SUMO and KSHV Replication

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. SUMO as a Signal Transducer

1.2. KSHV and SUMO
2. KSHV Proteins are Involved in Modulating Various SUMO Activities
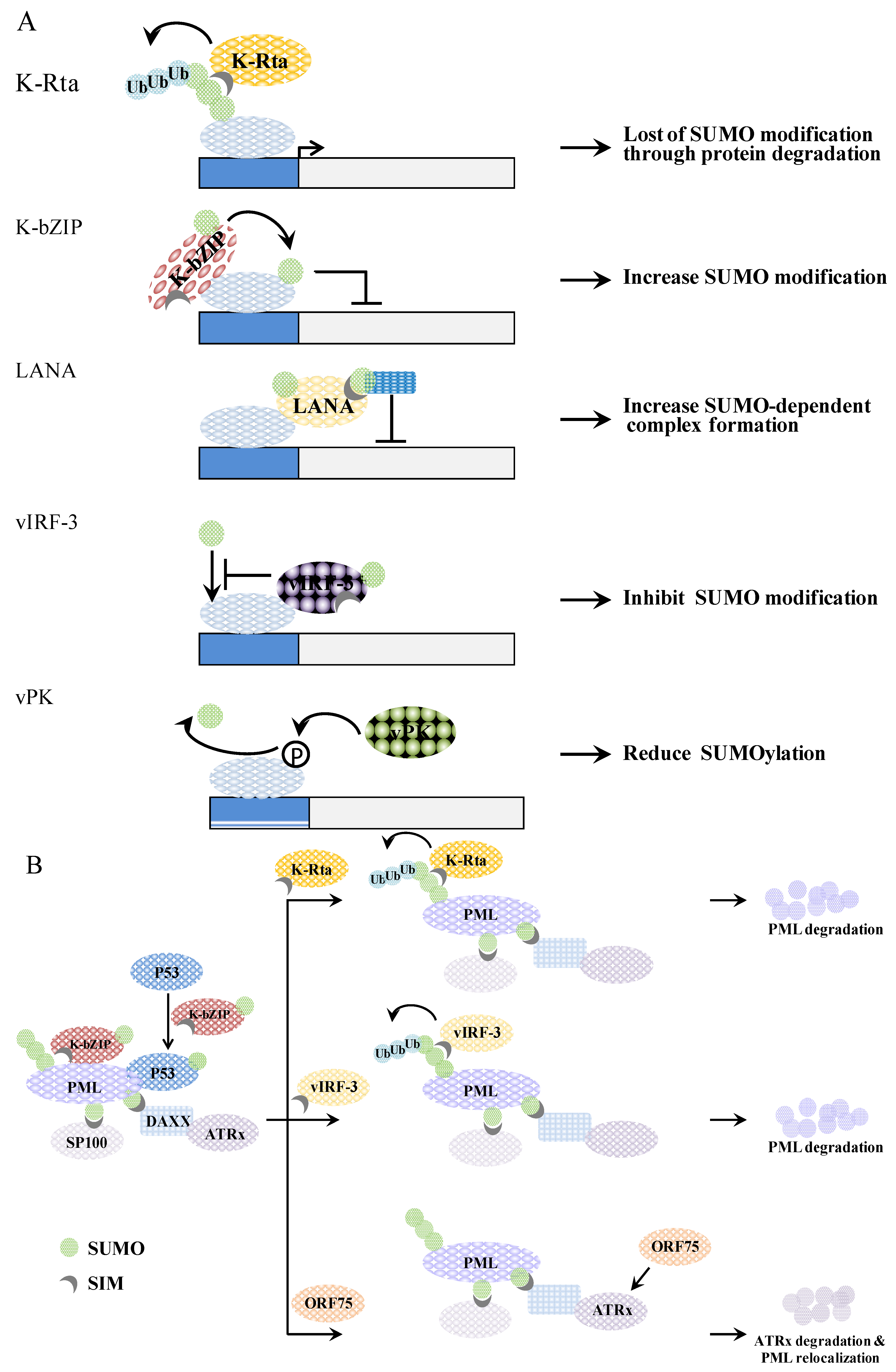
2.1. The KSHV IE Gene K-Rta (KSHV-Replication and Transcriptional Activator)

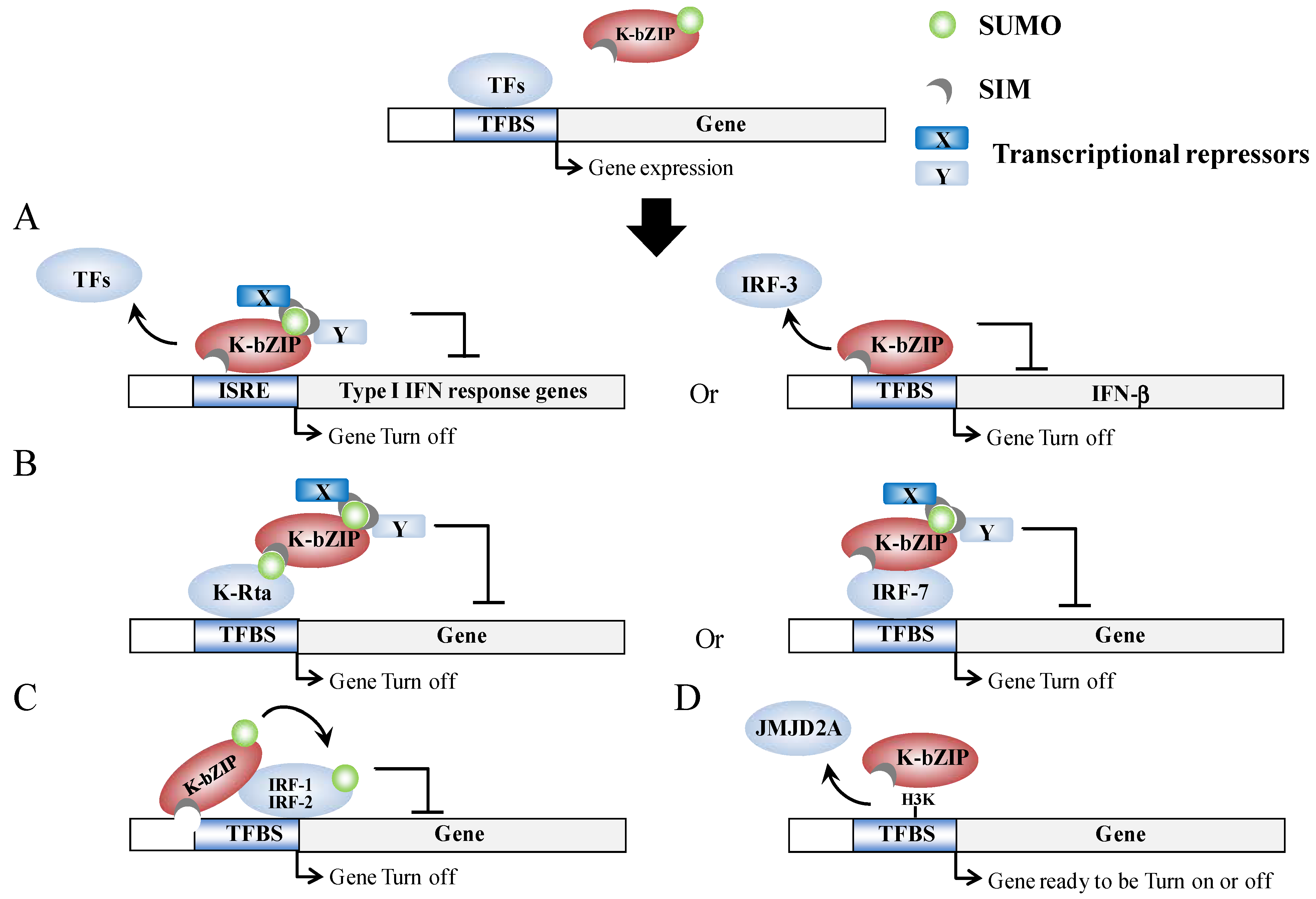
2.2. The KSHV IE Gene K-bZIP (KSHV Basic Leucine Zipper Protein)

2.3. The KSHV Latent Gene LANA (Latency-Associated Nuclear Antigen)
2.4. The KSHV Latent Gene vIRF-3/LANA2 (Viral Interferon Regulatory Factor-3/Latency-Associated Nuclear Antigen 2)

2.5. The KSHV Lytic Gene vPK (Viral Protein Kinase)
3. The SUMO Dynamics during KSHV Reactivation
4. Conclusions and Future Perspectives

Acknowledgements
Conflicts of Interest
References
- Gareau, J.R.; Lima, C.D. The sumo pathway: Emerging mechanisms that shape specificity, conjugation and recognition. Nat. Rev. Mol. Cell Biol. 2010, 11, 861–871. [Google Scholar] [PubMed]
- Hecker, C.M.; Rabiller, M.; Haglund, K.; Bayer, P.; Dikic, I. Specification of SUMO1- and SUMO2-interacting motifs. J. Biol. Chem. 2006, 281, 16117–16127. [Google Scholar] [PubMed]
- Lin, D.Y.; Huang, Y.S.; Jeng, J.C.; Kuo, H.Y.; Chang, C.C.; Chao, T.T.; Ho, C.C.; Chen, Y.C.; Lin, T.P.; Fang, H.I.; et al. Role of SUMO-interacting motif in Daxx SUMO modification, subnuclear localization, and repression of sumoylated transcription factors. Mol. Cell 2006, 24, 341–354. [Google Scholar] [PubMed]
- Kim, J.H.; Baek, S.H. Emerging roles of desumoylating enzymes. Biochim. Biophys. Acta 2009, 1792, 155–162. [Google Scholar] [PubMed]
- Meulmeester, E.; Kunze, M.; Hsiao, H.H.; Urlaub, H.; Melchior, F. Mechanism and consequences for paralog-specific sumoylation of ubiquitin-specific protease 25. Mol. Cell 2008, 30, 610–619. [Google Scholar] [PubMed]
- Zhu, J.; Zhu, S.; Guzzo, C.M.; Ellis, N.A.; Sung, K.S.; Choi, C.Y.; Matunis, M.J. Small ubiquitin-related modifier (SUMO) binding determines substrate recognition and paralog-selective SUMO modification. J. Biol. Chem. 2008, 283, 29405–29415. [Google Scholar] [PubMed]
- Zhu, S.; Goeres, J.; Sixt, K.M.; Bekes, M.; Zhang, X.D.; Salvesen, G.S.; Matunis, M.J. Protection from isopeptidase-mediated deconjugation regulates paralog-selective sumoylation of rangap1. Mol. Cell 2009, 33, 570–580. [Google Scholar] [PubMed]
- Boggio, R.; Colombo, R.; Hay, R.T.; Draetta, G.F.; Chiocca, S. A mechanism for inhibiting the SUMO pathway. Mol. Cell 2004, 16, 549–561. [Google Scholar] [PubMed]
- Yousef, A.F.; Fonseca, G.J.; Pelka, P.; Ablack, J.N.; Walsh, C.; Dick, F.A.; Bazett-Jones, D.P.; Shaw, G.S.; Mymryk, J.S. Identification of a molecular recognition feature in the E1A oncoprotein that binds the SUMO conjugase UBC9 and likely interferes with polySUMOylation. Oncogene 2010, 29, 4693–4704. [Google Scholar] [PubMed]
- Wimmer, P.; Schreiner, S.; Dobner, T. Human pathogens and the host cell sumoylation system. J. Virol. 2012, 86, 642–654. [Google Scholar] [PubMed]
- Mattoscio, D.; Segre, C.V.; Chiocca, S. Viral manipulation of cellular protein conjugation pathways: The SUMO lesson. World J. Virol. 2013, 2, 79–90. [Google Scholar] [PubMed]
- Everett, R.D.; Boutell, C.; Hale, B.G. Interplay between viruses and host sumoylation pathways. Nat. Rev. Microbiol. 2013, 11, 400–411. [Google Scholar] [PubMed]
- Endter, C.; Kzhyshkowska, J.; Stauber, R.; Dobner, T. SUMO-1 modification required for transformation by adenovirus type 5 early region 1B 55-kDa oncoprotein. Proc. Natl. Acad. Sci. USA 2001, 98, 11312–11317. [Google Scholar]
- Adamson, A.L.; Kenney, S. Epstein-Barr virus immediate-early protein BZLF1 is SUMO-1 modified and disrupts promyelocytic leukemia bodies. J. Virol. 2001, 75, 2388–2399. [Google Scholar] [PubMed]
- Chang, L.K.; Lee, Y.H.; Cheng, T.S.; Hong, Y.R.; Lu, P.J.; Wang, J.J.; Wang, W.H.; Kuo, C.W.; Li, S.S.; Liu, S.T. Post-translational modification of Rta of Epstein-Barr virus by SUMO-1. J. Biol. Chem. 2004, 279, 38803–38812. [Google Scholar] [PubMed]
- Cai, Q.; Cai, S.; Zhu, C.; Verma, S.C.; Choi, J.Y.; Robertson, E.S. A unique SUMO-2-interacting motif within LANA is essential for KSHV latency. PLoS Pathog. 2013, 9, e1003750. [Google Scholar] [PubMed]
- Izumiya, Y.; Ellison, T.J.; Yeh, E.T.; Jung, J.U.; Luciw, P.A.; Kung, H.J. Kaposi’s sarcoma-associated herpesvirus K-bZIP represses gene transcription via SUMO modification. J. Virol. 2005, 79, 9912–9925. [Google Scholar] [PubMed]
- Chang, P.C.; Izumiya, Y.; Wu, C.Y.; Fitzgerald, L.D.; Campbell, M.; Ellison, T.J.; Lam, K.S.; Luciw, P.A.; Kung, H.J. Kaposi’s sarcoma-associated herpesvirus (KSHV) encodes a SUMO E3 ligase that is SIM-dependent and SUMO-2/3-specific. J. Biol. Chem. 2010, 285, 5266–5273. [Google Scholar] [PubMed]
- Pennella, M.A.; Liu, Y.; Woo, J.L.; Kim, C.A.; Berk, A.J. Adenovirus E1B 55-kilodalton protein is a p53-SUMO1 E3 ligase that represses p53 and stimulates its nuclear export through interactions with promyelocytic leukemia nuclear bodies. J. Virol. 2010, 84, 12210–12225. [Google Scholar] [PubMed]
- Muller, S.; Dobner, T. The adenovirus E1B-55K oncoprotein induces SUMO modification of p53. Cell Cycle 2008, 7, 754–758. [Google Scholar] [PubMed]
- Boutell, C.; Cuchet-Lourenco, D.; Vanni, E.; Orr, A.; Glass, M.; McFarlane, S.; Everett, R.D. A viral ubiquitin ligase has substrate preferential SUMO targeted ubiquitin ligase activity that counteracts intrinsic antiviral defence. PLoS Pathog. 2011, 7, e1002245. [Google Scholar] [PubMed]
- Izumiya, Y.; Kobayashi, K.; Kim, K.Y.; Pochampalli, M.; Izumiya, C.; Shevchenko, B.; Wang, D.H.; Huerta, S.B.; Martinez, A.; Campbell, M.; et al. Kaposi’s sarcoma-associated herpesvirus K-Rta exhibits SUMO-targeting ubiquitin ligase (STUBL) like activity and is essential for viral reactivation. PLoS Pathog. 2013, 9, e1003506. [Google Scholar] [PubMed]
- Dupin, N.; Fisher, C.; Kellam, P.; Ariad, S.; Tulliez, M.; Franck, N.; van Marck, E.; Salmon, D.; Gorin, I.; Escande, J.P.; et al. Distribution of human herpesvirus-8 latently infected cells in Kaposi’s sarcoma, multicentric castleman’s disease, and primary effusion lymphoma. Proc. Natl. Acad. Sci. USA 1999, 96, 4546–4551. [Google Scholar]
- Moore, P.S.; Chang, Y. Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nat. Rev. Cancer 2010, 10, 878–889. [Google Scholar] [PubMed]
- Paulose-Murphy, M.; Ha, N.K.; Xiang, C.; Chen, Y.; Gillim, L.; Yarchoan, R.; Meltzer, P.; Bittner, M.; Trent, J.; Zeichner, S. Transcription program of human herpesvirus 8 (Kaposi’s sarcoma-associated herpesvirus). J. Virol. 2001, 75, 4843–4853. [Google Scholar] [PubMed]
- Jenner, R.G.; Alba, M.M.; Boshoff, C.; Kellam, P. Kaposi’s sarcoma-associated herpesvirus latent and lytic gene expression as revealed by DNA arrays. J. Virol. 2001, 75, 891–902. [Google Scholar] [PubMed]
- Parravicini, C.; Chandran, B.; Corbellino, M.; Berti, E.; Paulli, M.; Moore, P.S.; Chang, Y. Differential viral protein expression in Kaposi’s sarcoma-associated herpesvirus-infected diseases: Kaposi’s sarcoma, primary effusion lymphoma, and multicentric Castleman’s disease. Am. J. Pathol. 2000, 156, 743–749. [Google Scholar] [PubMed]
- Gunther, T.; Grundhoff, A. The epigenetic landscape of latent Kaposi sarcoma-associated herpesvirus genomes. PLoS Pathog. 2010, 6, e1000935. [Google Scholar] [PubMed]
- Toth, Z.; Maglinte, D.T.; Lee, S.H.; Lee, H.R.; Wong, L.Y.; Brulois, K.F.; Lee, S.; Buckley, J.D.; Laird, P.W.; Marquez, V.E.; et al. Epigenetic analysis of KSHV latent and lytic genomes. PLoS Pathog. 2010, 6, e1001013. [Google Scholar] [PubMed]
- Zhong, W.; Wang, H.; Herndier, B.; Ganem, D. Restricted expression of Kaposi sarcoma-associated herpesvirus (human herpesvirus 8) genes in Kaposi sarcoma. Proc. Natl. Acad. Sci. USA 1996, 93, 6641–6646. [Google Scholar]
- Staskus, K.A.; Zhong, W.; Gebhard, K.; Herndier, B.; Wang, H.; Renne, R.; Beneke, J.; Pudney, J.; Anderson, D.J.; Ganem, D.; et al. Kaposi’s sarcoma-associated herpesvirus gene expression in endothelial (spindle) tumor cells. J. Virol. 1997, 71, 715–719. [Google Scholar] [PubMed]
- Boshoff, C.; Schulz, T.F.; Kennedy, M.M.; Graham, A.K.; Fisher, C.; Thomas, A.; McGee, J.O.; Weiss, R.A.; O’Leary, J.J. Kaposi’s sarcoma-associated herpesvirus infects endothelial and spindle cells. Nat. Med. 1995, 1, 1274–1278. [Google Scholar] [PubMed]
- Chang, Y.; Moore, P.S. Kaposi’s sarcoma (KS)-associated herpesvirus and its role in KS. Infect. Agents Dis. 1996, 5, 215–222. [Google Scholar] [PubMed]
- Ambroziak, J.A.; Blackbourn, D.J.; Herndier, B.G.; Glogau, R.G.; Gullett, J.H.; McDonald, A.R.; Lennette, E.T.; Levy, J.A. Herpes-like sequences in HIV-infected and uninfected Kaposi’s sarcoma patients. Science 1995, 268, 582–583. [Google Scholar] [PubMed]
- Martin, D.F.; Kuppermann, B.D.; Wolitz, R.A.; Palestine, A.G.; Li, H.; Robinson, C.A. Roche ganciclovir study group. Oral ganciclovir for patients with cytomegalovirus retinitis treated with a ganciclovir implant. N. Engl. J. Med. 1999, 340, 1063–1070. [Google Scholar] [PubMed]
- Whitby, D.; Howard, M.R.; Tenant-Flowers, M.; Brink, N.S.; Copas, A.; Boshoff, C.; Hatzioannou, T.; Suggett, F.E.; Aldam, D.M.; Denton, A.S.; et al. Detection of Kaposi sarcoma associated herpesvirus in peripheral blood of HIV-infected individuals and progression to Kaposi’s sarcoma. Lancet 1995, 346, 799–802. [Google Scholar] [PubMed]
- Toth, Z.; Brulois, K.; Lee, H.R.; Izumiya, Y.; Tepper, C.; Kung, H.J.; Jung, J.U. Biphasic euchromatin-to-heterochromatin transition on the KSHV genome following de novo infection. PLoS Pathog. 2013, 9, e1003813. [Google Scholar] [PubMed]
- Chang, P.C.; Fitzgerald, L.D.; Hsia, D.A.; Izumiya, Y.; Wu, C.Y.; Hsieh, W.P.; Lin, S.F.; Campbell, M.; Lam, K.S.; Luciw, P.A.; et al. Histone demethylase JMJD2A regulates Kaposi’s sarcoma-associated herpesvirus replication and is targeted by a viral transcriptional factor. J. Virol. 2011, 85, 3283–3293. [Google Scholar] [PubMed]
- Everett, R.D. DNA viruses and viral proteins that interact with pml nuclear bodies. Oncogene 2001, 20, 7266–7273. [Google Scholar] [PubMed]
- Everett, R.D.; Parada, C.; Gripon, P.; Sirma, H.; Orr, A. Replication of ICP0-null mutant herpes simplex virus type 1 is restricted by both PML and Sp100. J. Virol. 2008, 82, 2661–2672. [Google Scholar] [PubMed]
- Everett, R.D.; Rechter, S.; Papior, P.; Tavalai, N.; Stamminger, T.; Orr, A. PML contributes to a cellular mechanism of repression of herpes simplex virus type 1 infection that is inactivated by ICP0. J. Virol. 2006, 80, 7995–8005. [Google Scholar] [PubMed]
- Cuchet-Lourenco, D.; Boutell, C.; Lukashchuk, V.; Grant, K.; Sykes, A.; Murray, J.; Orr, A.; Everett, R.D. SUMO pathway dependent recruitment of cellular repressors to herpes simplex virus type 1 genomes. PLoS Pathog. 2011, 7, e1002123. [Google Scholar] [PubMed]
- Sewatanon, J.; Liu, H.; Ling, P.D. Promyelocytic leukemia protein modulates establishment and maintenance of latent gammaherpesvirus infection in peritoneal cells. J. Virol. 2013, 87, 12151–12157. [Google Scholar] [PubMed]
- Zhong, S.; Muller, S.; Ronchetti, S.; Freemont, P.S.; Dejean, A.; Pandolfi, P.P. Role of SUMO-1-modified PML in nuclear body formation. Blood 2000, 95, 2748–2752. [Google Scholar] [PubMed]
- Bernardi, R.; Pandolfi, P.P. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat. Rev. Mol. Cell Biol. 2007, 8, 1006–1016. [Google Scholar] [PubMed]
- Erker, Y.; Neyret-Kahn, H.; Seeler, J.S.; Dejean, A.; Atfi, A.; Levy, L. Arkadia, a novel SUMO-targeted ubiquitin ligase involved in PML degradation. Mol. Cell. Biol. 2013, 33, 2163–2177. [Google Scholar] [PubMed]
- Tatham, M.H.; Geoffroy, M.C.; Shen, L.; Plechanovova, A.; Hattersley, N.; Jaffray, E.G.; Palvimo, J.J.; Hay, R.T. RNF4 is a poly-SUMO-specific E3 ubiquitin ligase required for arsenic-induced PML degradation. Nat. Cell Biol. 2008, 10, 538–546. [Google Scholar] [PubMed]
- Kubota, T.; Matsuoka, M.; Chang, T.H.; Tailor, P.; Sasaki, T.; Tashiro, M.; Kato, A.; Ozato, K. Virus infection triggers SUMOylation of IRF3 and IRF7, leading to the negative regulation of type I interferon gene expression. J. Biol. Chem. 2008, 283, 25660–25670. [Google Scholar] [PubMed]
- Bentz, G.L.; Shackelford, J.; Pagano, J.S. Epstein-Barr virus latent membrane protein 1 regulates the function of interferon regulatory factor 7 by inducing its sumoylation. J. Virol. 2012, 86, 12251–12261. [Google Scholar] [PubMed]
- Chang, T.H.; Kubota, T.; Matsuoka, M.; Jones, S.; Bradfute, S.B.; Bray, M.; Ozato, K. Ebola zaire virus blocks type I interferon production by exploiting the host SUMO modification machinery. PLoS Pathog. 2009, 5, e1000493. [Google Scholar] [PubMed]
- Campbell, M.; Izumiya, Y. Post-translational modifications of Kaposi’s sarcoma-associated herpesvirus regulatory proteins—SUMO and KSHV. Front. Microbiol. 2012, 3, e31. [Google Scholar]
- Yu, Y.; Wang, S.E.; Hayward, G.S. The KSHV immediate-early transcription factor RTA encodes ubiquitin E3 ligase activity that targets IRF7 for proteosome-mediated degradation. Immunity 2005, 22, 59–70. [Google Scholar] [PubMed]
- Yang, Z.; Yan, Z.; Wood, C. Kaposi’s sarcoma-associated herpesvirus transactivator RTA promotes degradation of the repressors to regulate viral lytic replication. J. Virol. 2008, 82, 3590–3603. [Google Scholar] [PubMed]
- Yu, Y.; Hayward, G.S. The ubiquitin E3 ligase RAUL negatively regulates type I interferon through ubiquitination of the transcription factors IRF7 and IRF3. Immunity 2010, 33, 863–877. [Google Scholar] [PubMed]
- Chang, P.C.; Cheng, C.Y.; Campbell, M.; Yang, Y.C.; Hsu, H.W.; Chang, T.Y.; Chu, C.H.; Lee, Y.W.; Hung, C.L.; Lai, S.M.; et al. The chromatin modification by SUMO-2/3 but not SUMO-1 prevents the epigenetic activation of key immune-related genes during Kaposi’s sarcoma associated herpesvirus reactivation. BMC Genomics 2013, 14, e824. [Google Scholar]
- Salsman, J.; Zimmerman, N.; Chen, T.; Domagala, M.; Frappier, L. Genome-wide screen of three herpesviruses for protein subcellular localization and alteration of PML nuclear bodies. PLoS Pathog. 2008, 4, e1000100. [Google Scholar] [PubMed]
- Yang, Y.C.; Yoshikai, Y.; Hsu, S.W.; Saitoh, H.; Chang, L.K. Role of RNF4 in the ubiquitination of Rta of Epstein-Barr virus. J. Biol. Chem. 2013, 288, 12866–12879. [Google Scholar] [PubMed]
- Izumiya, Y.; Lin, S.F.; Ellison, T.; Chen, L.Y.; Izumiya, C.; Luciw, P.; Kung, H.J. Kaposi’s sarcoma-associated herpesvirus K-bZIP is a coregulator of K-Rta: Physical association and promoter-dependent transcriptional repression. J. Virol. 2003, 77, 1441–1451. [Google Scholar] [PubMed]
- Glaunsinger, B.; Ganem, D. Lytic KSHV infection inhibits host gene expression by accelerating global mRNA turnover. Mol. Cell 2004, 13, 713–723. [Google Scholar] [PubMed]
- Richner, J.M.; Clyde, K.; Pezda, A.C.; Cheng, B.Y.; Wang, T.; Kumar, G.R.; Covarrubias, S.; Coscoy, L.; Glaunsinger, B. Global mRNA degradation during lytic gammaherpesvirus infection contributes to establishment of viral latency. PLoS Pathog. 2011, 7, e1002150. [Google Scholar] [PubMed]
- Glaunsinger, B.; Chavez, L.; Ganem, D. The exonuclease and host shutoff functions of the sox protein of Kaposi’s sarcoma-associated herpesvirus are genetically separable. J. Virol. 2005, 79, 7396–7401. [Google Scholar] [PubMed]
- Lefort, S.; Gravel, A.; Flamand, L. Repression of interferon-alpha stimulated genes expression by Kaposi’s sarcoma-associated herpesvirus K-bZIP protein. Virology 2010, 408, 14–30. [Google Scholar] [PubMed]
- Lefort, S.; Soucy-Faulkner, A.; Grandvaux, N.; Flamand, L. Binding of Kaposi’s sarcoma-associated herpesvirus K-bZIP to interferon-responsive factor 3 elements modulates antiviral gene expression. J. Virol. 2007, 81, 10950–10960. [Google Scholar] [PubMed]
- Kato-Noah, T.; Xu, Y.; Rossetto, C.C.; Colletti, K.; Papouskova, I.; Pari, G.S. Overexpression of the Kaposi’s sarcoma-associated herpesvirus transactivator K-Rta can complement a K-bZIP deletion bacmid and yields an enhanced growth phenotype. J. Virol. 2007, 81, 13519–13532. [Google Scholar] [PubMed]
- Wang, Y.; Sathish, N.; Hollow, C.; Yuan, Y. Functional characterization of Kaposi’s sarcoma-associated herpesvirus open reading frame k8 by bacterial artificial chromosome-based mutagenesis. J. Virol. 2011, 85, 1943–1957. [Google Scholar] [PubMed]
- Hagemeier, S.R.; Dickerson, S.J.; Meng, Q.; Yu, X.; Mertz, J.E.; Kenney, S.C. Sumoylation of the Epstein-Barr virus BZLF1 protein inhibits its transcriptional activity and is regulated by the virus-encoded protein kinase. J. Virol. 2010, 84, 4383–4394. [Google Scholar] [PubMed]
- Full, F.; Jungnickl, D.; Reuter, N.; Bogner, E.; Brulois, K.; Scholz, B.; Sturzl, M.; Myoung, J.; Jung, J.U.; Stamminger, T.; et al. Kaposi’s sarcoma associated herpesvirus tegument protein ORF75 is essential for viral lytic replication and plays a critical role in the antagonization of ND10-instituted intrinsic immunity. PLoS Pathog. 2014, 10, e1003863. [Google Scholar] [PubMed]
- Ballestas, M.E.; Chatis, P.A.; Kaye, K.M. Efficient persistence of extrachromosomal KSHV DNA mediated by latency-associated nuclear antigen. Science 1999, 284, 641–644. [Google Scholar] [PubMed]
- Garber, A.C.; Hu, J.; Renne, R. Latency-associated nuclear antigen (LANA) cooperatively binds to two sites within the terminal repeat, and both sites contribute to the ability of LANA to suppress transcription and to facilitate DNA replication. J. Biol. Chem. 2002, 277, 27401–27411. [Google Scholar] [PubMed]
- Hu, J.; Garber, A.C.; Renne, R. The latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus supports latent DNA replication in dividing cells. J. Virol. 2002, 76, 11677–11687. [Google Scholar] [PubMed]
- Lu, J.; Jha, H.C.; Verma, S.C.; Sun, Z.; Banerjee, S.; Dzeng, R.; Robertson, E.S. Kaposi’s sarcoma-associated herpesvirus-encoded LANA contributes to viral latent replication by activating phosphorylation of survivin. J. Virol. 2014, 88, 4204–4217. [Google Scholar] [PubMed]
- Lu, J.; Verma, S.C.; Murakami, M.; Cai, Q.; Kumar, P.; Xiao, B.; Robertson, E.S. Latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus (KSHV) upregulates survivin expression in KSHV-associated B-lymphoma cells and contributes to their proliferation. J. Virol. 2009, 83, 7129–7141. [Google Scholar] [PubMed]
- Garber, A.C.; Shu, M.A.; Hu, J.; Renne, R. DNA binding and modulation of gene expression by the latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2001, 75, 7882–7892. [Google Scholar] [PubMed]
- Renne, R.; Barry, C.; Dittmer, D.; Compitello, N.; Brown, P.O.; Ganem, D. Modulation of cellular and viral gene expression by the latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2001, 75, 458–468. [Google Scholar] [PubMed]
- Sun, R.; Liang, D.; Gao, Y.; Lan, K. Kaposi’s sarcoma-associated herpesvirus-encoded LANA interacts with host KAP1 to facilitate establishment of viral latency. J. Virol. 2014, 88, 7331–7344. [Google Scholar] [PubMed]
- Friborg, J., Jr.; Kong, W.; Hottiger, M.O.; Nabel, G.J. P53 inhibition by the LANA protein of KSHV protects against cell death. Nature 1999, 402, 889–894. [Google Scholar] [PubMed]
- Katano, H.; Sato, Y.; Sata, T. Expression of p53 and human herpesvirus-8 (HHV-8)-encoded latency-associated nuclear antigen with inhibition of apoptosis in HHV-8-associated malignancies. Cancer 2001, 92, 3076–3084. [Google Scholar] [PubMed]
- Chang, P.C.; Fitzgerald, L.D.; van Geelen, A.; Izumiya, Y.; Ellison, T.J.; Wang, D.H.; Ann, D.K.; Luciw, P.A.; Kung, H.J. Kruppel-associated box domain-associated protein-1 as a latency regulator for Kaposi’s sarcoma-associated herpesvirus and its modulation by the viral protein kinase. Cancer Res. 2009, 69, 5681–5689. [Google Scholar] [PubMed]
- Wies, E.; Mori, Y.; Hahn, A.; Kremmer, E.; Sturzl, M.; Fleckenstein, B.; Neipel, F. The viral interferon-regulatory factor-3 is required for the survival of KSHV-infected primary effusion lymphoma cells. Blood 2008, 111, 320–327. [Google Scholar] [PubMed]
- Marcos-Villar, L.; Lopitz-Otsoa, F.; Gallego, P.; Munoz-Fontela, C.; Gonzalez-Santamaria, J.; Campagna, M.; Shou-Jiang, G.; Rodriguez, M.S.; Rivas, C. Kaposi’s sarcoma-associated herpesvirus protein LANA2 disrupts PML oncogenic domains and inhibits PML-mediated transcriptional repression of the survivin gene. J. Virol. 2009, 83, 8849–8858. [Google Scholar] [PubMed]
- Marcos-Villar, L.; Campagna, M.; Lopitz-Otsoa, F.; Gallego, P.; Gonzalez-Santamaria, J.; Gonzalez, D.; Rodriguez, M.S.; Rivas, C. Covalent modification by SUMO is required for efficient disruption of PML oncogenic domains by Kaposi’s sarcoma-associated herpesvirus latent protein LANA2. J. Gen. Virol. 2011, 92, 188–194. [Google Scholar] [PubMed]
- Marcos-Villar, L.; Gallego, P.; Munoz-Fontela, C.; de la Cruz-Herrera, C.F.; Campagna, M.; Gonzalez, D.; Lopitz-Otsoa, F.; Rodriguez, M.S.; Rivas, C. Kaposi’s sarcoma-associated herpesvirus LANA2 protein interacts with the pocket proteins and inhibits their sumoylation. Oncogene 2014, 33, 495–503. [Google Scholar] [PubMed]
- Park, J.; Lee, D.; Seo, T.; Chung, J.; Choe, J. Kaposi’s sarcoma-associated herpesvirus (human herpesvirus-8) open reading frame 36 protein is a serine protein kinase. J. Gen. Virol. 2000, 81, 1067–1071. [Google Scholar] [PubMed]
- Izumiya, Y.; Izumiya, C.; van Geelen, A.; Wang, D.H.; Lam, K.S.; Luciw, P.A.; Kung, H.J. Kaposi’s sarcoma-associated herpesvirus-encoded protein kinase and its interaction with K-bZIP. J. Virol. 2007, 81, 1072–1082. [Google Scholar] [PubMed]
- Li, R.; Wang, L.; Liao, G.; Guzzo, C.M.; Matunis, M.J.; Zhu, H.; Hayward, S.D. SUMO binding by the Epstein-Barr virus protein kinase BGLF4 is crucial for BGLF4 function. J. Virol. 2012, 86, 5412–5421. [Google Scholar] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, P.-C.; Kung, H.-J. SUMO and KSHV Replication. Cancers 2014, 6, 1905-1924. https://doi.org/10.3390/cancers6041905
Chang P-C, Kung H-J. SUMO and KSHV Replication. Cancers. 2014; 6(4):1905-1924. https://doi.org/10.3390/cancers6041905
Chicago/Turabian StyleChang, Pei-Ching, and Hsing-Jien Kung. 2014. "SUMO and KSHV Replication" Cancers 6, no. 4: 1905-1924. https://doi.org/10.3390/cancers6041905
APA StyleChang, P. -C., & Kung, H. -J. (2014). SUMO and KSHV Replication. Cancers, 6(4), 1905-1924. https://doi.org/10.3390/cancers6041905