DNA Mismatch Repair and Oxidative DNA Damage: Implications for Cancer Biology and Treatment

Abstract
:1. Introduction

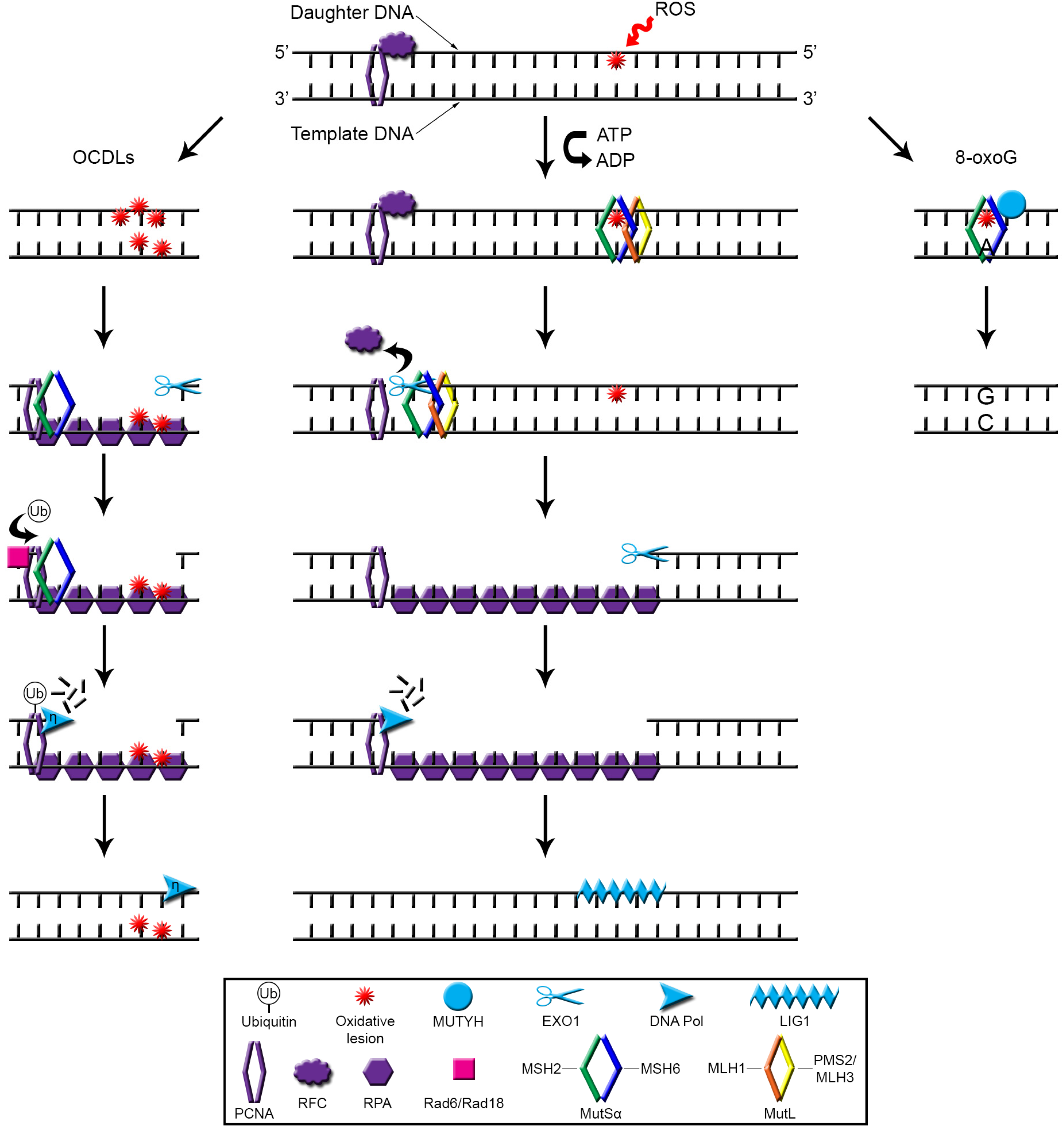{kind=link}
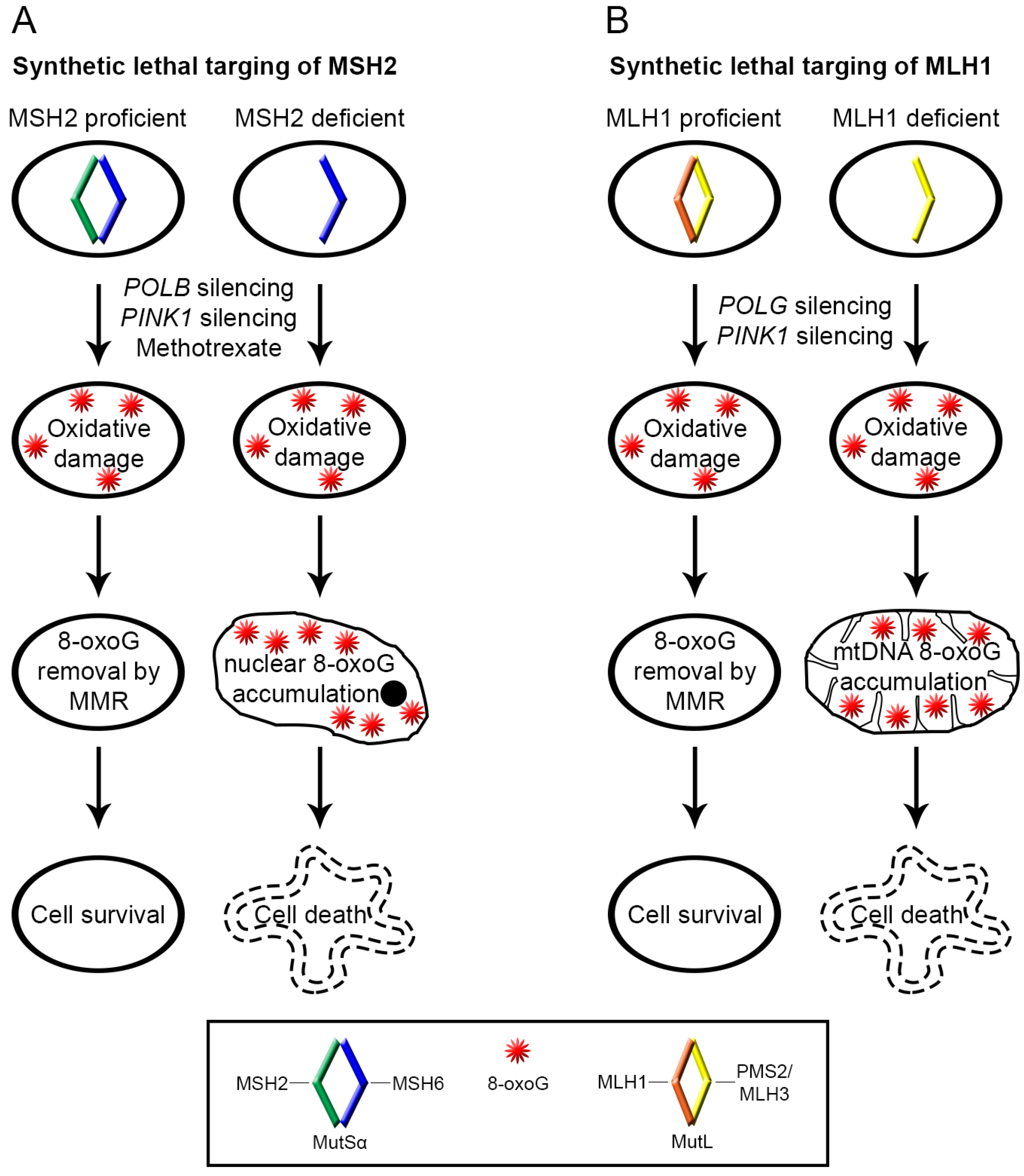{kind=link}
| Original DNA Base | Oxidatively Induced Product | Repaired by | Proteins Implicated |
|---|---|---|---|
Guanine | 8-Oxoguanine | BER | OGG1; MUTYH |
| MMR | MSH2; MSH6; MLH1 | ||
| NER | Multiple members | ||
| dNTP pool sanitisation | MTH1 | ||
2,6-Diamino-4-hydroxy-5-formamidopyrimidine  | BER | OGG1; NEIL1; NEIL3 | |
Spiroiminodihydantoin  | BER | NEIL1; NEIL3 | |
Guanidinohydantoin  | BER | NEIL1; NEIL3 | |
Cytosine  | 5-Hydroxycytosine  | BER | NEIL2; NEIL3; NTH1 |
5,6-Dihydroxycytosine  | BER | NTH1 | |
5-Hydroxyuracil  | BER | NEIL2; NEIL3; NTH1 | |
5,6-Dihydrouracil  | BER | NEIL2 | |
Adenine  | 8-Hydroxyadenine | dNTP pool sanitisation | MTH1 |
4,6-Diamino-5-formamidopyrimidine | BER | NEIL1; NEIL3 | |
2-Hydroxyadenine | BER | MUTYH | |
| MMR | MSH2; MSH6 | ||
| dNTP poolsanitisation | MTH1 | ||
Thymine | 5-Hydroxy-6-hydrothymine | BER | NTH1 |
Thymine glycol | BER | NEIL1; NEIL3; NTH1 | |
| MMR | MSH2 | ||
| NER | Multiple proteins | ||
5-Hydroxy-5-methylhydantoin | BER | NEIL1; NEIL3 |
2. DNA Repair of Oxidative DNA Damage
3. The DNA Mismatch Repair Pathway
4. The Role of MMR in Oxidative Damage Repair
5. Repair of Oxidative Damage to the Mitochondrial Genome

6. MMR and Oxidative Damage: Relevance to Carcinogenesis

7. Therapeutic Targeting of MMR-Deficient Tumours with Oxidative Damage
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dizdaroglu, M. Oxidatively induced DNA damage: Mechanisms, repair and disease. Cancer Lett. 2012, 327, 26–47. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Bazhin, A.V.; Werner, J.; Karakhanova, S. Reactive oxygen species in the immune system. Int. Rev. Immunol. 2013, 32, 249–270. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine, 4th ed.; Oxford University Press: New York, NY, USA, 2007. [Google Scholar]
- Storr, S.J.; Woolston, C.M.; Martin, S.G. Base excision repair, the redox environment and therapeutic implications. Curr. Mol. Pharmacol. 2012, 5, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.L.; Li, X.; Gu, Y.; Wright, P.M.; Chang, D.Y. Repair of oxidative DNA damage: Mechanisms and functions. Cell Biochem. Biophys. 2001, 35, 141–170. [Google Scholar] [CrossRef] [PubMed]
- Greenman, C.; Stephens, P.; Smith, R.; Dalgliesh, G.L.; Hunter, C.; Bignell, G.; Davies, H.; Teague, J.; Butler, A.; Stevens, C.; et al. Patterns of somatic mutation in human cancer genomes. Nature 2007, 446, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.A.; Lawrence, M.S.; Klimczak, L.J.; Grimm, S.A.; Fargo, D.; Stojanov, P.; Kiezun, A.; Kryukov, G.V.; Carter, S.L.; Saksena, G. An. APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet. 2013, 45, 970–976. [Google Scholar]
- Konturek, P.C.; Konturek, S.J.; Brzozowski, T. Gastric cancer and Helicobacter pylori infection. J. Physiol. Pharmacol. 2006, 57, S51–S65. [Google Scholar]
- Seril, D.N.; Liao, J.; Yang, G.; Yang, C.S. Oxidative stress and ulcerative colitis-associated carcinogenesis: Studies in humans and animal models. Carcinogenesis 2003, 24, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Kryston, T.B.; Georgiev, A.B.; Pissis, P.; Georgakilas, A.G. Role of oxidative stress and DNA damage in human carcinogenesis. Mutat. Res. 2011, 711, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Olinski, R.; Gackowski, D.; Rozalski, R.; Foksinski, M.; Bialkowski, K. Oxidative DNA damage in cancer patients: A cause or a consequence of the disease development? Mutat. Res. Fundam. Mol. Mech. Mut. 2003, 531, 177–190. [Google Scholar] [CrossRef]
- Kim, Y.J.; Wilson, D.M., 3rd. Overview of base excision repair biochemistry. Curr. Mol. Pharmacol. 2012, 5, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Kamileri, I.; Karakasilioti, I.; Garinis, G.A. Nucleotide excision repair: New tricks with old bricks. Trends Genet. 2012, 28, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.J.; Sancar, A. A new mechanism for repairing oxidative damage to DNA: (A)BC excinuclease removes AP sites and thymine glycols from DNA. Biochemistry 1989, 28, 7979–7984. [Google Scholar] [CrossRef] [PubMed]
- Reardon, J.T.; Bessho, T.; Kung, H.C.; Bolton, P.H.; Sancar, A. In vitro repair of oxidative DNA damage by human nucleotide excision repair system: Possible explanation for neurodegeneration in xeroderma pigmentosum patients. Proc. Natl. Acad. Sci. USA 1997, 94, 9463–9468. [Google Scholar] [CrossRef] [PubMed]
- Brooks, P.J.; Wise, D.S.; Berry, D.A.; Kosmoski, J.V.; Smerdon, M.J.; Somers, R.L.; Mackie, H.; Spoonde, A.Y.; Ackerman, E.J.; Coleman, K. The oxidative DNA lesion 8,5'-(S)-cyclo-2'-deoxyadenosine is repaired by the nucleotide excision repair pathway and blocks gene expression in mammalian cells. J. Biol. Chem. 2000, 275, 22355–22362. [Google Scholar] [CrossRef] [PubMed]
- Kuraoka, I.; Bender, C.; Romieu, A.; Cadet, J.; Wood, R.D.; Lindahl, T. Removal of oxygen free-radical-induced 5',8-purine cyclodeoxynucleosides from DNA by the nucleotide excision-repair pathway in human cells. Proc. Natl. Acad. Sci. USA 2000, 97, 3832–3837. [Google Scholar]
- Macpherson, P.; Barone, F.; Maga, G.; Mazzei, F.; Karran, P.; Bignami, M. 8-oxoguanine incorporation into DNA repeats in vitro and mismatch recognition by MutSalpha. Nucleic Acids Res. 2005, 33, 5094–5105. [Google Scholar] [CrossRef] [PubMed]
- Li, G.M. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008, 18, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Genschel, J.; Littman, S.J.; Drummond, J.T.; Modrich, P. Isolation of MutSbeta from human cells and comparison of the mismatch repair specificities of MutSbeta and MutSalpha. J. Biol. Chem. 1998, 273, 19895–19901. [Google Scholar] [CrossRef] [PubMed]
- Kolodner, R.D.; Marsischky, G.T. Eukaryotic DNA mismatch repair. Curr. Opin. Genet. Dev. 1999, 9, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Cannavo, E.; Marra, G.; Sabates-Bellver, J.; Menigatti, M.; Lipkin, S. M.; Fischer, F.; Cejka, P.; Jiricny, J. Expression of the MutL homologue hMLH3 in human cells and its role in DNA mismatch repair. Cancer Res. 2005, 65, 10759–10766. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, T.A.; Erie, D.A. DNA mismatch repair. Annu. Rev. Biochem. 2005, 74, 681–710. [Google Scholar] [CrossRef] [PubMed]
- Li, G.M.; Modrich, P. Restoration of mismatch repair to nuclear extracts of H6 colorectal tumor cells by a heterodimer of human MutL homologs. Proc. Natl. Acad. Sci. USA 1995, 92, 1950–1954. [Google Scholar] [CrossRef] [PubMed]
- Plotz, G.; Raedle, J.; Brieger, A.; Trojan, J.; Zeuzem, S. hMutSalpha forms an ATP-dependent complex with hMutLalpha and hMutLbeta on DNA. Nucleic Acids Res. 2002, 30, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Jiricny, J. The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell Biol. 2006, 7, 12. [Google Scholar] [CrossRef]
- Martin, S.A.; Lord, C.J.; Ashworth, A. Therapeutic targeting of the DNA mismatch repair pathway. Clin. Cancer Res. 2010, 16, 5107–5113. [Google Scholar] [CrossRef] [PubMed]
- Brierley, D.J.; Martin, S.A. Oxidative stress and the DNA mismatch repair pathway. Antioxid. Redox Signal. 2013, 18, 2420–2428. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.T.; De Luca, G.; Degan, P.; Bignami, M. Different DNA repair strategies to combat the threat from 8-oxoguanine. Mutat. Res. 2007, 614, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Yamamoto, H. Carcinogenesis and microsatellite instability: The interrelationship between genetics and epigenetics. Carcinogenesis 2008, 29, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Popat, S.; Hubner, R.; Houlston, R.S. Systematic review of microsatellite instability and colorectal cancer prognosis. J. Clin. Oncol. 2005, 23, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Resnick, K.E.; Frankel, W.L.; Morrison, C.D.; Fowler, J.M.; Copeland, L.J.; Stephens, J.; Kim, K.H.; Cohn, D.E. Mismatch repair status and outcomes after adjuvant therapy in patients with surgically staged endometrial cancer. Gynecol. Oncol. 2010, 117, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Melton, D.W.; Gourley, C. Mismatch repair deficiency in ovarian cancer—Molecular characteristics and clinical implications. Gynecol. Oncol. 2014, 132, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Hewish, M.; Lord, C.J.; Martin, S.A.; Cunningham, D.; Ashworth, A. Mismatch repair deficient colorectal cancer in the era of personalized treatment. Nat. Rev. Clin. Oncol. 2010, 7, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Karamurzin, Y.; Rutgers, J.K. DNA mismatch repair deficiency in endometrial carcinoma. Int. J. Gynecol. Pathol. 2009, 28, 239–255. [Google Scholar] [CrossRef] [PubMed]
- Lynch, H.T.; Lynch, J.F.; Lynch, P.M.; Attard, T. Hereditary colorectal cancer syndromes: Molecular genetics, genetic counseling, diagnosis and management. Fam. Cancer 2008, 7, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, N.P.; Skandalis, A.; Ganesh, A.; Groden, J.; Meuth, M. Mutator phenotypes in human colorectal carcinoma cell lines. Proc. Natl. Acad. Sci. USA 1994, 91, 6319–6323. [Google Scholar] [CrossRef] [PubMed]
- Parsons, R.; Li, G.M.; Longley, M.J.; Fang, W.H.; Papadopoulos, N.; Jen, J.; de la Chapelle, A.; Kinzler, K.W.; Vogelstein, B.; Modrich, P. Hypermutability and mismatch repair deficiency in RER+ tumor cells. Cell 1993, 75, 1227–1236. [Google Scholar]
- Johannsdottir, J.T.; Jonasson, J.G.; Bergthorsson, J.T.; Amundadottir, L.T.; Magnusson, J.; Egilsson, V.; Ingvarsson, S. The effect of mismatch repair deficiency on tumourigenesis; microsatellite instability affecting genes containing short repeated sequences. Int. J. Oncol. 2000, 16, 133–139. [Google Scholar] [PubMed]
- Colussi, C.; Parlanti, E.; Degan, P.; Aquilina, G.; Barnes, D.; Macpherson, P.; Karran, P.; Crescenzi, M.; Dogliotti, E.; Bignami, M. The mammalian mismatch repair pathway removes DNA 8-oxodGMP incorporated from the oxidized dNTP pool. Curr. Biol. 2002, 12, 912–918. [Google Scholar] [CrossRef] [PubMed]
- DeWeese, T.L.; Shipman, J.M.; Larrier, N.A.; Buckley, N.M.; Kidd, L.R.; Groopman, J.D.; Cutler, R.G.; te Riele, H.; Nelson, W.G. Mouse embryonic stem cells carrying one or two defective Msh2 alleles respond abnormally to oxidative stress inflicted by low-level radiation. Proc. Natl. Acad. Sci. USA 1998, 95, 11915–11920. [Google Scholar] [CrossRef] [PubMed]
- Sakumi, K.; Furuichi, M.; Tsuzuki, T.; Kakuma, T.; Kawabata, S.; Maki, H.; Sekiguchi, M. Cloning and expression of cDNA for a human enzyme that hydrolyzes 8-oxo-dGTP, a mutagenic substrate for DNA synthesis. J. Biol. Chem. 1993, 268, 23524–23530. [Google Scholar] [PubMed]
- Wang, G.; Alamuri, P.; Humayun, M.Z.; Taylor, D.E.; Maier, R.J. The Helicobacter pylori MutS protein confers protection from oxidative DNA damage. Mol. Microbiol. 2005, 58, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Mazurek, A.; Berardini, M.; Fishel, R. Activation of human MutS homologs by 8-oxo-guanine DNA damage. J. Biol. Chem. 2002, 277, 8260–8266. [Google Scholar] [CrossRef] [PubMed]
- Ni, T.T.; Marsischky, G.T.; Kolodner, R.D. MSH2 and MSH6 are required for removal of adenine misincorporated opposite 8-oxo-guanine in S. cerevisiae. Mol. Cell 1999, 4, 439–444. [Google Scholar] [CrossRef]
- Sage, E.; Harrison, L. Clustered DNA lesion repair in eukaryotes: Relevance to mutagenesis and cell survival. Mutat. Res. 2011, 711, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Holt, S.M.; Scemama, J.L.; Panayiotidis, M.I.; Georgakilas, A.G. Compromised repair of clustered DNA damage in the human acute lymphoblastic leukemia MSH2-deficient NALM-6 cells. Mutat. Res. 2009, 674, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Zlatanou, A.; Despras, E.; Braz-Petta, T.; Boubakour-Azzouz, I.; Pouvelle, C.; Stewart, G.S.; Nakajima, S.; Yasui, A.; Ishchenko, A.A.; Kannouche, P.L. The hMsh2-hMsh6 complex acts in concert with monoubiquitinated PCNA and Pol eta in response to oxidative DNA damage in human cells. Mol. Cell 2011, 43, 649–662. [Google Scholar] [CrossRef] [PubMed]
- Barone, F.; McCulloch, S.D.; Macpherson, P.; Maga, G.; Yamada, M.; Nohmi, T.; Minoprio, A.; Karran, P.; et al. Replication of 2-hydroxyadenine-containing DNA and recognition by human MutSα. DNA Repair 2007, 6, 355–366. [Google Scholar]
- Russo, M.T.; De Luca, G.; Casorelli, I.; Degan, P.; Molatore, S.; Barone, F.; Mazzei, F.; Pannellini, T.; Musiani, P.; Bignami, M. Role of MUTYH and MSH2 in the Control. of Oxidative DNA Damage, Genetic Instability, and Tumorigenesis. Cancer Res. 2009, 69, 4372–4379. [Google Scholar]
- Gu, Y.; Parker, A.; Wilson, T.M.; Bai, H.; Chang, D.Y.; Lu, A.L. Human MutY homolog, a DNA glycosylase involved in base excision repair, physically and functionally interacts with mismatch repair proteins human MutS homolog 2/human MutS homolog 6. J. Biol. Chem. 2002, 277, 11135–11142. [Google Scholar] [CrossRef] [PubMed]
- Tuppen, H.A.L.; Blakely, E.L.; Turnbull, D.M.; Taylor, R.W. Mitochondrial DNA mutations and human disease. Biochim. Biophys. Acta (BBA)-Bioenerg. 2010, 1797, 113–128. [Google Scholar]
- Svilar, D.; Goellner, E.M.; Almeida, K.H.; Sobol, R.W. Base excision repair and lesion-dependent subpathways for repair of oxidative DNA damage. Antioxid. Redox Signal. 2011, 14, 2491–2507. [Google Scholar] [CrossRef] [PubMed]
- Habano, W.; Nakamura, S.; Sugai, T. Microsatellite instability in the mitochondrial DNA of colorectal carcinomas: Evidence for mismatch repair systems in mitochondrial genome. Oncogene 1998, 17, 1931–1937. [Google Scholar] [CrossRef] [PubMed]
- Reenan, R.A.; Kolodner, R.D. Characterization of insertion mutations in the Saccharomyces cerevisiae MSH1 and MSH2 genes: Evidence for separate mitochondrial and nuclear functions. Genetics 1992, 132, 975–985. [Google Scholar] [PubMed]
- Hashiguchi, K.; Bohr, V.A.; de Souza-Pinto, N.C. Oxidative stress and mitochondrial DNA repair: Implications for NRTIs induced DNA damage. Mitochondrion 2004, 4, 215–222. [Google Scholar] [CrossRef] [PubMed]
- De Souza-Pinto, N.C.; Mason, P.A.; Hashiguchi, K.; Weissman, L.; Tian, J.; Guay, D.; Lebel, M.; Stevnsner, T.V.; Rasmussen, L.J.; Bohr, V.A. Novel DNA mismatch-repair activity involving YB-1 in human mitochondria. DNA Repair (Amst.) 2009, 8, 704–719. [Google Scholar] [CrossRef]
- Martin, S.A.; McCabe, N.; Mullarkey, M.; Cummins, R.; Burgess, D.J.; Nakabeppu, Y.; Oka, S.; Kay, E.; Lord, C.J.; Ashworth, A. DNA polymerases as potential therapeutic targets for cancers deficient in the DNA mismatch repair proteins MSH2 or MLH1. Cancer Cell 2010, 17, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Mootha, V.K.; Bunkenborg, J.; Olsen, J.V.; Hjerrild, M.; Wisniewski, J.R.; Stahl, E.; Bolouri, M.S.; Ray, H.N.; Sihag, S.; Kamal, M. Integrated analysis of protein composition, tissue diversity, and gene regulation in mouse mitochondria. Cell 2003, 115, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.A.; Hewish, M.; Sims, D.; Lord, C.J.; Ashworth, A. Parallel high-throughput RNA interference screens identify PINK1 as a potential therapeutic target for the treatment of DNA mismatch repair-deficient cancers. Cancer Res. 2011, 71, 1836–1848. [Google Scholar] [CrossRef] [PubMed]
- Glaab, W.E.; Hill, R.B.; Skopek, T.R. Suppression of spontaneous and hydrogen peroxide-induced mutagenesis by the antioxidant ascorbate in mismatch repair-deficient human colon cancer cells. Carcinogenesis 2001, 22, 1709–1713. [Google Scholar] [CrossRef] [PubMed]
- Hardman, R.A.; Afshari, C.A.; Barrett, J.C. Involvement of Mammalian MLH1 in the apoptotic response to peroxide-induced oxidative stress. Cancer Res. 2001, 61, 1392–1397. [Google Scholar] [PubMed]
- Martin, S.A.; McCarthy, A.; Barber, L.J.; Burgess, D.J.; Parry, S.; Lord, C.J.; Ashworth, A. Methotrexate induces oxidative DNA damage and is selectively lethal to tumour cells with defects in the DNA mismatch repair gene MSH2. EMBO Mol. Med. 2009, 1, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Piao, J.; Nakatsu, Y.; Ohno, M.; Taguchi, K.; Tsuzuki, T. Mismatch repair deficient mice show susceptibility to oxidative stress-induced intestinal carcinogenesis. Int. J. Biol. Sci. 2013, 10, 73–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Chiu, W.H.; Luo, S.J.; Chen, C.L.; Cheng, J.H.; Hsieh, C.Y.; Wang, C.Y.; Huang, W.C.; Su, W.C.; Lin, C.F. Vinca alkaloids cause aberrant ROS-mediated JNK activation, Mcl-1 downregulation, DNA damage, mitochondrial dysfunction, and apoptosis in lung adenocarcinoma cells. Biochem. Pharmacol. 2012, 83, 1159–1171. [Google Scholar] [CrossRef] [PubMed]
- Conklin, K.A. Chemotherapy-associated oxidative stress: Impact on chemotherapeutic effectiveness. Integr. Cancer Ther. 2004, 3, 294–300. [Google Scholar] [CrossRef] [PubMed]
- D’Anneo, A.; Carlisi, D.; Lauricella, M.; Puleio, R.; Martinez, R.; Di Bella, S.; Di Marco, P.; Emanuele, S.; di Fiore, R.; Guercio, A; et al. Parthenolide generates reactive oxygen species and autophagy in MDA-MB231 cells. A soluble parthenolide analogue inhibits tumour growth and metastasis in a xenograft model of breast cancer. Cell Death Dis 2013, 4, e891. [Google Scholar]
- Zhang, Y.; Zheng, S.; Zheng, J.S.; Wong, K.H.; Huang, Z.; Ngai, S.M.; Zheng, W.; Wong, Y.S.; Chen, T. Synergistic induction of apoptosis by methylseleninic acid and cisplatin, the role of ROS-ERK/AKT-p53 pathway. Mol. Pharm. 2014, 11, 1282–1293. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Dong, Y.; Bey, E.A.; Kilgore, J.A.; Bair, J.S.; Li, L.S.; Patel, M.; Parkinson, E.I.; Wang, Y.; Williams, N.S.; et al. An NQO1 substrate with potent antitumor activity that selectively kills by PARP1-induced programmed necrosis. Cancer Res. 2012, 72, 3038–3047. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.F.; Konstantinopoulos, P.A.; Matulonis, U.A. PARP inhibitors in ovarian cancer: Current status and future promise. Gynecol. Oncol. 2014, 133, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Hewish, M.; Martin, S.A.; Elliott, R.; Cunningham, D.; Lord, C.J.; Ashworth, A. Cytosine-Based nucleoside analogs are selectively lethal to DNA mismatch repair-deficient tumour cells by enhancing levels of intracellular oxidative stress. Br. J. Cancer 2013, 108, 983–992. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, D.J.; Belenky, P.A.; Yang, J.H.; MacDonald, I.C.; Martell, J.D.; Takahashi, N.; Chan, C.T.Y.; Lobritz, M.A.; Braff, D.; Schwarz, E.G.; et al. Antibiotics induce redox-related physiological alterations as part of their lethality. Proc. Natl. Acad. Sci. USA 2014. [Google Scholar] [CrossRef]
- Mendelsohn, A.R.; Larrick, J.W. Paradoxical effects of antioxidants on cancer. Rejuvenation Res. 2014, 17, 306–311. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bridge, G.; Rashid, S.; Martin, S.A. DNA Mismatch Repair and Oxidative DNA Damage: Implications for Cancer Biology and Treatment. Cancers 2014, 6, 1597-1614. https://doi.org/10.3390/cancers6031597
Bridge G, Rashid S, Martin SA. DNA Mismatch Repair and Oxidative DNA Damage: Implications for Cancer Biology and Treatment. Cancers. 2014; 6(3):1597-1614. https://doi.org/10.3390/cancers6031597
Chicago/Turabian StyleBridge, Gemma, Sukaina Rashid, and Sarah A. Martin. 2014. "DNA Mismatch Repair and Oxidative DNA Damage: Implications for Cancer Biology and Treatment" Cancers 6, no. 3: 1597-1614. https://doi.org/10.3390/cancers6031597
APA StyleBridge, G., Rashid, S., & Martin, S. A. (2014). DNA Mismatch Repair and Oxidative DNA Damage: Implications for Cancer Biology and Treatment. Cancers, 6(3), 1597-1614. https://doi.org/10.3390/cancers6031597