Characterization of a Dual CDC7/CDK9 Inhibitor in Multiple Myeloma Cellular Models

Abstract
:1. Introduction
2. Results and Discussion
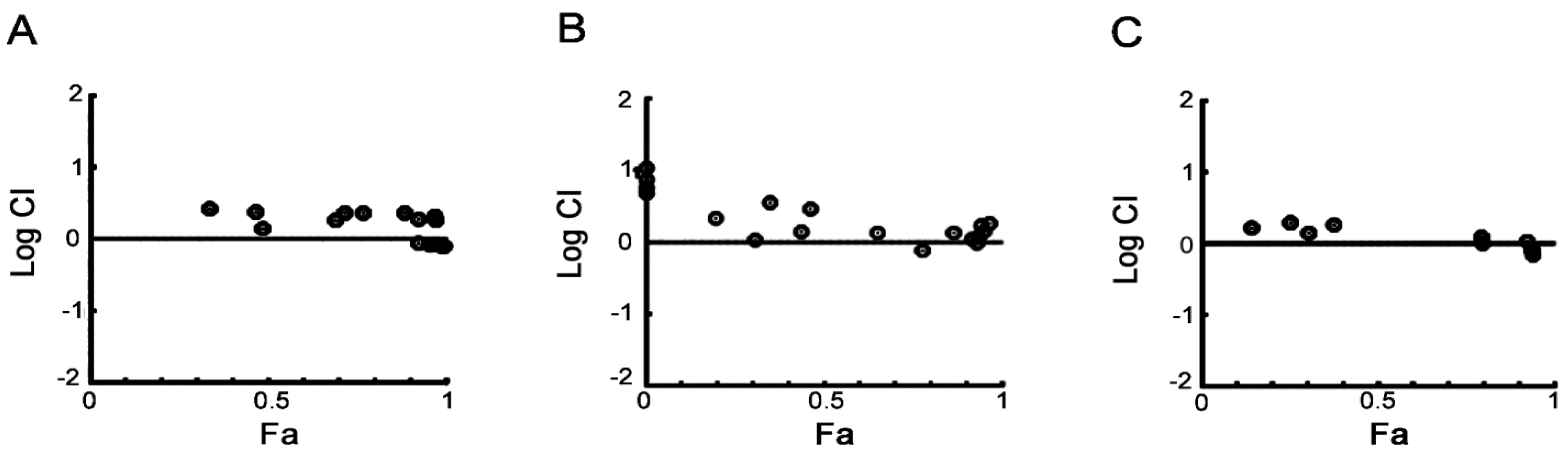
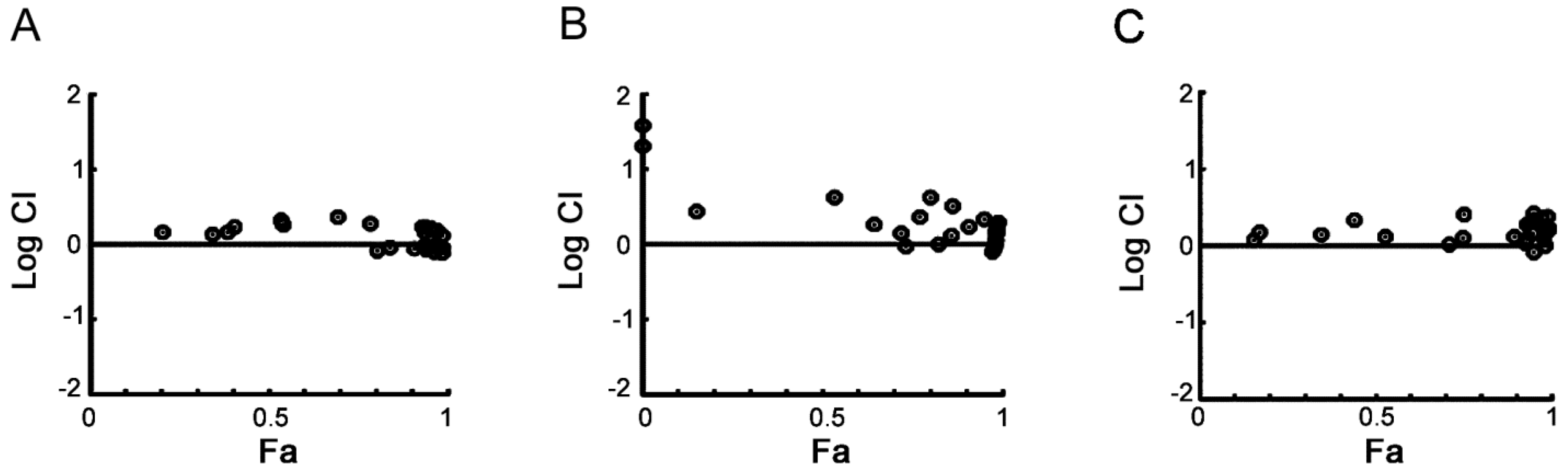
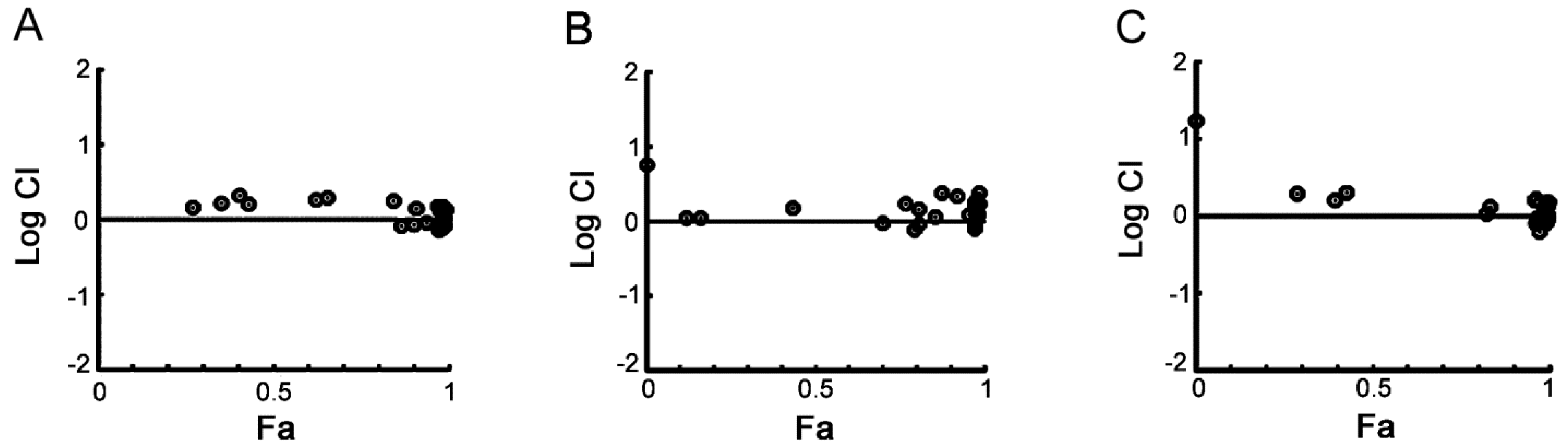
2.1. Cellular Responses of Myeloma Cells to Dual CDC7/CDK9 Inhibitors

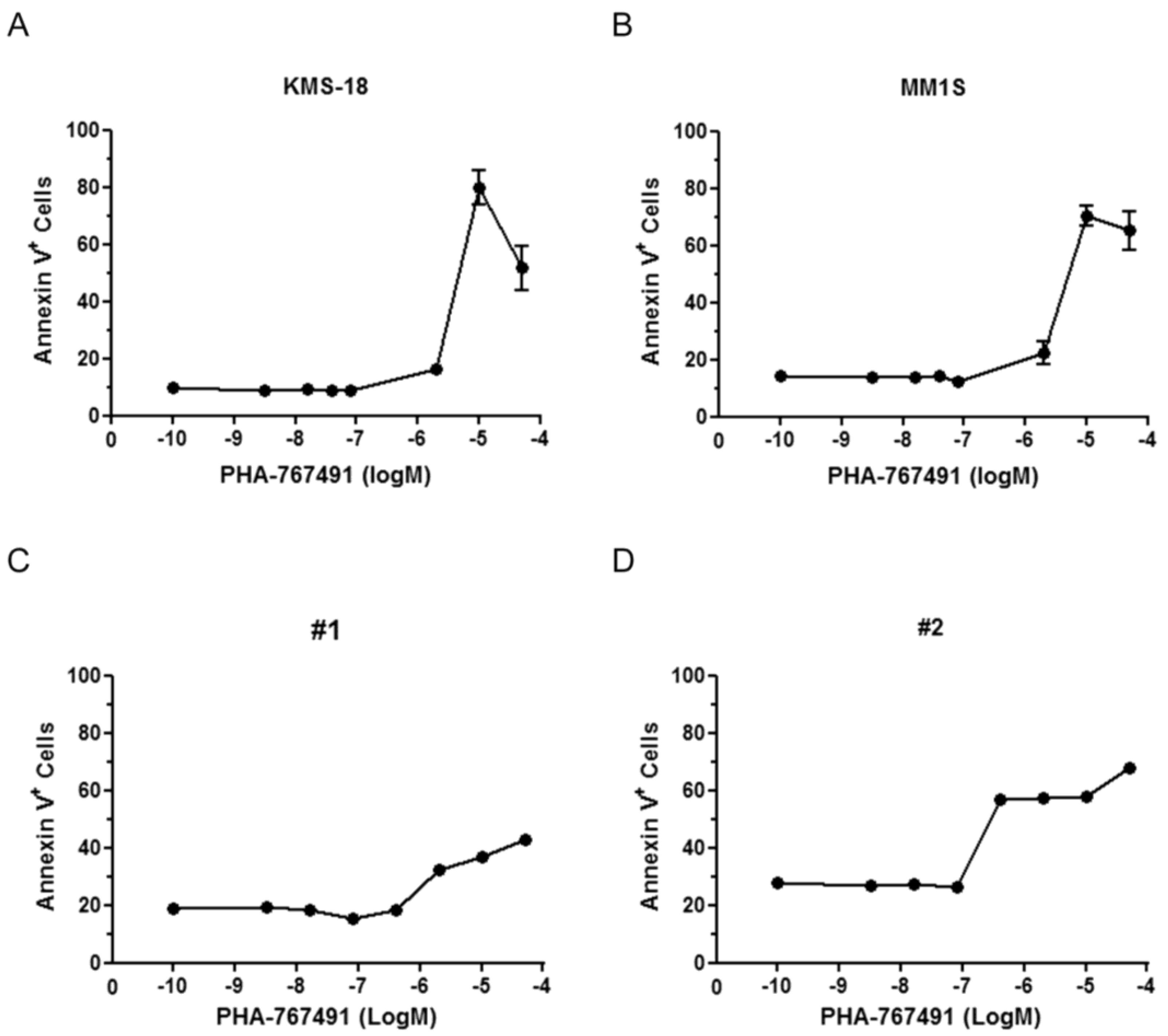{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | I⁰ Translocation | M-Spike | P53 Status | Copy Number (markers) | PHA-767491 IC50 (μM) |
|---|---|---|---|---|---|
| KMS-18 | t(4; 14) | IgG lambda | Not Known | 1.2 (613) | 1 |
| U-266 | Insertion IgH switch element on 11q13 | IgE lambda | Mutant [31,32] | 1.1 (614) | 1 |
| OCI-My5 | t(14; 16) | Not Known | WT and Mutant [33,34] | 2.0 (406) | 1.5 |
| RPMI-8226-Dox40 | t(14; 16) | IgG lambda | Mutant | Not Known | 2 |
| RPMI-8226-LR5 | t(14; 16) | IgG lambda | Deficient [32] | Not Known | 3.5 |
| MM1S | t(14; 16) | IgG lambda | WT [32] | 2.1 (337) | 2 |
| MM1R | t(14; 16) | IgG lambda | Not Known | 2.2 (336) | 1 |
| Primary Sample | Cytogenetics | ISS | mSMART | Prior Rx | PHA-767491 IC50 (μM) |
|---|---|---|---|---|---|
| GAL-MM-1 | Tetraploid, 13q | Stage 3 | High Risk | MPT/V/R/D | 3 |
| GAL-MM-2 | Clonal Evolution. t(11;14) | Stage 3 | High Risk | VMPT/ASCT | 2 |
| GAL-MM-3 | t(4; 14) | Stage 3 | High Risk | VRD/ASCT | 1.9 |
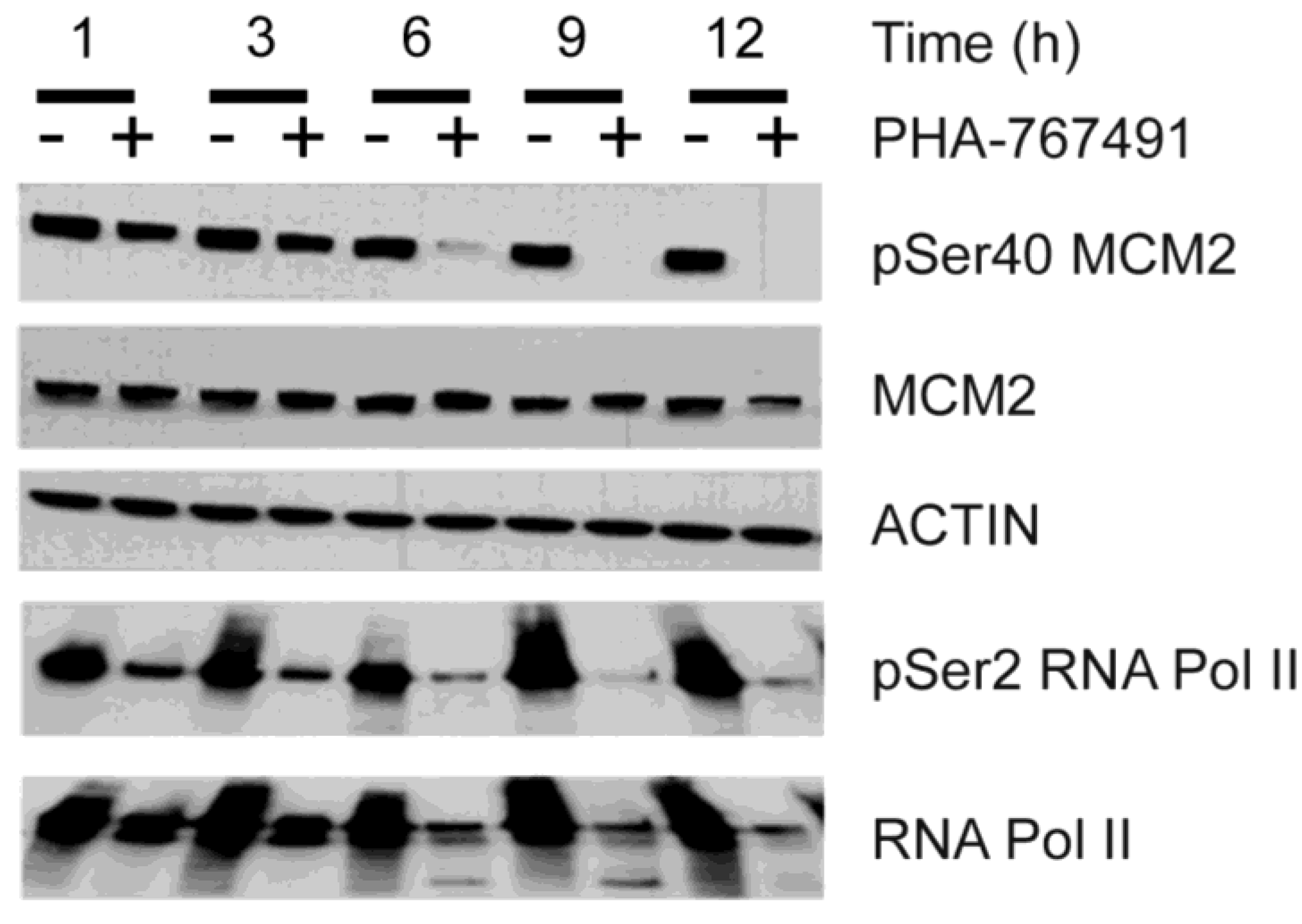
2.2. PHA-767491 Modulates Biomarkers of CDC7 and CDK9 Activity


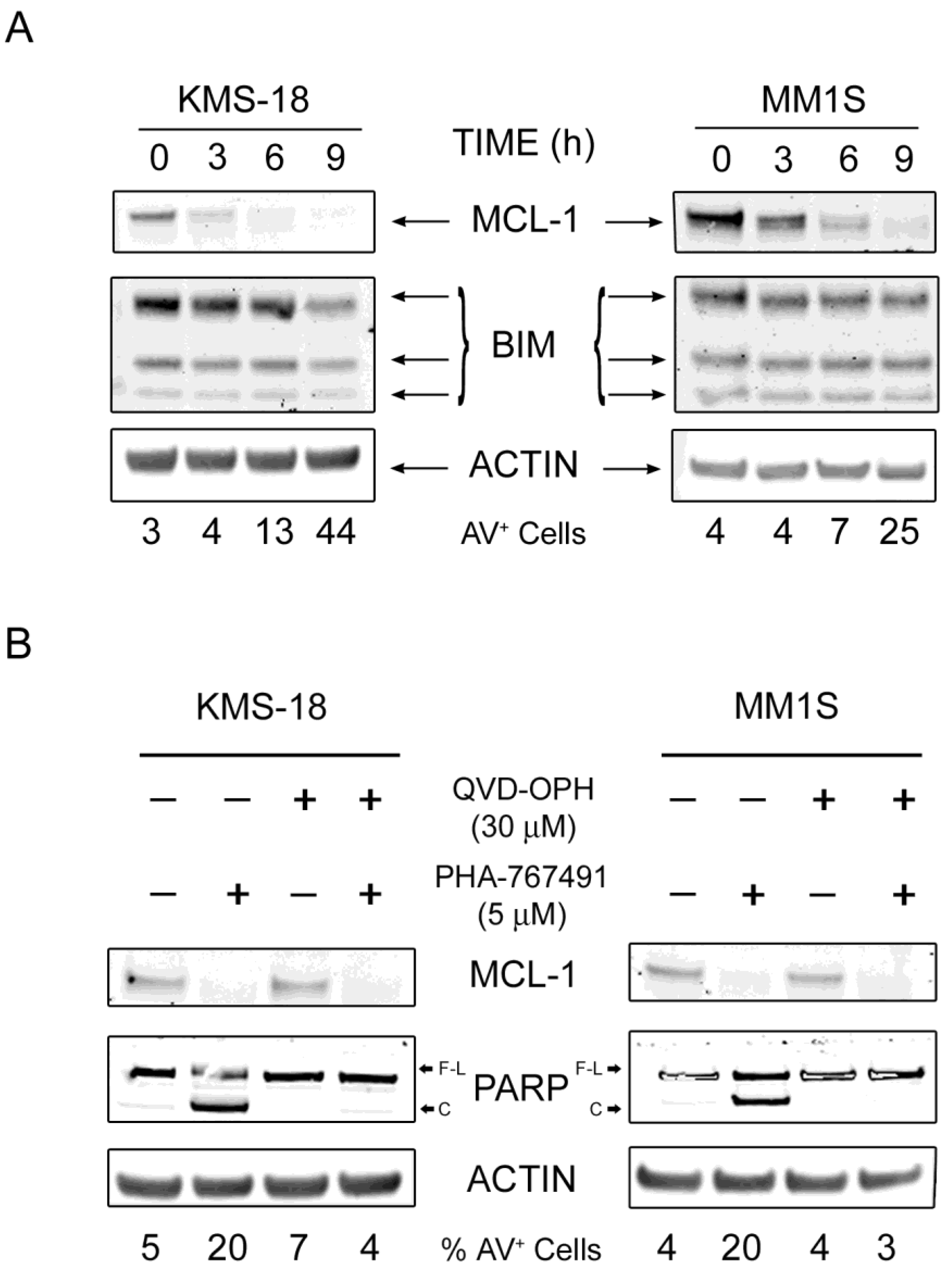
2.3. MCL-1 Is Potently Downregulated Following PHA-767491 Treatment

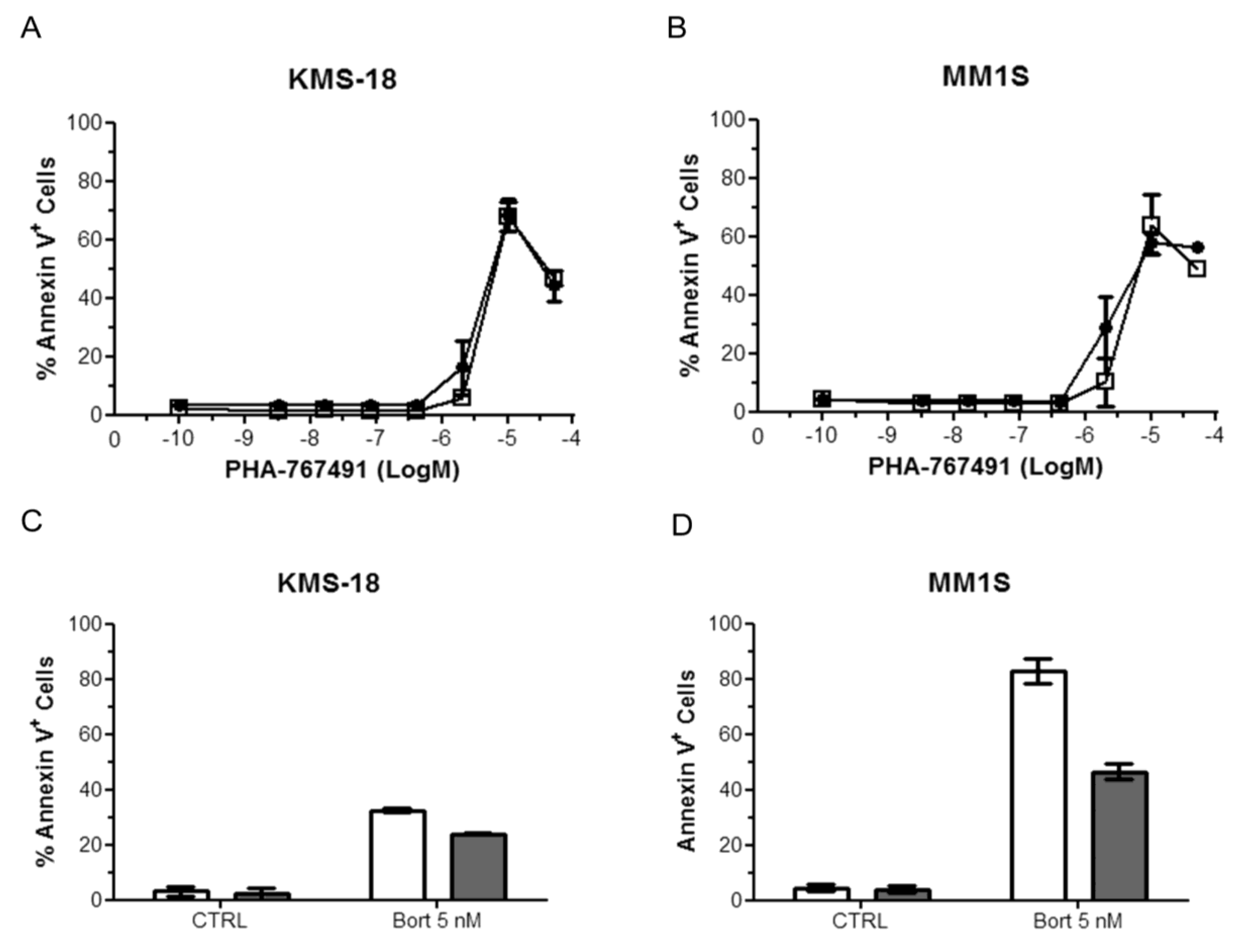
2.4. Effect of Stroma Cells on the Response of Myeloma Cells to PHA-767491


2.5. PHA-767491 Shows an Additive Effect with Melphalan, Bortezomib and Doxorubicin

2.6. Discussion
3. Experimental Section
3.1. Chemicals
3.2. Cell Culture
3.3. Cell Viability Assay
3.4. Immunoblotting
3.5. DNA Replication and Apoptosis Assays by Flow Cytometry
4. Conclusions
Acknowledgements
Conflict of Interest
References
- Altekruse, S.F.; Kosary, C.L.; Krapcho, M.; Neyman, N.; Aminou, R.; Waldron, W.; Ruhl, J.; Howlader, N.; Tatlovich, Z.; Cho, H.; Mariotto, A.; et al. SEER Cancer Statistics Review, 1975–2007; National Cancer Institute: Bethesda, MD, USA, 2009. [Google Scholar]
- Bergsagel, P.L.; Kuehl, W.M.; Zhan, F.; Sawyer, J.; Barlogie, B.; Shaughnessy, J., Jr. Cyclin D dysregulation: An early and unifying pathogenic event in multiple myeloma. Blood 2005, 106, 296–303. [Google Scholar] [CrossRef]
- Ely, S.; di Liberto, M.; Niesvizky, R.; Baughn, L.B.; Cho, H.J.; Hatada, E.N.; Knowles, D.M.; Lane, J.; Chen-Kiang, S. Mutually exclusive cyclin-dependent kinase 4/cyclin D1 and cyclin-dependent kinase 6/cyclin D2 pairing inactivates retinoblastoma protein and promotes cell cycle dysregulation in multiple myeloma. Cancer Res. 2005, 65, 11345–11353. [Google Scholar] [CrossRef]
- Zhan, F.; Huang, Y.; Colla, S.; Stewart, J.P.; Hanamura, I.; Gupta, S.; Epstein, J.; Yaccoby, S.; Sawyer, J.; Burington, B.; et al. The molecular classification of multiple myeloma. Blood 2006, 108, 2020–2028. [Google Scholar] [CrossRef]
- Shaughnessy, J.D., Jr.; Zhan, F.; Burington, B.E.; Huang, Y.; Colla, S.; Hanamura, I.; Stewart, J.P.; Kordsmeier, B.; Randolph, C.; Williams, D.R.; et al. A validated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome 1. Blood 2007, 109, 2276–2284. [Google Scholar] [CrossRef]
- Baughn, L.B.; di Liberto, M.; Wu, K.; Toogood, P.L.; Louie, T.; Gottschalk, R.; Niesvizky, R.; Cho, H.; Ely, S.; Moore, M.A.; et al. A novel orally active small molecule potently induces G1 arrest in primary myeloma cells and prevents tumor growth by specific inhibition of cyclin-dependent kinase 4/6. Cancer Res. 2006, 66, 7661–7667. [Google Scholar] [CrossRef]
- MacCallum, D.E.; Melville, J.; Frame, S.; Watt, K.; Anderson, S.; Gianella-Borradori, A.; Lane, D.P.; Green, S.R. Seliciclib (CYC202, R-Roscovitine) induces cell death in multiple myeloma cells by inhibition of RNA polymerase II-dependent transcription and down-regulation of Mcl-1. Cancer Res. 2005, 65, 5399–5407. [Google Scholar] [CrossRef]
- Raje, N.; Kumar, S.; Hideshima, T.; Roccaro, A.; Ishitsuka, K.; Yasui, H.; Shiraishi, N.; Chauhan, D.; Munshi, N.C.; Green, S.R.; et al. Seliciclib (CYC202 or R-roscovitine), a small-molecule cyclin-dependent kinase inhibitor, mediates activity via down-regulation of Mcl-1 in multiple myeloma. Blood 2005, 106, 1042–1047. [Google Scholar] [CrossRef]
- Conroy, A.; Stockett, D.E.; Walker, D.; Arkin, M.R.; Hoch, U.; Fox, J.A.; Hawtin, R.E. SNS-032 is a potent and selective CDK 2, 7 and 9 inhibitor that drives target modulation in patient samples. Cancer Chemother. Pharmacol. 2009, 64, 723–732. [Google Scholar] [CrossRef]
- Gojo, I.; Zhang, B.; Fenton, R.G. The cyclin-dependent kinase inhibitor flavopiridol induces apoptosis in multiple myeloma cells through transcriptional repression and down-regulation of Mcl-1. Clin. Cancer Res. 2002, 8, 3527–3538. [Google Scholar]
- Blow, J.J.; Gillespie, P.J. Replication licensing and cancer—A fatal entanglement? Nat. Rev. Cancer 2008, 8, 799–806. [Google Scholar] [CrossRef]
- Sclafani, R.A.; Holzen, T.M. Cell cycle regulation of DNA replication. Annu. Rev. Genet. 2007, 41, 237–280. [Google Scholar] [CrossRef]
- Labib, K. How do Cdc7 and cyclin-dependent kinases trigger the initiation of chromosome replication in eukaryotic cells? Genes Dev. 2010, 24, 1208–1219. [Google Scholar] [CrossRef]
- Montagnoli, A.; Valsasina, B.; Brotherton, D.; Troiani, S.; Rainoldi, S.; Tenca, P.; Molinari, A.; Santocanale, C. Identification of Mcm2 phosphorylation sites by S-phase-regulating kinases. J. Biol. Chem. 2006, 281, 10281–10290. [Google Scholar] [CrossRef]
- Tudzarova, S.; Trotter, M.W.; Wollenschlaeger, A.; Mulvey, C.; Godovac-Zimmermann, J.; Williams, G.H.; Stoeber, K. Molecular architecture of the DNA replication origin activation checkpoint. EMBO J. 2010, 29, 3381–3394. [Google Scholar] [CrossRef]
- Montagnoli, A.; Tenca, P.; Sola, F.; Carpani, D.; Brotherton, D.; Albanese, C.; Santocanale, C. Cdc7 inhibition reveals a p53-dependent replication checkpoint that is defective in cancer cells. Cancer Res. 2004, 64, 7110–7116. [Google Scholar] [CrossRef]
- Swords, R.; Mahalingam, D.; O’Dwyer, M.; Santocanale, C.; Kelly, K.; Carew, J.; Giles, F. CDC7 kinase—A new target for drug development. Eur. J. Cancer 2010, 46, 33–40. [Google Scholar] [CrossRef]
- Sawa, M.; Masai, H. Drug design with CDC7 kinase: A potential novel cancer therapy target. Drug Des. Dev. Ther. 2009, 2, 255–264. [Google Scholar]
- Montagnoli, A.; Valsasina, B.; Croci, V.; Menichincheri, M.; Rainoldi, S.; Marchesi, V.; Tibolla, M.; Tenca, P.; Brotherton, D.; Albanese, C.; et al. A CDC7 kinase inhibitor restricts initiation of DNA replication and has antitumor activity. Nat. Chem. Biol. 2008, 4, 357–365. [Google Scholar] [CrossRef]
- Hampsey, M.; Reinberg, D. RNA polymerase II as a control panel for multiple coactivator complexes. Curr. Opin. Genet. Dev. 1999, 9, 132–139. [Google Scholar] [CrossRef]
- Sharma, S.V.; Fischbach, M.A.; Haber, D.A.; Settleman, J. “Oncogenic shock”: Explaining oncogene addiction through differential signal attenuation. Clin. Cancer Res. 2006, 12, 4392s–4395s. [Google Scholar] [CrossRef]
- Derenne, S.; Monia, B.; Dean, N.M.; Taylor, J.K.; Rapp, M.J.; Harousseau, J.L.; Bataille, R.; Amiot, M. Antisense strategy shows that Mcl-1 rather than Bcl-2 or Bcl-x(L) is an essential survival protein of human myeloma cells. Blood 2002, 100, 194–199. [Google Scholar] [CrossRef]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef] [Green Version]
- Gertz, M.A.; Lacy, M.Q.; Dispenzieri, A.; Greipp, P.R.; Litzow, M.R.; Henderson, K.J.; van Wier, S.A.; Ahmann, G.J.; Fonseca, R. Clinical implications of t(11;14)(q13;q32), t(4;14)(p16.3;q32), and -17p13 in myeloma patients treated with high-dose therapy. Blood 2005, 106, 2837–2840. [Google Scholar] [CrossRef]
- Wuilleme-Toumi, S.; Robillard, N.; Gomez, P.; Moreau, P.; Le Gouill, S.; Avet-Loiseau, H.; Harousseau, J.L.; Amiot, M.; Bataille, R. Mcl-1 is overexpressed in multiple myeloma and associated with relapse and shorter survival. Leukemia 2005, 19, 1248–1252. [Google Scholar] [CrossRef]
- Natoni, A.; Murillo, L.S.; Kliszczak, A.E.; Catherwood, M.A.; Montagnoli, A.; Samali, A.; O’Dwyer, M.; Santocanale, C. Mechanisms of action of a dual Cdc7/Cdk9 kinase inhibitor against quiescent and proliferating CLL cells. Mol. Cancer Ther. 2011, 10, 1624–1634. [Google Scholar] [CrossRef]
- Garrido, S.M.; Appelbaum, F.R.; Willman, C.L.; Banker, D.E. Acute myeloid leukemia cells are protected from spontaneous and drug-induced apoptosis by direct contact with a human bone marrow stromal cell line (HS-5). Exp. Hematol. 2001, 29, 448–457. [Google Scholar] [CrossRef]
- Perez, L.E.; Parquet, N.; Meads, M.; Anasetti, C.; Dalton, W. Bortezomib restores stroma-mediated APO2L/TRAIL apoptosis resistance in multiple myeloma. Eur. J. Haematol. 2010, 84, 212–222. [Google Scholar] [CrossRef]
- Perez, L.E.; Parquet, N.; Shain, K.; Nimmanapalli, R.; Alsina, M.; Anasetti, C.; Dalton, W. Bone marrow stroma confers resistance to Apo2 ligand/TRAIL in multiple myeloma in part by regulating c-FLIP. J. Immunol. 2008, 180, 1545–1555. [Google Scholar]
- Xiong, W.; Wu, X.; Starnes, S.; Johnson, S.K.; Haessler, J.; Wang, S.; Chen, L.; Barlogie, B.; Shaughnessy, J.D., Jr.; Zhan, F. An analysis of the clinical and biologic significance of TP53 loss and the identification of potential novel transcriptional targets of TP53 in multiple myeloma. Blood 2008, 112, 4235–4246. [Google Scholar] [CrossRef]
- Mazars, G.R.; Portier, M.; Zhang, X.G.; Jourdan, M.; Bataille, R.; Theillet, C.; Klein, B. Mutations of the p53 gene in human myeloma cell lines. Oncogene 1992, 7, 1015–1018. [Google Scholar]
- Saha, M.N.; Jiang, H.; Mukai, A.; Chang, H. RITA inhibits multiple myeloma cell growth through induction of p53-mediated caspase-dependent apoptosis and synergistically enhances nutlin-induced cytotoxic responses. Mol. Cancer Ther. 2010, 9, 3041–3051. [Google Scholar] [CrossRef]
- Urashima, M.; Teoh, G.; Chauhan, D.; Hoshi, Y.; Ogata, A.; Treon, S.P.; Schlossman, R.L.; Anderson, K.C. Interleukin-6 overcomes p21WAF1 upregulation and G1 growth arrest induced by dexamethasone and interferon-gamma in multiple myeloma cells. Blood 1997, 90, 279–289. [Google Scholar]
- Teoh, G.; Urashima, M.; Ogata, A.; Chauhan, D.; DeCaprio, J.A.; Treon, S.P.; Schlossman, R.L.; Anderson, K.C. MDM2 protein overexpression promotes proliferation and survival of multiple myeloma cells. Blood 1997, 90, 1982–1992. [Google Scholar]
- Vanotti, E.; Amici, R.; Bargiotti, A.; Berthelsen, J.; Bosotti, R.; Ciavolella, A.; Cirla, A.; Cristiani, C.; D’Alessio, R.; Forte, B.; et al. Cdc7 kinase inhibitors: Pyrrolopyridinones as potential antitumor agents. 1. Synthesis and structure-activity relationships. J. Med. Chem. 2008, 51, 487–501. [Google Scholar] [CrossRef]
- Chen, L.; Willis, S.N.; Wei, A.; Smith, B.J.; Fletcher, J.I.; Hinds, M.G.; Colman, P.M.; Day, C.L.; Adams, J.M.; Huang, D.C.S. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol. Cell 2005, 17, 393–403. [Google Scholar] [CrossRef]
- McMillin, D.W.; Delmore, J.; Weisberg, E.; Negri, J.M.; Geer, D.C.; Klippel, S.; Mitsiades, N.; Schlossman, R.L.; Munshi, N.C.; Kung, A.L.; et al. Tumor cell-specific bioluminescence platform to identify stroma-induced changes to anticancer drug activity. Nat. Med. 2010, 16, 483–489. [Google Scholar] [CrossRef]
- Roecklein, B.A.; Torok-Storb, B. Functionally distinct human marrow stromal cell lines immortalized by transduction with the human papilloma virus E6/E7 genes. Blood 1995, 85, 997–1005. [Google Scholar]
- Chou, T.C. Preclinical versus clinical drug combination studies. Leuk. Lymphoma 2008, 49, 2059–2080. [Google Scholar] [CrossRef]
- Ito, S.; Taniyami, C.; Arai, N.; Masai, H. CDC7 as a potential new target for cancer therapy. Drug News Perspect. 2008, 21, 481–488. [Google Scholar] [CrossRef]
- Menichincheri, M.; Albanese, C.; Alli, C.; Ballinari, D.; Bargiotti, A.; Caldarelli, M.; Ciavolella, A.; Cirla, A.; Colombo, M.; Colotta, F.; et al. Cdc7 kinase inhibitors: 5-heteroaryl-3-carboxamido-2-aryl pyrroles as potential antitumor agents. 1. Lead finding. J. Med. Chem. 2010, 53, 7296–7315. [Google Scholar] [CrossRef]
- Menichincheri, M.; Bargiotti, A.; Berthelsen, J.; Bertrand, J.A.; Bossi, R.; Ciavolella, A.; Cirla, A.; Cristiani, C.; Croci, V.; D’Alessio, R.; et al. First Cdc7 kinase inhibitors: Pyrrolopyridinones as potent and orally active antitumor agents. 2. Lead discovery. J. Med. Chem. 2009, 52, 293–307. [Google Scholar] [CrossRef]
- Wickremasinghe, R.G.; Hoffbrand, A.V. Biochemical and genetic control of apoptosis: Relevance to normal hematopoiesis and hematological malignancies. Blood 1999, 93, 3587–3600. [Google Scholar]
- Baliga, B.C.; Kumar, S. Role of Bcl-2 family of proteins in malignancy. Hematol. Oncol. 2002, 20, 63–74. [Google Scholar] [CrossRef]
- Wei, G.; Margolin, A.A.; Haery, L.; Brown, E.; Cucolo, L.; Julian, B.; Shehata, S.; Kung, A.L.; Beroukhim, R.; Golub, T.R. Chemical genomics identifies small-molecule MCL1 repressors and BCL-xL as a predictor of MCL1 dependency. Cancer Cell 2012, 21, 547–562. [Google Scholar] [CrossRef]
- Baou, M.; Kohlhaas, S.L.; Butterworth, M.; Vogler, M.; Dinsdale, D.; Walewska, R.; Majid, A.; Eldering, E.; Dyer, M.J.; Cohen, G.M. Role of NOXA and its ubiquitination in proteasome inhibitor-induced apoptosis in chronic lymphocytic leukemia cells. Haematologica 2010, 95, 1510–1518. [Google Scholar] [CrossRef]
- Craxton, A.; Butterworth, M.; Harper, N.; Fairall, L.; Schwabe, J.; Ciechanover, A.; Cohen, G.M. NOXA, a sensor of proteasome integrity, is degraded by 26S proteasomes by an ubiquitin-independent pathway that is blocked by MCL-1. Cell Death Differ. 2012, 19, 1424–1434. [Google Scholar] [CrossRef]
- Senderowicz, A.M.; Headlee, D.; Stinson, S.F.; Lush, R.M.; Kalil, N.; Villalba, L.; Hill, K.; Steinberg, S.M.; Figg, W.D.; Tompkins, A.; et al. Phase I trial of continuous infusion flavopiridol, a novel cyclin-dependent kinase inhibitor, in patients with refractory neoplasms. J. Clin. Oncol. 1998, 16, 2986–2999. [Google Scholar]
- Mitsiades, C.S.; Mitsiades, N.S.; McMullan, C.J.; Poulaki, V.; Shringarpure, R.; Akiyama, M.; Hideshima, T.; Chauhan, D.; Joseph, M.; Libermann, T.A.; et al. Inhibition of the insulin-like growth factor receptor-1 tyrosine kinase activity as a therapeutic strategy for multiple myeloma, other hematologic malignancies, and solid tumors. Cancer Cell 2004, 5, 221–230. [Google Scholar] [CrossRef]
- Dalton, W.S.; Hazlehurst, L.; Shain, K.; Landowski, T.; Alsina, M. Targeting the bone marrow microenvironment in hematologic malignancies. Semin. Hematol. 2004, 41, 1–5. [Google Scholar]
- Logue, S.E.; Elgendy, M.; Martin, S.J. Expression, purification and use of recombinant annexin V for the detection of apoptotic cells. Nat. Protoc. 2009, 4, 1383–1395. [Google Scholar] [CrossRef]
Supplementary Materials


© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Natoni, A.; Coyne, M.R.E.; Jacobsen, A.; Rainey, M.D.; O'Brien, G.; Healy, S.; Montagnoli, A.; Moll, J.; O'Dwyer, M.; Santocanale, C. Characterization of a Dual CDC7/CDK9 Inhibitor in Multiple Myeloma Cellular Models. Cancers 2013, 5, 901-918. https://doi.org/10.3390/cancers5030901
Natoni A, Coyne MRE, Jacobsen A, Rainey MD, O'Brien G, Healy S, Montagnoli A, Moll J, O'Dwyer M, Santocanale C. Characterization of a Dual CDC7/CDK9 Inhibitor in Multiple Myeloma Cellular Models. Cancers. 2013; 5(3):901-918. https://doi.org/10.3390/cancers5030901
Chicago/Turabian StyleNatoni, Alessandro, Mark R. E. Coyne, Alan Jacobsen, Michael D. Rainey, Gemma O'Brien, Sandra Healy, Alessia Montagnoli, Jürgen Moll, Michael O'Dwyer, and Corrado Santocanale. 2013. "Characterization of a Dual CDC7/CDK9 Inhibitor in Multiple Myeloma Cellular Models" Cancers 5, no. 3: 901-918. https://doi.org/10.3390/cancers5030901
APA StyleNatoni, A., Coyne, M. R. E., Jacobsen, A., Rainey, M. D., O'Brien, G., Healy, S., Montagnoli, A., Moll, J., O'Dwyer, M., & Santocanale, C. (2013). Characterization of a Dual CDC7/CDK9 Inhibitor in Multiple Myeloma Cellular Models. Cancers, 5(3), 901-918. https://doi.org/10.3390/cancers5030901