The Role of PI3K/Akt/mTOR Signaling in Gastric Carcinoma

Abstract
:1. Introduction
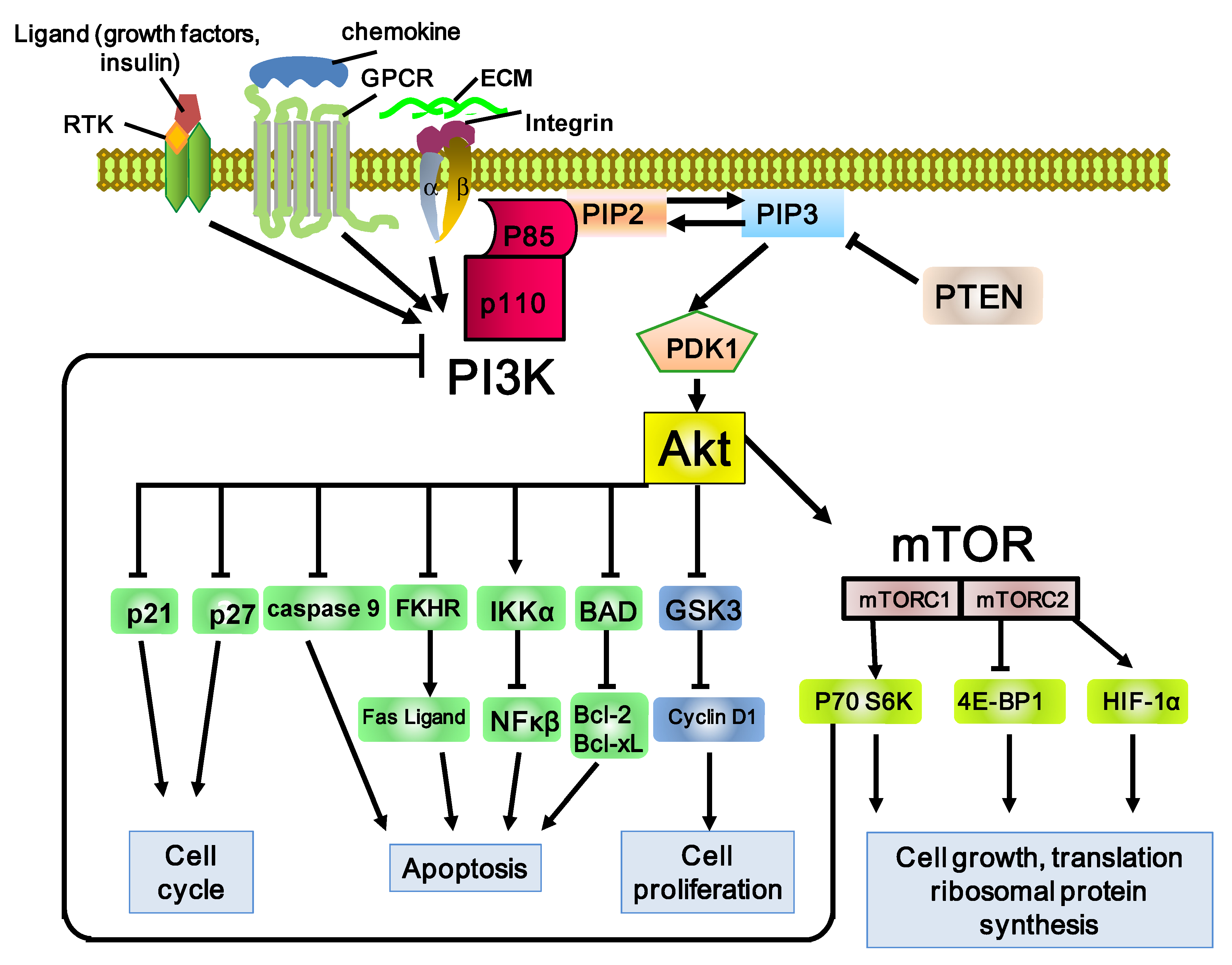
2. The PI3K/Akt/mTOR Pathway

3. Gene Mutation and Activation of PI3K/Akt/mTOR Pathway in Gastric Carcinoma

{kind=link}
{kind=link}
{kind=link}
| Study [Ref#] | PMID | Sample | Main Results |
|---|---|---|---|
| PI3K | |||
| Samuels et al. [11] | 15016963 | Tumor specimens | Mutations in PIK3CA were identified in 3 of 12 gastric cancers (25%). |
| Velho et al. [21] | 15994075 | Tumor specimens | PIK3CA mutations in exons 9 and 20 were present in 10.6% of gastric carcinomas. |
| Barbi et al. [23] | 20398348 | Tumor specimens | PIK3CA mutations were present in 16% of gastric carcinomas. No other association between PI3KCA mutations and their clinical pathological covariates was found. |
| Sukawa et al. [24] | 24458107 | Tumor specimens | The mutation incidence is high (21.4%) in T4 cancers and low (6.4%) in T2 cancers. |
| Corso et al. [25] | 20937558 | Tumor specimens | Mutations in PIK3CA gene occurred in 14.3% of the MSI gastric cancers. |
| Shi et al. [26] | 22292935 | Tumor specimens | PIK3CA mutations are rare, but their amplification is very common in gastric carcinoma. |
| Zhou et al. [9] | 22876838 | Cell lines/tumor specimens | PIK3R3 was significantly up-regulated in gastric cancer specimens, and 9.5% to 15% tumors showed more than 2 fold increase compare to the paired mucosa tissues. |
| Akt | |||
| Num et al. [27] | 14678019 | Tumor specimens | Akt expression was detected in 74% of the tumors and pAkt expression in 78%. |
| Cinti et al. [28] | 18841391 | Tumor specimens | There was a statistically significant correlation between pAkt expression and depth of infiltration of the tumor, number of infiltrated lymph nodes and p34/cdc2 expression. |
| Kobayashi et al. [29] | 16785763 | Cell lines/tumor specimens | pAkt expression was detected in 57% of the tumors, which was correlated with high clinicopathological parameters as well as a poor outcome. |
| Sukawa et al. [30] | 23236232 | Tumor specimens | pAkt expression was also significantly associated with HER2 overexpression but not with PIK3CA mutations |
| Staal et al. [31] | 3037531 | Cell lines/tumor specimens | A survey of 225 human tumors for changes involving AKT1 led to the discovery of a 20-fold amplification of this gene in one of the five gastric adenocarcinomas tested. |
| Cho et al. [32] | 20704706 | Cell lines/tumor specimens | High activity of GSK3β was found to be frequently present in early-stage gastric carcinoma and was positively associated with good prognosis. |
| PTEN | |||
| Wen et al. [33] | 20514448 | Tumor specimens | PTEN mutations were present in 55.6% of missense mutation, 33.3% of nonsense mutation, 7.4% of 1-bp deletion and 3.7% of a mutation within intron. |
| Mina et al. [34] | 22639407 | Tumor specimens | 4.4% of primary gastric cancer spots showed PTEN deletions. PTEN deletion was correlated with nodal and distant metastases. |
| Byun et al. [35] | 12569555 | Cell lines/tumor specimens | Frequent monoallelic deletions of PTEN phosphatase antagonism of PI3K/Akt in 47% of cases. |
| Kang et al. [36] | 11896207 | Tumor specimens | The promoter methylation frequency of PTEN was found to be present in 39% of cases examined, and 73% of gastric cancer tissues showing promoter methylation exhibited the loss of PTEN expression. |
| mTOR | |||
| Li et al. [37] | 23205120 | Tumor specimens | The expression levels of mTOR and PTEN were negatively correlated in the PI3K-AKT-mTOR signaling pathway. |
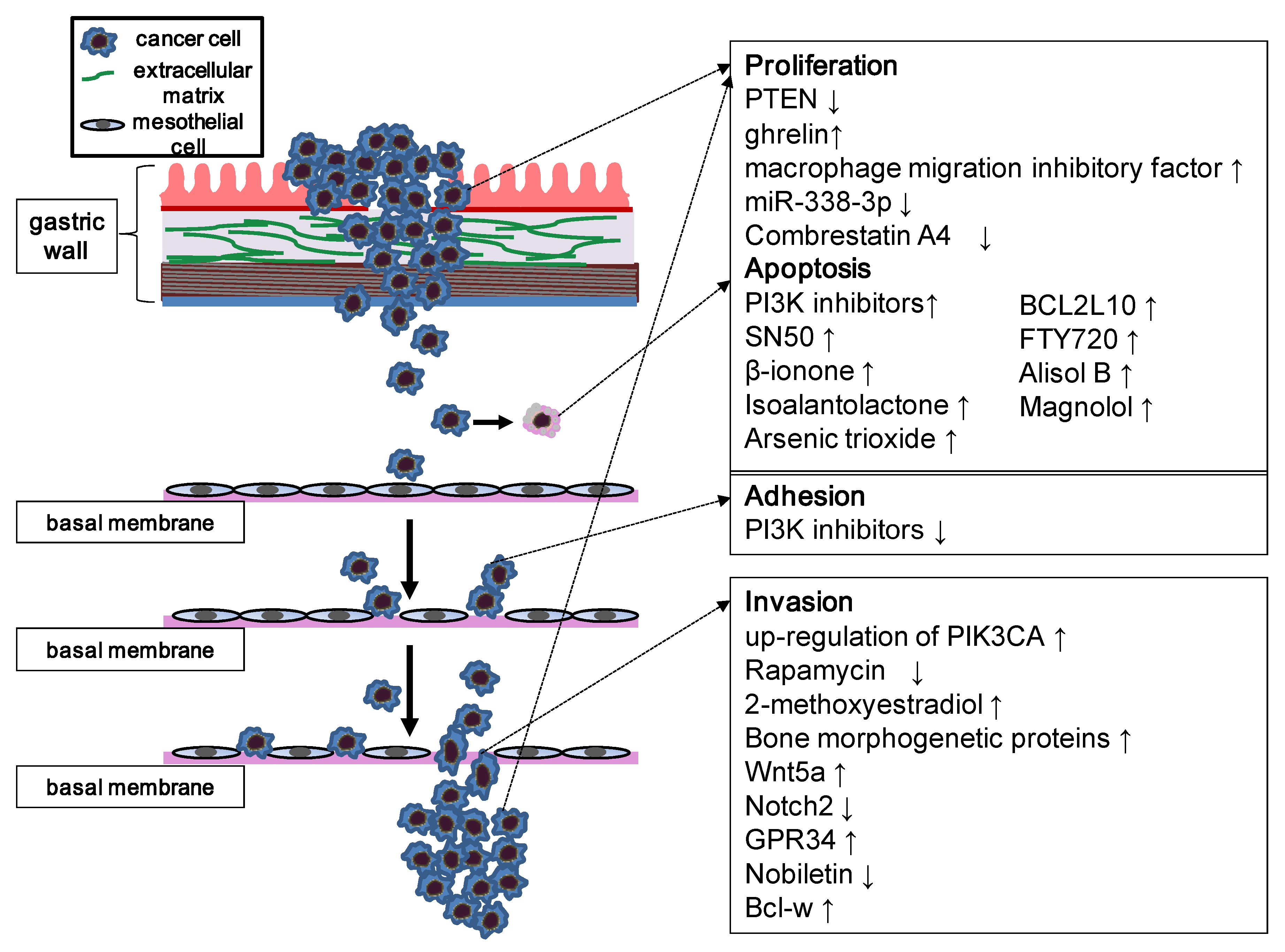
4. The Role of the PI3K/Akt/mTOR Pathway in the Biological Properties of Gastric Carcinoma
4.1. Apoptosis
4.2. Metastasis

5. The Role of the PI3K/Akt/mTOR Pathway in Resistance to Chemotherapy in Gastric Carcinoma
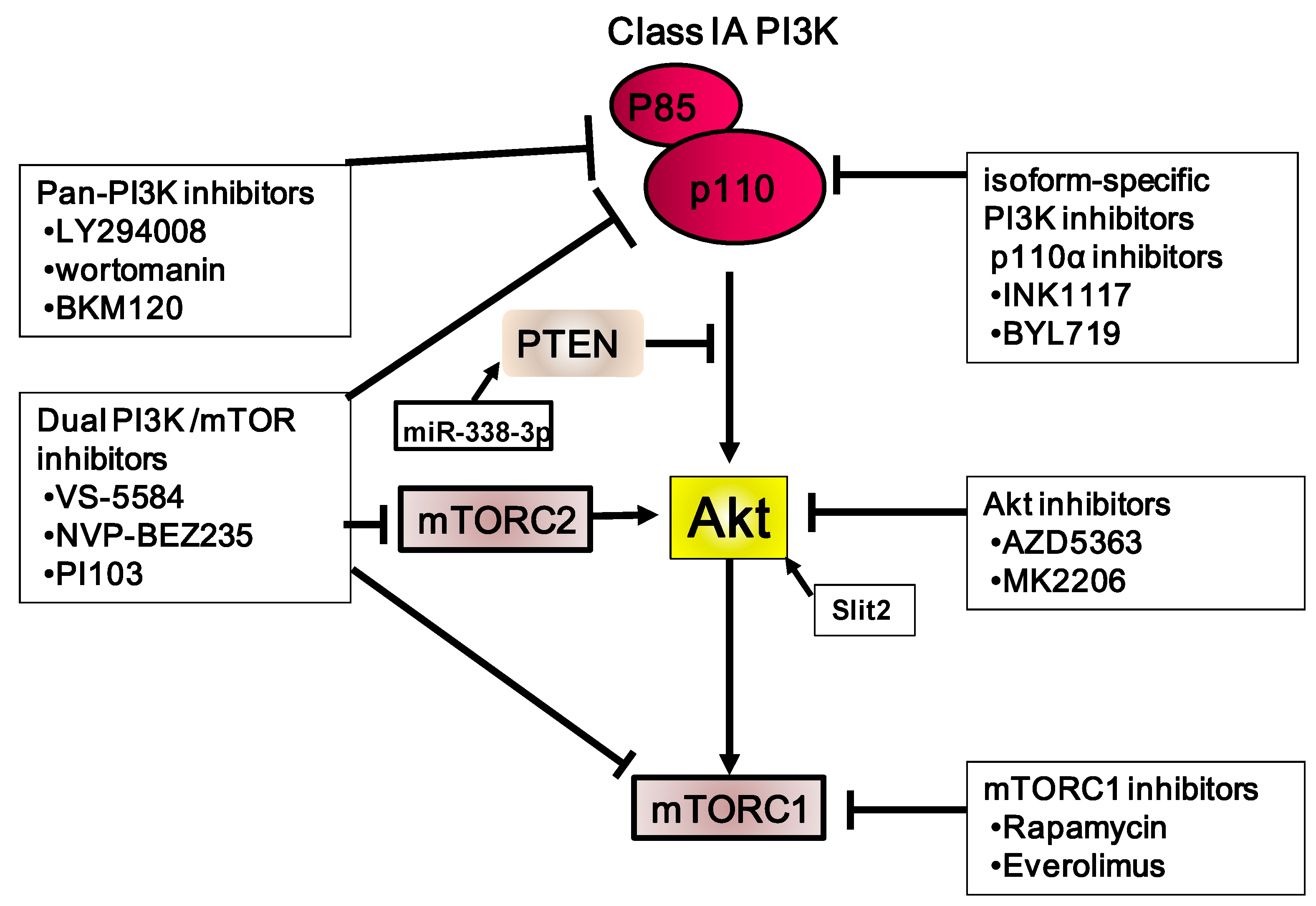
6. Targeting PI3K/Akt/mTOR as a Therapy for Gastric Carcinoma

6.1. PI3K Inhibitors
| Therapeutic Agent | Target | Clinical Trial | Efficacy | Year | Ref |
|---|---|---|---|---|---|
| Everolimus vs. placebo | mTORC1 | Phase III (GRANITE-1) | PFS 1.68 vs. 1.41, p < 0.0001
OS 1.68 vs. 1.41, p = 0.1244 | 2013 | [97] |
| Everolimus ± paclitaxel | mTORC1 | Phase III (AIO-STO-0111) | Enrolling | ||
| MK-2206 + Trastuzumab | Akt | Phase I | 1 of 4 patient archive SD | 2013 | [97] |
| BYL719 | p110α | Phase I | Enrolling | ||
| BKM120 | PI3K | Phase I | Enrolling |
6.2. Akt Inhibitors
6.3. mTOR Inhibitors
6.4. Dual mTORC1/2 Inhibitors
6.5. PI3K and mTOR Inhibitors
6.6. Other Therapeutic Approaches
7. Conclusions and Future Perspectives
Acknowledgments
Conflicts of Interest
References
- Guggenheim, D.E.; Shah, M.A. Gastric cancer epidemiology and risk factors. J. Surg. Oncol. 2013, 107, 230–236. [Google Scholar] [CrossRef]
- Bang, Y.J.; van Cutsem, E.; Feyereislova, A.; Chung, H.C.; Shen, L.; Sawaki, A.; Lordick, F.; Ohtsu, A.; Omuro, Y.; Satoh, T.; et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): A phase 3, open-label, randomised controlled trial. Lancet 2010, 376, 687–697. [Google Scholar] [CrossRef]
- Yashiro, M.; Shinto, O.; Nakamura, K.; Tendo, M.; Matsuoka, T.; Matsuzaki, T.; Kaizaki, R.; Miwa, A.; Hirakawa, K. Synergistic anti-tumor effects of FGFR2 inhibitor with 5-fluorouracil on scirrhous gastric carcinoma. Int. J. Cancer 2010, 126, 1004–1016. [Google Scholar]
- Nakamura, K.; Yashiro, M.; Matsuoka, T.; Tendo, M.; Shimizu, T.; Miwa, A.; Hirakawa, K. A novel molecular targeting compound as K-samII/FGF-R2 phosphorylation inhibitor, Ki23057, for Scirrhous gastric cancer. Gastroenterology 2006, 131, 1530–1541. [Google Scholar] [CrossRef]
- Deng, N.; Goh, L.K.; Wang, H.; Das, K.; Tao, J.; Tan, I.B.; Zhang, S.; Lee, M.; Wu, J.; Lim, K.H.; et al. A comprehensive survey of genomic alterations in gastric cancer reveals systematic patterns of molecular exclusivity and co-occurrence among distinct therapeutic targets. Gut 2012, 61, 673–684. [Google Scholar] [CrossRef]
- Wadhwa, R.; Song, S.; Lee, J.S.; Yao, Y.; Wei, Q.; Ajani, J.A. Gastric cancer-molecular and clinical dimensions. Nat. Rev. Clin. Oncol. 2013, 10, 643–655. [Google Scholar] [CrossRef]
- Willems, L.; Tamburini, J.; Chapuis, N.; Lacombe, C.; Mayeux, P.; Bouscary, D. PI3K and mTOR signaling pathways in cancer: New data on targeted therapies. Curr. Oncol. Rep. 2012, 14, 129–138. [Google Scholar] [CrossRef]
- Fresno Vara, J.A.; Casado, E.; de Castro, J.; Cejas, P.; Belda-Iniesta, C.; Gonzalez-Baron, M. PI3K/Akt signalling pathway and cancer. Cancer Treat. Rev. 2004, 30, 193–204. [Google Scholar] [CrossRef]
- Zhou, J.; Chen, G.B.; Tang, Y.C.; Sinha, R.A.; Wu, Y.; Yap, C.S.; Wang, G.; Hu, J.; Xia, X.; Tan, P.; et al. Genetic and bioinformatic analyses of the expression and function of PI3K regulatory subunit PIK3R3 in an Asian patient gastric cancer library. BMC Med. Genomics 2012, 5. [Google Scholar] [CrossRef]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef]
- Samuels, Y.; Wang, Z.; Bardelli, A.; Silliman, N.; Ptak, J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, S.M.; Riggins, G.J.; et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004, 304. [Google Scholar] [CrossRef]
- Siegfried, Z.; Bonomi, S.; Ghigna, C.; Karni, R. Regulation of the Ras-MAPK and PI3K-mTOR signalling pathways by alternative splicing in cancer. Int. J. Cell. Biol. 2013. [Google Scholar] [CrossRef]
- Staal, S.P.; Hartley, J.W.; Rowe, W.P. Isolation of transforming murine leukemia viruses from mice with a high incidence of spontaneous lymphoma. Proc. Natl. Acad. Sci. USA 1977, 74, 3065–3067. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef]
- Almhanna, K.; Strosberg, J.; Malafa, M. Targeting AKT protein kinase in gastric cancer. Anticancer Res. 2011, 31, 4387–4392. [Google Scholar]
- Ortega-Molina, A.; Serrano, M. PTEN in cancer, metabolism, and aging. Trends Endocrinol. Metab. 2013, 24, 184–189. [Google Scholar] [CrossRef]
- Dowling, R.J.; Topisirovic, I.; Fonseca, B.D.; Sonenberg, N. Dissecting the role of mTOR: Lessons from mTOR inhibitors. Biochim. Biophys. Acta 2010, 1804, 433–439. [Google Scholar] [CrossRef]
- Guertin, D.A.; Sabatini, D.M. Defining the role of mTOR in cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef]
- Brader, S.; Eccles, S.A. Phosphoinositide 3-kinase signalling pathways in tumor progression, invasion and angiogenesis. Tumori 2004, 90, 2–8. [Google Scholar]
- Zhou, H.; Huang, S. mTOR signaling in cancer cell motility and tumor metastasis. Crit. Rev. Eukaryot. Gene Expr. 2010, 20, 1–16. [Google Scholar] [CrossRef]
- Velho, S.; Oliveira, C.; Ferreira, A.; Ferreira, A.C.; Suriano, G.; Schwartz, S., Jr.; Duval, A.; Carneiro, F.; Machado, J.C.; Hamelin, R.; et al. The prevalence of PIK3CA mutations in gastric and colon cancer. Eur. J. Cancer 2005, 41, 1649–1654. [Google Scholar] [CrossRef]
- Garcia-Rostan, G.; Costa, A.M.; Pereira-Castro, I.; Salvatore, G.; Hernandez, R.; Hermsem, M.J.; Herrero, A.; Fusco, A.; Cameselle-Teijeiro, J.; Santoro, M. Mutation of the PIK3CA gene in anaplastic thyroid cancer. Cancer Res. 2005, 65, 10199–10207. [Google Scholar] [CrossRef]
- Barbi, S.; Cataldo, I.; de Manzoni, G.; Bersani, S.; Lamba, S.; Mattuzzi, S.; Bardelli, A.; Scarpa, A. The analysis of PIK3CA mutations in gastric carcinoma and metanalysis of literature suggest that exon-selectivity is a signature of cancer type. J. Exp. Clin. Cancer Res. 2010. [Google Scholar] [CrossRef]
- Sukawa, Y.; Yamamoto, H.; Nosho, K.; Ito, M.; Igarashi, H.; Naito, T.; Mitsuhashi, K.; Matsunaga, Y.; Takahashi, T.; Mikami, M.; et al. HER2 expression and PI3K-Akt pathway alterations in gastric cancer. Digestion 2014, 89, 12–17. [Google Scholar] [CrossRef]
- Corso, G.; Velho, S.; Paredes, J.; Pedrazzani, C.; Martins, D.; Milanezi, F.; Pascale, V.; Vindigni, C.; Pinheiro, H.; Leite, M.; et al. Oncogenic mutations in gastric cancer with microsatellite instability. Eur. J. Cancer 2011, 47, 443–451. [Google Scholar] [CrossRef]
- Shi, J.; Yao, D.; Liu, W.; Wang, N.; Lv, H.; Zhang, G.; Ji, M.; Xu, L.; He, N.; Shi, B.; et al. Highly frequent PIK3CA amplification is associated with poor prognosis in gastric cancer. BMC Cancer 2012, 12. [Google Scholar] [CrossRef]
- Nam, S.Y.; Lee, H.S.; Jung, G.A.; Choi, J.; Cho, S.J.; Kim, M.K.; Kim, W.H.; Lee, B.L. Akt/PKB activation in gastric carcinomas correlates with clinicopathologic variables and prognosis. Apmis 2003, 111, 1105–1113. [Google Scholar] [CrossRef]
- Cinti, C.; Vindigni, C.; Zamparelli, A.; La Sala, D.; Epistolato, M.C.; Marrelli, D.; Cevenini, G.; Tosi, P. Activated Akt as an indicator of prognosis in gastric cancer. Virchows Arch. 2008, 453, 449–455. [Google Scholar] [CrossRef]
- Kobayashi, I.; Semba, S.; Matsuda, Y.; Kuroda, Y.; Yokozaki, H. Significance of Akt phosphorylation on tumor growth and vascular endothelial growth factor expression in human gastric carcinoma. Pathobiology 2006, 73, 8–17. [Google Scholar] [CrossRef]
- Sukawa, Y.; Yamamoto, H.; Nosho, K.; Kunimoto, H.; Suzuki, H.; Adachi, Y.; Nakazawa, M.; Nobuoka, T.; Kawayama, M.; Mikami, M.; et al. Alterations in the human epidermal growth factor receptor 2-phosphatidylinositol 3-kinase-v-Akt pathway in gastric cancer. World J. Gastroenterol. 2012, 18, 6577–6586. [Google Scholar] [CrossRef]
- Staal, S.P. Molecular cloning of the akt oncogene and its human homologues AKT1 and AKT2: Amplification of AKT1 in a primary human gastric adenocarcinoma. Proc. Natl. Acad. Sci. USA 1987, 84, 5034–5037. [Google Scholar] [CrossRef]
- Cho, Y.J.; Kim, J.H.; Yoon, J.; Cho, S.J.; Ko, Y.S.; Park, J.W.; Lee, H.S.; Lee, H.E.; Kim, W.H.; Lee, B.L. Constitutive activation of glycogen synthase kinase-3beta correlates with better prognosis and cyclin-dependent kinase inhibitors in human gastric cancer. BMC Gastroenterol. 2010, 10. [Google Scholar] [CrossRef]
- Wen, Y.G.; Wang, Q.; Zhou, C.Z.; Qiu, G.Q.; Peng, Z.H.; Tang, H.M. Mutation analysis of tumor suppressor gene PTEN in patients with gastric carcinomas and its impact on PI3K/AKT pathway. Oncol. Rep. 2010, 24, 89–95. [Google Scholar]
- Mina, S.; Bohn, B.A.; Simon, R.; Krohn, A.; Reeh, M.; Arnold, D.; Bokemeyer, C.; Sauter, G.; Izbicki, J.R.; Marx, A.; et al. PTEN deletion is rare but often homogeneous in gastric cancer. J. Clin. Pathol. 2011, 65, 693–698. [Google Scholar]
- Byun, D.S.; Cho, K.; Ryu, B.K.; Lee, M.G.; Park, J.I.; Chae, K.S.; Kim, H.J.; Chi, S.G. Frequent monoallelic deletion of PTEN and its reciprocal associatioin with PIK3CA amplification in gastric carcinoma. Int. J. Cancer 2003, 104, 318–327. [Google Scholar] [CrossRef]
- Kang, Y.H.; Lee, H.S.; Kim, W.H. Promoter methylation and silencing of PTEN in gastric carcinoma. Lab. Investig. 2002, 82, 285–291. [Google Scholar] [CrossRef]
- Li, M.; Sun, H.; Song, L.; Gao, X.; Chang, W.; Qin, X. Immunohistochemical expression of mTOR negatively correlates with PTEN expression in gastric carcinoma. Oncol. Lett. 2012, 4, 1213–1218. [Google Scholar]
- Vasudevan, K.M.; Barbie, D.A.; Davies, M.A.; Rabinovsky, R.; McNear, C.J.; Kim, J.J.; Hennessy, B.T.; Tseng, H.; Pochanard, P.; Kim, S.Y.; et al. AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell 2009, 16, 21–32. [Google Scholar] [CrossRef]
- Marone, R.; Cmiljanovic, V.; Giese, B.; Wymann, M.P. Targeting phosphoinositide 3-kinase: Moving towards therapy. Biochim. Biophys. Acta 2008, 1784, 159–185. [Google Scholar] [CrossRef]
- Xing, C.G.; Zhu, B.S.; Liu, H.H.; Lin, F.; Yao, H.H.; Liang, Z.Q.; Qin, Z.H. LY294002 induces p53-dependent apoptosis of SGC7901 gastric cancer cells. Acta Pharmacol. Sin. 2008, 29, 489–498. [Google Scholar] [CrossRef]
- Xing, C.G.; Zhu, B.S.; Fan, X.Q.; Liu, H.H.; Hou, X.; Zhao, K.; Qin, Z.H. Effects of LY294002 on the invasiveness of human gastric cancer in vivo in nude mice. World J. Gastroenterol. 2009, 15, 5044–5052. [Google Scholar] [CrossRef]
- Chao, X.; Zao, J.; Xiao-Yi, G.; Li-Jun, M.; Tao, S. Blocking of PI3K/AKT induces apoptosis by its effect on NF-kappaB activity in gastric carcinoma cell line SGC7901. Biomed. Pharmacother. 2010, 64, 600–604. [Google Scholar] [CrossRef]
- Zhao, K.; Zhu, B.S.; Gong, W.; Zhu, M.L.; Gao, Z.T.; Wu, Y.Y.; Chen, Q.; Yang, X.D.; Xing, C.G. SN50 enhances the effects of LY294002 on cell death induction in gastric cancer cell line SGC7901. Arch. Med. Sci. 2011, 9, 990–998. [Google Scholar]
- Liu, Q.; Dong, H.W.; Sun, W.G.; Liu, M.; Ibla, J.C.; Liu, L.X.; Parry, J.W.; Han, X.H.; Li, M.S.; Liu, J.R. Apoptosis initiation of beta-ionone in SGC-7901 gastric carcinoma cancer cells via a PI3K-AKT pathway. Arch. Toxicol. 2013, 87, 481–490. [Google Scholar] [CrossRef]
- Gao, Y.H.; Zhang, H.P.; Yang, S.M.; Yang, Y.; Ma, Y.Y.; Zhang, X.Y.; Yang, Y.M. Inactivation of Akt by arsenic trioxide induces cell death via mitochondrial-mediated apoptotic signaling in SGC-7901 human gastric cancer cells. Oncol. Rep. 2014, 31, 1645–1652. [Google Scholar]
- Xu, J.D.; Cao, X.X.; Long, Z.W.; Liu, X.P.; Furuya, T.; Xu, J.W.; Liu, X.L.; de Xu, Z.; Sasaki, K.; Li, Q.Q. BCL2L10 protein regulates apoptosis/proliferation through differential pathways in gastric cancer cells. J. Pathol. 2011, 223, 400–409. [Google Scholar] [CrossRef]
- Zheng, T.; Meng, X.; Wang, J.; Chen, X.; Yin, D.; Liang, Y.; Song, X.; Pan, S.; Jiang, H.; Liu, L. PTEN- and p53-mediated apoptosis and cell cycle arrest by FTY720 in gastric cancer cells and nude mice. J. Cell. Biochem. 2010, 111, 218–228. [Google Scholar] [CrossRef]
- Xu, Y.H.; Zhao, L.J.; Li, Y. Alisol B acetate induces apoptosis of SGC7901 cells via mitochondrial and phosphatidylinositol 3-kinases/Akt signaling pathways. World J. Gastroenterol. 2009, 15, 2870–2877. [Google Scholar] [CrossRef]
- Rasul, A.; Khan, M.; Yu, B.; Ali, M.; Bo, Y.J.; Yang, H.; Ma, T. Isoalantolactone, a sesquiterpene lactone, induces apoptosis in SGC-7901 cells via mitochondrial and phosphatidylinositol 3-kinase/Akt signaling pathways. Arch. Pharm. Res. 2013, 36, 1262–1269. [Google Scholar]
- Xu, W.T.; Yang, Z.; Lu, N.H. Roles of PTEN (phosphatase and tensin homolog) in gastric cancer development and progression. Asian Pac. J. Cancer Prev. 2014, 15, 17–24. [Google Scholar] [CrossRef]
- Liu, J.F.; Zhou, X.K.; Chen, J.H.; Yi, G.; Chen, H.G.; Ba, M.C.; Lin, S.Q.; Qi, Y.C. Up-regulation of PIK3CA promotes metastasis in gastric carcinoma. World J. Gastroenterol. 2010, 16, 4986–4991. [Google Scholar] [CrossRef]
- Zhou, X.D.; Chen, H.X.; Guan, R.N.; Lei, Y.P.; Shu, X.; Zhu, Y.; Lv, N.H. Protein kinase B phosphorylation correlates with vascular endothelial growth factor A and microvessel density in gastric adenocarcinoma. J. Int. Med. Res. 2012, 40, 2124–2134. [Google Scholar] [CrossRef]
- Han, Z.; Wu, K.; Shen, H.; Li, C.; Han, S.; Hong, L.; Shi, Y.; Liu, N.; Guo, C.; Xue, Y.; et al. Akt1/protein kinase B alpha is involved in gastric cancer progression and cell proliferation. Dig. Dis. Sci. 2008, 53, 1801–1810. [Google Scholar] [CrossRef]
- Ye, B.; Jiang, L.L.; Xu, H.T.; Zhou, D.W.; Li, Z.S. Expression of PI3K/AKT pathway in gastric cancer and its blockade suppresses tumor growth and metastasis. Int. J. Immunopathol. Pharmacol. 2012, 25, 627–636. [Google Scholar]
- Lin, H.L.; Chiou, S.H.; Wu, C.W.; Lin, W.B.; Chen, L.H.; Yang, Y.P.; Tsai, M.L.; Uen, Y.H.; Liou, J.P.; Chi, C.W. Combretastatin A4-induced differential cytotoxicity and reduced metastatic ability by inhibition of AKT function in human gastric cancer cells. J. Pharmacol. Exp. Ther. 2007, 323, 365–373. [Google Scholar] [CrossRef]
- Matsuoka, T.; Yashiro, M.; Nishioka, N.; Hirakawa, K.; Olden, K.; Roberts, J.D. PI3K/Akt signalling is required for the attachment and spreading, and growth in vivo of metastatic scirrhous gastric carcinoma. Br. J. Cancer 2012, 106, 1535–1542. [Google Scholar] [CrossRef]
- Zhou, X.K.; Tang, S.S.; Yi, G.; Hou, M.; Chen, J.H.; Yang, B.; Liu, J.F.; He, Z.M. RNAi knockdown of PIK3CA preferentially inhibits invasion of mutant PIK3CA cells. World J. Gastroenterol. 2011, 17, 3700–3708. [Google Scholar] [CrossRef]
- Xue, Y.; Li, N.L.; Yang, J.Y.; Chen, Y.; Yang, L.L.; Liu, W.C. Phosphatidylinositol 3'-kinase signaling pathway is essential for Rac1-induced hypoxia-inducible factor-1(alpha) and vascular endothelial growth factor expression. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H2169–H2176. [Google Scholar] [CrossRef]
- Zhang, L.L.; Liu, J.; Lei, S.; Zhang, J.; Zhou, W.; Yu, H.G. PTEN inhibits the invasion and metastasis of gastric cancer via downregulation of FAK expression. Cell. Signal. 2014, 26, 1011–1020. [Google Scholar] [CrossRef]
- Chen, G.; Chen, S.M.; Wang, X.; Ding, X.F.; Ding, J.; Meng, L.H. Inhibition of chemokine (CXC motif) ligand 12/chemokine (CXC motif) receptor 4 axis (CXCL12/CXCR4)-mediated cell migration by targeting mammalian target of rapamycin (mTOR) pathway in human gastric carcinoma cells. J. Biol. Chem. 2012, 287, 12132–12141. [Google Scholar]
- Yu, W.; Ma, S.; Wang, L.; Zuo, B.; Li, M.; Qiao, Z.; Pan, X.; Liu, Y.; Wang, J. Upregulation of GPR34 expression affects the progression and prognosis of human gastric adenocarcinoma by PI3K/PDK1/AKT pathway. Histol. Histopathol. 2013, 28, 1629–1638. [Google Scholar]
- Tian, P.Y.; Fan, X.M. The proliferative effects of ghrelin on human gastric cancer AGS cells. J. Dig. Dis. 2012, 13, 453–458. [Google Scholar] [CrossRef]
- Lee, Y.C.; Cheng, T.H.; Lee, J.S.; Chen, J.H.; Liao, Y.C.; Fong, Y.; Wu, C.H.; Shih, Y.W. Nobiletin, a citrus flavonoid, suppresses invasion and migration involving FAK/PI3K/Akt and small GTPase signals in human gastric adenocarcinoma AGS cells. Mol. Cell. Biochem. 2011, 347, 103–115. [Google Scholar] [CrossRef]
- Li, G.Q.; Xie, J.; Lei, X.Y.; Zhang, L. Macrophage migration inhibitory factor regulates proliferation of gastric cancer cells via the PI3K/Akt pathway. World J. Gastroenterol. 2009, 15, 5541–5548. [Google Scholar] [CrossRef]
- Bae, I.H.; Park, M.J.; Yoon, S.H.; Kang, S.W.; Lee, S.S.; Choi, K.M.; Um, H.D. Bcl-w promotes gastric cancer cell invasion by inducing matrix metalloproteinase-2 expression via phosphoinositide 3-kinase, Akt, and Sp1. Cancer Res. 2006, 66, 4991–4995. [Google Scholar] [CrossRef]
- Lin, H.L.; Yang, M.H.; Wu, C.W.; Chen, P.M.; Yang, Y.P.; Chu, Y.R.; Kao, C.L.; Ku, H.H.; Lo, J.F.; Liou, J.P.; et al. 2-Methoxyestradiol attenuates phosphatidylinositol 3-kinase/Akt pathway-mediated metastasis of gastric cancer. Int. J. Cancer 2007, 121, 2547–2555. [Google Scholar] [CrossRef]
- Kang, M.H.; Oh, S.C.; Lee, H.J.; Kang, H.N.; Kim, J.L.; Kim, J.S.; Yoo, Y.A. Metastatic function of BMP-2 in gastric cancer cells: The role of PI3K/AKT, MAPK, the NF-kappaB pathway, and MMP-9 expression. Exp. Cell Res. 2011, 317, 1746–1762. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, Y.; Xu, R.; Du, J.; Hu, Z.; Yang, L.; Chen, Y.; Zhu, Y.; Gu, L. PI3K/Akt-dependent phosphorylation of GSK3beta and activation of RhoA regulate Wnt5a-induced gastric cancer cell migration. Cell. Signal. 2013, 25, 447–456. [Google Scholar] [CrossRef]
- Guo, L.Y.; Li, Y.M.; Qiao, L.; Liu, T.; Du, Y.Y.; Zhang, J.Q.; He, W.T.; Zhao, Y.X.; He, D.Q. Notch2 regulates matrix metallopeptidase 9 via PI3K/AKT signaling in human gastric carcinoma cell MKN-45. World J. Gastroenterol. 2012, 18, 7262–7270. [Google Scholar] [CrossRef]
- Guo, B.; Liu, L.; Yao, J.; Ma, R.; Chang, D.; Li, Z.; Song, T.; Huang, C. miR-338–3p suppresses progression of gastric cancer through PTEN-AKT signaling pathways by targeting P-REX2a. Mol. Cancer Res. 2014, 12, 313–321. [Google Scholar] [CrossRef]
- Fodale, V.; Pierobon, M.; Liotta, L.; Petricoin, E. Mechanism of cell adaptation: When and how do cancer cells develop chemoresistance? Cancer J. 2011, 17, 89–95. [Google Scholar] [CrossRef]
- Ang, K.L.; Shi, D.L.; Keong, W.W.; Epstein, R.J. Upregulated Akt signaling adjacent to gastric cancers: Implications for screening and chemoprevention. Cancer Lett. 2005, 225, 53–59. [Google Scholar] [CrossRef]
- Oki, E.; Baba, H.; Tokunaga, E.; Nakamura, T.; Ueda, N.; Futatsugi, M.; Mashino, K.; Yamamoto, M.; Ikebe, M.; Kakeji, Y.; et al. Akt phosphorylation associates with LOH of PTEN and leads to chemoresistance for gastric cancer. Int. J. Cancer 2005, 117, 376–380. [Google Scholar] [CrossRef]
- Osaki, M.; Kase, S.; Adachi, K.; Takeda, A.; Hashimoto, K.; Ito, H. Inhibition of the PI3K-Akt signaling pathway enhances the sensitivity of Fas-mediated apoptosis in human gastric carcinoma cell line, MKN-45. J. Cancer Res. Clin. Oncol. 2004, 130, 8–14. [Google Scholar] [CrossRef]
- Xie, X.; Tang, B.; Zhou, J.; Gao, Q.; Zhang, P. Inhibition of the PI3K/Akt pathway increases the chemosensitivity of gastric cancer to vincristine. Oncol. Rep. 2013, 30, 773–782. [Google Scholar]
- Zhang, C.; Awasthi, N.; Schwarz, M.A.; Schwarz, R.E. The dual PI3K/mTOR inhibitor NVP-BEZ235 enhances nab-paclitaxel antitumor response in experimental gastric cancer. Int. J. Oncol. 2013, 43, 1627–1635. [Google Scholar]
- Yu, H.G.; Ai, Y.W.; Yu, L.L.; Zhou, X.D.; Liu, J.; Li, J.H.; Xu, X.M.; Liu, S.; Chen, J.; Liu, F.; et al. Phosphoinositide 3-kinase/Akt pathway plays an important role in chemoresistance of gastric cancer cells against etoposide and doxorubicin induced cell death. Int. J. Cancer 2008, 122, 433–443. [Google Scholar] [CrossRef]
- Yu, L.L.; Dai, N.; Yu, H.G.; Sun, L.M.; Si, J.M. Akt associates with nuclear factor kappaB and plays an important role in chemoresistance of gastric cancer cells. Oncol. Rep. 2012, 24, 113–119. [Google Scholar]
- Liang, J.; Ge, F.; Guo, C.; Luo, G.; Wang, X.; Han, G.; Zhang, D.; Wang, J.; Li, K.; Pan, Y.; et al. Inhibition of PI3K/Akt partially leads to the inhibition of PrP(C)-induced drug resistance in gastric cancer cells. FEBS J. 2009, 276, 685–694. [Google Scholar] [CrossRef]
- Han, Z.; Hong, L.; Han, Y.; Wu, K.; Han, S.; Shen, H.; Li, C.; Yao, L.; Qiao, T.; Fan, D. Phospho Akt mediates multidrug resistance of gastric cancer cells through regulation of P-gp, Bcl-2 and Bax. J. Exp. Clin. Cancer Res. 2007, 26, 261–268. [Google Scholar]
- Zhou, W.; Fu, X.Q.; Liu, J.; Yu, H.G. RNAi knockdown of the Akt1 gene increases the chemosensitivity of gastric cancer cells to cisplatin both in vitro and in vivo. Regul. Pept. 2012, 176, 13–21. [Google Scholar] [CrossRef]
- Steelman, L.S.; Navolanic, P.M.; Sokolosky, M.L.; Taylor, J.R.; Lehmann, B.D.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Stadelman, K.M.; Terrian, D.M.; et al. Suppression of PTEN function increases breast cancer chemotherapeutic drug resistance while conferring sensitivity to mTOR inhibitors. Oncogene 2008, 27, 4086–4095. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Montalto, G.; Cervello, M.; Nicoletti, F.; Fagone, P.; Malaponte, G.; Mazzarino, M.C.; et al. Mutations and deregulation of Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascades which alter therapy response. Oncotarget 2012, 3, 954–987. [Google Scholar]
- Tomioka, H.; Mukohara, T.; Kataoka, Y.; Ekyalongo, R.C.; Funakoshi, Y.; Imai, Y.; Kiyota, N.; Fujiwara, Y.; Minami, H. Inhibition of the mTOR/S6K signal is necessary to enhance fluorouracil-induced apoptosis in gastric cancer cells with HER2 amplification. Int. J. Oncol. 2012, 41, 551–558. [Google Scholar]
- Yang, S.M.; Huang, C.; Li, X.F.; Yu, M.Z.; He, Y.; Li, J. miR-21 confers cisplatin resistance in gastric cancer cells by regulating PTEN. Toxicology 2013, 306, 162–168. [Google Scholar] [CrossRef]
- Wang, F.; Li, T.; Zhang, B.; Li, H.; Wu, Q.; Yang, L.; Nie, Y.; Wu, K.; Shi, Y.; Fan, D. MicroRNA-19a/b regulates multidrug resistance in human gastric cancer cells by targeting PTEN. Biochem. Biophys. Res. Commun. 2013, 434, 688–694. [Google Scholar]
- Fu, X.; Feng, J.; Zeng, D.; Ding, Y.; Yu, C.; Yang, B. PAK4 confers cisplatin resistance in gastric cancer cells via PI3K/Akt- and MEK/Erk-dependent pathways. Biosci. Rep. 2014. [Google Scholar] [CrossRef]
- Park, J.; Ko, Y.S.; Yoon, J.; Kim, M.A.; Park, J.W.; Kim, W.H.; Choi, Y.; Kim, J.H.; Cheon, Y.; Lee, B.L. The forkhead transcription factor FOXO1 mediates cisplatin resistance in gastric cancer cells by activating phosphoinositide 3-kinase/Akt pathway. Gastric Cancer 2013. [Google Scholar] [CrossRef]
- Mao, Z.; Zhou, J.; Luan, J.; Sheng, W.; Shen, X.; Dong, X. Tamoxifen reduces P-gp-mediated multidrug resistance via inhibiting the PI3K/Akt signaling pathway in ER-negative human gastric cancer cells. Biomed. Pharmacother. 2014, 68, 179–183. [Google Scholar]
- Liu, J.; Qu, X.; Xu, L.; Zhang, Y.; Qu, J.; Hou, K.; Liu, Y. Phosphoinositide 3-kinase/Akt and nuclear factor kappaB pathways are involved in tumor necrosis factor-related apoptosis-inducing ligand resistance in human gastric cancer cells. Mol. Med. Rep. 2010, 3, 491–496. [Google Scholar]
- Pal, I.; Mandal, M. PI3K and Akt as molecular targets for cancer therapy: Current clinical outcomes. Acta Pharmacol. Sin. 2012, 33, 1441–1458. [Google Scholar] [CrossRef]
- Vlahos, C.J.; Matter, W.F.; Hui, K.Y.; Brown, R.F. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J. Biol. Chem. 1994, 269, 5241–5248. [Google Scholar]
- Powis, G.; Bonjouklian, R.; Berggren, M.M.; Gallegos, A.; Abraham, R.; Ashendel, C.; Zalkow, L.; Matter, W.F.; Dodge, J.; Grindey, G.; et al. Wortmannin, a potent and selective inhibitor of phosphatidylinositol-3-kinase. Cancer Res. 1994, 54, 2419–2423. [Google Scholar]
- Park, E.; Park, J.; Han, S.W.; Im, S.A.; Kim, T.Y.; Oh, D.Y.; Bang, Y.J. NVP-BKM120, a novel PI3K inhibitor, shows synergism with a STAT3 inhibitor in human gastric cancer cells harboring KRAS mutations. Int. J. Oncol. 2012, 40, 1259–1266. [Google Scholar]
- Garrett, J.T.; Sutton, C.R.; Kurupi, R.; Bialucha, C.U.; Ettenberg, S.A.; Collins, S.D.; Sheng, Q.; Wallweber, J.; Defazio-Eli, L.; Arteaga, C.L. Combination of antibody that inhibits ligand-independent HER3 dimerization and a p110alpha inhibitor potently blocks PI3K signaling and growth of HER2+ breast cancers. Cancer Res. 2013, 73, 6013–6023. [Google Scholar] [CrossRef]
- Ohtsu, A.; Ajani, J.A.; Bai, Y.X.; Bang, Y.J.; Chung, H.C.; Pan, H.M.; Sahmoud, T.; Shen, L.; Yeh, K.H.; Chin, K.; et al. Everolimus for previously treated advanced gastric cancer: Results of the randomized, double-blind, phase III GRANITE-1 study. J. Clin. Oncol. 2013, 31, 3935–3943. [Google Scholar]
- Hudis, C.; Swanton, C.; Janjigian, Y.Y.; Lee, R.; Sutherland, S.; Lehman, R.; Chandarlapaty, S.; Hamilton, N.; Gajria, D.; Knowles, J.; et al. A phase 1 study evaluating the combination of an allosteric AKT inhibitor (MK-2206) and trastuzumab in patients with HER2-positive solid tumors. Breast Cancer Res. 2013, 15. [Google Scholar] [CrossRef]
- Li, J.; Davies, B.R.; Han, S.; Zhou, M.; Bai, Y.; Zhang, J.; Xu, Y.; Tang, L.; Wang, H.; Liu, Y.J.; et al. The AKT inhibitor AZD5363 is selectively active in PI3KCA mutant gastric cancer, and sensitizes a patient-derived gastric cancer xenograft model with PTEN loss to Taxotere. J. Transl. Med. 2013, 11. [Google Scholar] [CrossRef]
- 99 Rhodes, N.; Heerding, D.A.; Duckett, D.R.; Eberwein, D.J.; Knick, V.B.; Lansing, T.J.; McConnell, R.T.; Gilmer, T.M.; Zhang, S.Y.; Robell, K.; et al. Characterization of an Akt kinase inhibitor with potent pharmacodynamic and antitumor activity. Cancer Res. 2008, 68, 2366–2374. [Google Scholar] [CrossRef]
- Matsuzaki, T.; Yashiro, M.; Kaizaki, R.; Yasuda, K.; Doi, Y.; Sawada, T.; Ohira, M.; Hirakawa, K. Synergistic antiproliferative effect of mTOR inhibitors in combination with 5-fluorouracil in scirrhous gastric cancer. Cancer Sci. 2009, 100, 2402–2410. [Google Scholar] [CrossRef]
- Doi, T.; Muro, K.; Boku, N.; Yamada, Y.; Nishina, T.; Takiuchi, H.; Komatsu, Y.; Hamamoto, Y.; Ohno, N.; Fujita, Y.; et al. Multicenter phase II study of everolimus in patients with previously treated metastatic gastric cancer. J. Clin. Oncol. 2010, 28, 1904–1910. [Google Scholar] [CrossRef]
- Ji, D.; Zhang, Z.; Cheng, L.; Chang, J.; Wang, S.; Zheng, B.; Zheng, R.; Sun, Z.; Wang, C.; Zhang, Z.; et al. The combination of RAD001 and MK-2206 exerts synergistic cytotoxic effects against PTEN mutant gastric cancer cells: Involvement of MAPK-dependent autophagic, but not apoptotic cell death pathway. PLoS One 2014, 9, e85116. [Google Scholar]
- O’Reilly, K.E.; Rojo, F.; She, Q.B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006, 66, 1500–1508. [Google Scholar] [CrossRef]
- Lang, S.A.; Hackl, C.; Moser, C.; Fichtner-Feigl, S.; Koehl, G.E.; Schlitt, H.J.; Geissler, E.K.; Stoeltzing, O. Implication of RICTOR in the mTOR inhibitor-mediated induction of insulin-like growth factor-I receptor (IGF-IR) and human epidermal growth factor receptor-2 (Her2) expression in gastrointestinal cancer cells. Biochim. Biophys. Acta 2010, 1803, 435–442. [Google Scholar] [CrossRef]
- Hart, S.; Novotny-Diermayr, V.; Goh, K.C.; Williams, M.; Tan, Y.C.; Ong, L.C.; Cheong, A.; Ng, B.K.; Amalini, C.; Madan, B.; et al. VS-5584, a novel and highly selective PI3K/mTOR kinase inhibitor for the treatment of cancer. Mol. Cancer Ther. 2013, 12, 151–161. [Google Scholar]
- Bhattacharya, B.; Akram, M.; Balasubramanian, I.; Tam, K.K.; Koh, K.X.; Yee, M.Q.; Soong, R. Pharmacologic synergy between dual phosphoinositide-3-kinase and mammalian target of rapamycin inhibition and 5-fluorouracil in PIK3CA mutant gastric cancer cells. Cancer Biol. Ther. 2012, 13, 34–42. [Google Scholar] [CrossRef]
- Mueller, A.; Bachmann, E.; Linnig, M.; Khillimberger, K.; Schimanski, C.C.; Galle, P.R.; Moehler, M. Selective PI3K inhibition by BKM120 and BEZ235 alone or in combination with chemotherapy in wild-type and mutated human gastrointestinal cancer cell lines. Cancer Chemother. Pharmacol. 2012, 69, 1601–1615. [Google Scholar] [CrossRef]
- Engelman, J.A.; Chen, L.; Tan, X.; Crosby, K.; Guimaraes, A.R.; Upadhyay, R.; Maira, M.; McNamara, K.; Perera, S.A.; Song, Y.; et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat. Med. 2008, 14, 1351–1356. [Google Scholar] [CrossRef]
- Fu, Y.; Zhang, Q.; Kang, C.; Zhang, J.; Zhang, K.; Pu, P.; Wang, G.; Wang, T. Inhibitory effects of adenovirus mediated Akt1 and PIK3R1 shRNA on the growth of malignant tumor cells in vitro and in vivo. Cancer Biol. Ther. 2009, 8, 1002–1009. [Google Scholar] [CrossRef]
- Zhu, B.S.; Yu, L.Y.; Zhao, K.; Wu, Y.Y.; Cheng, X.L.; Wu, Y.; Zhong, F.Y.; Gong, W.; Chen, Q.; Xing, C.G. Effects of small interfering RNA inhibit Class I phosphoinositide 3-kinase on human gastric cancer cells. World J. Gastroenterol. 2013, 19, 1760–1769. [Google Scholar] [CrossRef]
- Yashiro, M.; Nishii, T.; Hasegawa, T.; Matsuzaki, T.; Morisaki, T.; Fukuoka, T.; Hirakawa, K. A c-Met inhibitor increases the chemosensitivity of cancer stem cells to the irinotecan in gastric carcinoma. Br. J. Cancer 2013, 109, 2619–2628. [Google Scholar] [CrossRef]
- Christensen, J.G.; Schreck, R.; Burrows, J.; Kuruganti, P.; Chan, E.; Le, P.; Chen, J.; Wang, X.; Ruslim, L.; Blake, R.; et al. A selective small molecule inhibitor of c-Met kinase inhibits c-Met-dependent phenotypes in vitro and exhibits cytoreductive antitumor activity in vivo. Cancer Res. 2003, 63, 7345–7355. [Google Scholar]
- Shinomiya, N.; Gao, C.F.; Xie, Q.; Gustafson, M.; Waters, D.J.; Zhang, Y.W.; Vande Woude, G.F. RNA interference reveals that ligand-independent met activity is required for tumor cell signaling and survival. Cancer Res. 2004, 64, 7962–7970. [Google Scholar] [CrossRef]
- Hong, S.W.; Jung, K.H.; Park, B.H.; Zheng, H.M.; Lee, H.S.; Choi, M.J.; Yun, J.I.; Kang, N.S.; Lee, J.; Hong, S.S. KRC-408, a novel c-Met inhibitor, suppresses cell proliferation and angiogenesis of gastric cancer. Cancer Lett. 2013, 332, 74–82. [Google Scholar] [CrossRef]
- Lu, Y.; Muller, M.; Smith, D.; Dutta, B.; Komurov, K.; Iadevaia, S.; Ruths, D.; Tseng, J.T.; Yu, S.; Yu, Q.; et al. Kinome siRNA-phosphoproteomic screen identifies networks regulating AKT signaling. Oncogene 2011, 30, 4567–4577. [Google Scholar] [CrossRef]
- Wong, H.; Yau, T. Targeted therapy in the management osf advanced gastric cancer: Are we making progress in the era of personalized medicine? Oncologist 2012, 17, 346–358. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Matsuoka, T.; Yashiro, M. The Role of PI3K/Akt/mTOR Signaling in Gastric Carcinoma. Cancers 2014, 6, 1441-1463. https://doi.org/10.3390/cancers6031441
Matsuoka T, Yashiro M. The Role of PI3K/Akt/mTOR Signaling in Gastric Carcinoma. Cancers. 2014; 6(3):1441-1463. https://doi.org/10.3390/cancers6031441
Chicago/Turabian StyleMatsuoka, Tasuku, and Masakazu Yashiro. 2014. "The Role of PI3K/Akt/mTOR Signaling in Gastric Carcinoma" Cancers 6, no. 3: 1441-1463. https://doi.org/10.3390/cancers6031441
APA StyleMatsuoka, T., & Yashiro, M. (2014). The Role of PI3K/Akt/mTOR Signaling in Gastric Carcinoma. Cancers, 6(3), 1441-1463. https://doi.org/10.3390/cancers6031441