Situational Awareness: Regulation of the Myb Transcription Factor in Differentiation, the Cell Cycle and Oncogenesis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
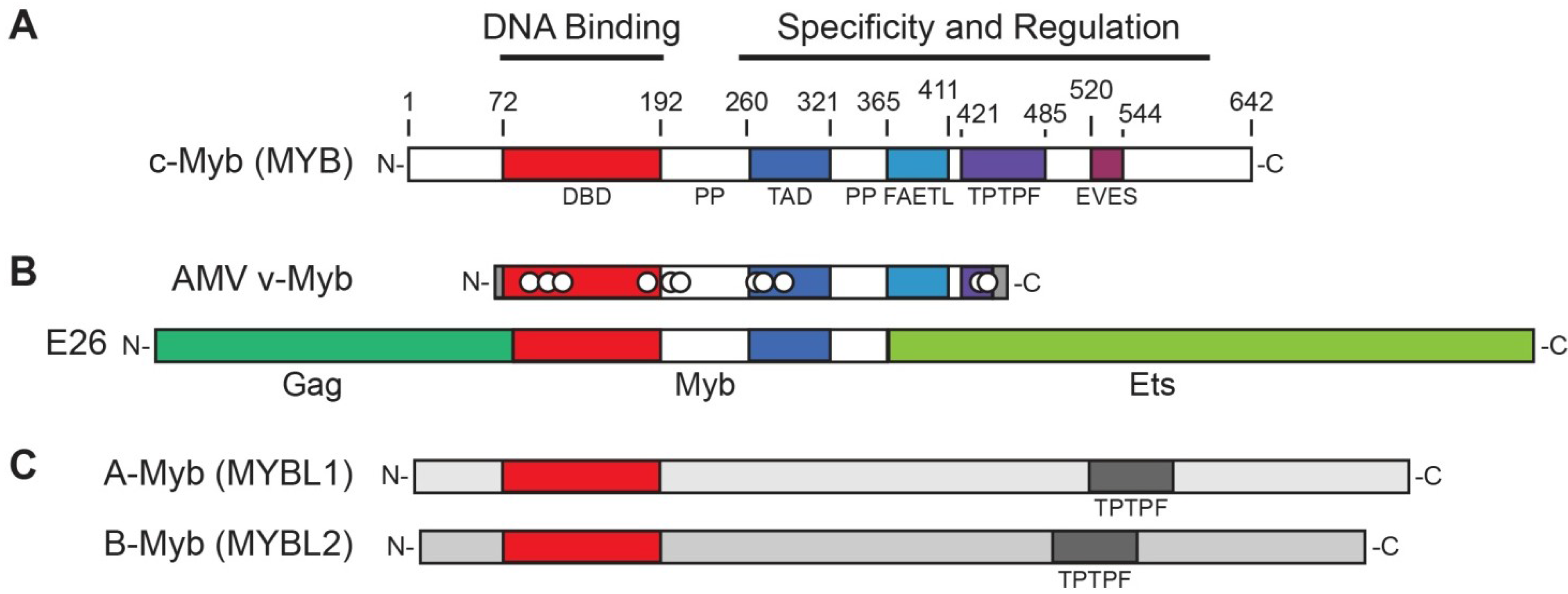
1.1. Structures and Functions of Myb Proteins

1.1.1. Effects of DNA Binding Domain Mutations in v-Myb
1.1.2. Microarray Assays Uncover the Complexity of Myb Activities

1.2. Mechanisms of Myb Activation in Cancer

2. Myb as a Cell Cycle-Regulated Transcription Factor
2.1. Links between Myb and Cell Cycle Regulation
2.1.1. Myb Proteins, Cyclins and CDKs
2.1.2. Myb Regulation of Genes that Regulate the Cell Cycle
2.1.3. Retargeting Myb to Different Promoters during the Cell Cycle

2.2. Regulation of B-Myb in the Cell Cycle
2.3. Contrasting Roles of B-Myb and c-Myb in Cell Cycle Regulation
3. Situation-Specific Activities of Myb Proteins
3.1. Context-Specific Activities of Myb
3.1.1. Myb Activities during Differentiation
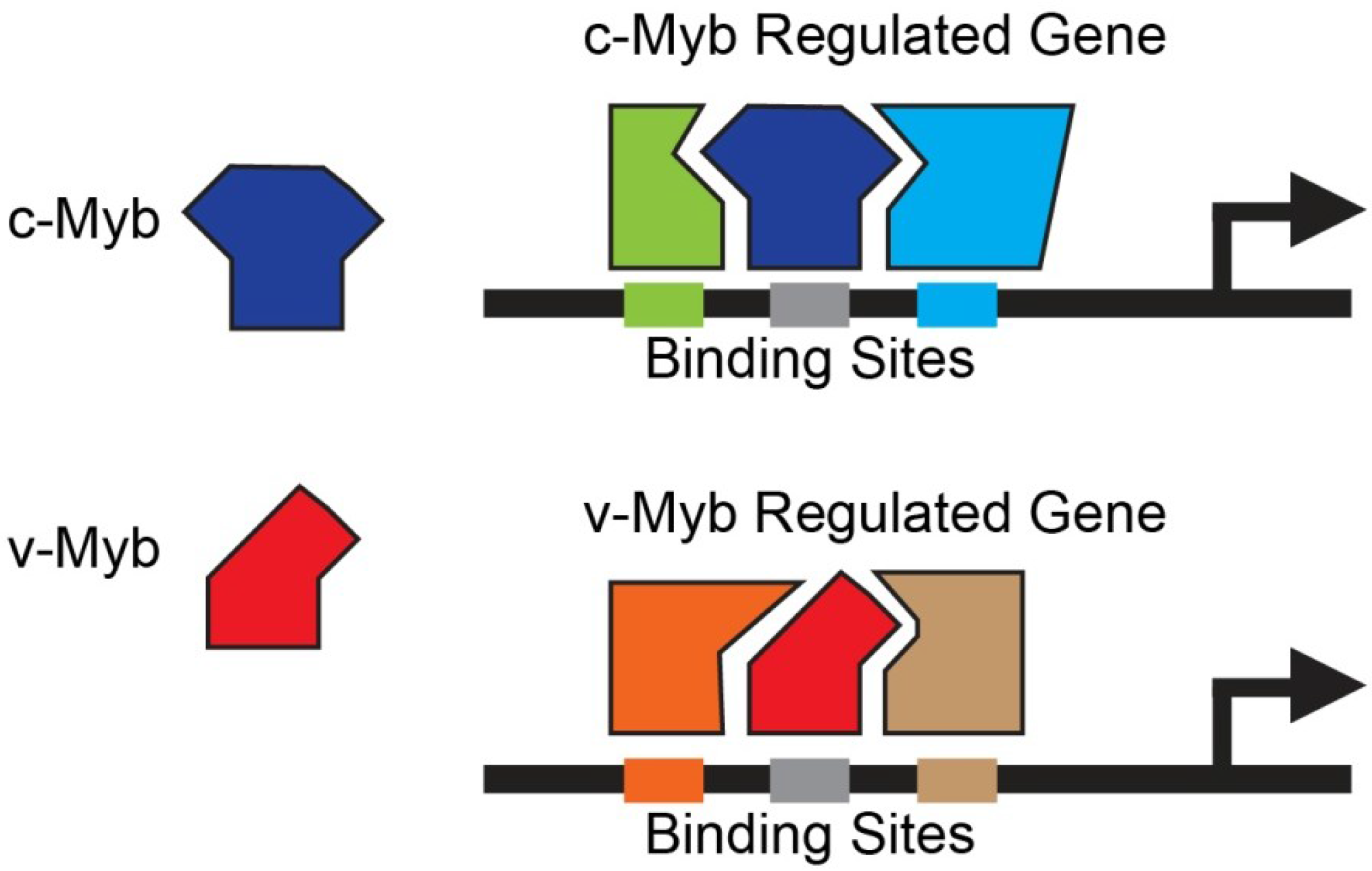
3.1.2. Combinatorial Interactions between Myb and Other Transcription Factors
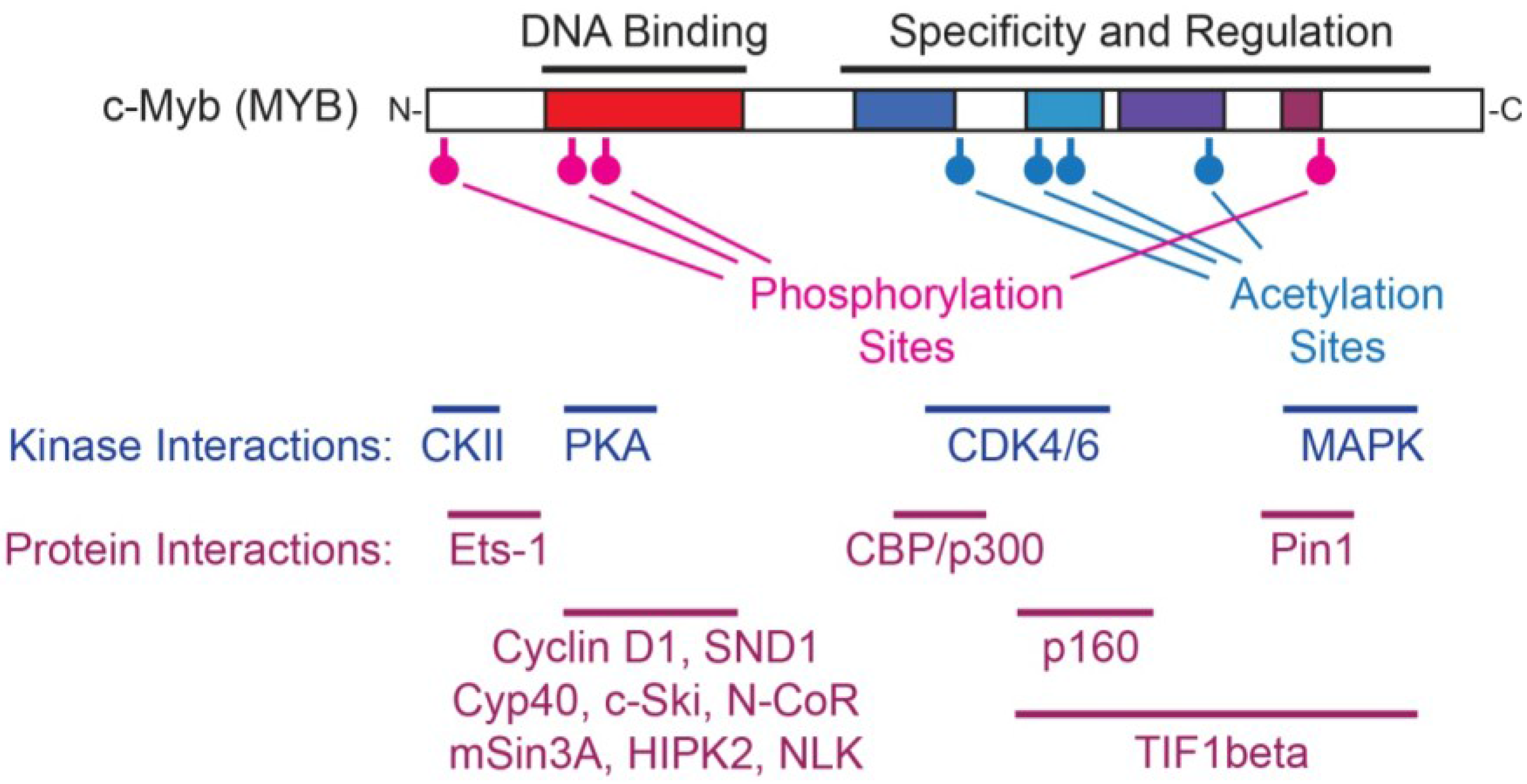
3.1.3. Myb Interactions with CBP and p300
3.2. Protein-Protein Interactions Regulate Myb Activity

4. Conclusions: Protein Interactions Could Provide Novel Therapeutic Targets
Acknowledgments
Conflicts of Interest
References
- Zhou, Y.; Ness, S.A. Myb proteins: Angels and demons in normal and transformed cells. Front Biosci. Landmark Ed. 2011, 16, 1109–1131. [Google Scholar]
- Feller, A.; Machemer, K.; Braun, E.L.; Grotewold, E. Evolutionary and comparative analysis of MYB and bHLH plant transcription factors. Plant J. 2011, 66, 94–116. [Google Scholar]
- Du, H.; Zhang, L.; Liu, L.; Tang, X.F.; Yang, W.J.; Wu, Y.M.; Huang, Y.B.; Tang, Y.X. Biochemical and molecular characterization of plant MYB transcription factor family. Biochem. Biokhimiia 2009, 74, 1–11. [Google Scholar]
- Ramsay, R.G.; Gonda, T.J. MYB function in normal and cancer cells. Nat. Rev. Cancer 2008, 8, 523–534. [Google Scholar]
- Ness, S.A. Myb protein specificity: Evidence of a context-specific transcription factor code. Blood Cells Mol. Dis. 2003, 31, 192–200. [Google Scholar]
- Lipsick, J.S.; Manak, J.; Mitiku, N.; Chen, C.K.; Fogarty, P.; Guthrie, E. Functional evolution of the Myb oncogene family. Blood Cells Mol. Dis. 2001, 27, 456–458. [Google Scholar]
- Ness, S.A. The myb oncoprotein: Regulating a regulator. Biochim. Biophys. Acta 1996, 1288, F123–F139. [Google Scholar]
- Lipsick, J.S. One billion years of Myb. Oncogene 1996, 13, 223–235. [Google Scholar]
- Park, D.J.; Vuong, P.T.; de Vos, S.; Douer, D.; Koeffler, H.P. Comparative analysis of genes regulated by PML/RAR alpha and PLZF/RAR alpha in response to retinoic acid using oligonucleotide arrays. Blood 2003, 102, 3727–3736. [Google Scholar]
- Minucci, S.; Monestiroli, S.; Giavara, S.; Ronzoni, S.; Marchesi, F.; Insinga, A.; Diverio, D.; Gasparini, P.; Capillo, M.; Colombo, E.; et al. PML-RAR induces promyelocytic leukemias with high efficiency following retroviral gene transfer into purified murine hematopoietic progenitors. Blood 2002, 100, 2989–2995. [Google Scholar]
- Rushton, J.J.; Davis, L.M.; Lei, W.; Mo, X.; Leutz, A.; Ness, S.A. Distinct changes in gene expression induced by A-Myb, B-Myb and c-Myb proteins. Oncogene 2003, 22, 308–313. [Google Scholar]
- Rushton, J.J.; Ness, S.A. The conserved DNA binding domain mediates similar regulatory interactions for A-Myb, B-Myb, and c-Myb transcription factors. Blood Cells Mol. Dis. 2001, 27, 459–463. [Google Scholar]
- Dini, P.; Eltman, J.; Lipsick, J. Mutations in the DNA-binding and transcriptional activation domains of v-Myb cooperate in transformation. J. Virol. 1995, 69, 2515–2524. [Google Scholar]
- Dini, P.W.; Lipsick, J.S. Oncogenic truncation of the first repeat of c-Myb decreases DNA binding in vitro and in vivo. Mol. Cell. Biol. 1993, 13, 7334–7348. [Google Scholar]
- Lei, W.; Rushton, J.J.; Davis, L.M.; Liu, F.; Ness, S.A. Positive and negative determinants of target gene specificity in Myb transcription factors. J. Biol. Chem. 2004, 279, 29519–29527. [Google Scholar]
- Gonda, T.J.; Cory, S.; Sobieszczuk, P.; Holtzman, D.; Adams, J.M. Generation of altered transcripts by retroviral insertion within the c-Myb gene in two murine monocytic leukemias. J. Virol. 1987, 61, 2754–2763. [Google Scholar]
- Gonda, T.J.; Buckmaster, C.; Ramsay, R.G. Activation of c-Myb by carboxy-terminal truncation: Relationship to transformation of murine haemopoietic cells in vitro. EMBO J. 1989, 8, 1777–1783. [Google Scholar]
- Dubendorff, J.W.; Whittaker, L.J.; Eltman, J.T.; Lipsick, J.S. Carboxy-terminal elements of c-Myb negatively regulate transcriptional activation in cis and in trans. Genes Dev. 1992, 6, 2524–2535. [Google Scholar]
- Wang, D.M.; Lipsick, J.S. Mutational analysis of the transcriptional activation domains of v-Myb. Oncogene 2002, 21, 1611–1615. [Google Scholar]
- Fu, S.L.; Lipsick, J.S. FAETL motif required for leukemic transformation by v-Myb. J. Virol. 1996, 70, 5600–5610. [Google Scholar]
- Dash, A.B.; Orrico, F.C.; Ness, S.A. The EVES motif mediates both intermolecular and intramolecular regulation of c-Myb. Genes Dev. 1996, 10, 1858–1869. [Google Scholar]
- Leverson, J.D.; Ness, S.A. Point mutations in v-Myb disrupt a cyclophilin-catalyzed negative regulatory mechanism. Mol. Cell 1998, 1, 203–211. [Google Scholar]
- Pani, E.; Menigatti, M.; Schubert, S.; Hess, D.; Gerrits, B.; Klempnauer, K.H.; Ferrari, S. Pin1 interacts with c-Myb in a phosphorylation-dependent manner and regulates its transactivation activity. Biochim. Biophys. Acta 2008, 1783, 1121–1128. [Google Scholar]
- Ness, S.A.; Marknell, A.; Graf, T. The v-Myb oncogene product binds to and activates the promyelocyte-specific mim-1 gene. Cell 1989, 59, 1115–1125. [Google Scholar]
- Introna, M.; Golay, J.; Frampton, J.; Nakano, T.; Ness, S.; Graf, T. Mutations in v-Myb alter the differentiation of myelomonocytic cells transformed by the oncogene. Cell 1990, 63, 1289–1297. [Google Scholar]
- Ogata, K.; Kanai, H.; Inoue, T.; Sekikawa, A.; Sasaki, M.; Nagadoi, A.; Sarai, A.; Ishii, S.; Nishimura, Y. Solution structures of Myb DNA-binding domain and its complex with DNA. Nucleic Acids Symp. Ser. 1993, 29, 201–202. [Google Scholar]
- Ogata, K.; Hojo, H.; Aimoto, S.; Nakai, T.; Nakamura, H.; Sarai, A.; Ishii, S.; Nishimura, Y. Solution structure of a DNA-binding unit of Myb: A helix-turn-helix-related motif with conserved tryptophans forming a hydrophobic core. Proc. Natl. Acad. Sci. USA 1992, 89, 6428–6432. [Google Scholar]
- Tahirov, T.H.; Morii, H.; Uedaira, H.; Sarai, A.; Ogata, K. Crystallization and preliminary X-ray analysis of wild-type and V103L mutant Myb R2 DNA-binding domain. Acta Crystallogr. D Biol. Crystallogr. 1999, 55, 1345–1347. [Google Scholar]
- Boyer, L.A.; Langer, M.R.; Crowley, K.A.; Tan, S.; Denu, J.M.; Peterson, C.L. Essential role for the SANT domain in the functioning of multiple chromatin remodeling enzymes. Mol. Cell 2002, 10, 935–942. [Google Scholar]
- Sterner, D.E.; Wang, X.; Bloom, M.H.; Simon, G.M.; Berger, S.L. The SANT domain of Ada2 is required for normal acetylation of histones by the yeast SAGA complex. J. Biol. Chem. 2002, 277, 8178–8186. [Google Scholar]
- Boyer, L.A.; Latek, R.R.; Peterson, C.L. The SANT domain: A unique histone-tail-binding module? Nat. Rev. Mol. Cell Biol. 2004, 5, 158–163. [Google Scholar]
- Humphrey, G.W.; Wang, Y.; Russanova, V.R.; Hirai, T.; Qin, J.; Nakatani, Y.; Howard, B.H. Stable histone deacetylase complexes distinguished by the presence of SANT domain proteins CoREST/kiaa0071 and Mta-L1. J. Biol. Chem. 2001, 276, 6817–6824. [Google Scholar]
- Zaret, K.S.; Watts, J.; Xu, J.; Wandzioch, E.; Smale, S.T.; Sekiya, T. Pioneer factors, genetic competence, and inductive signaling: Programming liver and pancreas progenitors from the endoderm. Cold Spring Harb. Symp. Quant. Biol. 2008, 73, 119–126. [Google Scholar]
- Zaret, K.S.; Carroll, J.S. Pioneer transcription factors: Establishing competence for gene expression. Genes Dev. 2011, 25, 2227–2241. [Google Scholar]
- Mo, X.; Kowenz-Leutz, E.; Laumonnier, Y.; Xu, H.; Leutz, A. Histone H3 tail positioning and acetylation by the c-Myb but not the v-Myb DNA-binding SANT domain. Genes Dev. 2005, 19, 2447–2457. [Google Scholar]
- Nomura, T.; Tanikawa, J.; Akimaru, H.; Kanei-Ishii, C.; Ichikawa-Iwata, E.; Khan, M.M.; Ito, H.; Ishii, S. Oncogenic activation of c-Myb correlates with a loss of negative regulation by TIF1beta and Ski. J. Biol. Chem. 2004, 279, 16715–16726. [Google Scholar]
- Bies, J.; Markus, J.; Wolff, L. Covalent attachment of the SUMO-1 protein to the negative regulatory domain of the c-Myb transcription factor modifies Its stability and transactivation capacity. J. Biol. Chem. 2002, 277, 8999–9009. [Google Scholar]
- Hu, Y.L.; Ramsay, R.G.; Kanei-Ishii, C.; Ishii, S.; Gonda, T.J. Transformation by carboxyl-deleted Myb reflects increased transactivating capacity and disruption of a negative regulatory domain. Oncogene 1991, 6, 1549–1553. [Google Scholar]
- Liu, F.; Lei, W.; OʼRourke, J.P.; Ness, S.A. Oncogenic mutations cause dramatic, qualitative changes in the transcriptional activity of c-Myb. Oncogene 2006, 25, 795–805. [Google Scholar]
- Clappier, E.; Cuccuini, W.; Kalota, A.; Crinquette, A.; Cayuela, J.M.; Dik, W.A.; Langerak, A.W.; Montpellier, B.; Nadel, B.; Walrafen, P.; et al. The C-MYB locus is involved in chromosomal translocation and genomic duplications in human T-cell acute leukemia (T-ALL), the translocation defining a new T-ALL subtype in very young children. Blood 2007, 110, 1251–1261. [Google Scholar]
- Lahortiga, I.; de Keersmaecker, K.; van Vlierberghe, P.; Graux, C.; Cauwelier, B.; Lambert, F.; Mentens, N.; Beverloo, H.B.; Pieters, R.; Speleman, F.; et al. Duplication of the MYB oncogene in T cell acute lymphoblastic leukemia. Nat. Genet. 2007, 39, 593–595. [Google Scholar]
- Drabsch, Y.; Hugo, H.; Zhang, R.; Dowhan, D.; Miao, Y.; Gewirtz, A.; Barry, S.; Ramsay, R.; Gonda, T. Mechanism of and requirement for estrogen-regulated MYB expression in estrogen-receptor-positive breast cancer cells. Proc. Natl. Acad. Sci. USA 2007, 104, 13762–13767. [Google Scholar]
- Thorner, A.R.; Parker, J.S.; Hoadley, K.A.; Perou, C.M. Potential tumor suppressor role for the c-Myb oncogene in luminal breast cancer. PLoS One 2010, 5, e13073. [Google Scholar]
- Beug, H.; Blundell, P.; Graf, T. Reversibility of differentiation and proliferative capacity in avian myelomonocytic cells transformed by ts E26 leukemia virus. Genes Dev. 1987, 1, 277–286. [Google Scholar]
- Ness, S.A.; Beug, H.; Graf, T. v-Myb dominance over v-Myc in doubly transformed chick myelomonocytic cells. Cell 1987, 51, 41–50. [Google Scholar]
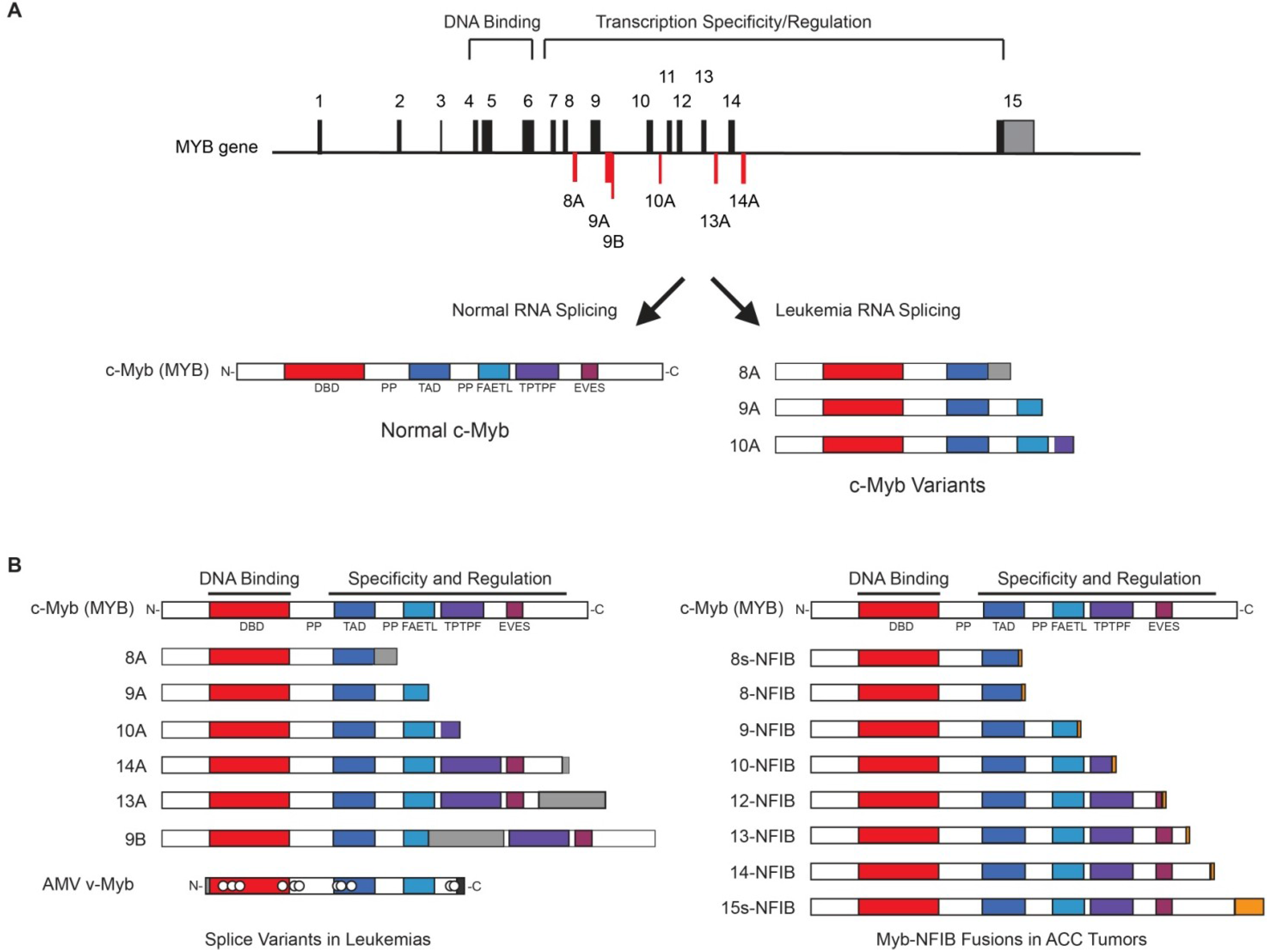
- Zhou, Y.E.; O’Rourke, J.P.; Edwards, J.S.; Ness, S.A. Single molecule analysis of c-Myb alternative splicing reveals novel classifiers for precursor B-ALL. PLoS One 2011, 6, e22880. [Google Scholar]
- O’Rourke, J.P.; Ness, S.A. Alternative RNA splicing produces multiple forms of c-Myb with unique transcriptional activities. Mol. Cell. Biol. 2008, 28, 2091–2101. [Google Scholar]
- Schuur, E.R.; Rabinovich, J.M.; Baluda, M.A. Distribution of alternatively spliced chicken c-Myb exon 9A among hematopoietic tissues. Oncogene 1994, 9, 3363–3365. [Google Scholar]
- Shen-Ong, G.L.; Skurla, R.M.; Owens, J.D.; Mushinski, J.F. Alternative splicing of RNAs transcribed from the human c-Myb gene. Mol. Cell. Biol. 1990, 10, 2715–2722. [Google Scholar]
- Westin, E.H.; Gorse, K.M.; Clarke, M.F. Alternative splicing of the human c-Myb gene. Oncogene 1990, 5, 1117–1124. [Google Scholar]
- Brill, L.B., 2nd; Kanner, W.A.; Fehr, A.; Andren, Y.; Moskaluk, C.A.; Loning, T.; Stenman, G.; Frierson, H.F., Jr. Analysis of MYB expression and MYB-NFIB gene fusions in adenoid cystic carcinoma and other salivary neoplasms. Mod. Pathol. 2011, 24, 1169–1176. [Google Scholar]
- Persson, M.; Andren, Y.; Mark, J.; Horlings, H.M.; Persson, F.; Stenman, G. Recurrent fusion of MYB and NFIB transcription factor genes in carcinomas of the breast and head and neck. Proc. Natl. Acad. Sci. USA 2009, 106, 18740–18744. [Google Scholar]
- Ganter, B.; Fu, S.; Lipsick, J.S. D-type cyclins repress transcriptional activation by the v-Myb but not the c-Myb DNA-binding domain. EMBO J. 1998, 17, 255–268. [Google Scholar]
- Lei, W.; Liu, F.; Ness, S.A. Positive and negative regulation of c-Myb by Cyclin D1, Cyclin-Dependent kinases and p27 Kip1. Blood 2005, 105, 3855–3861. [Google Scholar]
- Nakata, Y.; Shetzline, S.; Sakashita, C.; Kalota, A.; Rallapalli, R.; Rudnick, S.; Zhang, Y.; Emerson, S.; Gewirtz, A. c-Myb contributes to G2/M cell cycle transition in human hematopoietic cells by direct regulation of cyclin B1 expression. Mol. Cell. Biol. 2007, 27, 2048–2058. [Google Scholar]
- Frampton, J.; Ramqvist, T.; Graf, F. v-Myb of E26 leukemia virus up-regulates bcl-2 and suppresses apoptosis in myeloid cells. Genes Dev. 1996, 10, 2720–2731. [Google Scholar]
- Wasner, M.; Haugwitz, U.; Reinhard, W.; Tschop, K.; Spiesbach, K.; Lorenz, J.; Mossner, J.; Engeland, K. Three CCAAT-boxes and a single cell cycle genes homology region (CHR) are the major regulating sites for transcription from the human cyclin B2 promoter. Gene 2003, 312, 225–237. [Google Scholar]
- Wasner, M.; Tschop, K.; Spiesbach, K.; Haugwitz, U.; Johne, C.; Mossner, J.; Mantovani, R.; Engeland, K. Cyclin B1 transcription is enhanced by the p300 coactivator and regulated during the cell cycle by a CHR-dependent repression mechanism. FEBS Lett. 2003, 536, 66–70. [Google Scholar]
- Spender, L.C.; Inman, G.J. Developments in Burkitt’s lymphoma: Novel cooperations in oncogenic MYC signaling. Cancer Manag. Res. 2014, 6, 27–38. [Google Scholar]
- Bretones, G.; Delgado, M.D.; Leon, J. Myc and cell cycle control. Biochim. Biophys. Acta 2014. [Google Scholar] [CrossRef]
- Nakagoshi, H.; Kanei-Ishii, C.; Sawazaki, T.; Mizuguchi, G.; Ishii, S. Transcriptional activation of the c-myc gene by the c-Myb and B-Myb gene products. Oncogene 1992, 7, 1233–1240. [Google Scholar]
- Cogswell, J.P.; Cogswell, P.C.; Kuehl, W.M.; Cuddihy, A.M.; Bender, T.M.; Engelke, U.; Marcu, K.B.; Ting, J.P. Mechanism of c-Myc regulation by c-Myb in different cell lineages. Mol. Cell. Biol. 1993, 13, 2858–2869. [Google Scholar]
- Berge, T.; Matre, V.; Brendeford, E.M.; Saether, T.; Luscher, B.; Gabrielsen, O.S. Revisiting a selection of target genes for the hematopoietic transcription factor c-Myb using chromatin immunoprecipitation and c-Myb knockdown. Blood Cells Mol. Dis. 2007, 39, 278–286. [Google Scholar]
- Ciznadija, D.; Tothill, R.; Waterman, M.L.; Zhao, L.; Huynh, D.; Yu, R.M.; Ernst, M.; Ishii, S.; Mantamadiotis, T.; Gonda, T.J.; et al. Intestinal adenoma formation and MYC activation are regulated by cooperation between MYB and Wnt signaling. Cell Death Differ. 2009, 16, 1530–1538. [Google Scholar]
- Quintana, A.M.; Liu, F.; O’Rourke, J.P.; Ness, S.A. Identification and regulation of c-Myb target genes in MCF-7 cells. BMC Cancer 2011, 11, 30. [Google Scholar]
- Shi, J.; Whyte, W.A.; Zepeda-Mendoza, C.J.; Milazzo, J.P.; Shen, C.; Roe, J.S.; Minder, J.L.; Mercan, F.; Wang, E.; Eckersley-Maslin, M.A.; et al. Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Genes Dev. 2013, 27, 2648–2662. [Google Scholar]
- Loven, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar]
- Wright, J.B.; Brown, S.J.; Cole, M.D. Upregulation of c-MYC in cis through a large chromatin loop linked to a cancer risk-associated single-nucleotide polymorphism in colorectal cancer cells. Mol. Cell. Biol. 2010, 30, 1411–1420. [Google Scholar]
- Cheasley, D.; Pereira, L.; Lightowler, S.; Vincan, E.; Malaterre, J.; Ramsay, R.G. Myb controls intestinal stem cell genes and self-renewal. Stem Cells 2011, 29, 2042–2050. [Google Scholar]
- Malaterre, J.; Carpinelli, M.; Ernst, M.; Alexander, W.; Cooke, M.; Sutton, S.; Dworkin, S.; Heath, J.K.; Frampton, J.; McArthur, G.; et al. c-Myb is required for progenitor cell homeostasis in colonic crypts. Proc. Natl. Acad. Sci. USA 2007, 104, 3829–3834. [Google Scholar]
- Quintana, A.M.; Zhou, Y.E.; Pena, J.J.; O’Rourke, J.P.; Ness, S.A. Dramatic repositioning of c-Myb to different promoters during the cell cycle observed by combining cell sorting with chromatin immunoprecipitation. PLoS One 2011, 6, e17362. [Google Scholar]
- Bartusel, T.; Schubert, S.; Klempnauer, K.H. Regulation of the cyclin D1 and cyclin A1 promoters by B-Myb is mediated by Sp1 binding sites. Gene 2005, 351, 171–180. [Google Scholar]
- Joaquin, M.; Watson, R.J. Cell cycle regulation by the B-Myb transcription factor. Cell. Mol. Life Sci. 2003, 60, 2389–2401. [Google Scholar]
- Gonda, T.J.; Leo, P.; Ramsay, R.G. Estrogen and MYB in breast cancer: Potential for new therapies. Expert Opin. Biol. Ther. 2008, 8, 713–717. [Google Scholar]
- Greig, K.T.; Carotta, S.; Nutt, S.L. Critical roles for c-Myb in hematopoietic progenitor cells. Semin. Immunol. 2008, 20, 247–256. [Google Scholar]
- Ramsay, R.G.; Barton, A.L.; Gonda, T.J. Targeting c-Myb expression in human disease. Expert Opin. Ther. Targets 2003, 7, 235–248. [Google Scholar]
- Sala, A.; Watson, R. B-Myb protein in cellular proliferation, transcription control, and cancer: Latest developments. J. Cell. Physiol. 1999, 179, 245–250. [Google Scholar]
- Martinez, I.; Dimaio, D. B-Myb, cancer, senescence, and microRNAs. Cancer Res. 2011, 71, 5370–5373. [Google Scholar]
- Zhu, W.; Giangrande, P.H.; Nevins, J.R. E2Fs link the control of G1/S and G2/M transcription. EMBO J. 2004, 23, 4615–4626. [Google Scholar]
- Johnson, L.R.; Johnson, T.K.; Desler, M.; Luster, T.A.; Nowling, T.; Lewis, R.E.; Rizzino, A. Effects of B-Myb on gene transcription: Phosphorylation-dependent activity and acetylation by p300. J. Biol. Chem. 2002, 277, 4088–4097. [Google Scholar]
- Robinson, C.; Light, Y.; Groves, R.; Mann, D.; Marias, R.; Watson, R. Cell-cycle regulation of B-Myb protein expression: Specific phosphorylation during the S phase of the cell cycle. Oncogene 1996, 12, 1855–1864. [Google Scholar]
- Knight, A.S.; Notaridou, M.; Watson, R.J. A Lin-9 complex is recruited by B-Myb to activate transcription of G2/M genes in undifferentiated embryonal carcinoma cells. Oncogene 2009, 28, 1737–1747. [Google Scholar]
- Mannefeld, M.; Klassen, E.; Gaubatz, S. B-Myb is required for recovery from the DNA damage-induced G2 checkpoint in p53 mutant cells. Cancer Res. 2009, 69, 4073–4080. [Google Scholar]
- Schmit, F.; Cremer, S.; Gaubatz, S. LIN54 is an essential core subunit of the DREAM/LINC complex that binds to the cdc2 promoter in a sequence-specific manner. FEBS J. 2009, 276, 5703–5716. [Google Scholar]
- Sadasivam, S.; Duan, S.; DeCaprio, J.A. The MuvB complex sequentially recruits B-Myb and FoxM1 to promote mitotic gene expression. Genes Dev. 2012, 26, 474–489. [Google Scholar]
- Bar-Shira, A.; Pinthus, J.H.; Rozovsky, U.; Goldstein, M.; Sellers, W.R.; Yaron, Y.; Eshhar, Z.; Orr-Urtreger, A. Multiple genes in human 20q13 chromosomal region are involved in an advanced prostate cancer xenograft. Cancer Res. 2002, 62, 6803–6807. [Google Scholar]
- Raschella, G.; Cesi, V.; Amendola, R.; Negroni, A.; Tanno, B.; Altavista, P.; Tonini, G.P.; de Bernardi, B.; Calabretta, B. Expression of B-Myb in neuroblastoma tumors is a poor prognostic factor independent from MYCN amplification. Cancer Res. 1999, 59, 3365–3368. [Google Scholar]
- Thorner, A.R.; Hoadley, K.A.; Parker, J.S.; Winkel, S.; Millikan, R.C.; Perou, C.M. In vitro and in vivo analysis of B-Myb in basal-like breast cancer. Oncogene 2009, 28, 742–751. [Google Scholar]
- Sala, A.; Calabretta, B. Regulation of BALB/c 3T3 fibroblast proliferation by B-Myb is accompanied by selective activation of cdc2 and cyclin D1 expression. Proc. Natl. Acad. Sci. USA 1992, 89, 10415–10419. [Google Scholar]
- Sala, A.; de Luca, A.; Giordano, A.; Peschle, C. The retinoblastoma family member p107 binds to B-Myb and suppresses its autoregulatory activity. J. Biol. Chem. 1996, 271, 28738–28740. [Google Scholar]
- Marhamati, D.J.; Bellas, R.E.; Arsura, M.; Kypreos, K.E.; Sonenshein, G.E. A-Myb is expressed in bovine vascular smooth muscle cells during the late G1-to-S phase transition and cooperates with c-myc to mediate progression to S phase. Mol. Cell. Biol. 1997, 17, 2448–2457. [Google Scholar]
- Mucenski, M.L.; McLain, K.; Kier, A.B.; Swerdlow, S.H.; Schreiner, C.M.; Miller, T.A.; Pietryga, D.W.; Scott, W.J.; Potter, S.S. A functional c-Myb gene is required for normal murine fetal hepatic hematopoiesis. Cell 1991, 65, 677–689. [Google Scholar]
- Emambokus, N.; Vegiopoulos, A.; Harman, B.; Jenkinson, E.; Anderson, G.; Frampton, J. Progression through key stages of haemopoiesis is dependent on distinct threshold levels of c-Myb. EMBO J. 2003, 22, 4478–4488. [Google Scholar]
- Hess, J.L.; Bittner, C.B.; Zeisig, D.T.; Bach, C.; Fuchs, U.; Borkhardt, A.; Frampton, J.; Slany, R.K. c-Myb is an essential downstream target for homeobox-mediated transformation of hematopoietic cells. Blood 2006, 108, 297–304. [Google Scholar]
- Vegiopoulos, A.; Garcia, P.; Emambokus, N.; Frampton, J. Coordination of erythropoiesis by the transcription factor c-Myb. Blood 2006, 107, 4703–4710. [Google Scholar]
- Allen, R.D. c-Myb is essential for early T cell development. Genes Dev. 1999, 13, 1073–1078. [Google Scholar]
- Bender, T.P.; Kremer, C.S.; Kraus, M.; Buch, T.; Rajewsky, K. Critical functions for c-Myb at three checkpoints during thymocyte development. Nat. Immunol. 2004, 5, 721–729. [Google Scholar]
- Fahl, S.P.; Crittenden, R.B.; Allman, D.; Bender, T.P. c-Myb is required for pro-B cell differentiation. J. Immunol. 2009, 183, 5582–5592. [Google Scholar]
- Thomas, M.D.; Kremer, C.S.; Ravichandran, K.S.; Rajewsky, K.; Bender, T.P. c-Myb is critical for B cell development and maintenance of follicular B cells. Immunity 2005, 23, 275–286. [Google Scholar]
- Sandberg, M.L.; Sutton, S.E.; Pletcher, M.T.; Wiltshire, T.; Tarantino, L.M.; Hogenesch, J.B.; Cooke, M.P. c-Myb and p300 regulate hematopoietic stem cell proliferation and differentiation. Dev. Cell 2005, 8, 153–166. [Google Scholar]
- Lu, B.C.; Cebrian, C.; Chi, X.; Kuure, S.; Kuo, R.; Bates, C.M.; Arber, S.; Hassell, J.; MacNeil, L.; Hoshi, M.; et al. Etv4 and Etv5 are required downstream of GDNF and Ret for kidney branching morphogenesis. Nat. Genet. 2009, 41, 1295–1302. [Google Scholar]
- Zorbas, M.; Sicurella, C.; Bertoncello, I.; Venter, D.; Ellis, S.; Mucenski, M.L.; Ramsay, R.G. c-Myb is critical for murine colon development. Oncogene 1999, 18, 5821–5830. [Google Scholar]
- Kolodziejska, K.M.; Noyan-Ashraf, M.H.; Nagy, A.; Bacon, A.; Frampton, J.; Xin, H.B.; Kotlikoff, M.I.; Husain, M. c-Myb-dependent smooth muscle cell differentiation. Circ. Res. 2008, 102, 554–561. [Google Scholar]
- Malaterre, J.; Mantamadiotis, T.; Dworkin, S.; Lightowler, S.; Yang, Q.; Ransome, M.I.; Turnley, A.M.; Nichols, N.R.; Emambokus, N.R.; Frampton, J.; et al. c-Myb is required for neural progenitor cell proliferation and maintenance of the neural stem cell niche in adult brain. Stem Cells 2008, 26, 173–181. [Google Scholar]
- Hofmann, J.W.; McBryan, T.; Adams, P.D.; Sedivy, J.M. The effects of aging on the expression of Wnt pathway genes in mouse tissues. Age 2014, 36, 9618. [Google Scholar]
- Ness, S.A.; Kowenz-Leutz, E.; Casini, T.; Graf, T.; Leutz, A. Myb and NF-M: Combinatorial activators of myeloid genes in heterologous cell types. Genes Dev. 1993, 7, 749–759. [Google Scholar]
- Chayka, O.; Kintscher, J.; Braas, D.; Klempnauer, K.H. v-Myb mediates cooperation of a cell-specific enhancer with the mim-1 promoter. Mol. Cell. Biol. 2005, 25, 499–511. [Google Scholar]
- Yamkamon, V.; Ivanova, O.; Braas, D.; Chayka, O.; Patmasiriwat, P.; Klempnauer, K.H. A dual activation mechanism for Myb-responsive genes in myelomonocytic cells. Blood Cells Mol. Dis. 2008, 40, 219–226. [Google Scholar]
- Wilczek, C.; Chayka, O.; Plachetka, A.; Klempnauer, K.H. Myb-induced chromatin remodeling at a dual enhancer/promoter element involves non-coding rna transcription and is disrupted by oncogenic mutations of v-Myb. J. Biol. Chem. 2009, 284, 35314–35324. [Google Scholar]
- Tomita, A.; Towatari, M.; Tsuzuki, S.; Hayakawa, F.; Kosugi, H.; Tamai, K.; Miyazaki, T.; Kinoshita, T.; Saito, H. c-Myb acetylation at the carboxyl-terminal conserved domain by transcriptional co-activator p300. Oncogene 2000, 19, 444–451. [Google Scholar]
- Dai, P.; Akimaru, H.; Tanaka, Y.; Hou, D.X.; Yasukawa, T.; Kanei-Ishii, C.; Takahashi, T.; Ishii, S. CBP as a transcriptional coactivator of c-Myb. Genes Dev. 1996, 10, 528–540. [Google Scholar]
- Kasper, L.H.; Fukuyama, T.; Lerach, S.; Chang, Y.; Xu, W.; Wu, S.; Boyd, K.L.; Brindle, P.K. Genetic interaction between mutations in c-Myb and the KIX domains of CBP and p300 affects multiple blood cell lineages and influences both gene activation and repression. PLoS One 2013, 8, e82684. [Google Scholar]
- Zor, T.; de Guzman, R.N.; Dyson, H.J.; Wright, P.E. Solution structure of the KIX domain of CBP bound to the transactivation domain of c-Myb. J. Mol. Biol. 2004, 337, 521–534. [Google Scholar]
- Bayly, R.; Chuen, L.; Currie, R.A.; Hyndman, B.D.; Casselman, R.; Blobel, G.A.; LeBrun, D.P. E2A-PBX1 interacts directly with the KIX domain of CBP/p300 in the induction of proliferation in primary hematopoietic cells. J. Biol. Chem. 2004, 279, 55362–55371. [Google Scholar]
- Denis, C.M.; Chitayat, S.; Plevin, M.J.; Wang, F.; Thompson, P.; Liu, S.; Spencer, H.L.; Ikura, M.; LeBrun, D.P.; Smith, S.P. Structural basis of CBP/p300 recruitment in leukemia induction by E2A-PBX1. Blood 2012, 120, 3968–3977. [Google Scholar]
- Mink, S.; Haenig, B.; Klempnauer, K.H. Interaction and functional collaboration of p300 and C/EBPbeta. Mol. Cell. Biol. 1997, 17, 6609–6617. [Google Scholar]
- Mukherjee, S.P.; Behar, M.; Birnbaum, H.A.; Hoffmann, A.; Wright, P.E.; Ghosh, G. Analysis of the RelA:CBP/p300 interaction reveals its involvement in NF-kappaB-driven transcription. PLoS Biol. 2013, 11, e1001647. [Google Scholar]
- Wang, F.; Marshall, C.B.; Li, G.Y.; Yamamoto, K.; Mak, T.W.; Ikura, M. Synergistic interplay between promoter recognition and CBP/p300 coactivator recruitment by FOXO3a. ACS Chem. Biol. 2009, 4, 1017–1027. [Google Scholar]
- Yang, C.; Shapiro, L.H.; Rivera, M.; Kumar, A.; Brindle, P.K. A role for CREB binding protein and p300 transcriptional coactivators in Ets-1 transactivation functions. Mol. Cell. Biol. 1998, 18, 2218–2229. [Google Scholar]
- Toto, A.; Giri, R.; Brunori, M.; Gianni, S. The mechanism of binding of the KIX domain to the Mixed Lineage Leukemia protein and its allosteric role in the recognition of c-Myb. Protein Sci. 2014, 23, 962–969. [Google Scholar]
- Goto, N.K.; Zor, T.; Martinez-Yamout, M.; Dyson, H.J.; Wright, P.E. Cooperativity in transcription factor binding to the coactivator CREB-binding protein (CBP). The mixed lineage leukemia protein (MLL) activation domain binds to an allosteric site on the KIX domain. J. Biol. Chem. 2002, 277, 43168–43174. [Google Scholar]
- Arai, M.; Dyson, H.J.; Wright, P.E. Leu628 of the KIX domain of CBP is a key residue for the interaction with the MLL transactivation domain. FEBS Lett. 2010, 584, 4500–4504. [Google Scholar]
- Luscher, B.; Christenson, E.; Litchfield, D.W.; Krebs, E.G.; Eisenman, R.N. Myb DNA binding inhibited by phosphorylation at a site deleted during oncogenic activation. Nature 1990, 344, 517–522. [Google Scholar]
- Lüscher, B.; Eisenman, R.N. Mitosis-specific phosphorylation of the nuclear oncoproteins Myc and Myb. J. Cell Biol. 1992, 118, 775–784. [Google Scholar]
- Aziz, N.; Wu, J.; Dubendorff, J.W.; Lipsick, J.S.; Sturgill, T.W.; Bender, T.P. c-Myb and v-Myb are differentially phosphorylated by p42mapk in vitro. Oncogene 1993, 8, 2259–2265. [Google Scholar]
- Bousset, K.; Oelgeschlager, M.H.; Henriksson, M.; Schreek, S.; Burkhardt, H.; Litchfield, D.W.; Luscher-Firzlaff, J.M.; Luscher, B. Regulation of transcription factors c-Myc, Max, and c-Myb by casein kinase II. Cell. Mol. Biol. Res. 1994, 40, 501–511. [Google Scholar]
- Aziz, N.; Miglarese, M.R.; Hendrickson, R.C.; Shabanowitz, J.; Sturgill, T.W.; Hunt, D.F.; Bender, T.P. Modulation of c-Myb-induced transcription activation by a phosphorylation site near the negative regulatory domain. Proc. Natl. Acad. Sci. USA 1995, 92, 6429–6433. [Google Scholar]
- Oelgeschlager, M.; Krieg, J.; Luscher-Firzlaff, J.M.; Luscher, B. Casein kinase II phosphorylation site mutations in c-Myb affect DNA binding and transcriptional cooperativity with NF-M. Mol. Cell. Biol. 1995, 15, 5966–5974. [Google Scholar]
- Ramsay, R.G.; Morrice, N.; van Eeden, P.; Kanagasundaram, V.; Nomura, T.; de Blaquiere, J.; Ishii, S.; Wettenhall, R. Regulation of c-Myb through protein phosphorylation and leucine zipper interactions. Oncogene 1995, 11, 2113–2120. [Google Scholar]
- Winn, L.M.; Lei, W.; Ness, S.A. Pim-1 phosphorylates the DNA binding domain of c-Myb. Cell Cycle 2003, 2, 258–262. [Google Scholar]
- Bies, J.; Sramko, M.; Wolff, L. Stress-induced Phosphorylation of Thr486 in c-Myb by p38MAPKs Attenuates Conjugation of SUMO-2/3. J. Biol. Chem. 2013, 288, 36983–36993. [Google Scholar]
- Kitagawa, K.; Kotake, Y.; Hiramatsu, Y.; Liu, N.; Suzuki, S.; Nakamura, S.; Kikuchi, A.; Kitagawa, M. GSK3 regulates the expressions of human and mouse c-Myb via different mechanisms. Cell Div. 2010, 5, 27. [Google Scholar]
- Sramko, M.; Markus, J.; Kabát, J.; Wolff, L.; Bies, J. Stress-induced inactivation of the c-Myb transcription factor through conjugation of SUMO-2/3 proteins. J. Biol. Chem. 2006, 281, 40065–40075. [Google Scholar]
- Best, J.L.; Amezcua, C.A.; Mayr, B.; Flechner, L.; Murawsky, C.M.; Emerson, B.; Zor, T.; Gardner, K.H.; Montminy, M. Identification of small-molecule antagonists that inhibit an activator: Coactivator interaction. Proc. Natl. Acad. Sci. USA 2004, 101, 17622–17627. [Google Scholar]
- Sun, H.; Chung, W.C.; Ryu, S.H.; Ju, Z.; Tran, H.T.; Kim, E.; Kurie, J.M.; Koo, J.S. Cyclic AMP-responsive element binding protein- and nuclear factor-kappaB-regulated CXC chemokine gene expression in lung carcinogenesis. Cancer Prev. Res. (Phila) 2008, 1, 316–328. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
George, O.L.; Ness, S.A. Situational Awareness: Regulation of the Myb Transcription Factor in Differentiation, the Cell Cycle and Oncogenesis. Cancers 2014, 6, 2049-2071. https://doi.org/10.3390/cancers6042049
George OL, Ness SA. Situational Awareness: Regulation of the Myb Transcription Factor in Differentiation, the Cell Cycle and Oncogenesis. Cancers. 2014; 6(4):2049-2071. https://doi.org/10.3390/cancers6042049
Chicago/Turabian StyleGeorge, Olivia L., and Scott A. Ness. 2014. "Situational Awareness: Regulation of the Myb Transcription Factor in Differentiation, the Cell Cycle and Oncogenesis" Cancers 6, no. 4: 2049-2071. https://doi.org/10.3390/cancers6042049
APA StyleGeorge, O. L., & Ness, S. A. (2014). Situational Awareness: Regulation of the Myb Transcription Factor in Differentiation, the Cell Cycle and Oncogenesis. Cancers, 6(4), 2049-2071. https://doi.org/10.3390/cancers6042049