Standard of Care and Promising New Agents for Triple Negative Metastatic Breast Cancer

Abstract
:1. Introduction
2. Genetic Subtyping of TNBC: Differential Sensitivity to Chemotherapy According to Gene Prophile, Current Treatment and Novel Strategies
2.1. BL1 Subtype

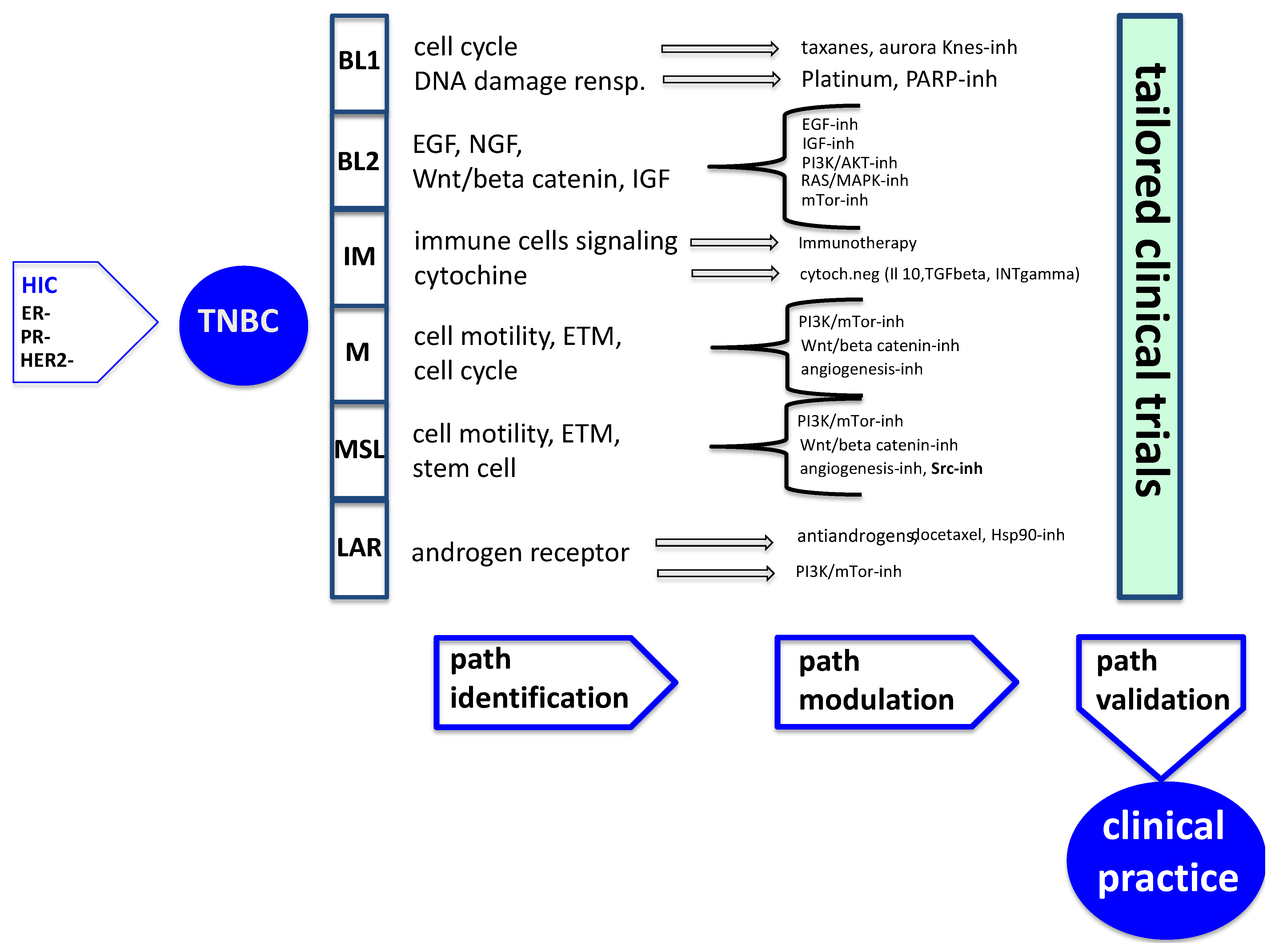{kind=link}
| Gene Expression | Therapeutic Agents | pCR to CT | CT | |
|---|---|---|---|---|
| BL1 | Cell cycle pathway, cell division pathway | Taxanes/Anthracicline Platinum | 52% | +++ |
| Proliferation pathway (AURKA, AURKB, MYC, NRAS) | Aurora kinases inhibitor (AMG900, AS703569) | |||
| DNA damage pathway (ATR/BRCA pathway) | PARP-inhibitor (iniparib, olaparib, veliparib) | |||
| RNA Polymerase | ||||
| BL2 | EGF pathway | Cetuximab, erlotinib, gefitinib | 0 | − |
| IGFIR pathway | BMS-754807 | |||
| MET pathway | ||||
| NGF pathway | ||||
| WNT/B-catenin pathway | ||||
| Glicolisis/Gluconeogenesis | ||||
| IM | Taxanes/Anthracicline | 30% | + | |
| TH1/TH2 pathway, T cell receptor signaling | Platinum/Lambrolizumab (MK-3475)/Nivolumab | |||
| Cytokine signaling | PLX3397 (CSF-1 inhibitor) | |||
| DC pathway, NK cell pathway | Indoximod (IDO inhibitor ) | |||
| B cell receptor signaling pathway | ||||
| NFκB, TNF, JAK/STAT signaling | ||||
| CTL4, IL12, IL7 pathway | Ipilimumab | |||
| Antigen processing/presentation | ||||
| DNA damage pathway (ATR/BRCA pathway) | Platinum | |||
| M | Taxanes/Anthracicline | 31% | + | |
| Cell motility pathway (Regulation of Actin by RHO) | Dasatinib | |||
| Cell differentiation pathway (WNT/B-catenin, ALK, TGFβ) | Windorphen | |||
| IGF/mTOR pathway | NVP-BEZ235 | |||
| ECM receptor interaction pathway | ||||
| MSL | Taxanes/Anthracicline | 23% | + | |
| Cell motility pathway (Regulation of Actin by RHO, RAC1) EMT-associated genes/low expression of claudin low 3,4,7 Smooth muscle contraction | Dasatinib | |||
| Cell differentiation pathway (WNT/B-catenin, ALK, TGFβ) | Windorphen | |||
| Angiogenesis-associated genes | Bevacizumab | |||
| Growth factor signaling pathway (EGF, PDGF, calcium signaling, GPCR, ERK1/2, ABC transporter, adipocytokine signaling, PI3K-AKT-mTOR pathway) | NVP-BEZ235 | |||
| ECM receptor interaction pathway | ||||
| T cell receptor signaling/NK cell pathway/NFκB signaling | ||||
| LAR | Taxanes | 10% | +/− | |
| Hormonally regulated pathways (steroid synthesis, porphiryn metabolism, androgen/estrogen metabolism) | Abiraterone/bicalutamide/enzalutamide | |||
| PI3K/mTOR/AKT pathway; HSP90 | NVP-BEZ235; 17-DMAG (HSP90 inhibitor) |
2.2. BL2 Subtype
2.3. IM Subtype
2.4. M and MSL Subtypes
2.5. LAR Subtype
3. Conclusions

Abbreviations
| BL1 | basal-like 1 |
| BL2 | basal-like 2 |
| IM | immunomodulatory |
| M | mesenchymal |
| MSL | mesenchymal stem-like |
| LAR | luminal androgen receptor |
| AURKA | aurora kinase A |
| AURKB | aurora kinase B |
| MYC | myelocytomatosis oncogene |
| NRAS | neuroblastoma RAS (Rat sarcoma) viral (v-ras) oncogene homolog |
| DNA | deoxyribonucleic acid |
| ATR | ataxia telangiectasia and Rad3-related |
| BRCA | breast cancer susceptibility protein |
| RNA | ribonucleic acid |
| EGF | epidermal growth factor |
| IGF1R | insulin-like growth factor 1 receptor |
| MET | N-methyl-N’-nitroso-guanidine human osteosarcoma transforming gene |
| NGF | nerve growth factor |
| WNT | wingless-related integration site |
| TH1/TH2 | helper T 1/helper T 2 cells |
| DC | dendritic cell |
| NK | natural killer |
| NFκB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| TNF | tumor necrosis factor |
| JAK | Janus kinase |
| STAT | signal transducers and activators of transcription |
| CTL4 | cytotoxic T-lymphocyte antigen 4 |
| IL12 | interleukin 12 |
| IL7 | interleukin 7 |
| RHO | Ras homologous |
| ALK | anaplastic lymphoma kinase |
| TGFβ | transforming growth factor beta |
| IGF | insulin-like growth factor |
| mTOR | mammalian target of rapamycin |
| ECM | extracellular matrix |
| RAC1 | Ras-related C3 botulinum toxin substrate 1 |
| EMT | epithelial mesenchymal transition |
| PDGF | platelet-derived growth factor |
| GPCR | G protein-coupled receptor |
| ERK1/2 | extracellular-signal-regulated kinases 1/2 |
| ABC transporter | ATP-binding cassette transporter |
| PI3K | phosphatidylinositol 3-kinase |
| AKT | protein kinase B |
| HSP90 | heat shock protein 90 |
| PARP | Poly (ADP-ribose) polymerase |
| CSF-1 | colony stimulating factor 1 |
| IDO | indoleamine 2,3-dioxygenase |
| pCR | pathological complete response |
| CT | chemotherapy |
Conflicts of Interest
References
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [PubMed]
- Sasa, M.; Bando, Y.; Takahashi, M.; Hirose, T.; Nagao, T. Screening for basal marker expression is necessary for decision of therapeutic strategy for triple-negative breast cancer. J. Surg. Oncol. 2008, 97, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Viale, G.; Rotmensz, N.; Maisonneuve, P.; Bottiglieri, L.; Montagna, E.; Luini, A.; Veronesi, P.; Intra, M.; Torrisi, R.; Cardillo, A.; et al. Invasive ductal carcinoma of the breast with the “triple-negative” phenotype: Prognostic implications of EGFR immunoreactivity. Breast Cancer Res. Treat. 2009, 116, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Cheang, M.C.; Voduc, D.; Bajdik, C.; Leung, S.; McKinney, S.; Chia, S.K.; Perou, C.M.; Nielsen, T.O. Basal-like breast cancer defined by five biomarkers has superior prognostic value than triple-negative phenotype. Clin. Cancer Res. 2008, 14, 1368–1376. [Google Scholar] [CrossRef] [PubMed]
- Bostrom, P.; Söderström, M.; Palokangas, T.; Vahlberg, T.; Collan, Y.; Carpen, O.; Hirsimäki, P. Analysis of cyclins A, B1, D1 and E in breast cancer in relation to tumour grade and other prognostic factors. BMC Res. Notes 2009, 2. [Google Scholar] [CrossRef]
- Voduc, D.; Nielsen, T.O.; Cheang, M.C.; Foulkes, W.D. The combination of high cyclin E and Skp2 expression in breast cancer is associated with a poor prognosis and the basal phenotype. Hum. Pathol. 2008, 39, 1431–1437. [Google Scholar] [CrossRef] [PubMed]
- Geyer, F.C.; Lacroix-Triki, M.; Savage, K.; Arnedos, M.; Lambros, M.B.; MacKay, A.; Natrajan, R.; Reis-Filho, J.S. β-Catenin pathway activation in breast cancer. Mod. Pathol. 2011, 24, 209–231. [Google Scholar] [CrossRef] [PubMed]
- Rakha, E.A.; El-Sayed, M.E.; Green, A.R.; Lee, A.H.; Robertson, J.F.; Ellis, I.O. Prognostic markers in triple-negative breast cancer. Cancer 2007, 109, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Parker, J.S.; Karginova, O.; Fan, C.; Livasy, C.; Herschkowitz, J.I.; He, X.; Perou, C.M. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010, 12, R68. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M. Molecular stratification of triple-negative breast cancers. Oncologist 2011, 16, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, T.O.; Hsu, F.D.; Jensen, K.; Cheang, M.; Karaca, G.; Hu, Z.; Hernandez-Boussard, T.; Livasy, C.; Cowan, D.; Dressler, L.; et al. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin. Cancer Res. 2004, 10, 5367–5374. [Google Scholar] [CrossRef] [PubMed]
- Bertucci, F.; Finetti, P.; Cervera, N.; Esterni, B.; Hermitte, F.; Viens, P.; Birnbaum, D. How basal are triple-negative breast cancers? Int. J. Cancer 2008, 123, 236–240. [Google Scholar]
- Kreike, B.; van Kouwenhove, M.; Horlings, H.; Weigelt, B.; Peterse, H.; Bartelink, H.; van de Vijver, M.J. Gene expression profiling and histopathological characterization of triple-negative/basal-like breast carcinomas. Breast Cancer Res. 2007, 9, R65. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Perou, C.M. Deconstructing the molecular portraits of breast cancer. Mol. Oncol. 2011, 5, 5–23. [Google Scholar] [CrossRef] [PubMed]
- Martín, M.; Rodríguez-Lescure, A.; Ruiz, A.; Alba, E.; Calvo, L.; Ruiz-Borrego, M.; Munárriz, B.; Rodríguez, C.A.; Crespo, C.; de Alava, E.; et al. Randomized phase 3 trial of fluorouracil, epirubicin, and cyclophosphamide alone or followed by Paclitaxel for early breast cancer. J. Natl. Cancer Inst. 2008, 100, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Bramwell, V.H.C.; Pritchard, K.I.; Tu, D.; Tonkin, K.; Vachhrajani, H.; Vandenberg, T.A.; Robert, J.; Arnold, A.; O’Reilly, S.E.; Graham, B.; et al. A randomized placebo-controlled study of tamoxifen after adjuvant chemotherapy in premenopausal women with early breast cancer (National Cancer Institute of Canada—Clinical Trials Group Trial, MA.12). Ann. Oncol. 2010, 21, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.N.; Pritchard, K.I.; Bramwell, V.H.C.; Shepherd, L.E.; Tu, D.; Paul, N. Randomized trial comparing cyclophosphamide, epirubicin, and fluorouracil with cyclophosphamide, methotrexate, and fluorouracil in premenopausal women with node-positive breast cancer: Update of National Cancer Institute of Canada Clinical Trials Group Trial MA5. J. Clin. Oncol. 2005, 23, 5166–5170. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Adamo, B.; Cheang, M.C.; Anders, C.K.; Carey, L.A.; Perou, C.M. Molecular characterization of basal-like and non-basal-like triple-negative breast cancer. Oncologist 2013, 18, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.J.; Naidu, S.; Topham, A.K.; Guiles, F.; Xu, Y.; McCue, P.; Schwartz, G.F.; Park, P.K.; Rosenberg, A.L.; Brill, K.; et al. Differences in breast carcinoma characteristics in newly diagnosed African-American and Caucasian patients: A single-institution compilation compared with the National Cancer Institute’s Surveillance, Epidemiology, and End Results database. Cancer 2007, 110, 876–884. [Google Scholar] [CrossRef] [PubMed]
- Carey, L.A.; Perou, C.M.; Livasy, C.A.; Dressler, L.G.; Cowan, D.; Conway, K.; Karaca, G.; Troester, M.A.; Tse, C.K.; Edmiston, S.; et al. Race, breast cancer subtypes, and survival in the Carolina breast cancer study. JAMA 2006, 295, 2492–2502. [Google Scholar] [CrossRef] [PubMed]
- Tavassoli, F.A.; Devilee, P. World Health Organization classification of tumours. In Pathology and Genetics of Tumours of the Breast and Female Genital Organs; IARC Press: Lyon, France, 2003; pp. 10–112. [Google Scholar]
- Denkert, C.; Loibl, S.; Salat, C.; Sinn, B.V.; Schem, C.; Endris, V.; Klare, P.; Schmitt, W.D.; Blohmer, J.-U.; Weichert, W.; et al. Increased tumor-associated lymphocytes predict benefit from addition of carboplatin to neoadjuvant therapy for triple-negative and HER2-positive early breast cancer in the GeparSixto trial (GBG 66). Cancer Res. 2013, 73. [Google Scholar] [CrossRef]
- Atchley, D.P.; Albarracin, C.T.; Lopez, A.; Valero, V.; Amos, C.I.; Gonzalez-Angulo, A.M.; Hortobagyi, G.N.; Arun, B.K. Clinical and pathologic characteristics of patients with BRCA-positive and BRCA-negative breast cancer. J. Clin. Oncol. 2008, 26, 4282–4288. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Angulo, A.M.; Timms, K.M.; Liu, S.; Chen, H.; Litton, J.K.; Potter, J.; Lanchbury, J.S.; Stemke-Hale, K.; Hennessy, B.T.; Arun, B.K.; et al. Incidence and outcome of BRCA mutations in unselected patients with triple receptor-negative breast cancer. Clin. Cancer Res. 2011, 17, 1082–1089. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Tutt, A.; Ashworth, A. Hallmarks of “BRCAness” in sporadic cancers. Nat. Rev. Cancer 2004, 4, 814–819. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.C.; Reis-Filho, J.S.; Russell, A.M.; Springall, R.J.; Ryder, K.; Steele, D.; Savage, K.; Gillett, C.E.; Schmitt, F.C.; Ashworth, A.; et al. BRCA1 dysfunction in sporadic basal-like breast cancer. Oncogene 2007, 26, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.A.; Kaklamani, V.G. Metabolic syndrome and triple-negative breast cancer: A new paradigm. Int. J. Breast Cancer 2012, 2012, e809291. [Google Scholar] [CrossRef]
- Jardé, T.; Perrier, S.; Vasson, M.P.; Caldefie-Chézet, F. Molecular mechanisms of leptin and adiponectin in breast cancer. Eur. J. Cancer 2011, 47, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Eliassen, A.H.; Missmer, S.A.; Tworoger, S.S.; Spiegelman, D.; Barbieri, R.L.; Dowsett, M.; Hankinson, S.E. Endogenous steroid hormone concentrations and risk of breast cancer among premenopausal women. J. Natl. Cancer Inst. 2006, 98, 1406–1415. [Google Scholar] [CrossRef] [PubMed]
- Missmer, S.A.; Eliassen, A.H.; Barbieri, R.L.; Hankinson, S.E. Endogenous estrogen, androgen, and progesterone concentrations and breast cancer risk among postmenopausal women. J. Natl. Cancer Inst. 2004, 96, 1856–1865. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Hamilton-Fairley, D.; Koistinen, R.; Seppälä, M.; James, V.H.; Franks, S.; Reed, M.J. Effect of insulin-like growth factor-type I (IGF-I) and insulin on the secretion of sex hormone binding globulin and IGF-I binding protein (IBP-I) by human hepatoma cells. J. Endocrinol. 1990, 124, R1–R3. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, C.; Hasegawa, T.; Tani, Y.; Takahashi, F.; Takeuchi, M.; Watanabe, T.; Ando, M.; Katsumata, N.; Fujiwara, Y. Expression of insulin-like growth factor 1 receptor in primary breast cancer: Immunohistochemical analysis. Hum. Pathol. 2004, 35, 1537–1542. [Google Scholar] [CrossRef] [PubMed]
- Davison, Z.; de Blacquière, G.E.; Westley, B.R.; May, F.E. Insulin-like growth factor-dependent proliferation and survival of triple-negative breast cancer cells: Implications for therapy. Neoplasia 2011, 13, 504–515. [Google Scholar] [PubMed]
- Carey, L.A. Directed therapy of subtypes of triple-negative breast cancer. Oncologist 2010, 15, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Liedtke, C.; Mazouni, C.; Hess, K.R.; André, F.; Tordai, A.; Mejia, J.A.; Symmans, W.F.; Gonzalez-Angulo, A.M.; Hennessy, B.; Green, M.; et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J. Clin. Oncol. 2008, 26, 1275–1281. [Google Scholar] [PubMed]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef] [PubMed]
- Smid, M.; Wang, Y.; Zhang, Y.; Sieuwerts, A.M.; Yu, J.; Klijn, J.G.; Foekens, J.A.; Martens, J.W. Subtypes of breast cancer show preferential site of relapse. Cancer Res. 2008, 68, 3108–3114. [Google Scholar] [CrossRef] [PubMed]
- Masuda, H.; Baggerly, K.A.; Wang, Y.; Zhang, Y.; Gonzalez-Angulo, A.M.; Meric-Bernstam, F.; Valero, V.; Lehmann, B.D.; Pietenpol, J.A.; Hortobagyi, G.N.; et al. Differential response to neoadjuvant chemotherapy among 7 triple-negative breast cancer molecular subtypes. Clin. Cancer Res. 2013, 19, 5533–5540. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Pietenpol, J.A. Identification and use of biomarkers in treatment strategies for triple-negative breast cancer subtypes. J. Pathol. 2014, 232, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Bauer, J.A.; Chakravarthy, A.B.; Rosenbluth, J.M.; Mi, D.; Seeley, E.H.; de Matos Granja-Ingram, N.; Olivares, M.G.; Kelley, M.C.; Mayer, I.A.; Meszoely, I.M.; et al. Identification of markers of taxane sensitivity using proteomic and genomic analyses of breast tumors from patients receiving neoadjuvant paclitaxel and radiation. Clin. Cancer Res. 2010, 16, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Harris, L.N.; Broadwater, G.; Lin, N.U.; Miron, A.; Schnitt, S.J.; Cowan, D.; Lara, J.; Bleiweiss, I.; Berry, D.; Ellis, M.; et al. Molecular subtypes of breast cancer in relation to paclitaxel response and outcomes in women with metastatic disease: Results from CALGB 9342. Breast Cancer Res. 2006, 8, R66. [Google Scholar] [CrossRef] [PubMed]
- Rugo, H.S.; Barry, W.T.; Moreno-Aspitia, A.; Lyss, A.P.; Cirrincione, C.; Mayer, E.L.; Naughton, M.; Layman, R.M.; Lisa, A.; Carey, L.A.; et al. CALGB 40502/NCCTG N063H: Randomized phase III trial of weekly paclitaxel (P) compared to weekly nanoparticle albumin bound nab-paclitaxel (NP) or ixabepilone (Ix) with or without bevacizumab (B) as first-line therapy for locally recurrent or metastatic breast cancer (MBC). J. Clin. Oncol. 2012, 30 (Suppl. 15). Abstract CRA1002. [Google Scholar]
- Byrski, T.; Huzarski, T.; Dent, R.; Gronwald, J.; Zuziak, D.; Cybulski, C.; Kladny, J.; Gorski, B.; Lubinski, J.; Narod, S.A. Response to neo-adjuvant chemotherapy in women with BRCA1-positive breast cancers. Breast Cancer Res. Treat. 2008, 108, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Wysocki, P.J.; Korski, K.; Lamperska, K.; Zaluski, J.; Mackiewicz, A. Primary resistance to docetaxel-based chemotherapy in metastatic breast cancer patients correlates with a high frequency of BRCA1 mutations. Med. Sci. Monit. 2008, 14, SC7–SC10. [Google Scholar] [PubMed]
- Thompson, L.H.; Schild, D. Homologous recombinational repair of DNA ensures mammalian chromosome stability. Mutat. Res. 2001, 477, 131–153. [Google Scholar] [CrossRef] [PubMed]
- Dantzer, F.; de la Rubia, G.; Menissier-De Murcia, J.; Hostomsky, Z.; de Murcia, G.; Schreiber, V. Base excision repair is impaired in mammalian cells lacking Poly(ADP-ribose) polymerase-1. Biochemistry 2000, 39, 7559–7569. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, A.; Ear, U.S.; Koller, B.H.; Weichselbaum, R.R.; Bishop, D.K. The breast cancer susceptibility gene BRCA1 is required for subnuclear assembly of Rad51 and survival following treatment with the DNA cross-linking agent cisplatin. J. Biol. Chem. 2000, 275, 23899–23903. [Google Scholar] [CrossRef] [PubMed]
- Moynahan, M.E.; Cui, T.Y.; Jasin, M. Homology-directed DNA repair, mitomycin-c resistance, and chromosome stability is restored with correction of a Brca1 mutation. Cancer Res. 2001, 61, 4842–4850. [Google Scholar] [PubMed]
- Quinn, J.E.; Kennedy, R.D.; Mullan, P.B.; Gilmore, P.M.; Carty, M.; Johnston, P.G.; Harkin, D.P. BRCA1 functions as a differential modulator of chemotherapy-induced apoptosis. Cancer Res. 2003, 63, 6221–6228. [Google Scholar]
- Bergamaschi, A.; Kim, Y.H.; Wang, P.; Sørlie, T.; Hernandez-Boussard, T.; Lonning, P.E.; Tibshirani, R.; Børresen-Dale, A.L.; Pollack, J.R. Distinct patterns of DNA copy number alteration are associated with different clinicopathological features and gene-expression subtypes of breast cancer. Genes Chromosomes Cancer 2006, 45, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Byrski, T.; Gronwald, J.; Huzarski, T.; Grzybowska, E.; Budryk, M.; Stawicka, M.; Mierzwa, T.; Szwiec, M.; Wisniowski, R.; Siolek, M.; et al. Pathologic complete response rates in young women with BRCA1-positive breast cancers after neoadjuvant chemotherapy. J. Clin. Oncol. 2010, 28, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Silver, D.P.; Richardson, A.L.; Eklund, A.C.; Wang, Z.C.; Szallasi, Z.; Li, Q.; Juul, N.; Leong, C.O.; Calogrias, D.; Buraimoh, A.; et al. Effıcacy of neoadjuvant cisplatin in triple-negative breast cancer. J. Clin. Oncol. 2010, 28, 1145–1153. [Google Scholar] [CrossRef] [PubMed]
- Decatris, M.P.; Sundar, S.; O’Byrne, K.J. Platinum-based chemotherapy in metastatic breast cancer: Current status. Cancer Treat. Rev. 2004, 30, 53–81. [Google Scholar] [CrossRef] [PubMed]
- Carey, L.A.; Dees, E.C.; Sawyer, L.; Gatti, L.; Moore, D.T.; Collichio, F.; Ollila, D.W.; Sartor, C.I.; Graham, M.L.; Perou, C.M. The triple negative paradox: Primary tumor chemosensitivity of breast cancer subtypes. Clin. Cancer Res. 2007, 13, 2329–2334. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Gomez, P.; Awada, A. The addition of cetuximab to cisplatin increases overall response rate and progression-free survival in metastatic triple-negative breast cancer: Results of a randomized phase II study (BALI-1). Ann. Oncol. 2010, 21, viii96–viii121. [Google Scholar] [CrossRef]
- Isakoff, S.J.; Goss, P.E.; Mayer, E.L.; Traina, T.A.; Carey, L.A.; Krag, K.; Rugo, H.S.; Liu, M.C.; Stearns, V.; Come, S.E.; et al. TBCRC009: A multicenter phase II study of cisplatin or carboplatin for metastatic triple-negative breast cancer and evaluation of p63/p73 as a biomarker of response. J. Clin. Oncol. 2011, 29. Abstract 1025. [Google Scholar] [CrossRef]
- Maisano, R.; Zavettieri, M.; Azzarello, D.; Raffaele, M.; Maisano, M.; Bottari, M.; Nardi, M. Carboplatin and gemcitabine combination in metastatic triple-negative anthracycline- and taxane-pretreated breast cancer patients: A phase II study. J. Chemother. 2011, 23, 40–43. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Hu, X.; Chen, L.; Wang, J.; Wang, H.; Wang, L.; Liu, G.; Hu, Z.; Wu, J.; Zhimin, S. Efficacy of gemcitabine and cisplatin (GP) as first-line combination therapy in patients with triple-negative metastatic breast cancer: Preliminary results report of a phase II trial. J. Clin. Oncol. 2010, 28 (Suppl. 15). Abstract 1100. [Google Scholar] [CrossRef]
- Kim, T.; Lee, H.; Han, S.; Oh, D.; Im, S.; Bang, Y. The comparison of the benefits obtained from platinum-containing chemotherapy between triple-negative and non-triple-negative metastatic breast cancer. J. Clin. Oncol. 2010, 28 (Suppl. 15). Abstract 1071. [Google Scholar]
- Liu, M.; Mo, Q.G.; Wei, C.Y.; Qin, Q.H.; Huang, Z.; He, J. Platinum-based chemotherapy in triple-negative breast cancer: A meta-analysis. Oncol. Lett. 2013, 5, 983–991. [Google Scholar] [PubMed]
- Isakoff, S.J.; He, L.; Mayer, E.L.; Goss, P.E.; Traina, T.A.; Carey, L.A.; Krag, K.; Liu, M.C.; Rugo, H.S.; Stearns, V.; et al. Identification of biomarkers to predict response to single-agent platinum chemotherapy in metastatic triple-negative breast cancer (mTNBC): Correlative studies from TBCRC009. J. Clin. Oncol. 2014, 32 (Suppl. 15). Abstract 1020. [Google Scholar] [PubMed]
- Leong, C.O.; Vidnovic, N.; Deyoung, M.P.; Sgroi, D.; Ellisen, L.W. The p63/p73 network mediates chemosensitivity to cisplatin in a biologically defined subset of primary breast cancers. J. Clin. Investig. 2007, 117, 1370–1380. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, E.; Kimmick, G.; Hopkins, J.; Marcom, P.K.; Rocha, G.; Welch, R.; Broadwater, G.; Blackwell, K. nNab-paclitaxel/bevacizumab/carboplatin chemotherapy in first-line triple negative metastatic breast cancer. Clin. Breast Cancer 2013, 13, 416–420. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Stemmer, S.; Pego, A.; Chan, A.; Goeminne, A.J.; Graas, C.M.; Kennedy, P.J.; Ciruelos Gil, E.M.; Zubel, A.; Groos, J.; et al. Cetuximab+ cisplatin in estrogen receptor-negative, progesterone receptor-negative, HER2-negative (triple-negative) metastatic breast cancer: Results of the randomized phase II BALI-1 trial. Cancer Res. 2010, 70. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Khan, Q.J.; Kimler, B.F.; Klemp, J.R.; Connor, C.S.; McGinness, M.K.; Mammen, J.W.M.; Tawfik, O.W.; Fan, F.; Fabian, C.J. Results of a phase II study of neoadjuvant platinum/taxane based chemotherapy and erlotinib for triple negative breast cancer. Cancer Res. 2010, 70. [Google Scholar] [CrossRef] [PubMed]
- O’Shaughnessy, J.; Osborne, C.; Pippen, J.E.; Yoffe, M.; Patt, D.; Rocha, C.; Koo, I.C.; Sherman, B.M.; Bradley, C. Iniparib plus chemotherapy in metastatic triple-negative breast cancer. N. Engl. J. Med. 2011, 364, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Carey, L.A.; Rugo, H.S.; Marcom, P.K.; Irvin, W., Jr.; Ferraro, M.; Burrows, E.; He, X.; Perou, C.M. TBCRC 001: EGFR inhibition with cetuximab added to carboplatin in metastatic triple-negative (basal-like) breast cancer. J. Clin. Oncol. 2008, 26, S1009. [Google Scholar]
- Tutt, A.; Ashworth, A. The relationship between the roles of BRCA genes in DNA repair and cancer predisposition. Trends Mol. Med. 2002, 8, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Byrski, T.; Huzarski, T.; Dent, R.; Gronwald, J.; Zuziak, D.; Cybulski, C.; Kladny, J.; Gorski, B.; Lubinski, J.; Narod, S.A. Response to neoadjuvant therapy with cisplatin in BRCA1-positive breast cancer patients. Breast Cancer Res. Treat. 2009, 115, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Tentori, L.; Portarena, I.; Graziani, G. Potential clinical applications of poly(ADP-ribose) polymerase (PARP) inhibitors. Pharmacol. Res. 2002, 45, 73–85. [Google Scholar] [CrossRef] [PubMed]
- He, J.X.; Yang, C.H.; Miao, Z.H. Poly(ADP-ribose) polymerase inhibitors as promising cancer therapeutics. Acta Pharmacol. Sin. 2010, 31, 1172–1180. [Google Scholar] [CrossRef] [PubMed]
- Tutt, A.; Robson, M.; Garber, J.E.; Domchek, S.M.; Audeh, M.W.; Weitzel, J.N.; Friedlander, M.; Arun, B.; Loman, N.; Schmutzler, R.K.; et al. Oral poly(ADP- ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: A proof-of- concept trial. Lancet 2010, 376, 235–244. [Google Scholar] [CrossRef] [PubMed]
- O’Shaughnessy, J.; Schwartzberg, L.S.; Danso, M.A.; Rugo, H.S.; Miller, K.; Yardley, D.A.; Carlson, R.W.; Finn, R.S.; Charpentier, E.; Freese, M.; et al. A randomized phase III study of iniparib (BSI-201) in combination with gemcitabine/carboplatin (G/C) in metastatic triple negative breast cancer (TNBC). J. Clin. Oncol. 2011, 29 (Suppl. 15). Abstract 1007. [Google Scholar]
- Yang, S.X.; Kummar, S.; Steinberg, S.M.; Murgo, A.J.; Gutierrez, M.; Rubinstein, L.; Nguyen, D.; Kaur, G.; Chen, A.P.; Giranda, V.L.; et al. Immunohistochemical detection of poly(ADP-ribose) polymerase inhibition by ABT-888 in patients with refractory solid tumors and lymphomas. Cancer Biol. Ther. 2009, 8, 2004–2009. [Google Scholar] [CrossRef] [PubMed]
- Donawho, C.K.; Luo, Y.; Luoetal, Y. ABT-888, anorallyactive poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin. Cancer Res. 2007, 13, 2728–2737. [Google Scholar] [CrossRef] [PubMed]
- Isakoff, S.J.; Overmoyer, B.; Tung, N.M.; Gelman, R.S.; Giranda, V.L.; Bernhard, K.M.; Habin, K.R.; Ellisen, L.W.; Winer, E.P.; Goss, P.E. A phase II trial of the PARP inhibitor veliparib (ABT888) and temozolomide for metastatic breast cancer. J. Clin. Oncol. 2010, 28, S1019. [Google Scholar]
- Rugo, H.; Olopade, O.; de Michele, A.; van’t Veer, L.; Buxton, M.; Hylton, N.; Yee, N.; Chien, D.M.J.; Wallace, A. Veliparib/carboplatin plus standard neoadjuvant therapy for high-risk breast cancer: First efficacy results from the I-SPY 2 TRIAL. Cancer Res. 2013, 73. [Google Scholar] [CrossRef]
- Somlo, G.; Frankel, P.H.; Luu, T.H.; Ma, C.; Arun, B.; Garcia, A.; Cigler, T.; Cream, L.; Harvey, H.A.; Sparano, J.A.; et al. Phase II trial of single agent PARP inhibitor ABT-888 (veliparib [vel]) followed by postprogression therapy of vel with carboplatin (carb) in patients (pts) with stage BRCA-associated metastatic breast caner (MBC): California Cancer Consortium trial PHII-96. J. Clin. Oncol. 2014, 32, S1021. [Google Scholar]
- Mina, L.A.; Ramanathan, R.K.; Wainberg, Z.A. BMN 673 is a PARP inhibitor in clinical development for the treatment of breast cancer patients with deleterious germline BRCA 1 and 2 mutations. In Proceedings of the SABCS, San Antonio, TX, USA, 10–14 December 2013. P2-09-02.
- Fu, J.; Bian, M.; Jiang, Q.; Zhang, C. Roles of Aurora kinases in mitosis and tumorigenesis. Mol. Cancer Res. 2007, 5, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Marumoto, T.; Zhang, D.; Saya, H. Aurora-AA guardian of poles. Nat. Rev. Cancer 2005, 5, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Giet, R.; Petretti, C.; Prigent, C. Aurora kinases, aneuploidy and cancer, a coincidence or a real link? Trends Cell Biol. 2005, 15, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Sasai, K.; Katayama, H.; Stenoien, D.L.; Fujii, S.; Honda, R.; Kimura, M.; Okano, Y.; Tatsuka, M.; Suzuki, F.; Nigg, E.A.; et al. Aurora-C kinase is a novel chromosomal passenger protein that can complement Aurora-B kinase function in mitotic cells. Cell Motil. Cytoskelet. 2004, 59, 249–263. [Google Scholar] [CrossRef]
- Yan, X.; Cao, L.; Li, Q.; Wu, Y.; Zhang, H.; Saiyin, H.; Liu, X.; Zhang, X.; Shi, Q.; Yu, L. Aurora C is directly associated with Survivin and required for cytokinesis. Genes Cells 2005, 10, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Bayliss, R.; Sardon, T.; Vernos, I.; Conti, E. Structural basis of Aurora—A activation by TPX2 at the mitotic spindle. Mol. Cell 2003, 12, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Eyers, P.A.; Maller, J.L. Regulation of Xenopus Aurora a activation by TPX2. J. Biol. Chem. 2004, 279, 9008–9015. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wu, X.; Zhou, W.H.; Liu, A.W.; Wu, J.B.; Deng, J.Y.; Yue, C.F.; Yang, S.B.; Wang, J.; Yuan, Z.Y.; et al. Aurora-A identifies early recurrence and poor prognosis and promises a potential therapeutic target in triple negative breast cancer. PLoS One 2013, 8, e56919. [Google Scholar] [CrossRef] [PubMed]
- Carmena, M.; Earnshaw, W.C. The cellular geography of aurora kinases. Nat. Rev. Mol. Cell Biol. 2003, 4, 842–854. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.; Wordeman, L. C-terminus of mitotic centromere-associated kinesin (MCAK) inhibits its lattice-stimulated ATPase activity. Biochem. J. 2004, 383, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Romanelli, A.; Clark, A.; Assayag, F.; Chateau-Joubert, S.; Poupon, M.F.; Servely, J.L.; Fontaine, J.J.; Liu, X.; Spooner, E.; Goodstal, S.; et al. Inhibiting Aurora kinases reduces tumor growth and suppresses tumor recurrence after chemotherapy in patient-derived Triple-Negative Breast Cancer xenografts. Mol. Cancer Ther. 2012, 11, 2693–2703. [Google Scholar] [PubMed]
- Sebastian, S.; Settleman, J.; Reshkin, S.J.; Azzariti, A.; Bellizzi, A.; Paradiso, A. The complexity of targeting EGFR signaling in cancer: From expression to turnover. Biochim. Biophys. Acta 2006, 1766, 120–139. [Google Scholar] [PubMed]
- Shiu, K.K.; Tan, D.S.; Reis-Filho, J.S. Development of therapeutic approaches to “triple negative” phenotype breast cancer. Expert Opin. Ther. Targets 2008, 12, 1123–1137. [Google Scholar] [CrossRef] [PubMed]
- Corkery, B.; Crown, J.; Clynes, M.; O’Donovan, N. Epidermal growth factor receptor as a potential therapeutic target in triple-negative breast cancer. Ann. Oncol. 2009, 20, 862–867. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; O’Donovan, N.; Crown, J. Use of molecular markers for predicting therapy response in cancer patients. Cancer Treat. Rev. 2011, 37, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Hoadley, K.A.; Weigman, V.J.; Fan, C.; Sawyer, L.R.; He, X.; Troester, M.A.; Sartor, C.I.; Rieger-House, T.; Bernard, P.S.; Carey, L.A.; et al. EGFR associated expression profiles vary with breast tumor subtype. BMC Genomics 2007, 8, e258. [Google Scholar] [CrossRef]
- O’Shaughnessy, J.; Weckstein, D.; Vukelja, S. Preliminary results of a randomized phase II study of weekly irinotecan/carboplatin with or without cetuximab in patients with metastatic breast cancer. In Proceedings of the SABCS, San Antonio, TX, USA, 13–16 December 2007. Abstract 308.
- Carey, L.A.; Rugo, H.S.; Marcom, P.K.; Mayer, E.L.; Esteva, F.J.; Ma, C.X.; Liu, M.C.; Storniolo, A.M.; Rimawi, M.F.; Forero-Torres, A.; et al. TBCRC 001: Randomized phase II study of cetuximab in combination with carboplatin in stage IV triple-negative breast cancer. J. Clin. Oncol. 2012, 30, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Masuda, H.; Zhang, D.; Bartholomeusz, C.; Doihara, H.; Hortobagyi, G.N.; Ueno, N.T. Role of epidermal growth factor receptor in breast cancer. Breast Cancer Res. Treat. 2012, 136, 331–345. [Google Scholar] [CrossRef] [PubMed]
- Gelmon, K.; Dent, R.; Mackey, J.R.; Laing, K.; McLeod, D.; Verma, S. Targeting triple-negative breast cancer: Optimising therapeutic outcomes. Ann. Oncol. 2012, 23, 2223–2234. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A.; Gutteridge, E.; Gee, J.M.; Nicholson, R.I.; Robertson, J.F. Overview of tyrosine kinase inhibitors in clinical breast cancer. Endocr. Relat. Cancer 2005, 12, S135–S144. [Google Scholar] [CrossRef] [PubMed]
- Twelves, C.; Trigo, J.M.; Jones, R.; de Rosa, F.; Rakhit, A.; Fettner, S.; Wright, T.; Baselga, J. Erlotinib in combination with capecitabine and docetaxel in patients with metastatic breast cancer: A dose-escalation study. Eur. J. Cancer 2008, 44, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Ciardiello, F.; Troiani, T.; Caputo, F.; de Laurentiis, M.; Tortora, G.; Palmieri, G.; de Vita, F.; Diadema, M.R.; Orditura, M.; Colantuoni, G.; et al. Phase II study of gefitinib in combination with docetaxel as first-line therapy in metastatic breast cancer. Br. J. Cancer. 2006, 94, 1604–1609. [Google Scholar] [PubMed]
- Litzenburger, B.C.; Creighton, C.J.; Tsimelzon, A.; Chan, B.T.; Hilsenbeck, S.G.; Wang, T.; Carboni, J.M.; Gottardis, M.M.; Huang, F.; Chang, J.C.; et al. High IGF-IR activity in triple-negative breast cancer cell lines and tumorgrafts correlates with sensitivity to anti-IGF-IR therapy. Clin. Cancer Res. 2011, 17, 2314–2327. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yee, D. Targeting insulin and insulin-like growth factor signaling in breast cancer. J. Mammary Gland Biol. Neoplasia 2012, 17, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Greer, A.; Hurlburt, W.; Han, X.; Hafezi, R.; Wittenberg, G.M.; Reeves, K.; Chen, J.; Robinson, D.; Li, A.; et al. The mechanisms of differential sensitivity to an insulin-like growth factor-1 receptor inhibitor (BMS-536924) and rationale for combining with EGFR/HER2 inhibitors. Cancer Res. 2009, 69, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Rensing, K.L.; Houttuijn Bloemendaal, F.M.; Weijers, E.M.; Richel, D.J.; Büller, H.R.; Koolwijk, P.; van der Loos, C.M.; Twickler, T.B.; von der Thüsen, J.H. Could recombinant insulin compounds contribute to adenocarcinoma progression by stimulating local angiogenesis? Diabetologia 2010, 53, 966–970. [Google Scholar] [CrossRef]
- Rozengurt, E.; Sinnett-Smith, J.; Kisfalvi, K. Crosstalk between insulin/insulin-like growth factor-1 receptors and G protein-coupled receptor signaling systems: A novel target for the antidiabetic drug metformin in pancreatic cancer. Clin. Cancer Res. 2010, 16, 2505–2511. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Yan, H.; Frost, P.; Gera, J.; Lichtenstein, A. Mammalian target of rapamycin inhibitors activate the AKT kinase in multiple myeloma cells by up-regulating the insulin-like growth factor receptor/insulin receptor substrate-1/phosphatidylinositol 3-kinase cascade. Mol. Cancer Ther. 2005, 4, 1533–1540. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, K.E.; Rojo, F.; She, Q.B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006, 66, 1500–1508. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Harkavy, B.; Shen, N.; Grohar, P.; Helman, L.J. Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene 2007, 26, 1932–1940. [Google Scholar] [CrossRef]
- Clark, A.S.; West, K.; Streicher, S.; Dennis, P.A. Constitutive and inducible Akt activity promotes resistance to chemotherapy, trastuzumab, or tamoxifen in breast cancer cells. Mol. Cancer Ther. 2002, 1, 707–717. [Google Scholar] [PubMed]
- Becker, M.A.; Ibrahim, Y.H.; Cui, X.; Lee, A.V.; Yee, D. The IGF pathway regulates ER through a S6K1-dependent mechanism in breast cancer cells. Mol. Endocrinol. 2011, 25, 516–528. [Google Scholar] [CrossRef] [PubMed]
- Fukazawa, H.; Noguchi, K.; Murakami, Y.; Uehara, Y. Mitogen-activated protein/extracellular signal-regulated kinase kinase (MEK) inhibitors restore anoikis sensitivity in human breast cancer cell lines with a constitutively activated extracellular-regulated kinase (ERK) pathway. Mol. Cancer Ther. 2002, 1, 303–309. [Google Scholar] [PubMed]
- Brachmann, S.M.; Hofmann, I.; Schnell, C.; Fritsch, C.; Wee, S.; Lane, H.; Wang, S.; Garcia-Echeverria, C.; Maira, S.M. Specific apoptosis induction by the dual PI3K/mTor inhibitor NVP-BEZ235 in HER2 amplified and PIK3CA mutant breast cancer cells. Proc. Natl. Acad. Sci. USA 2009, 106, 22299–22304. [Google Scholar] [CrossRef] [PubMed]
- Bertucci, F.; Finetti, P.; Cervera, N.; Charafe-Jauffret, E.; Mamessier, E.; Adélaïde, J.; Debono, S.; Houvenaeghel, G.; Maraninchi, D.; Viens, P.; et al. Gene expression profiling shows medullary breast cancer is a subgroup of basal breast cancers. Cancer Res. 2006, 66, 4636–4644. [Google Scholar] [CrossRef] [PubMed]
- Vinayak, S.; Gray, R.J.; Adams, S.; Jensen, K.C.; Manola, J.; Afghahi, A.; Goldstein, L.J.; Ford, J.M.; Badve, S.S.; Telli, M.L. Association of increased tumor-infiltrating lymphocytes (TILs) with immunomodulatory (IM) triple-negative breast cancer (TNBC) subtype and response to neoadjuvant platinum-based therapy in PrECOG0105. J. Clin. Oncol. 2014, 32, S1000. [Google Scholar]
- Ghiotto, M.; Gauthier, L.; Serriari, N.; Pastor, S.; Truneh, A.; Nunès, J.A.; Olive, D. PD-L1 and PD-L2 differ in their molecular mechanisms of interaction with PD-1. Int. Immunol. 2010, 22, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Muenst, S.; Schaerli, A.R.; Gao, F.; Däster, S.; Trella, E.; Droeser, R.A.; Muraro, M.G.; Zajac, P.; Zanetti, R.; Gillanders, W.E.; et al. Expression of programmed death ligand 1 (PD-L1) is associated with poor prognosis in human breast cancer. Breast Cancer Res. Treat. 2014, 146, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Basu, G.D.; Ghazalpour, A.; Gatalica, Z.; Anderson, K.S.; McCullough, A.E.; Spetzer, D.B.; Pockaj, B.A. Expression of novel immunotherapeutic targets in triple-negative breast cancer. J. Clin. Oncol. 2014, 32, S1001. [Google Scholar]
- Gibson, G.R.; Qian, D.; Ku, J.K.; Lai, L.L. Metaplastic breast cancer: Clinical features and outcomes. Am. Surg. 2005, 71, 725–730. [Google Scholar] [PubMed]
- Hennessy, B.T.; Gonzalez-Angulo, A.M.; Stemke-Hale, K.; Gilcrease, M.Z.; Krishnamurthy, S.; Lee, J.S.; Fridlyand, J.; Sahin, A.; Agarwal, R.; Joy, C.; et al. Characterization of a naturally occurring breast cancer subset enriched in epithelial-to-mesenchymal transition and stem cell characteristics. Cancer Res. 2009, 69, 4116–4124. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.Y.; Rath, O.; Zebisch, A.; Choo, S.M.; Kolch, W.; Cho, K.H. Functional roles of multiple feedback loops in extracellular signal-regulated kinase and Wnt signaling pathways that regulate epithelial-mesenchymal transition. Cancer Res. 2010, 70, 6715–6724. [Google Scholar] [CrossRef] [PubMed]
- Hayes, M.J.; Thomas, D.; Emmons, A.; Giordano, T.J.; Kleer, C.G. Genetic changes of Wnt pathway genes are common events in metaplastic carcinomas of the breast. Clin. Cancer Res. 2008, 14, 4038–4044. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Ao, A.; Zhou, L.; Murphy, C.K.; Frist, A.Y.; Keel, J.J.; Thorne, C.A.; Kim, K.; Lee, E.; Hong, C.C. Selective small molecule targeting β-catenin function discovered by in vivo chemical genetic screen. Cell Rep. 2013, 4, 898–904. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Verbeek, B.S.; Vroom, T.M.; Adriaansen-Slot, S.S.; Ottenhoff-Kalff, A.E.; Geertzema, J.G.; Hennipman, A.; Rijksen, G. c-Src protein expression is increased in human breast cancer. An immunohistochemical and biochemical analysis. J. Pathol. 1996, 180, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Tryfonopoulos, D.; Walsh, S.; Collins, D.M.; Flanagan, L.; Quinn, C.; Corkery, B.; McDermott, E.W.; Evoy, D.; Pierce, A.; O’Donovan, N.; et al. Src: A potential target for the treatment of triple-negative breast cancer. Ann. Oncol. 2011, 22, 2234–2240. [Google Scholar] [CrossRef] [PubMed]
- Talpaz, M.; Shah, N.P.; Kantarjian, H.; Donato, N.; Nicoll, J.; Paquette, R.; Cortes, J.; O’Brien, S.; Nicaise, C.; Bleickardt, E.; et al. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N. Engl. J. Med. 2006, 354, 2531–2541. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Dering, J.; Ginther, C.; Wilson, C.A.; Glaspy, P.; Tchekmedyian, N.; Slamon, D.J. Dasatinib, an orally active small molecule inhibitor of both the src and abl kinases, selectively inhibits growth of basal-type/“triple-negative” breast cancer cell lines growing in vitro. Breast Cancer Res. Treat. 2007, 105, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Reeves, K.; Han, X.; Fairchild, C.; Platero, S.; Wong, T.W.; Lee, F.; Shaw, P.; Clark, E. Identification of candidate molecular markers predicting sensitivity in solid tumors to dasatinib: Rationale for patient selection. Cancer Res. 2007, 67, 2226–2238. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Bengala, C.; Ibrahim, N.; Roché, H.; Sparano, J.; Strauss, L.C.; Fairchild, J.; Sy, O.; Goldstein, L.J. Dasatinib as a single agent in triple-negative breast cancer: Results of an open-label phase 2 study. Clin. Cancer Res. 2011, 17, 6905–6913. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.M.; Mueller, K.; Gartner, E.; Boerner, J. Dasatinib is synergistic with cetuximab and cisplatin in triple-negative breast cancer cells. J. Surg. Res. 2013, 185, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Mezi, S.; Todi, L.; Orsi, E.; Angeloni, A.; Mancini, P. Involvement of the Src-cortactin pathway in migration induced by IGF-1 and EGF in human breast cancer cells. Int. J. Oncol. 2012, 41, 2128–2138. [Google Scholar] [PubMed]
- Fornier, M.N.; Morris, P.G.; Abbruzzi, A.; D’Andrea, G.; Gilewski, T.; Bromberg, J.; Dang, C.; Dickler, M.; Modi, S.; Seidman, A.D.; et al. A phase I study of dasatinib and weekly paclitaxel for metastatic breast cancer. Ann. Oncol. 2011, 22, 2575–2581. [Google Scholar] [CrossRef] [PubMed]
- Somlo, G.; Atzori, F.; Strauss, L.C.; Geese, W.J.; Specht, J.M.; Gradishar, W.J.; Rybicki, A.; Sy, O.; Vahdat, L.T.; Cortes, J. Dasatinib plus capecitabine for advanced breast cancer: Safety and efficacy in phase I study CA180004. Clin. Cancer Res. 2013, 19, 1884–1893. [Google Scholar] [CrossRef] [PubMed]
- Ueno, N.T.; Zhang, D. Targeting EGFR in triple negative breast cancer. J. Cancer 2011, 2, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Rydén, L.; Ferno, M.; Stal, O.; Linderholm, B.; Ostman, A.; Jirstrom, K. Vascular endothelial growth factor receptor 2 is a significant negative prognostic biomarker in triple-negative breast cancer: Results from a controlled randomised trial of premenopausal breast cancer. Cancer Res. 2009, 6. [Google Scholar] [CrossRef]
- Jayson, G.C.; Haas, S.D.; Delmar, P.; Miles, D.W.; Shah, M.A.; van Cutsem, E.; Carmeliet, P.; Hegde, P.; Wild, N.; Scherer, S.J. Evaluation of plasma VEGFA as a potential predictive pan-tumour biomarker for bevacizumab. In Presented at the European Multidisciplinary Cancer Congress, Stockholm, Sweden, 23–27 September 2011. Abstract 804.
- Yang, S.X.; Steinberg, S.M.; Nguyen, D.; Wu, T.D.; Modrusan, Z.; Swain, S.M. Gene expression profile and angiogenic marker correlates with response to neoadjuvant bevacizumab followed by bevacizumab plus chemotherapy in breast cancer. Clin. Cancer Res. 2008, 14, 5893–5899. [Google Scholar] [CrossRef] [PubMed]
- Linderholm, B.K.; Hellborg, H.; Johansson, U.; Elmberger, G.; Skoog, L.; Lehtiö, J.; Lewensohn, R. Significantly higher levels of vascular endothelial growth factor (VEGF) and shorter survival times for patients with primary operable triple-negative breast cancer. Ann. Oncol. 2009, 20, 1639–1646. [Google Scholar] [CrossRef] [PubMed]
- Lindholm, E.M.; Kristian, A.; Nalwoga, H.; Krüger, K.; Nygård, S.; Akslen, L.A.; Mælandsmo, G.M.; Engebraaten, O. Effect of antiangiogenic therapy on tumor growth, vasculature and kinase activity in basal- and luminal-like breast cancer xenografts. Mol. Oncol. 2012, 6, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.; Wang, M.; Gralow, J.; Dickler, M.; Cobleigh, M.; Perez, E.A.; Shenkier, T.; Cella, D.; Davidson, N.E. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N. Engl. J. Med. 2007, 357, 2666–2676. [Google Scholar] [CrossRef] [PubMed]
- Miles, D.W.; Chan, A.; Dirix, L.Y.; Cortés, J.; Pivot, X.; Tomczak, P.; Delozier, T.; Sohn, J.H.; Provencher, L.; Puglisi, F.; et al. Phase III study of bevacizumab plus docetaxel compared with placebo plus docetaxel for the first-line treatment of human epidermal growth factor receptor 2-negative metastatic breast cancer. J. Clin. Oncol. 2010, 28, 3239–3247. [Google Scholar] [CrossRef] [PubMed]
- Robert, N.J.; Diéras, V.; Glaspy, J.; Brufsky, A.M.; Bondarenko, I.; Lipatov, O.N.; Perez, E.A.; Yardley, D.A.; Chan, S.Y.; Zhou, X.; et al. RIBBON-1: Randomized, double-blind, placebo-controlled, phase III trial of chemotherapy with or without bevacizumab for first-line treatment of human epidermal growth factor receptor 2-negative, locally recurrent or metastatic breast cancer. J. Clin. Oncol. 2011, 29, 1252–1260. [Google Scholar] [CrossRef] [PubMed]
- Miles, D.W.; Diéras, V.; Cortés, J.; Duenne, A.A.; Yi, J.; O’Shaughnessy, J. First-line bevacizumab in combination with chemotherapy for HER2-negative metastatic breast cancer: Pooled and subgroup analyses of data from 2447 patients. Ann. Oncol. 2013, 24, 2773–2780. [Google Scholar] [CrossRef] [PubMed]
- Rossari, J.R.; Metzger-Filho, O.; Paesmans, M.; Saini, K.S.; Gennari, A.; de Azambuja, E.; Piccart-Gebhart, M. Bevacizumab and breast cancer: A meta-analysis of first-line phase III studies and a critical reappraisal of available evidence. J. Oncol. 2012, 2012. PMCID: PMC3447373. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar]
- Stemke-Hale, K.; Gonzalez-Angulo, A.M.; Lluch, A.; Neve, R.M.; Kuo, W.L.; Davies, M.; Carey, M.; Hu, Z.; Guan, Y.; Sahin, A.; et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008, 68, 6084–6091. [Google Scholar] [CrossRef]
- Sohn, J.; Do, K.A.; Liu, S.; Chen, H.; Mills, G.B.; Hortobagyi, G.N.; Meric-Bernstam, F.; Gonzalez-Angulo, A.M. Functional proteomics characterization of residual triple-negative breast cancer after standard neoadjuvant chemotherapy. Ann. Oncol. 2013, 24, 2522–2526. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Semiglazov, V.; van Dam, P.; Manikhas, A.; Bellet, M.; Mayordomo, J.; Campone, M.; Kubista, E.; Greil, R.; Bianchi, G.; et al. Phase II randomized study of neoadjuvant everolimus plus letrozole compared with placebo plus letrozole in patients with estrogen receptor-positive breast cancer. J. Clin. Oncol. 2009, 27, 2630–2637. [Google Scholar] [CrossRef] [PubMed]
- Jerusalem, G.; Fasolo, A.; Dieras, V.; Cardoso, F.; Bergh, J.; Vittori, L.; Zhang, Y.; Massacesi, C.; Sahmoud, T.; Gianni, L. Phase I trial of oral mTOR inhibitor everolimus in combination with trastuzumab and vinorelbine in pre-treated patients with HER2-overexpressing metastatic breast cancer. Breast Cancer Res. Treat. 2011, 125, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Yunokawa, M.; Koizumi, F.; Kitamura, Y.; Katanasaka, Y.; Okamoto, N.; Kodaira, M.; Yonemori, K.; Shimizu, C.; Ando, M.; Masutomi, K.; et al. Efficacy of everolimus, a novel mTOR inhibitor, against basal-like triple-negative breast cancer cells. Cancer Sci. 2012, 103, 1665–1671. [Google Scholar] [CrossRef] [PubMed]
- Serra, V.; Markman, B.; Scaltriti, M.; Eichhorn, P.J.; Valero, V.; Guzman, M.; Botero, M.L.; Llonch, E.; Atzori, F.; di Cosimo, S.; et al. NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res. 2008, 68, 8022–8030. [Google Scholar] [CrossRef] [PubMed]
- Montero, J.C.; Esparís-Ogando, A.; Re-Louhau, M.F.; Seoane, S.; Abad, M.; Calero, R.; Ocana, A.; Pandiella, A. Active kinase profiling, genetic and pharmacological data define mTOR as an important common target in triple-negative breast cancer. Oncogene 2014, 33, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Juvekar, A.; Burga, L.N.; Hu, H.; Lunsford, E.P.; Ibrahim, Y.H.; Balmañà, J.; Rajendran, A.; Papa, A.; Spencer, K.; Lyssiotis, C.A.; et al. Combining a PI3K inhibitor with a PARP inhibitor provides an effective therapy for BRCA1-related breast cancer. Cancer Discov. 2012, 2, 1048–1063. [Google Scholar] [CrossRef] [PubMed]
- Doane, A.S.; Danso, M.; Lal, P.; Donaton, M.; Zhang, L.; Hudis, C.; Gerald, W.L. An estrogen receptor-negative breast cancer subset characterized by a hormonally regulated transcriptional program and response to androgen. Oncogene 2006, 25, 3994–4008. [Google Scholar] [CrossRef] [PubMed]
- Farmer, P.; Bonnefoi, H.; Becette, V.; Tubiana-Hulin, M.; Fumoleau, P.; Larsimont, D.; Macgrogan, G.; Bergh, J.; Cameron, D.; Goldstein, D.; et al. Identification of molecular apocrine breast tumours by microarray analysis. Oncogene 2005, 24, 4660–4671. [Google Scholar] [CrossRef] [PubMed]
- Rouzier, R.; Perou, C.M.; Symmans, W.F.; Ibrahim, N.; Cristofanilli, M.; Anderson, K.; Hess, K.R.; Stec, J.; Ayers, M.; Wagner, P.; et al. Breast cancer molecular subtypes respond differently to preoperative chemotherapy. Clin. Cancer Res. 2005, 11, 5678–5685. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.S.; Mullins, M.; Cheang, M.C.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 2009, 27, 1160–1167. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Scher, H.I.; Molina, A.; Logothetis, C.J.; Chi, K.N.; Jones, R.J.; Staffurth, J.N.; North, S.; Vogelzang, N.J.; Saad, F.; et al. Abiraterone acetate for treatment of metastatic castration-resistant prostate cancer: Final overall survival analysis of the COU-AA-301 randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. 2012, 13, 983–992. [Google Scholar] [CrossRef] [PubMed]
- Ryan, C.J.; Smith, M.R.; de Bono, J.S.; Molina, A.; Logothetis, C.J.; de Souza, P.; Fizazi, K.; Mainwaring, P.; Piulats, J.M.; Ng, S.; et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N. Engl. J. Med. 2013, 368, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Nadal, R.; Taplin, M.E.; Bellmunt, J. Enzalutamide for the treatment of prostate cancer: Results and implications of the AFFIRM trial. Future Oncol. 2014, 10, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Beer, T.M.; Armstrong, A.J.; Sternberg, C.N.; Higano, C.S.; Iversen, P.; Loriot, Y.; Rathkopf, D.E.; Bhattacharya, S.; Carles, J.; de Bono, J.S.; et al. Enzalutamide in men with chemotherapy-naive metastatic prostate cancer (mCRPC): Results of phase III PREVAIL study. J. Clin. Oncol. 2014, 32 (Suppl. 4). Abstract LBA1. [Google Scholar] [PubMed]
- Tannock, I.F.; de Wit, R.; Berry, W.R.; Horti, J.; Pluzanska, A.; Chi, K.N.; Oudard, S.; Théodore, C.; James, N.D.; Turesson, I.; et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N. Engl. J. Med. 2004, 351, 1502–1512. [Google Scholar] [CrossRef] [PubMed]
- Petrylak, D.P.; Tangen, C.M.; Hussain, M.H.; Lara, P.N.; Jones, J.A.; Taplin, M.E.; Burch, P.A.; Berry, D.; Moinpour, C.; Kohli, M.; et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N. Engl. J. Med. 2004, 351, 1513–1520. [Google Scholar] [CrossRef] [PubMed]
- De Bono, J.S.; Oudard, S.; Ozguroglu, M.; Hansen, S.; Machiels, J.P.; Kocak, I.; Gravis, G.; Bodrogi, I.; Mackenzie, M.J.; Shen, L.; et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: A randomised open-label trial. Lancet 2010, 376, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Solit, D.B.; Zheng, F.F.; Drobnjak, M.; Münster, P.N.; Higgins, B.; Verbel, D.; Heller, G.; Tong, W.; Cordon-Cardo, C.; Agus, D.B.; et al. 17-Allylamino-17-demethoxygeldanamycin induces the degradation of androgen receptor and HER2/neu and inhibits the growth of prostate cancer xenografts. Clin. Cancer Res. 2002, 8, 986–993. [Google Scholar] [PubMed]
- Bukau, B.; Weissman, J.; Horwich, A. Molecular chaperones and protein quality control. Cell 2006, 125, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Calderwood, S.K. Heat shock proteins in breast cancer progression—A suitable case for treatment? Int. J. Hyperth. 2010, 26, 681–685. [Google Scholar] [CrossRef]
- Beliakoff, J.; Whitesell, L. Hsp90: An emerging target for breast cancer therapy. Anticancer Drugs 2004, 15, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Powers, M.V.; Workman, P. Targeting of multiple signalling pathways by heat shock protein 90 molecular chaperone inhibitors. Endocr. Relat. Cancer 2006, 13, S125–S135. [Google Scholar] [CrossRef] [PubMed]
- Zagouri, F.; Bournakis, E.; Koutsoukos, K.; Papadimitriou, C.A. Heat shock protein 90 (hsp90) expression and breast cancer. Pharmaceuticals (Basel) 2012, 5, 1008–1020. [Google Scholar] [CrossRef]
- Sequist, L.; Janne, P.; Sweney, J.; Walker, J.; Grayzel, D.; Lynch, T. Phase 1/2 trial of the novel Hsp90 inhibitor, IPI-504, in patients with relapsed and/or refractory stage IIIb or stage IV non-small cell lung cancer (NSCLC) stratified by EGFR mutation status. In Proceedings of the International Conference on Molecular Targets and Cancer Therapeutics, San Francisco, CA, USA, 22–26 October 2007.
- Bryson, J.; Infante, J.; Ramanathan, R.K.; Jones, S.F.; von Hoff, D.D.; Burris, H.A. A phase I dose-escalation study of the safety and pharmacokinetics (PK) of the oral Hsp90 inhibitor SNX-5422. J. Clin. Oncol. 2008, 26 (Suppl. 15). Abstract 14613. [Google Scholar] [PubMed]
- Wagner, A.; Morgan, J.; Chugh, R.; Rosen, L.S.; George, S.; Gordon, M.S.; Devine, C.M.; van den Abbeele, A.D.; Grayzel, D.; Demetri, G.D. Inhibition of heat shock protein 90 (Hsp90) with the novel agent IPI-504 in metastatic GIST following failure of tyrosine kinase inhibitors (TKIs) or other sarcomas: Clinical results from a phase I trial. J. Clin. Oncol. 2008, 26 (Suppl. 15). Abstract 10503. [Google Scholar] [CrossRef] [PubMed]
- Elfiky, A.; Saif, M.W.; Beeram, M.; O’Brien, S.; Lammanna, N.; Castro, J.E.; Woodworth, J.; Perea, R.; Storgard, C.; von Hoff, D.D. BIIB021, an oral, synthetic non-ansamycin Hsp90 inhibitor: Phase I experience. J. Clin. Oncol. 2008, 26 (Suppl. 15). Abstract 2503. [Google Scholar] [PubMed]
- Ide, S.; Motwani, M.; Jensen, M.R.; Wang, J.; Huseinovic, N.; Stiegler, P.; Wang, X.; Quadt, C. Pharmacodynamics and pharmacokinetics of AUY922 in a phase I study of solid tumor patients. J. Clin. Oncol. 2009, 27 (Suppl. 15). Abstract 3533. [Google Scholar] [PubMed]
- Ramanathan, R.K.; Egorin, M.J.; Eiseman, J.L.; Ramalingam, S.; Friedland, D.; Agarwala, S.S.; Ivy, S.P.; Potter, D.M.; Chatta, G.; Zuhowski, E.G.; et al. Phase I and pharmacodynamic study of 17-(allylamino)-17-demethoxygeldanamycin in adult patients with refractory advanced cancers. Clin. Cancer Res. 2007, 13, 1769–1774. [Google Scholar] [CrossRef] [PubMed]
- Caldas-Lopes, E.; Cerchietti, L.; Ahn, J.H.; Clement, C.C.; Robles, A.I.; Rodina, A.; Moulick, K.; Taldone, T.; Gozman, A.; Guo, Y.; et al. Hsp90 inhibitor PU-H71, a multimodal inhibitor of malignancy, induces complete responses in triple-negative breast cancer models. Proc. Natl. Acad. Sci. USA 2009, 106, 8368–8373. [Google Scholar] [CrossRef] [PubMed]
- Proia, D.A.; Zhang, C.; Sequeira, M.; Jimenez, J.P.; He, S.; Spector, N.; Shapiro, G.I.; Tolaney, S.; Nagai, M.; Acquaviva, J.; et al. Preclinical activity profile and therapeutic efficacy of the HSP90 inhibitor ganetespib in triple-negative breast cancer. Clin. Cancer Res. 2014, 20, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Ying, W.; Du, Z.; Sun, L.; Foley, K.P.; Proia, D.A.; Blackman, R.K.; Zhou, D.; Inoue, T.; Tatsuta, N.; Sang, J.; et al. Ganetespib, a unique triazolone-containing Hsp90 inhibitor, exhibits potent antitumor activity and a superior safety profile for cancer therapy. Mol. Cancer Ther. 2012, 11, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Gomez-Pinillos, A.; Ferrari, A.C. Simultaneous targeting of the androgen receptor and PI3K/mTOR pathway in androgen-dependent and androgen-independent prostate cancer cells. J. Clin. Oncol. 2010, 28. Abstract e15049. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mancini, P.; Angeloni, A.; Risi, E.; Orsi, E.; Mezi, S. Standard of Care and Promising New Agents for Triple Negative Metastatic Breast Cancer. Cancers 2014, 6, 2187-2223. https://doi.org/10.3390/cancers6042187
Mancini P, Angeloni A, Risi E, Orsi E, Mezi S. Standard of Care and Promising New Agents for Triple Negative Metastatic Breast Cancer. Cancers. 2014; 6(4):2187-2223. https://doi.org/10.3390/cancers6042187
Chicago/Turabian StyleMancini, Patrizia, Antonio Angeloni, Emanuela Risi, Errico Orsi, and Silvia Mezi. 2014. "Standard of Care and Promising New Agents for Triple Negative Metastatic Breast Cancer" Cancers 6, no. 4: 2187-2223. https://doi.org/10.3390/cancers6042187
APA StyleMancini, P., Angeloni, A., Risi, E., Orsi, E., & Mezi, S. (2014). Standard of Care and Promising New Agents for Triple Negative Metastatic Breast Cancer. Cancers, 6(4), 2187-2223. https://doi.org/10.3390/cancers6042187