Mouse Genetic Models Reveal Surprising Functions of IkB Kinase Alpha in Skin Development and Skin Carcinogenesis

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Why Is It Surprising that IKKα Has a Role in Embryonic Skin Development?
3. Loss and Gain of IKKα Function in Skin Carcinogenesis
3.1. Chemical Carcinogen-Induced Skin Carcinogenesis
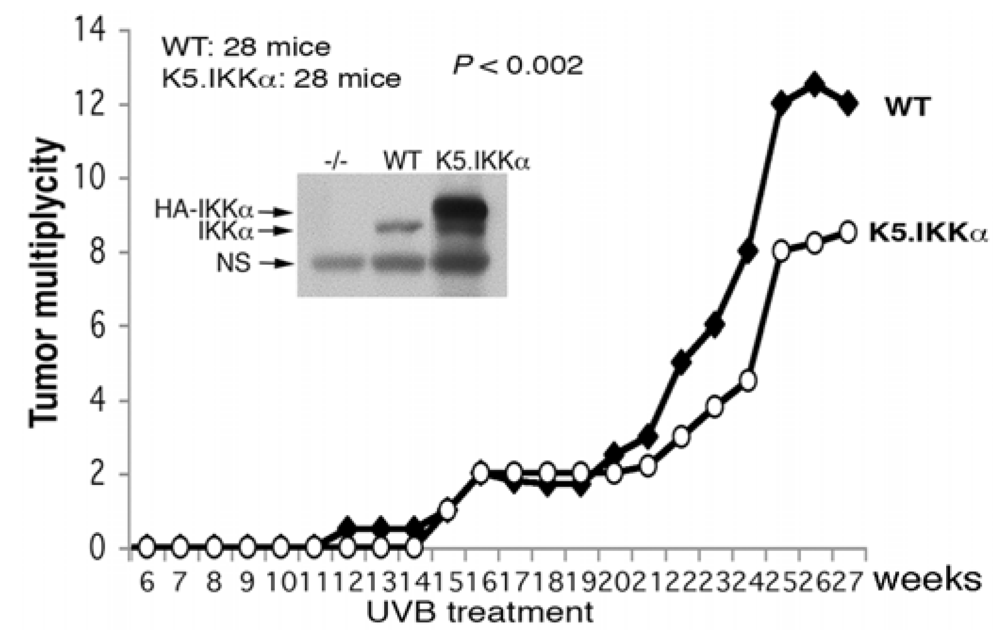
3.2. UVB-Induced Skin Carcinogenesis

4. Keratinocyte-Specific IKKα Deletion Induces Epidermal Hyperplasia and Spontaneous Skin SCCs; EGFR Reduction, but not TNFR Loss, Inhibits IKKα Loss-Mediated Epidermal Hyperplasia
5. IKKα Regulates the Cell Cycle in Keratinocytes

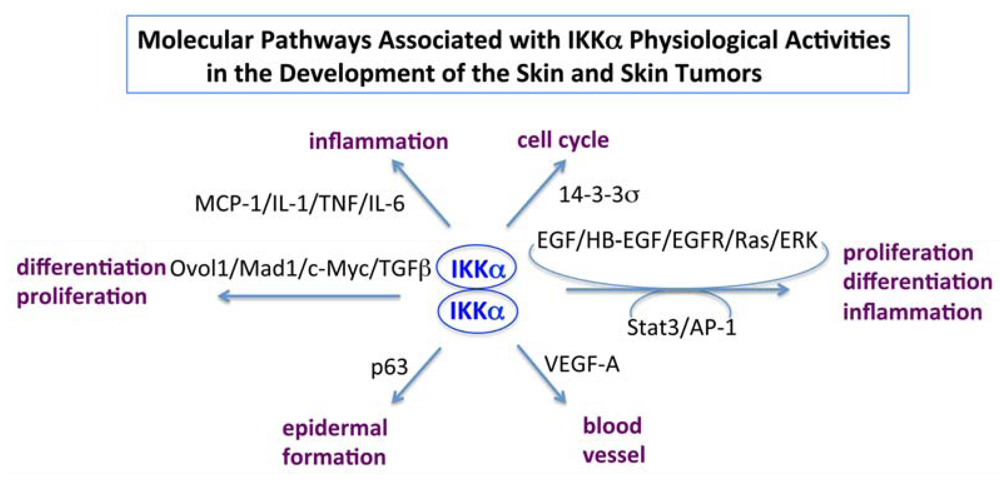
6. Conclusions and Puzzles
References
- Connelly, M.A.; Marcu, K.B. CHUK, a new member of the helix-loop-helix and leucine zipper families of interacting proteins, contains a serine-threonine kinase catalytic domain. Cell. Mol. Biol. Res. 1995, 41, 537–549. [Google Scholar]
- Mock, B.A.; Connelly, M.A.; McBride, O.W.; Kozak, C.A.; Marcu, K.B. CHUK, a conserved helix-loop-helix ubiquitous kinase, maps to human chromosome 10 and mouse chromosome 19. Genomics 1995, 27, 348–351. [Google Scholar] [CrossRef]
- Balkhi, M.Y.; Willette-Brown, J.; Zhu, F.; Chen, Z.; Liu, S.; Guttridge, D.C.; Karin, M.; Hu, Y. IKKalpha-mediated signaling circuitry regulates early B lymphopoiesis during hematopoiesis. Blood 2012, 119, 5467–5477. [Google Scholar] [CrossRef]
- Ghosh, S.; Karin, M. Missing pieces in the NF-kappaB puzzle. Cell 2002, 109, S81–S96. [Google Scholar] [CrossRef]
- Liu, B.; Xia, X.; Zhu, F.; Park, E.; Carbajal, S.; Kiguchi, K.; DiGiovanni, J.; Fischer, S.M.; Hu, Y. IKKalpha is required to maintain skin homeostasis and prevent skin cancer. Cancer Cell 2008, 14, 212–225. [Google Scholar] [CrossRef]
- Descargues, P.; Sil, A.K.; Sano, Y.; Korchynskyi, O.; Han, G.; Owens, P.; Wang, X.J.; Karin, M. IKKalpha is a critical coregulator of a Smad4-independent TGFbeta-Smad2/3 signaling pathway that controls keratinocyte differentiation. Proc. Natl. Acad. Sci. USA 2008, 105, 2487–2492. [Google Scholar]
- Marinari, B.; Moretti, F.; Botti, E.; Giustizieri, M.L.; Descargues, P.; Giunta, A.; Stolfi, C.; Ballaro, C.; Papoutsaki, M.; Alema, S.; et al. The tumor suppressor activity of IKKalpha in stratified epithelia is exerted in part via the TGF-beta antiproliferative pathway. Proc. Natl. Acad. Sci. USA 2008, 105, 17091–17096. [Google Scholar]
- Maeda, G.; Chiba, T.; Kawashiri, S.; Satoh, T.; Imai, K. Epigenetic inactivation of IkappaB Kinase-alpha in oral carcinomas and tumor progression. Clin. Cancer Res. 2007, 13, 5041–5047. [Google Scholar] [CrossRef]
- DiDonato, J.A.; Hayakawa, M.; Rothwarf, D.M.; Zandi, E.; Karin, M. A cytokine-responsive IκB kinase that activates the transcription factor NF-κB. Nature 1997, 388, 548–554. [Google Scholar] [CrossRef]
- Mercurio, F.; Zhu, H.; Murray, B.W.; Shevchenko, A.; Bennett, B.L.; Li, J.; Young, D.B.; Barbosa, M.; Mann, M.; Manning, A.; et al. A. KK-1 and IKK-2: Cytokine-activated IκB kinases essential for NF-κB activation. Science 1997, 278, 860–866. [Google Scholar] [CrossRef]
- Beg, A.A.; Sha, W.C.; Bronson, R.T.; Ghosh, S.; Baltimore, D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-κB. Nature 1995, 376, 167–170. [Google Scholar]
- Li, Z.W.; Chu, W.; Hu, Y.; Delhase, M.; Deerinck, T.; Ellisman, M.; Johnson, R.; Karin, M. The IKKβ subunit of IκB kinase (IKK) is essential for nuclear factor κB activation and prevention of apoptosis. J. Exp. Med. 1999, 189, 1839–1845. [Google Scholar] [CrossRef]
- Rudolph, D.; Yeh, W.C.; Wakeham, A.; Rudolph, B.; Nallainathan, D.; Potter, J.; Elia, A.J.; Mak, T.W. Severe liver degeneration and lack of NF-κB activation in NEMO/IKKgamma-deficient mice. Genes Dev. 2000, 14, 854–862. [Google Scholar]
- Makris, C.; Godfrey, V.L.; Krahn-Senftleben, G.; Takahashi, T.; Roberts, J.L.; Schwarz, T.; Feng, L.; Johnson, R.S.; Karin, M. Female mice heterozygous for IKK gamma/NEMO deficiencies develop a dermatopathy similar to the human X-linked disorder incontinentia pigmenti. Mol. Cell 2000, 5, 969–979. [Google Scholar] [CrossRef]
- Li, Q.; Lu, Q.; Hwang, J.Y.; Buscher, D.; Lee, K.F.; Izpisua-Belmonte, J.C.; Verma, I.M. IKK1-deficient mice exhibit abnormal development of skin and skeleton. Genes Dev. 1999, 13, 1322–1328. [Google Scholar] [CrossRef]
- Li, Q.; van Antwerp, D.; Mercurio, F.; Lee, K.F.; Verma, I.M. Severe liver degeneration in mice lacking the IkappaB kinase 2 gene. Science 1999, 284, 321–325. [Google Scholar] [CrossRef]
- Schmidt-Supprian, M.; Bloch, W.; Courtois, G.; Addicks, K.; Israel, A.; Rajewsky, K.; Pasparakis, M. NEMO/IKK gamma-deficient mice model incontinentia pigmenti. Mol. Cell 2000, 5, 981–992. [Google Scholar] [CrossRef]
- Doi, T.S.; Marino, M.W.; Takahashi, T.; Yoshida, T.; Sakakura, T.; Old, L.J.; Obata, Y. Absence of tumor necrosis factor rescues RelA-deficient mice from embryonic lethality. Proc. Natl. Acad. Sci. USA 1999, 96, 2994–2999. [Google Scholar]
- Alcamo, E.; Mizgerd, J.P.; Horwitz, B.H.; Bronson, R.; Beg, A.A.; Scott, M.; Doerschuk, C.M.; Hynes, R.O.; Baltimore, D. Targeted mutation of TNF receptor I rescues the RelA-deficient mouse and reveals a critical role for NF-kappa B in leukocyte recruitment. J. Immunol. 2001, 167, 1592–1600. [Google Scholar]
- Zandi, E.; Rothwarf, D.M.; Delhase, M.; Hayakawa, M.; Karin, M. The IκB kinase complex (IKK) contains two kinase subunits, IKKα and IKKβ, necessary for IκB phosphorylation and NF-κB activation. Cell 1997, 91, 243–252. [Google Scholar] [CrossRef]
- Hu, Y.; Baud, V.; Delhase, M.; Zhang, P.; Deerinck, T.; Ellisman, M.; Johnson, R.; Karin, M. Abnormal morphogenesis but intact IKK activation in mice lacking the IKKα subunit of IκB kinase. Science 1999, 284, 316–320. [Google Scholar] [CrossRef]
- Takeda, K.; Takeuchi, O.; Tsujimura, T.; Itami, S.; Adachi, O.; Kawai, T.; Sanjo, H.; Yoshikawa, K.; Terada, N.; Akira, S. Limb and skin abnormalities in mice lacking IKKα. Science 1999, 284, 313–316. [Google Scholar]
- Fuchs, E.; Byrne, C. The epidermis: Rising to the surface. Curr. Opin. Genet. Dev. 1994, 4, 725–736. [Google Scholar] [CrossRef]
- Hu, Y.; Baud, V.; Oga, T.; Kim, K.I.; Yoshida, K.; Karin, M. IKKα controls formation of the epidermis independently of NF-κB. Nature 2001, 410, 710–714. [Google Scholar] [CrossRef]
- Richardson, R.J.; Dixon, J.; Malhotra, S.; Hardman, M.J.; Knowles, L.; Boot-Handford, R.P.; Shore, P.; Whitmarsh, A.; Dixon, M.J. IRF6 is a key determinant of the keratinocyte proliferation-differentiation switch. Nat. Genet. 2006, 38, 1329–1334. [Google Scholar]
- Herron, B.J.; Liddell, R.A.; Parker, A.; Grant, S.; Kinne, J.; Fisher, J.K.; Siracusa, L.D. A mutation in stratifin is responsible for the repeated epilation (Er) phenotype in mice. Nat. Genet. 2005, 37, 1210–1212. [Google Scholar] [CrossRef]
- Li, Q.; Lu, Q.; Estepa, G.; Verma, I.M. Identification of 14-3-3{sigma} mutation causing cutaneous abnormality in repeated-epilation mutant mouse. Proc. Natl. Acad. Sci. USA 2005, 102, 15977–15982. [Google Scholar]
- Kondo, S.; Schutte, B.C.; Richardson, R.J.; Bjork, B.C.; Knight, A.S.; Watanabe, Y.; Howard, E.; de Lima, R.L.; Daack-Hirsch, S.; Sander, A.; et al. Mutations in IRF6 cause Van der Woude and popliteal pterygium syndromes. Nat. Genet. 2002, 32, 285–289. [Google Scholar] [CrossRef]
- Blackburn, J.; Ohazama, A.; Kawasaki, K.; Otsuka-Tanaka, Y.; Liu, B.; Honda, K.; Rountree, R.B.; Hu, Y.; Kawasaki, M.; Birchmeier, W.; et al. The role of Irf6 in tooth epithelial invagination. Dev. Biol. 2012, 365, 61–70. [Google Scholar] [CrossRef]
- Urano, T.; Saito, T.; Tsukui, T.; Fujita, M.; Hosoi, T.; Muramatsu, M.; Ouchi, Y.; Inoue, S. EFP targets 14-3-3σ for proteolysis and promotes breast tumour growth. Nature 2002, 417, 871–875. [Google Scholar] [CrossRef]
- Zhu, F.; Xia, X.; Liu, B.; Shen, J.; Hu, Y.; Person, M.; Hu, Y. IKKalpha Shields 14-3-3sigma, a G(2)/M Cell Cycle Checkpoint Gene, from Hypermethylation, Preventing Its Silencing. Mol. Cell 2007, 27, 214–227. [Google Scholar] [CrossRef]
- Hermeking, H.; Lengauer, C.; Polyak, K.; He, T.C.; Zhang, L.; Thiagalingam, S.; Kinzler, K.W.; Vogelstein, B. 14-3-3σ is a p53-regulated inhibitor of G2/M progression. Mol. Cell 1997, 1, 3–11. [Google Scholar] [CrossRef]
- Liu, B.; Zhu, F.; Xia, X.; Park, E.; Hu, Y. A tale of terminal differentiation: IKKalpha, the master keratinocyte regulator. Cell Cycle 2009, 8, 527–531. [Google Scholar]
- Mills, A.A.; Zheng, B.; Wang, X.J.; Vogel, H.; Roop, D.R.; Bradley, A. p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature 1999, 398, 708–713. [Google Scholar] [CrossRef]
- Candi, E.; Terrinoni, A.; Rufini, A.; Chikh, A.; Lena, A.M.; Suzuki, Y.; Sayan, B.S.; Knight, R.A.; Melino, G. p63 is upstream of IKK alpha in epidermal development. J. Cell Sci. 2006, 119, 4617–4622. [Google Scholar] [CrossRef]
- Koster, M.I.; Dai, D.; Marinari, B.; Sano, Y.; Costanzo, A.; Karin, M.; Roop, D.R. p63 induces key target genes required for epidermal morphogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 3255–3260. [Google Scholar]
- Marinari, B.; Ballaro, C.; Koster, M.I.; Giustizieri, M.L.; Moretti, F.; Crosti, F.; Papoutsaki, M.; Karin, M.; Alema, S.; Chimenti, S.; et al. IKKalpha is a p63 transcriptional target involved in the pathogenesis of ectodermal dysplasias. J. Invest. Dermatol. 2009, 129, 60–69. [Google Scholar] [CrossRef]
- Liu, B.; Willette-Brown, J.; Liu, S.; Chen, X.; Fischer, S.M.; Hu, Y. IKKalpha represses a network of inflammation and proliferation pathways and elevates c-Myc antagonists and differentiation in a dose-dependent manner in the skin. Cell Death Differ. 2011, 18, 1854–1864. [Google Scholar] [CrossRef]
- Lahtela, J.; Nousiainen, H.O.; Stefanovic, V.; Tallila, J.; Viskari, H.; Karikoski, R.; Gentile, M.; Saloranta, C.; Varilo, T.; Salonen, R.; et al. Mutant CHUK and severe fetal encasement malformation. N. Engl. J. Med. 2010, 363, 1631–1637. [Google Scholar] [CrossRef]
- Sil, A.K.; Maeda, S.; Sano, Y.; Roop, D.R.; Karin, M. IKKα acts in the epidermis to control skeletal and craniofacial morphogenesis. Nature 2004, 428, 660–664. [Google Scholar]
- Park, E.; Zhu, F.; Liu, B.; Xia, X.; Shen, J.; Bustos, T.; Fischer, S.M.; Hu, Y. Reduction in IkappaB kinase alpha expression promotes the development of skin papillomas and carcinomas. Cancer Res. 2007, 67, 9158–9168. [Google Scholar] [CrossRef]
- Quintanilla, M.; Brown, K.; Ramsden, M.; Balmain, A. Carcinogen-specific mutation and amplification of Ha-ras during mouse skin carcinogenesis. Nature 1986, 322, 78–80. [Google Scholar] [CrossRef]
- Slaga, T.J.; O’Connell, J.; Rotstein, J.; Patskan, G.; Morris, R.; Aldaz, C.M.; Conti, C.J. Critical genetic determinants and molecular events in multistage skin carcinogenesis. Symp. Fundam. Cancer Res. 1986, 39, 31–44. [Google Scholar]
- Ziegler, A.; Jonason, A.S.; Leffell, D.J.; Simon, J.A.; Sharma, H.W.; Kimmelman, J.; Remington, L.; Jacks, T.; Brash, D.E. Sunburn and p53 in the onset of skin cancer. Nature 1994, 372, 773–776. [Google Scholar] [CrossRef]
- Melnikova, V.O.; Ananthaswamy, H.N. Cellular and molecular events leading to the development of skin cancer. Mutat. Res. 2005, 571, 91–106. [Google Scholar] [CrossRef]
- Halliday, G.M. Inflammation, gene mutation and photoimmunosuppression in response to UVR-induced oxidative damage contributes to photocarcinogenesis. Mutat. Res. 2005, 571, 107–120. [Google Scholar] [CrossRef]
- Liu, B.; Park, E.; Zhu, F.; Bustos, T.; Liu, J.; Shen, J.; Fischer, S.M.; Hu, Y. A critical role for I{kappa}B kinase {alpha} in the development of human and mouse squamous cell carcinomas. Proc. Natl. Acad. Sci. USA 2006, 103, 17202–17207. [Google Scholar]
- Jonason, A.S.; Kunala, S.; Price, G.J.; Restifo, R.J.; Spinelli, H.M.; Persing, J.A.; Leffell, D.J.; Tarone, R.E.; Brash, D.E. Frequent clones of p53-mutated keratinocytes in normal human skin. Proc. Natl. Acad. Sci. USA 1996, 93, 14025–14029. [Google Scholar]
- Xia, X.; Park, E.; Liu, B.; Willette-Brown, J.; Gong, W.; Wang, J.; Mitchell, D.; Fischer, S.M.; Hu, Y. Reduction of IKKalpha expression promotes chronic ultraviolet B exposure-induced skin inflammation and carcinogenesis. Am. J. Pathol. 2010, 176, 2500–2508. [Google Scholar] [CrossRef]
- Ouhtit, A.; Gorny, A.; Muller, H.K.; Hill, L.L.; Owen-Schaub, L.; Ananthaswamy, H.N. Loss of Fas-ligand expression in mouse keratinocytes during UV carcinogenesis. Am. J. Pathol. 2000, 157, 1975–1981. [Google Scholar] [CrossRef]
- Li, Q.; Sambandam, S.A.; Lu, H.J.; Thomson, A.; Kim, S.H.; Lu, H.; Xin, Y.; Lu, Q. 14-3-3sigma and p63 play opposing roles in epidermal tumorigenesis. Carcinogenesis 2011, 32, 1782–1788. [Google Scholar]
- Restivo, G.; Nguyen, B.C.; Dziunycz, P.; Ristorcelli, E.; Ryan, R.J.; Ozuysal, O.Y.; di Piazza, M.; Radtke, F.; Dixon, M.J.; Hofbauer, G.F.; et al. IRF6 is a mediator of Notch pro-differentiation and tumour suppressive function in keratinocytes. EMBO J. 2011, 30, 4571–4585. [Google Scholar] [CrossRef]
- Botti, E.; Spallone, G.; Moretti, F.; Marinari, B.; Pinetti, V.; Galanti, S.; de Meo, P.D.; de Nicola, F.; Ganci, F.; Castrignano, T.; et al. Developmental factor IRF6 exhibits tumor suppressor activity in squamous cell carcinomas. Proc. Natl. Acad. Sci. USA 2011, 108, 13710–13715. [Google Scholar]
- Pasparakis, M.; Courtois, G.; Hafner, M.; Schmidt-Supprian, M.; Nenci, A.; Toksoy, A.; Krampert, M.; Goebeler, M.; Gillitzer, R.; Israel, A.; et al. TNF-mediated inflammatory skin disease in mice with epidermis-specific deletion of IKK2. Nature 2002, 417, 861–866. [Google Scholar] [CrossRef]
- Nenci, A.; Huth, M.; Funteh, A.; Schmidt-Supprian, M.; Bloch, W.; Metzger, D.; Chambon, P.; Rajewsky, K.; Krieg, T.; Haase, I.; et al. Skin lesion development in a mouse model of incontinentia pigmenti is triggered by NEMO deficiency in epidermal keratinocytes and requires TNF signaling. Hum. Mol. Genet. 2006, 15, 531–542. [Google Scholar] [CrossRef]
- Moore, R.J.; Owens, D.M.; Stamp, G.; Arnott, C.; Burke, F.; East, N.; Holdsworth, H.; Turner, L.; Rollins, B.; Pasparakis, M.; et al. Mice deficient in tumor necrosis factor-alpha are resistant to skin carcinogenesis. Nat. Med. 1999, 5, 828–831. [Google Scholar] [CrossRef]
- Ramirez, A.; Page, A.; Gandarillas, A.; Zanet, J.; Pibre, S.; Vidal, M.; Tusell, L.; Genesca, A.; Whitaker, D.A.; Melton, D.W.; et al. A keratin K5Cre transgenic line appropriate for tissue-specific or generalized Cre-mediated recombination. Genesis 2004, 39, 52–57. [Google Scholar] [CrossRef]
- Hafner, M.; Wenk, J.; Nenci, A.; Pasparakis, M.; Scharffetter-Kochanek, K.; Smyth, N.; Peters, T.; Kess, D.; Holtkotter, O.; Shephard, P.; et al. Keratin 14 Cre transgenic mice authenticate keratin 14 as an oocyte-expressed protein. Genesis 2004, 38, 176–181. [Google Scholar] [CrossRef]
- Gareus, R.; Huth, M.; Breiden, B.; Nenci, A.; Rosch, N.; Haase, I.; Bloch, W.; Sandhoff, K.; Pasparakis, M. Normal epidermal differentiation but impaired skin-barrier formation upon keratinocyte-restricted IKK1 ablation. Nat. Cell Biol. 2007, 9, 461–469. [Google Scholar] [CrossRef]
- Zenz, R.; Eferl, R.; Scheinecker, C.; Redlich, K.; Smolen, J.; Schonthaler, H.B.; Kenner, L.; Tschachler, E.; Wagner, E.F. Activator protein 1 (Fos/Jun) functions in inflammatory bone and skin disease. Arthritis Res. Ther. 2008, 10, 201. [Google Scholar]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Page, A.; Navarro, M.; Garin, M.; Perez, P.; Casanova, M.L.; Moreno, R.; Jorcano, J.L.; Cascallana, J.L.; Bravo, A.; Ramirez, A. IKKbeta leads to an inflammatory skin disease resembling interface dermatitis. J. Invest. Dermatol. 2010, 130, 1598–1610. [Google Scholar] [CrossRef]
- Alameda, J.P.; Moreno-Maldonado, R.; Fernandez-Acenero, M.J.; Navarro, M.; Page, A.; Jorcano, J.L.; Bravo, A.; Ramirez, A.; Casanova, M.L. Increased IKKalpha expression in the basal layer of the epidermis of transgenic mice enhances the malignant potential of skin tumors. PLoS One 2011, 6, e21984. [Google Scholar]
- Senftleben, U.; Cao, Y.; Xiao, G.; Greten, F.R.; Krahn, G.; Bonizzi, G.; Chen, Y.; Hu, Y.; Fong, A.; Sun, S.C.; et al. Activation by IKKα of a second, evolutionary conserved, NF-κB signaling pathway. Science 2001, 293, 1495–1499. [Google Scholar] [CrossRef]
- Moreno-Maldonado, R.; Ramirez, A.; Navarro, M.; Fernandez-Acenero, M.J.; Villanueva, C.; Page, A.; Jorcano, J.L.; Bravo, A.; Llanos Casanova, M. IKKalpha enhances human keratinocyte differentiation and determines the histological variant of epidermal squamous cell carcinomas. Cell Cycle 2008, 7, 2021–2029. [Google Scholar] [CrossRef]
- Descargues, P.; Sil, A.K.; Karin, M. IKKalpha, a critical regulator of epidermal differentiation and a suppressor of skin cancer. EMBO J. 2008, 27, 2639–2647. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Xia, X.; Park, E.; Fischer, S.M.; Hu, Y. Mouse Genetic Models Reveal Surprising Functions of IkB Kinase Alpha in Skin Development and Skin Carcinogenesis. Cancers 2013, 5, 170-183. https://doi.org/10.3390/cancers5010170
Xia X, Park E, Fischer SM, Hu Y. Mouse Genetic Models Reveal Surprising Functions of IkB Kinase Alpha in Skin Development and Skin Carcinogenesis. Cancers. 2013; 5(1):170-183. https://doi.org/10.3390/cancers5010170
Chicago/Turabian StyleXia, Xiaojun, Eunmi Park, Susan M. Fischer, and Yinling Hu. 2013. "Mouse Genetic Models Reveal Surprising Functions of IkB Kinase Alpha in Skin Development and Skin Carcinogenesis" Cancers 5, no. 1: 170-183. https://doi.org/10.3390/cancers5010170
APA StyleXia, X., Park, E., Fischer, S. M., & Hu, Y. (2013). Mouse Genetic Models Reveal Surprising Functions of IkB Kinase Alpha in Skin Development and Skin Carcinogenesis. Cancers, 5(1), 170-183. https://doi.org/10.3390/cancers5010170