepatic cholangiocarcinoma patient with a PIK3CA mutation. World J. Gastroenterol. 2017, 23, 4311–4316. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Tang, S. WNT/β-catenin signaling in the development of liver cancers. Biomed. Pharmacother. 2020, 132, 110851. [Google Scholar] [CrossRef] [PubMed]
- Perugorria, M.J.; Olaizola, P.; Labiano, I.; Esparza-Baquer, A.; Marzioni, M.; Marin, J.J.G.; Bujanda, L.; Banales, J.M. Wnt-β-catenin signalling in liver development, health and disease. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Smits, R.; Hao, H.; He, C. Wnt/β-Catenin Signaling in Liver Cancers. Cancers 2019, 11, 926. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.F.; Qiu, L.; Yang, S.L.; Wu, J.C.; Liu, T.J. Wnt/β-catenin signaling as an emerging potential key pharmacological target in cholangiocarcinoma. Biosci. Rep. 2020, 40, BSR20193353. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.D.; Yu, X.H.; Wu, W.R.; Xu, X.L.; Wang, J.Y.; Xu, L.B.; Zhang, R.; Liu, C. Dickkopf-1 expression is associated with tumorigenity and lymphatic metastasis in human hilar cholangiocarcinoma. Oncotarget 2016, 7, 70378–70387. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Lee, H.S.; Bang, S.M.; Han, D.H.; Hwang, H.K.; Choi, G.H.; Chung, M.J.; Kim, S.U. Serum Dickkopf-1 in Combined with CA 19-9 as a Biomarker of Intrahepatic Cholangiocarcinoma. Cancers 2021, 13, 1828. [Google Scholar] [CrossRef] [PubMed]
- Arsenijevic, T.; Coulonval, K.; Raspé, E.; Demols, A.; Roger, P.P.; Van Laethem, J.L. CDK4/6 Inhibitors in Pancreatobiliary Cancers: Opportunities and Challenges. Cancers 2023, 15, 968. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta 2007, 1773, 1263–1284. [Google Scholar] [CrossRef]
- Thein, K.Z.; Biter, A.B.; Banks, K.C.; Duda, A.W.; Saam, J.; Roszik, J.; Janku, F.; Skoulidis, F.; Heymach, J.V.; Kopetz, S.; et al. Identification of KRAS(G12C) Mutations in Circulating Tumor DNA in Patients with Cancer. JCO Precis. Oncol. 2022, 6, e2100547. [Google Scholar] [CrossRef]
- Pant, S.; Yaeger, R.; Spira, A.I.; Pelster, M.; Sabari, J.K.; Hafez, N.; Barve, M.A.; Velastegui, K.; Yan, X.; Der-Torossian, H.; et al. KRYSTAL-1: Activity and safety of adagrasib (MRTX849) in patients with advanced solid tumors harboring a KRASG12C mutation. J. Clin. Oncol. 2023, 41, 425082. [Google Scholar] [CrossRef]
- Strickler, J.H.; Satake, H.; George, T.J.; Yaeger, R.; Hollebecque, A.; Garrido-Laguna, I.; Schuler, M.; Burns, T.F.; Coveler, A.L.; Falchook, G.S.; et al. Sotorasib in KRAS p.G12C-Mutated Advanced Pancreatic Cancer. N. Engl. J. Med. 2023, 388, 33–43. [Google Scholar] [CrossRef]
- Bekaii-Saab, T.; Phelps, M.A.; Li, X.; Saji, M.; Goff, L.; Kauh, J.S.; O’Neil, B.H.; Balsom, S.; Balint, C.; Liersemann, R.; et al. Multi-institutional phase II study of selumetinib in patients with metastatic biliary cancers. J. Clin. Oncol. 2011, 29, 2357–2363. [Google Scholar] [CrossRef]
- Doherty, M.; Tam, V.C.; McNamara, M.G.; Hedley, D.W.; Dhani, N.C.; Chen, E.X.; Jang, R.W.-J.; Tang, P.A.; Sim, H.-W.; O’Kane, G.M.; et al. Selumetinib (Sel) and cisplatin/gemcitabine (CisGem) for advanced biliary tract cancer (BTC): A randomized trial. J. Clin. Oncol. 2018, 36, 4084. [Google Scholar] [CrossRef]
- Shroff, R.T.; Yarchoan, M.; O’Connor, A.; Gallagher, D.; Zahurak, M.L.; Rosner, G.; Ohaji, C.; Sartorius-Mergenthaler, S.; Parkinson, R.; Subbiah, V.; et al. The oral VEGF receptor tyrosine kinase inhibitor pazopanib in combination with the MEK inhibitor trametinib in advanced cholangiocarcinoma. Br. J. Cancer 2017, 116, 1402–1407. [Google Scholar] [CrossRef]
- Kim, J.W.; Lee, K.H.; Kim, J.W.; Suh, K.J.; Nam, A.R.; Bang, J.H.; Bang, Y.J.; Oh, D.Y. Enhanced antitumor effect of binimetinib in combination with capecitabine for biliary tract cancer patients with mutations in the RAS/RAF/MEK/ERK pathway: Phase Ib study. Br. J. Cancer 2019, 121, 332–339. [Google Scholar] [CrossRef]
- Tang, D.; Nagano, H.; Yamamoto, H.; Wada, H.; Nakamura, M.; Kondo, M.; Ota, H.; Yoshioka, S.; Kato, H.; Damdinsuren, B.; et al. Angiogenesis in cholangiocellular carcinoma: Expression of vascular endothelial growth factor, angiopoietin-1/2, thrombospondin-1 and clinicopathological significance. Oncol. Rep. 2006, 15, 525–532. [Google Scholar] [CrossRef]
- Zhu, A.X.; Meyerhardt, J.A.; Blaszkowsky, L.S.; Kambadakone, A.R.; Muzikansky, A.; Zheng, H.; Clark, J.W.; Abrams, T.A.; Chan, J.A.; Enzinger, P.C.; et al. Efficacy and safety of gemcitabine, oxaliplatin, and bevacizumab in advanced biliary-tract cancers and correlation of changes in 18-fluorodeoxyglucose PET with clinical outcome: A phase 2 study. Lancet Oncol. 2010, 11, 48–54. [Google Scholar] [CrossRef]
- Bréchon, M.; Dior, M.; Dréanic, J.; Brieau, B.; Guillaumot, M.A.; Brezault, C.; Mir, O.; Goldwasser, F.; Coriat, R. Addition of an antiangiogenic therapy, bevacizumab, to gemcitabine plus oxaliplatin improves survival in advanced biliary tract cancers. Investig. New Drugs 2018, 36, 156–162. [Google Scholar] [CrossRef]
- Guion-Dusserre, J.F.; Lorgis, V.; Vincent, J.; Bengrine, L.; Ghiringhelli, F. FOLFIRI plus bevacizumab as a second-line therapy for metastatic intrahepatic cholangiocarcinoma. World J. Gastroenterol. 2015, 21, 2096–2101. [Google Scholar] [CrossRef]
- Iyer, R.V.; Pokuri, V.K.; Groman, A.; Ma, W.W.; Malhotra, U.; Iancu, D.M.; Grande, C.; Saab, T.B. A Multicenter Phase II Study of Gemcitabine, Capecitabine, and Bevacizumab for Locally Advanced or Metastatic Biliary Tract Cancer. Am J. Clin. Oncol. 2018, 41, 649–655. [Google Scholar] [CrossRef]
- Lubner, S.J.; Mahoney, M.R.; Kolesar, J.L.; Loconte, N.K.; Kim, G.P.; Pitot, H.C.; Philip, P.A.; Picus, J.; Yong, W.P.; Horvath, L.; et al. Report of a multicenter phase II trial testing a combination of biweekly bevacizumab and daily erlotinib in patients with unresectable biliary cancer: A phase II Consortium study. J. Clin. Oncol. 2010, 28, 3491–3497. [Google Scholar] [CrossRef]
- Amin, N.E.L.; Hansen, T.F.; Fernebro, E.; Ploen, J.; Eberhard, J.; Lindebjerg, J.; Jensen, L.H. Randomized Phase II trial of combination chemotherapy with panitumumab or bevacizumab for patients with inoperable biliary tract cancer without KRAS exon 2 mutations. Int. J. Cancer 2021, 149, 119–126. [Google Scholar] [CrossRef]
- Lee, S.; Shroff, R.T.; Makawita, S.; Xiao, L.; Danner De Armas, A.; Bhosale, P.; Reddy, K.; Shalaby, A.; Raghav, K.; Pant, S.; et al. Phase II Study of Ramucirumab in Advanced Biliary Tract Cancer Previously Treated By Gemcitabine-Based Chemotherapy. Clin. Cancer Res. 2022, 28, 2229–2236. [Google Scholar] [CrossRef]
- Herbst, R.S.; Arkenau, H.T.; Santana-Davila, R.; Calvo, E.; Paz-Ares, L.; Cassier, P.A.; Bendell, J.; Penel, N.; Krebs, M.G.; Martin-Liberal, J.; et al. Ramucirumab plus pembrolizumab in patients with previously treated advanced non-small-cell lung cancer, gastro-oesophageal cancer, or urothelial carcinomas (JVDF): A multicohort, non-randomised, open-label, phase 1a/b trial. Lancet Oncol. 2019, 20, 1109–1123. [Google Scholar] [CrossRef]
- Valle, J.W.; Vogel, A.; Denlinger, C.S.; He, A.R.; Bai, L.Y.; Orlova, R.; Van Cutsem, E.; Adeva, J.; Chen, L.T.; Obermannova, R.; et al. Addition of ramucirumab or merestinib to standard first-line chemotherapy for locally advanced or metastatic biliary tract cancer: A randomised, double-blind, multicentre, phase 2 study. Lancet Oncol. 2021, 22, 1468–1482. [Google Scholar] [CrossRef]
- Camera, S.; Deleporte, A.; Bregni, G.; Trevisi, E.; Pretta, A.; Telli, T.A.; Polastro, L.; Gombos, A.; Kayumba, A.; Ameye, L.; et al. MOMENTUM: A Phase I Trial Investigating 2 Schedules of Capecitabine with Aflibercept in Patients with Gastrointestinal and Breast Cancer. Clin Colorectal Cancer 2020, 19, 311–318.e1. [Google Scholar] [CrossRef]
- Bengala, C.; Bertolini, F.; Malavasi, N.; Boni, C.; Aitini, E.; Dealis, C.; Zironi, S.; Depenni, R.; Fontana, A.; Del Giovane, C.; et al. Sorafenib in patients with advanced biliary tract carcinoma: A phase II trial. Br. J. Cancer 2010, 102, 68–72. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Rankin, C.J.; Ben-Josef, E.; Lenz, H.J.; Gold, P.J.; Hamilton, R.D.; Govindarajan, R.; Eng, C.; Blanke, C.D. SWOG 0514: A phase II study of sorafenib in patients with unresectable or metastatic gallbladder carcinoma and cholangiocarcinoma. Investig. New Drugs 2012, 30, 1646–1651. [Google Scholar] [CrossRef]
- Moehler, M.; Maderer, A.; Schimanski, C.; Kanzler, S.; Denzer, U.; Kolligs, F.T.; Ebert, M.P.; Distelrath, A.; Geissler, M.; Trojan, J.; et al. Gemcitabine plus sorafenib versus gemcitabine alone in advanced biliary tract cancer: A double-blind placebo-controlled multicentre phase II AIO study with biomarker and serum programme. Eur. J. Cancer 2014, 50, 3125–3135. [Google Scholar] [CrossRef]
- Lee, J.K.; Capanu, M.; O’Reilly, E.M.; Ma, J.; Chou, J.F.; Shia, J.; Katz, S.S.; Gansukh, B.; Reidy-Lagunes, D.; Segal, N.H.; et al. A phase II study of gemcitabine and cisplatin plus sorafenib in patients with advanced biliary adenocarcinomas. Br. J. Cancer 2013, 109, 915–919. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, X.; Wang, D.; Yang, X.; Wang, Y.; Long, J.; Zhou, J.; Lu, Z.; Mao, Y.; Sang, X.; et al. Lenvatinib Beyond First-Line Therapy in Patients with Advanced Biliary Tract Carcinoma. Front. Oncol. 2022, 12, 785535. [Google Scholar] [CrossRef]
- Shi, G.-M.; Jian, Z.; Fan, J.; Huang, X.-Y.; Wu, D.; Liang, F.; Lu, J.-C.; Yang, G.-H.; Chen, Y.; Ge, N.-L.; et al. Phase II study of lenvatinib in combination with GEMOX chemotherapy for advanced intrahepatic cholangiocarcinoma. J. Clin. Oncol. 2021, 39, e16163. [Google Scholar] [CrossRef]
- Lin, J.; Yang, X.; Long, J.; Zhao, S.; Mao, J.; Wang, D.; Bai, Y.; Bian, J.; Zhang, L.; Yang, X.; et al. Pembrolizumab combined with lenvatinib as non-first-line therapy in patients with refractory biliary tract carcinoma. Hepatobiliary Surg. Nutr. 2020, 9, 414–424. [Google Scholar] [CrossRef]
- Villanueva, L.; Lwin, Z.; Chung, H.C.; Gomez-Roca, C.; Longo, F.; Yanez, E.; Senellart, H.; Doherty, M.; García-Corbacho, J.; Hendifar, A.E.; et al. Lenvatinib plus pembrolizumab for patients with previously treated biliary tract cancers in the multicohort phase II LEAP-005 study. J. Clin. Oncol. 2021, 39, 321. [Google Scholar] [CrossRef]
- Galle, P.R.; Dufour, J.F.; Peck-Radosavljevic, M.; Trojan, J.; Vogel, A. Systemic therapy of advanced hepatocellular carcinoma. Future Oncol. 2021, 17, 1237–1251. [Google Scholar] [CrossRef]
- Sun, W.; Patel, A.; Normolle, D.; Patel, K.; Ohr, J.; Lee, J.J.; Bahary, N.; Chu, E.; Streeter, N.; Drummond, S. A phase 2 trial of regorafenib as a single agent in patients with chemotherapy-refractory, advanced, and metastatic biliary tract adenocarcinoma. Cancer 2019, 125, 902–909. [Google Scholar] [CrossRef]
- Kim, D.W.; Sanoff, H.K.; Poklepovic, A.S.; Tariq, F.; Nixon, A.B.; Liu, Y.; Kim, R.D. Final analysis of phase II trial of regorafenib (REG) in refractory advanced biliary cancers (BC). J. Clin. Oncol. 2019, 37, 4083. [Google Scholar] [CrossRef]
- Demols, A.; Borbath, I.; Van den Eynde, M.; Houbiers, G.; Peeters, M.; Marechal, R.; Delaunoit, T.; Goemine, J.C.; Laurent, S.; Holbrechts, S.; et al. Regorafenib after failure of gemcitabine and platinum-based chemotherapy for locally advanced/metastatic biliary tumors: REACHIN, a randomized, double-blind, phase II trial. Ann. Oncol. 2020, 31, 1169–1177. [Google Scholar] [CrossRef]
- Morabito, A.; Piccirillo, M.C.; Costanzo, R.; Sandomenico, C.; Carillio, G.; Daniele, G.; Giordano, P.; Bryce, J.; Carotenuto, P.; La Rocca, A.; et al. Vandetanib: An overview of its clinical development in NSCLC and other tumors. Drugs Today 2010, 46, 683–698. [Google Scholar] [CrossRef] [PubMed]
- Kessler, E.R.; Eckhardt, S.G.; Pitts, T.M.; Bradshaw-Pierce, E.L.; O’Byrant, C.L.; Messersmith, W.A.; Nallapreddy, S.; Weekes, C.; Spratlin, J.; Lieu, C.H.; et al. Phase I trial of vandetanib in combination with gemcitabine and capecitabine in patients with advanced solid tumors with an expanded cohort in pancreatic and biliary cancers. Investig. New Drugs 2016, 34, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Santoro, A.; Gebbia, V.; Pressiani, T.; Testa, A.; Personeni, N.; Arrivas Bajardi, E.; Foa, P.; Buonadonna, A.; Bencardino, K.; Barone, C.; et al. A randomized, multicenter, phase II study of vandetanib monotherapy versus vandetanib in combination with gemcitabine versus gemcitabine plus placebo in subjects with advanced biliary tract cancer: The VanGogh study. Ann. Oncol. 2015, 26, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Valle, J.W.; Wasan, H.; Lopes, A.; Backen, A.C.; Palmer, D.H.; Morris, K.; Duggan, M.; Cunningham, D.; Anthoney, D.A.; Corrie, P.; et al. Cediranib or placebo in combination with cisplatin and gemcitabine chemotherapy for patients with advanced biliary tract cancer (ABC-03): A randomised phase 2 trial. Lancet Oncol. 2015, 16, 967–978. [Google Scholar] [CrossRef] [PubMed]
- Neuzillet, C.; Seitz, J.-F.; Fartoux, L.; Malka, D.; Lledo, G.; Tijeras-Raballand, A.; Gramont, A.D.; Ronot, M.; Bouattour, M.; Dreyer, C.; et al. Sunitinib as second-line treatment in patients with advanced intrahepatic cholangiocarcinoma (SUN-CK phase II trial): Safety, efficacy, and updated translational results. J. Clin. Oncol. 2015, 33, 343. [Google Scholar] [CrossRef]
- OKANO, N.; KASUGA, A.; KAWAI, K.; KOBAYASHI, T.; NARUGE, D.; NAGASHIMA, F.; FURUSE, J. Axitinib for Gemcitabine-refractory Advanced Biliary Tract Cancer: Report of 5 Cases. Anticancer Res. 2017, 37, 3711–3715. [Google Scholar] [PubMed]
- Xu, J.; Bai, Y.; Sun, H.; Bai, C.; Jia, R.; Li, Y.; Zhang, W.; Liu, L.; Huang, C.; Guan, M.; et al. A single-arm, multicenter, open-label phase 2 trial of surufatinib in patients with unresectable or metastatic biliary tract cancer. Cancer 2021, 127, 3975–3984. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.-W.; Yang, X.-R.; Xu, Y.; Huang, X.-W.; Qiu, S.-J.; Sun, H.-C.; Fan, J.; Zhou, J. The clinical efficacy and safety of sintilimab plus anlotinib for unresectable intrahepatic cholangiocarcinoma (ICC): A prospective, single-arm phase II study. J. Clin. Oncol. 2023, 41, e16170. [Google Scholar] [CrossRef]
- Zhou, J.; Sun, Y.; Zhang, W.; Yuan, J.; Peng, Z.; Wang, W.; Gong, J.; Yang, L.; Cao, Y.; Zhao, H.; et al. Phase Ib study of anlotinib combined with TQB2450 in pretreated advanced biliary tract cancer and biomarker analysis. Hepatology 2023, 77, 65–76. [Google Scholar] [CrossRef]
- Maio, M.; Ascierto, P.A.; Manzyuk, L.; Motola-Kuba, D.; Penel, N.; Cassier, P.A.; Bariani, G.; Jesus-Acosta, A.D.; Doi, T.; Longo, F.; et al. Pembrolizumab in microsatellite instability high (MSI-H)/mismatch repair deficient (dMMR) cancers: Updated analysis from phase 2 KEYNOTE-158 study. J. Clin. Oncol. 2021, 39, 2565. [Google Scholar] [CrossRef]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients with Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Boilève, A.; Hilmi, M.; Smolenschi, C.; Ducreux, M.; Hollebecque, A.; Malka, D. Immunotherapy in Advanced Biliary Tract Cancers. Cancers 2021, 13, 1569. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, R.; Ali, S.M.; Borad, M.J.; Shroff, R.T.; Kwong, L.; Vauthey, J.-N.; Koay, E.J.; Zuo, M.; Rashid, A.; Schrock, A.B.; et al. Variations in DNA repair genomic alterations and tumor mutation burden in biliary tract cancer (BTC) subtypes. J. Clin. Oncol. 2018, 36, 263. [Google Scholar] [CrossRef]
- Piha-Paul, S.A.; Oh, D.Y.; Ueno, M.; Malka, D.; Chung, H.C.; Nagrial, A.; Kelley, R.K.; Ros, W.; Italiano, A.; Nakagawa, K.; et al. Efficacy and safety of pembrolizumab for the treatment of advanced biliary cancer: Results from the KEYNOTE-158 and KEYNOTE-028 studies. Int. J. Cancer 2020, 147, 2190–2198. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Yoo, C.; Jeong, J.H.; Kang, B.; Hong, Y.; Kim, K.-P.; Ryoo, B.-Y. Efficacy and safety of pembrolizumab in patients with PD-L1 positive advanced biliary tract cancer (BTC): A prospective cohort study. J. Clin. Oncol. 2019, 37, 4082. [Google Scholar] [CrossRef]
- Gou, M.; Zhang, Y.; Si, H.; Dai, G. Efficacy and safety of nivolumab for metastatic biliary tract cancer. OncoTargets Ther. 2019, 12, 861–867. [Google Scholar] [CrossRef]
- Ueno, M.; Ikeda, M.; Morizane, C.; Kobayashi, S.; Ohno, I.; Kondo, S.; Okano, N.; Kimura, K.; Asada, S.; Namba, Y.; et al. Nivolumab alone or in combination with cisplatin plus gemcitabine in Japanese patients with unresectable or recurrent biliary tract cancer: A non-randomised, multicentre, open-label, phase 1 study. Lancet Gastroenterol. Hepatol. 2019, 4, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.D.; Chung, V.; Alese, O.B.; El-Rayes, B.F.; Li, D.; Al-Toubah, T.E.; Schell, M.J.; Zhou, J.M.; Mahipal, A.; Kim, B.H.; et al. A Phase 2 Multi-institutional Study of Nivolumab for Patients with Advanced Refractory Biliary Tract Cancer. JAMA Oncol. 2020, 6, 888–894. [Google Scholar] [CrossRef]
- Ioka, T.; Ueno, M.; Oh, D.-Y.; Fujiwara, Y.; Chen, J.-S.; Doki, Y.; Mizuno, N.; Park, K.; Asagi, A.; Hayama, M.; et al. Evaluation of safety and tolerability of durvalumab (D) with or without tremelimumab (T) in patients (pts) with biliary tract cancer (BTC). J. Clin. Oncol. 2019, 37, 387. [Google Scholar] [CrossRef]
- Doki, Y.; Ueno, M.; Hsu, C.H.; Oh, D.Y.; Park, K.; Yamamoto, N.; Ioka, T.; Hara, H.; Hayama, M.; Nii, M.; et al. Tolerability and efficacy of durvalumab, either as monotherapy or in combination with tremelimumab, in patients from Asia with advanced biliary tract, esophageal, or head-and-neck cancer. Cancer Med. 2022, 11, 2550–2560. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.-Y.; He, A.R.; Qin, S.; Chen, L.-T.; Okusaka, T.; Vogel, A.; Kim, J.W.; Suksombooncharoen, T.; Lee, M.A.; Kitano, M.; et al. Durvalumab plus Gemcitabine and Cisplatin in Advanced Biliary Tract Cancer. NEJM Evid. 2022, 1, EVIDoa2200015. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.; Boeck, S.; Waidmann, O.; Bitzer, M.; Wenzel, P.; Belle, S.; Springfeld, C.; Schulze, K.; Weinmann, A.; Lindig, U.; et al. 52MO A randomized phase II trial of durvalumab and tremelIMUmab with gemcitabine or gemcitabine and cisplatin compared to gemcitabine and cisplatin in treatment-naïve patients with CHolangio- and gallbladdEr Carcinoma (IMMUCHEC). Ann. Oncol. 2022, 33, S563. [Google Scholar] [CrossRef]
- Monge, C.; Pehrsson, E.C.; Xie, C.; Duffy, A.G.; Mabry, D.; Wood, B.J.; Kleiner, D.E.; Steinberg, S.M.; Figg, W.D.; Redd, B.; et al. A Phase II Study of Pembrolizumab in Combination with Capecitabine and Oxaliplatin with Molecular Profiling in Patients with Advanced Biliary Tract Carcinoma. Oncologist 2022, 27, e273–e285. [Google Scholar] [CrossRef] [PubMed]
- Sahai, V.; Griffith, K.A.; Beg, M.S.; Shaib, W.L.; Mahalingam, D.; Zhen, D.B.; Deming, D.A.; Zalupski, M.M. A randomized phase 2 trial of nivolumab, gemcitabine, and cisplatin or nivolumab and ipilimumab in previously untreated advanced biliary cancer: BilT-01. Cancer 2022, 128, 3523–3530. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wu, X.; Wu, H.; Shao, Q.; Zhu, F.; Qian, X.; Zhao, F.; Mao, W.; Sun, J.; Wang, J.; et al. SHR-1210 combined with GEMOX as first-line treatment in patients with advanced biliary tract cancer. J. Clin. Oncol. 2020, 38, 535. [Google Scholar] [CrossRef]
- Li, W.; Yu, Y.; Xu, X.; Guo, X.; Wang, Y.; Li, Q.; Wang, Y.; Cui, Y.; Liu, H.; Hao, Q.; et al. Toripalimab with chemotherapy as first-line treatment for advanced biliary tract tumors: Update analytic results of an open-label phase II clinical study (JS001-ZS-BC001). J. Clin. Oncol. 2021, 39, e16170. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Ren, Z.; Chon, H.; Park, J.O.; Kim, J.W.; Pressiani, T.; Li, D.; Zhukova, L.; Chen, M.-H.; Hack, S.P.; et al. IMbrave151: A phase 2, randomized, double-blind, placebo-controlled study of atezolizumab with or without bevacizumab in combination with cisplatin plus gemcitabine in patients with untreated, advanced biliary tract cancer. J. Clin. Oncol. 2023, 41, 491. [Google Scholar] [CrossRef]
- Shi, G.M.; Huang, X.Y.; Wu, D.; Sun, H.C.; Liang, F.; Ji, Y.; Chen, Y.; Yang, G.H.; Lu, J.C.; Meng, X.L.; et al. Toripalimab combined with lenvatinib and GEMOX is a promising regimen as first-line treatment for advanced intrahepatic cholangiocarcinoma: A single-center, single-arm, phase 2 study. Signal Transduct. Target. Ther. 2023, 8, 106. [Google Scholar] [CrossRef]
- Yoo, C.; Oh, D.Y.; Choi, H.J.; Kudo, M.; Ueno, M.; Kondo, S.; Chen, L.T.; Osada, M.; Helwig, C.; Dussault, I.; et al. 73P Long-term follow-up of bintrafusp alfa, a bifunctional fusion protein targeting TGF-β and PD-L1, in patients with pretreated biliary tract cancer. Ann. Oncol. 2020, 31, S268–S269. [Google Scholar] [CrossRef]
- Feng, K.; Liu, Y.; Guo, Y.; Qiu, J.; Wu, Z.; Dai, H.; Yang, Q.; Wang, Y.; Han, W. Phase I study of chimeric antigen receptor modified T cells in treating HER2-positive advanced biliary tract cancers and pancreatic cancers. Protein Cell 2018, 9, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Feng, K.; Liu, Y.; Wu, Z.; Dai, H.; Yang, Q.; Wang, Y.; Jia, H.; Han, W. Phase I Study of Chimeric Antigen Receptor-Modified T Cells in Patients with EGFR-Positive Advanced Biliary Tract Cancers. Clin. Cancer Res. 2018, 24, 1277–1286. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Ueno, T.; Kawaoka, T.; Hazama, S.; Fukui, M.; Suehiro, Y.; Hamanaka, Y.; Ikematsu, Y.; Imai, K.; Oka, M.; et al. MUC1 peptide vaccination in patients with advanced pancreas or biliary tract cancer. Anticancer Res. 2005, 25, 3575–3579. [Google Scholar]
- Kaida, M.; Morita-Hoshi, Y.; Soeda, A.; Wakeda, T.; Yamaki, Y.; Kojima, Y.; Ueno, H.; Kondo, S.; Morizane, C.; Ikeda, M.; et al. Phase 1 trial of Wilms tumor 1 (WT1) peptide vaccine and gemcitabine combination therapy in patients with advanced pancreatic or biliary tract cancer. J. Immunother. 2011, 34, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Aruga, A.; Takeshita, N.; Kotera, Y.; Okuyama, R.; Matsushita, N.; Ohta, T.; Takeda, K.; Yamamoto, M. Phase I clinical trial of multiple-peptide vaccination for patients with advanced biliary tract cancer. J. Transl. Med. 2014, 12, 61. [Google Scholar] [CrossRef]
- Aruga, A.; Takeshita, N.; Kotera, Y.; Okuyama, R.; Matsushita, N.; Ohta, T.; Takeda, K.; Yamamoto, M. Long-term Vaccination with Multiple Peptides Derived from Cancer-Testis Antigens Can Maintain a Specific T-cell Response and Achieve Disease Stability in Advanced Biliary Tract Cancer. Clin. Cancer Res. 2013, 19, 2224–2231. [Google Scholar] [CrossRef] [PubMed]
- Shirahama, T.; Muroya, D.; Matsueda, S.; Yamada, A.; Shichijo, S.; Naito, M.; Yamashita, T.; Sakamoto, S.; Okuda, K.; Itoh, K.; et al. A randomized phase II trial of personalized peptide vaccine with low dose cyclophosphamide in biliary tract cancer. Cancer Sci. 2017, 108, 838–845. [Google Scholar] [CrossRef] [PubMed]
- Parasramka, M.; Yan, I.K.; Wang, X.; Nguyen, P.; Matsuda, A.; Maji, S.; Foye, C.; Asmann, Y.; Patel, T. BAP1 dependent expression of long non-coding RNA NEAT-1 contributes to sensitivity to gemcitabine in cholangiocarcinoma. Mol. Cancer 2017, 16, 22. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Qin, X.; Zhou, Y.; Li, G.; Liu, Z.; Geng, X.; Yue, H. Long non-coding RNA LINC00665 promotes gemcitabine resistance of Cholangiocarcinoma cells via regulating EMT and stemness properties through miR-424-5p/BCL9L axis. Cell Death Dis. 2021, 12, 72. [Google Scholar] [CrossRef]
- Lu, Q.; Fang, T. Circular RNA SMARCA5 correlates with favorable clinical tumor features and prognosis, and increases chemotherapy sensitivity in intrahepatic cholangiocarcinoma. J. Clin. Lab. Anal. 2020, 34, e23138. [Google Scholar] [CrossRef]
- Banales, J.M.; Cardinale, V.; Carpino, G.; Marzioni, M.; Andersen, J.B.; Invernizzi, P.; Lind, G.E.; Folseraas, T.; Forbes, S.J.; Fouassier, L.; et al. Cholangiocarcinoma: Current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 261–280. [Google Scholar] [CrossRef]
- Jimeno, A.; Rubio-Viqueira, B.; Amador, M.L.; Oppenheimer, D.; Bouraoud, N.; Kulesza, P.; Sebastiani, V.; Maitra, A.; Hidalgo, M. Epidermal growth factor receptor dynamics influences response to epidermal growth factor receptor targeted agents. Cancer Res. 2005, 65, 3003–3010. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yee, D. The therapeutic potential of agents targeting the type I insulin-like growth factor receptor. Expert Opin. Investig. Drugs 2004, 13, 1569–1577. [Google Scholar] [CrossRef]
- Sachdev, D.; Yee, D. Disrupting insulin-like growth factor signaling as a potential cancer therapy. Mol. Cancer Ther. 2007, 6, 1–12. [Google Scholar] [CrossRef]
- Namwat, N.; Amimanan, P.; Loilome, W.; Jearanaikoon, P.; Sripa, B.; Bhudhisawasdi, V.; Tassaneeyakul, W. Characterization of 5-fluorouracil-resistant cholangiocarcinoma cell lines. Chemotherapy 2008, 54, 343–351. [Google Scholar] [CrossRef]
- Hwang, I.G.; Jang, J.S.; Do, J.H.; Kang, J.H.; Lee, G.W.; Oh, S.Y.; Kwon, H.C.; Jun, H.J.; Lim, H.Y.; Lee, S.; et al. Different relation between ERCC1 overexpression and treatment outcomes of two platinum agents in advanced biliary tract adenocarcinoma patients. Cancer Chemother. Pharmacol. 2011, 68, 935–944. [Google Scholar] [CrossRef]
- Obama, K.; Satoh, S.; Hamamoto, R.; Sakai, Y.; Nakamura, Y.; Furukawa, Y. Enhanced expression of RAD51 associating protein-1 is involved in the growth of intrahepatic cholangiocarcinoma cells. Clin. Cancer Res. 2008, 14, 1333–1339. [Google Scholar] [CrossRef]
- Momoi, H.; Itoh, T.; Nozaki, Y.; Arima, Y.; Okabe, H.; Satoh, S.; Toda, Y.; Sakai, E.; Nakagawara, K.; Flemming, P.; et al. Microsatellite instability and alternative genetic pathway in intrahepatic cholangiocarcinoma. J. Hepatol. 2001, 35, 235–244. [Google Scholar] [CrossRef]
- Yang, T.; Deng, Z.; Xu, L.; Li, X.; Yang, T.; Qian, Y.; Lu, Y.; Tian, L.; Yao, W.; Wang, J. Macrophages-aPKC(ɩ)-CCL5 Feedback Loop Modulates the Progression and Chemoresistance in Cholangiocarcinoma. J Exp Clin. Cancer Res. 2022, 41, 23. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
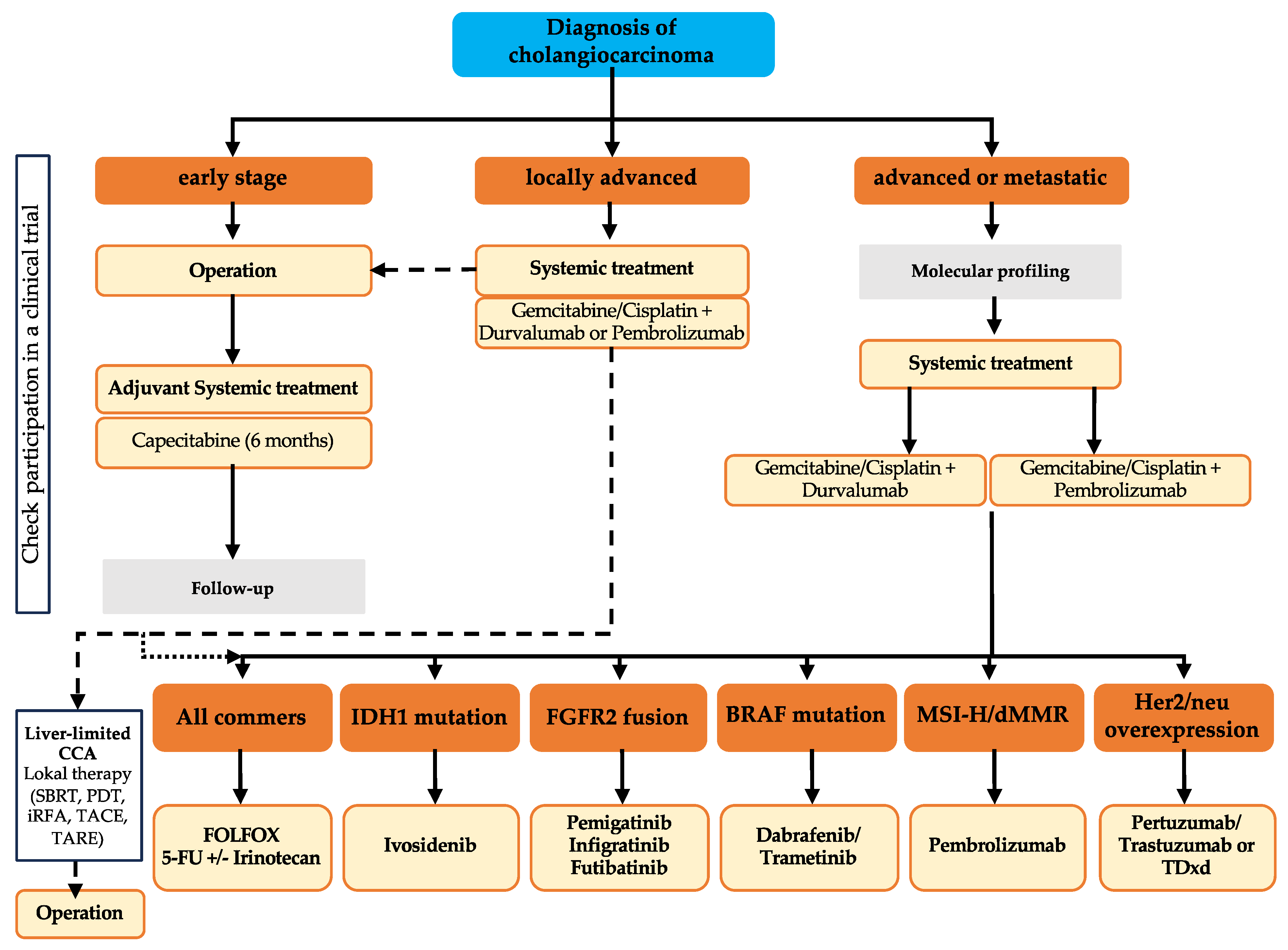{kind=link}
{kind=link}
{kind=link}
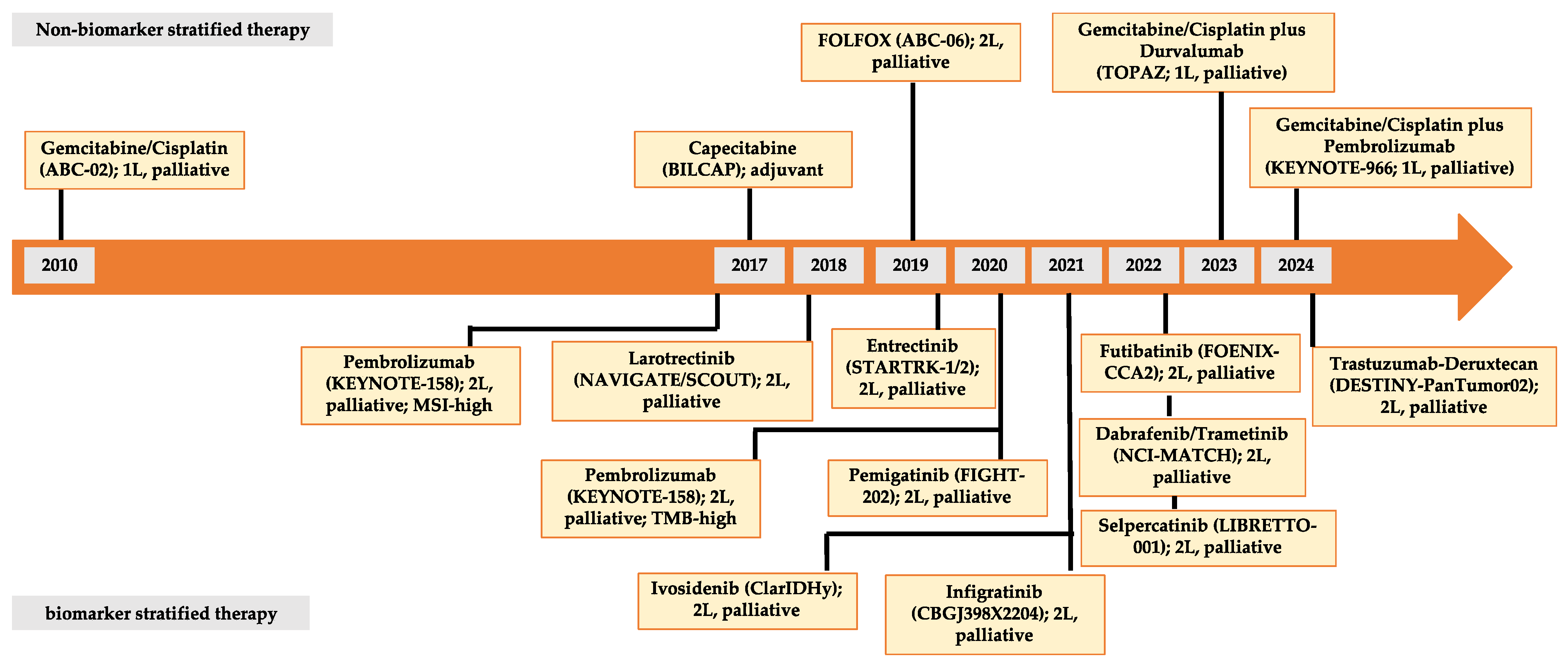{kind=link}
| Risk Factor | Study Design | Risk Estimate (95% CI) | References |
|---|---|---|---|
| Alcohol abuse | Case–control | iCCA: 5.9 (2.1–17.4); 7.4 (4.3–12.8); 3.1 (1.3–7.5) eCCA: 3.6 (1.5–9.4); 4.5 (2.2–9.1) | [7,11,12,13,14] |
| Case–control | iCCA: 3.72 (3.17–4.35) eCCA: 2.60 (2.23–3.04) | ||
| Meta-analysis | 2.81 (1.52–5.21) | ||
| Pooled analysis | iCCA: 2.35 (1.46–3.78) eCCA: 1.82 (0.98–3.39) | ||
| Asbestos | Cohort | 1.50 (1.16–1.94) | [15] |
| Bile duct cysts | Case–control | iCCA: 15.66 (11.58–21.18); 36.9 (22.7–59.7) eCCA: 27.12 (22.06–33.34); 47.1 (30.4–73.2) | [14,16,17,18] |
| Caroli’s disease | Case-control | iCCA: 38.13 (14.20–102.38) eCCA: 96.81 (51.02–183.68) | [14,19] |
| Cholangitis | Case–control | iCCA: 21.52 (17.21–26.90); 6.32 (2.3–17.5); 8.8 (4.9–16.0); 64.2 (47.7–86.5) eCCA: 40.80 (34.96–47.60); 45.7 (32.9–63.3) | [14,18,20,21] |
| Cholelithiasis | Case–control | iCCA: 3.93 (3.49–4.43) eCCA: 5.29 (4.83–5.80) | [7,13,14] |
| Choledocholithiasis | Case–control | iCCA: 6.94 (5.64–8.54); 23.97 (2.9–198.9); 4.0 (1.9–8.5); 22.5 (16.9–30.0) eCCA: 14.22 (12.48–16.20); 34.0 (26.6–43.6) | [13,14,18,20,21] |
| Chronic inflammatory bowel disease | Case–control | iCCA: 4.67 (1.6–13.9) | [18,22] |
| Chronic hepatitis B viral infection | Case–control | iCCA: 2.97 (1.97–4.46); 28.6 (3.9–1268.1); 0.8 (0.1–5.9); 2.7 (0.4–18.5); 2.3 (1.6–3.3); 8.9 (5.97–13.2) eCCA: 2.38 (1.65–3.44); 3.2 (0.6–382) | [7,11,14,20,23,24,25,26] |
| Chronic hepatitis C viral infection | Case–control | iCCA: 4.67 (3.57–6.11); 6.02 (1.5–24.1); 7.9 (1.3–84.5); 5.2 (2.1–12.8); 2.2 (1.4–14.0); 9.7 (1.6–58.9); 0.93 (0.3–3.1) eCCA: 3.18 (2.43–4.16); 2.8 (0.3–35.1); 1–5 (0.2–11.0) | [7,11,14,18,20,23,24,26,27,28] |
| Cohort | iCCA: 2.55 (1.3–4.9) eCCA: 1.05 (0.6–1.9) | ||
| Chronic pancreatitis | Case–control | iCCA: 2.66 (1.72–4.10) eCCA: 6.61 (5.21–8.40) | [14,29] |
| Cirrhosis of the liver | Case–control | iCCA: 5.03 (0.045–56.82); 27.2 (19.9–37.1); 10.0 (6.1–16.4); 13.6 (6.5–28.5) eCCA: 5.4 (2.9–10.2) | [7,11,18,20,25,28,30] |
| Crohn’s disease | Case–control | iCCA: 1.77 (1.13–2.75); 2.0 (0.6–6.3); 2.4 (1.0–5.9) eCCA: 1.71 (1.17–2.51); 2.8 (1.3–6.4 | [14,18,20,31] |
| Cohort | iCCA/eCCA: 3.0 (0.9–8.6) | ||
| Diabetes mellitus type 1 | Case–control | iCCA: 1.43 (1.25–1.63) eCCA: 1.30 (1.16–1.46) | [7,11,14,32,33] |
| Diabetes mellitus type 2 | Case–control | iCCA: 1.54 (1.41–1.68) eCCA: 1.45 (1.34–1.56) | [7,11,14,32,33] |
| Duodenal/gastric ulcer | Case–control | iCCA: 1.14 (1.21–1.66) eCCA: 1.46 (1.29–1.66) | [14] |
| Hemochromatosis | Case–control | iCCA: 2.07 (1.33–3.22) | [14,34,35,36] |
| liver flukes | N/A | N/A | [37,38,39] |
| Non-alcoholic fatty liver disease | Case–control | iCCA: 3.52 (2.87–4.32) eCCA: 2.93 (2.42–3.55) | [7,14,40] |
| Obesity | Case–control | iCCA: 1.42 (1.21–1.66); 1.7 (1.1–2.6); 2.05 (0.7–5.6) eCCA: 1.17 (1.01–1.35); 1.1 (0.7–1.8) | [11,14,18,21,32] |
| Primary biliary cholangitis | Case–control | iCCA: 9.84 (6.24–15.52) eCCA: 8.34 (5.44–12.78) | [14] |
| Primary sclerosing cholangitis | Case–control | iCCA 93.4 (27.1–322) distal eCCA 34.0 (3.6–323) perihilar eCCA 453 (104–999) | [7,41,42,43,44,45] |
| Smoking | Case–control | iCCA: 1.46 (1.28–1.66); 1.8 (1.0–3.2); 1.8 (1.2–2.7) eCCA: 1.77 (1.59–1.96); 1.7 (1.0–3.0) | [7,12,14,18,23,46] |
| Thorotrast | N/A | N/A | [47] |
| Ulcerative colitis | Case–control | iCCA: 2.18 (1.61–2.95); 2.2 (1.2–3.9); 4.5 (2.6–7.9) eCCA: 1.75 (1.32–2.33); 1.7 (0.7–4.0) | [14,18,20,31] |
| Cohort | iCCA/eCCA: 4.1 (2.4–6.8) |
| Risk Factor | Study Design | Risk Estimate (95% CI) | References |
|---|---|---|---|
| Aflatoxin | Case–control | 2.0 (1.0–3.9) | [48,49] |
| Age | N/A | N/A | [48] |
| Anatomical anomalies of the intra- and extrahepatic bile ducts | N/A | N/A | [48] |
| Arsen | Retrospective analysis | 1.72 (1.54–1.91); 1.45 (1.30–1.62) | [48,50] |
| Cholecystolithiasis | Case–control | 5.3 (1.5–18.9) | [48,51,52] |
| Crohn’s disease | Case–control | 1.83 (1.23–2.71) | [48] |
| Diabetes mellitus | Case–control | 2.7 (1.2–6.4) | [48,52] |
| Female sex | Case–control | 2.4 (1.3–4.3) | [48,52] |
| Gallbladder polyps | Retrospective analysis | 8.147 (2.56–23.40 | [48,51,53] |
| Liver flukes/infections | N/A | Helicobacter pilis OR of 6.5 in Japanese patients and 5.86 in Thai patients; S. typhi carriers have an 8 to 12-fold increased risk of developing GB | [48,54] |
| Obesity | Meta-analysis | 1.10 (1.02–1.1); 1.69 (1.54–1.86) | [48,51] |
| Porcelain gallbladder | Retrospective analysis | 8.0 (1.0–63.0); if symptoms 83.6 (2.3–2979.1); if gallbladder mass 3226.6 (17.2–603884.8) | [48,55,56] |
| Primary sclerosing cholangitis | Case–control | 2.06 (1.27–3.33) | [48,51] |
| Tobacco | Case–control | 3.8 (1.7–8.1) | [52] |
| Target | iCCA | eCCA | GB |
|---|---|---|---|
| FGFR2 alterration | 9–13% | 0% | 2–7% |
| IDH1/2 mutation | 10–29% | 3–5% | 0–2% |
| BRAF mutation | 5% | 2–3% | 0–1% |
| HER2/ERBB2 overexpression | 3–8% | 1.3–11% | 6–15% |
| KRAS mutation | 15–22% | 38–57% | 7–10% |
| ARID1A mutation | 18–23% | 12–20% | 12–17% |
| PIK3CA mutation | 3–7% | 5–7% | 9–10% |
| CDKN2A/B mutation | 9–27% | 9–28% | 12–25% |
| MET overexpression | 2–4% | 0% | 1–2% |
| BAP1 mutation | 15–19% | 0% | 3–13% |
| RET fusion | 0–5% | 0–5% | 0–5% |
| NRTK fusion | <1–2% | <1–2% | <1–2% |
| MSI-high | Up to 5% | Up to 5% | Up to 5% |
| BRCA1/2 mutation | Up to 3–5% | Up to 3–5% | Up to 3–5% |
| EGFR overexpression | 11–27% | 5–19% | N/A |
| Study | Phase | Setting | Treatment Arm A | Treatment Arm B | Primary Endpoint | NCT |
|---|---|---|---|---|---|---|
| FIGHT-302 | III | 1L Palliative | Pemigatinib | Gemcitabine/cisplatin | PFS | NCT03656536 |
| PEARLDIFER | II | Adjuvant | Pemigatinib | - | ORR | NCT05565794 |
| - | II | 1L Palliative | Pemigatinib plus sintilimab | - | ORR | NCT05913661 |
| - | I | Palliative | Gemcitabine/cisplatin plus pemigatinib (FGFR2 alteration) | Gemcitabine/cisplatin plus Ivosidenib (IDH1 mutation) | safety | NCT04088188 |
| Study | Phase | Setting | Treatment Arm A | Treatment Arm B | Primary Endpoint | NCT |
|---|---|---|---|---|---|---|
| FOENIX-CCA3 | III | 1L Palliative | Futibatinib | Gemcitabine/cisplatin | PFS | NCT04093362 |
| FOENIX-CCA4 | II | Palliative | Futibatinib (20 mg) | Futibatinib (16 mg) | ORR | NCT05727176 |
| Study | Phase | Setting | Treatment Arm A | Treatment Arm B | Primary Endpoint | NCT |
|---|---|---|---|---|---|---|
| OPTIC | II | Neoadjuvant | Gemcitabine/cisplatin/nab-Paclitaxel plus infigratinib | Gemcitabine/cisplatin/nab-paclitaxel | Safety | NCT05514912 |
| - | III | 1L, Palliative | Infigratinib | Gemcitabine/cisplatin | PFS | NCT03773302 |
| Study | Phase | Setting | Treatment Arm A | Treatment Arm B | Primary Endpoint | NCT |
|---|---|---|---|---|---|---|
| ADVANCE_2020 | II | Palliative | Atezolizumab plus derazantinib | - | Safety | NCT05174650 |
| Study | Phase | Setting | Treatment Arm A | Treatment Arm B | Primary Endpoint | NCT |
|---|---|---|---|---|---|---|
| ADVANCE_2020 | I | Palliative | Bemarituzumab (dose-finding) | - | Safety | NCT02318329 |
| REFOCUS | I/II | Palliative | RLY-4008 | - | Safety | NCT04526106 |
| - | II | Palliative | Tinengotinib | - | ORR | NCT06057571 |
| FIRST-308 | III | Palliative | Tinengotinib | Physician’s choice | Safety, PFS | NCT05948475 |
| Study | Phase | Setting | Treatment Arm A | Treatment Arm B | Primary Endpoint | NCT |
|---|---|---|---|---|---|---|
| ProvIDHe | III | Palliative | Ivosidenib | - | Safety | NCT05876754 |
| - | II | Palliative | Ivosidenib plus nivolumab | - | OR | NCT04056910 |
| - | I/II | Palliative | Ivosidenib plus nivolumab plus ipilimumab | - | Safety, OR | NCT05921760 |
| - | I | Palliative | Gemcitabine/cisplatin plus pemigatinib (FGFR2 alteration) | Gemcitabine/cisplatin plus ivosidenib (IDH1 mutation) | Safety | NCT04088188 |
| Study | Phase | Setting | Treatment Arm A | Treatment Arm B | Primary Endpoint | NCT |
|---|---|---|---|---|---|---|
| - | II | Palliative | Dasatinib | - | ORR | NCT02428855 |
| - | I/II | Palliative | Olutasidenib | Olutasidenib plus azacitidine or nivolumab or gemcitabine/cisplatin | Safety, ORR | NCT03684811 |
| - | I/II | Palliative | Enasidenib | - | Safety | NCT02273739 |
| - | II | Palliative | BAY-1436032 | - | Safety | NCT02746081 |
| - | I | Palliative | LY3410738 | LY3410738 plus gemcitabine/cisplatin plus durvalumab | Safety | NCT04521686 |
| - | I | Palliative | IDH305 | - | Safety | NCT02381886 |
| - | I | Palliative | Vorasidenib | - | Safety | NCT02481154 |
| Study | Phase | Setting | Treatment Arm A | Treatment Arm B | Primary Endpoint | NCT |
|---|---|---|---|---|---|---|
| - | I/II | Palliative | Zanidatamab plus evorpacept | - | Safety, ORR | NCT05027139 |
| - | II | Palliative | Zanidatamab plus cisplatin/5-FU or mFOLFOX6 or XELOX or mFOLFOX6 plus bevacizumab or Gemcitabine/cisplatin | - | Safety, ORR | NCT03929666 |
| - | I | Palliative | Tipifarnib plus trastuzumab | - | Safety | NCT00005842 |
| DPT02 | II | Palliative | TDxd | - | ORR | NCT04482309 |
| - | I | Palliative | ZN-A-1041 (dose-finding) | ZN-A-1041 plus TDxd or TD-M1 or pertuzumab/trastuzumab | Safety | NCT05593094 |
| - | II | Palliative | TD-M1 | - | ORR | NCT02675829 |
| TAPISTRY | II | Palliative | TD-M1 | - | ORR | NCT04589845 |
| Study | Phase | Setting | Treatment Arm A | Treatment Arm B | Primary Endpoint | NCT |
|---|---|---|---|---|---|---|
| - | II | Palliative | Larotrectinib | - | OR | NCT04879121 |
| TAPISTRY | II | Palliative | Entrectinib | - | Safety | NCT04589845 |
| Study | Phase | Setting | Treatment Arm A | Treatment Arm B | Primary Endpoint | NCT |
|---|---|---|---|---|---|---|
| - | II | Palliative | Selumetinib plus gemcitabine/cisplatin | Gemcitabine/cisplatin | Response | NCT02151084 |
| BEAVER | II | Palliative | Binimetinib plus encorafenib | - | ORR | NCT03839342 |
| - | I | Palliative | ABM-1310 | ABM-1310 plus cobimetinib | safety | NCT04190628 |
| TAPISTRY | II | Palliative | Belvarafenib | - | ORR | NCT04589845 |
| - | - | Palliative | Ulixertinib | - | - | NCT04566393 |
| - | II | Palliative | LY3214996 plus abemaciclib | - | ORR | NCT04534283 |
| Study | Phase | Setting | Treatment Arm A | Treatment Arm B | Primary Endpoint | NCT |
|---|---|---|---|---|---|---|
| - | I | Palliative | Niraparib plus anlotinib | - | Safety | NCT02151084 |
| UF-STO-ETI-001 | II | Palliative | Niraparib | - | ORR | NCT03207347 |
| - | II | Palliative | Olaparib | - | PFS | NCT04042831 |
| - | II | Palliative | Pembrolizumab plus olaparib | - | ORR | NCT04306367 |
| - | II | Palliative | Nivolumab plus olaparib | - | Response | NCT03639935 |
| - | I/II | Palliative | Liposomal irinotecan plus 5-FU plus rucaparib | - | Response, safety | NCT03337087 |
| - | I | Palliative | Veliparib plus gemcitabine/cisplatin | - | Safety | NCT01282333 |
| DDR-Umbrella | II | Palliative | Ceralasertib plus durvalumab | Ceralasertib plus Durvalumab | DCR | NCT04298021 |
| - | II | Palliative | Rucaparib plus nivolumab | - | Response | NCT03639935 |
| - | II | Palliative | Adavosertib | - | OR | NCT02465060 |
| - | II | Palliative | Olaparib | - | ORR | NCT03212274 |
| - | II | Palliative | AZD6738 plus durvalumab | - | DCR | NCT04298008 |
| SOLID | II | Palliative | Olaparib plus durvalumab | - | ORR, DCR | NCT03991832 |
| - | II | Palliative | Toripalimab | - | ORR | NCT03810339 |
| - | II | Palliative | Pembrolizumab | - | Response | NCT03428802 |
| OPTIMUM | II | Palliative | Olaparib plus durvalumab | Olaparib | PFS | NCT05222971 |
| - | I | palliative | Copanlisib plus olaparib plus durvalumab | - | Safety | NCT03842228 |
| TAPISTRY | II | palliative | Camonsertib | - | ORR | NCT04589845 |
| Study | Phase | Setting | Treatment Arm A | Treatment Arm B | Primary endpoint | NCT |
|---|---|---|---|---|---|---|
| TalaCom | I | Palliative | Talazoparib plus crizotinib | - | Safety | NCT04693468 |
| STARTRK-2 | II | Palliative | Entrectinib | - | ORR | NCT02568267 |
| TAPISTRY | II | Palliative | Alectinib | - | ORR | NCT04589845 |
| TAPISTRY | II | Palliative | Entrectinib | - | ORR | NCT04589845 |
| Study | Phase | Setting | Treatment Arm A | Treatment Arm B | Primary Endpoint | NCT |
|---|---|---|---|---|---|---|
| TAPISTRY | II | Palliative | Pralsetinib | - | ORR | NCT04589845 |
| Study | Phase | Setting | Treatment Arm A | Treatment Arm B | Primary Endpoint | NCT |
|---|---|---|---|---|---|---|
| TAPISTRY | II | Palliative | Inavolisib | - | ORR | NCT04589845 |
| TAPISTRY | II | Palliative | Ipatasertib | - | ORR | NCT04589845 |
| - | I | Palliative | Copanlisib plus olaparib plus durvalumab | - | Safety | NCT03842228 |
| Study | Phase | Setting | Treatment Arm A | Treatment Arm B | Primary Endpoint | NCT |
|---|---|---|---|---|---|---|
| KEYNOTE 596 | I | Palliative | CGX1321 | - | Safety | NCT02675946 |
| - | I | Palliative | DKN-01 plus gemcitabine/cisplatin | - | Safety | NCT02375880 |
| - | II | Palliative | DKN-01 plus nivolumab | - | ORR | NCT04057365 |
| Study | Phase | Setting | Treatment Arm A | Treatment Arm B | Primary Endpoint | NCT |
|---|---|---|---|---|---|---|
| - | I/II | Palliative | Abemaciclib plus paclitaxel | - | Safety, ORR | NCT04594005 |
| - | II | Palliative | Abemaciclib | - | PFR | NCT03310879 |
| Study | Phase | Setting | Treatment Arm A | Treatment Arm B | Primary Endpoint | NCT |
|---|---|---|---|---|---|---|
| - | II | Palliative | LY3214996 plus abemaciclib | - | ORR | NCT04534283 |
| - | - | Palliative | Ulixertinib | - | - | NCT04566393 |
| TAPISTRY | II | Palliative | GDC-6036 | - | ORR | NCT04589845 |
| - | I | Palliative | LY3537982 plus abemaciclib | - | Safety | NCT04956640 |
| - | I/II | Palliative | GFH925 | - | Safety, ORR | NCT05005234 |
| KRYSTAL-16 | I | Palliative | Adagrasib plus palbociclib | - | Safety | NCT05178888 |
| - | II | Palliative | Sotorasib plus panitumumab | - | ORR | NCT05993455 |
| CodeBreak 101 | II | Palliative | Sotorasib | - | Safety, ORR | NCT04185883 |
| CodeBreak 101 | I/II | Palliative | Sotorasib | - | Safety, ORR, DOR, TTR | NCT03600883 |
| KRYSTAL-19 | I/II | Palliative | Adagrasib plus nab-sirolimus | - | Safety, ORR | NCT05840510 |
| - | I/II | Palliative | JAB-21822 | - | Safety, ORR, DOR | NCT05002270 |
| - | I/II | Palliative | JAB-21822 plus cetuximab | - | Safety, ORR | NCT05194995 |
| - | I/II | Palliative | YL-15293 | - | ORR | NCT05119933 |
| - | I | Palliative | HBI-2438 | - | Safety | NCT05485974 |
| - | I/II | Palliative | HS-10370 | - | Safety | NCT05367778 |
| - | I | Palliative | MRTX849 | - | Safety | NCT05263986 |
| - | I/II | Palliative | BMS-986466 plus adagrasib | BMS-986466 plus Adagrasib plus Cetuximab | Safety, ORR | NCT06024174 |
| - | I/II | Palliative | MRTX0902 | MRTX0902 plus Adagrasib | ORR, DOR, OS, PFS | NCT05578092 |
| - | I | Palliative | MK-1084 | - | Safety | NCT05067283 |
| KontRASt-01 | I/II | Palliative | JDQ443 | JDQ443 plus TNO155 or tislelizumab or both | Safety, ORR | NCT04699188 |
| - | I | Palliative | RMC-6291 | - | Safety | NCT05462717 |
| - | I | Palliative | SY-5933 | - | Safety | NCT06006793 |
| - | I | Palliative | D3S-001 | - | Safety | NCT05410145 |
| - | I | Palliative | GEC255 | - | Safety | NCT05768321 |
| AMPLIFY-7P | I/II | Palliative | ELI-002 7P | - | Safety, DFS | NCT05726864 |
| Study | Phase | Setting | Treatment Arm A | Treatment Arm B | Primary Endpoint | NCT |
|---|---|---|---|---|---|---|
| - | II | Palliative | Sintilimab plus IBI305 plus GEMOX | Sintilimab plus GEMOX (Arm C: GEMOX) | ORR | NCT05251662 |
| - | II | Palliative | Atezolizumab plus bevacizumab plus Gemcitabine/cisplatin | Atezolizumab plus gemcitabine/cisplatin | PFS | NCT05211323 |
| Study | Phase | Setting | Treatment Arm A | Treatment Arm B | Primary Endpoint | NCT |
|---|---|---|---|---|---|---|
| - | II | Palliative | Lenvatinib plus gemcitabine/cisplatin | - | ORR | NCT04527679 |
| - | II | Palliative | Sintilimab plus lenvatinib | - | ORR | NCT05010681 |
| - | - | - | GEMOX plus lenvatinib plus toripalimab | GEMOX plus toripalimab (Arm C: lenvatinib plus toripalimab) | ORR, safety | NCT05215665 |
| - | III | Palliative | GEMOX or gemcitabine/cisplatin plus lenvatinib plus toripalimab | GEMOX or gemcitabine/cisplatin plus toripalimab (Arm C: GEMOX or gemcitabine/cisplatin) | OS | NCT05342194 |
| - | II | Palliative | Lenvatinib plus tislelizumab plus gemcitabine/cisplatin | Gemcitabine/cisplatin | ORR | NCT05532059 |
| - | II | Palliative | Lenvatinib plus tislelizumab plus gemcitabine/oxaliplatin | Tislelizumab plus gemcitabine/oxaliplatin | OR | NCT05620498 |
| - | II | Palliative | Toripalimab plus lenvatinib | - | ORR, safety | NCT04211168 |
| - | II/III | Neoadjuvant | Toripalimab plus lenvatinib plus GEMOX | - | Event-free survival | NCT04669496 |
| Study | Phase | Setting | Treatment Arm A | Treatment Arm B | Primary Endpoint | NCT |
|---|---|---|---|---|---|---|
| BREGO | II | Palliative | Regorafenib plus GEMOX | GEMOX | Safety | NCT02386397 |
| Study | Phase | Setting | Treatment Arm A | Treatment Arm B | Primary Endpoint | NCT |
|---|---|---|---|---|---|---|
| - | II | Palliative | Anlotinib plus sintilimab plus GEMOX | - | ORR | NCT06033118 |
| Study | Phase | Setting | Treatment Arm A | Treatment Arm B | Primary Endpoint | NCT |
|---|---|---|---|---|---|---|
| KEYNOTE-158 | II | Palliative | Pembrolizumab | - | ORR | NCT02628067 |
| Study | Phase | Setting | Treatment Arm A | Treatment Arm B | Primary Endpoint | NCT |
|---|---|---|---|---|---|---|
| DART | II | Palliative | Nivolumab plus ipilimumab | Nivolumab | ORR | NCT02834013 |
| - | II | Palliative | Nivolumab plus ipilimumab | - | Efficacy | NCT02923934 |
| IMMUNO-BIL | II | Palliative | Durvalumab plus tremelimumab | - | OS | NCT03704480 |
| Study | Phase | Setting | Treatment Arm A | Treatment Arm B | Primary Endpoint | NCT |
|---|---|---|---|---|---|---|
| - | II | Palliative | Pembrolizumab plus GC | - | PFS | NCT03260712 |
| - | II | Perioperative | Pembrolizumab plus GC | - | Safety | NCT05967182 |
| - | II | Palliative | Toripalimab plus GC | - | PFS, OS | NCT03796429 |
| - | II | Palliative | Toripalimab plus gemcitabine plus 5-FU | - | Safety, PFS | NCT03982680 |
| - | II | Palliative | Toripalimab plus S1 plus nab-paclitaxel | - | ORR | NCT04027764 |
| - | II | Palliative | Lenvatinib plus toripalimab | Lenvatinib plus toripalimab plus GEMOX (Arm C: GEMOX plus toripalimab) | ORR, safety | NCT05215665 |
| - | III | Palliative | Lenvatinib plus toripalimab plus GC or GEMOX | Toripalimab plus GC or GEMOX | OS | NCT05342194 |
| - | II | Palliative | Lenvatinib plus toripalimab | - | ORR, safety | NCT04211168 |
| - | II/III | Neoadjuvant | Lenvatinib plus toripalimab plus GEMOX | - | Event free survival | NCT04669496 |
| - | II | Palliative | Axitinib plus toripalimab | - | ORR, PFS | NCT04010071 |
| - | III | Palliative | Envafolimab plus GEMOX | - | OS | NCT03478488 |
| DEBATE | II | Neoadjuvant | Durvalumab plus GC | GC | R0 resection | NCT04308174 |
| - | II | Neoadjuvant | Durvalumab plus GC | - | Relapse-free survival rate | NCT05672537 |
| - | II | Neoadjuvant | Durvalumab plus GC | - | Complete treatment | NCT06050252 |
| - | II | Neoadjuvant | Durvalumab plus tremelimumab plus GC | - | Conversion from unresectable to resectable | NCT06017297 |
| - | I/II | Neoadjuvant | Durvalumab plus tremelimumab plus GC | - | ORR | NCT04989218 |
| ADJUBIL | II | Adjuvant | Durvalumab plus tremelimumab plus capecitabine | Durvalumab plus tremelimumab | RFS | NCT05239169 |
| NeoTreme | II | Neoadjuvant | Durvalumab plus tremelimumab plus GC | - | - | - |
| - | I | Palliative | Durvalumab plus guadecitabine | - | Safety, response | NCT03257761 |
| BATTALION | II | Palliative | Botensilimab plus balstilimab plus GC | - | ORR | - |
| - | II | Palliative | Atezolizumab | Atezolizumab plus cobimetinib | PFS | NCT03201458 |
| - | II | 2L, Palliative | SHR1316 plus IBI310 | - | ORR | NCT04634058 |
| Study | Phase | Setting | Treatment Arm A | Treatment Arm B | Primary Endpoint | NCT |
|---|---|---|---|---|---|---|
| - | II/III | Palliative | Bintrafusp alfa plus gemcitabine/cisplatin | Gemcitabine/cisplatin | Safety, OS | NCT04066491 |
| - | II | Palliative | Bintrafusp alfa | - | Response | NCT03833661 |
| Study | Phase | Setting | Treatment Arm A | Treatment Arm B | Primary Endpoint | NCT |
|---|---|---|---|---|---|---|
| - | I/II | Palliative | MUC-1 CART | - | DCR | NCT03633773 |
| Study | Phase | Setting | Drug | Target | FDA | EMA | NCT |
|---|---|---|---|---|---|---|---|
| FIGHT-202 | III | 2L, Palliative | Pemigatinib | FGFR | x | x | NCT02924376 |
| FOENIX-CCA2 | II | 2L, Palliative | Futibatinib | FGFR | x | x | NCT02052778 |
| CBGJ398X2204 | II | 2L, Palliative | Infigratinib | FGFR | x | - | NCT02150967 |
| Keynote-158 | II | 2L, Palliative | Pembrolizumab | MSI-high | x | x | NCT02628067 |
| Keynote-158 | II | 2L, Palliative | Pembrolizumab | TMB-high | x | - | NCT02628067 |
| ClarIDHy | III | 2L, Palliative | Ivosidenib | IDH1 | x | x | NCT02989857 |
| STARTRK-1/2 | I/II | 2L, Palliative | Entrectinib | NTRK | x | x | NCT02097810 NCT02568267 |
| NAVIGATE SCOUT | I/II | 2L, Palliative | Larotrectinib | NTRK | x | x | NCT02576431 NCT02637687 |
| NCI-MATCH | II | 2L, Palliative | Dabrafenib and Trametinib | BRAFV600E | x | - | NCT03155620 |
| LIBRETTO-001 | I/II | 2L, Palliative | Selpercatinib | RET | x | - | NCT03157128 |
| DESTINY-PanTumor02 | II | 2L, Palliative | Trastuzumab–deruxtecan | HER2+ | x | - | NCT04482309 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heumann, P.; Albert, A.; Gülow, K.; Tümen, D.; Müller, M.; Kandulski, A. Current and Future Therapeutic Targets for Directed Molecular Therapies in Cholangiocarcinoma. Cancers 2024, 16, 1690. https://doi.org/10.3390/cancers16091690
Heumann P, Albert A, Gülow K, Tümen D, Müller M, Kandulski A. Current and Future Therapeutic Targets for Directed Molecular Therapies in Cholangiocarcinoma. Cancers. 2024; 16(9):1690. https://doi.org/10.3390/cancers16091690
Chicago/Turabian StyleHeumann, Philipp, Andreas Albert, Karsten Gülow, Denis Tümen, Martina Müller, and Arne Kandulski. 2024. "Current and Future Therapeutic Targets for Directed Molecular Therapies in Cholangiocarcinoma" Cancers 16, no. 9: 1690. https://doi.org/10.3390/cancers16091690
APA StyleHeumann, P., Albert, A., Gülow, K., Tümen, D., Müller, M., & Kandulski, A. (2024). Current and Future Therapeutic Targets for Directed Molecular Therapies in Cholangiocarcinoma. Cancers, 16(9), 1690. https://doi.org/10.3390/cancers16091690