
Collagenolytic Matrix Metalloproteinases in Chronic Obstructive Lung Disease and Cancer

{kind=link}
Abstract
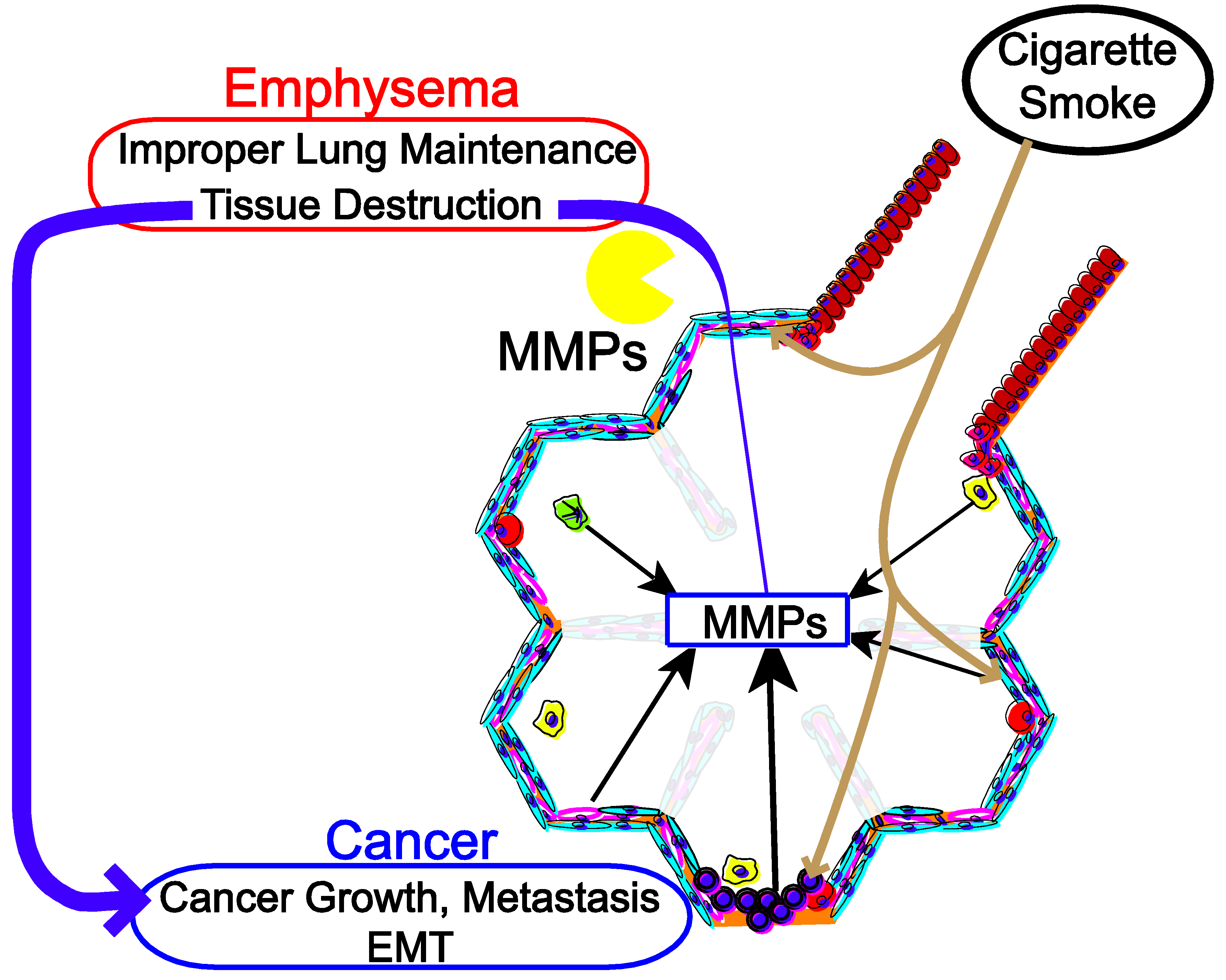
:1. Introduction

2. Lung Diseases and Collagenases
2.1. COPD and Collagenases
2.2. Cancer and Collagenases
3. Cigarette Smoke in COPD, Lung Cancer through the Regulation of Collagenase
4. Limitations of Collagenase Research
5. Future Directions
Acknowledgments
Conflicts of Interest
References
- Lopez, A.D.; Shibuya, K.; Rao, C.; Mathers, C.D.; Hansell, A.L.; Held, L.S.; Schmid, V.; Buist, S. Chronic obstructive pulmonary disease: Current burden and future projections. Eur. Respir. J. 2006, 27, 397–412. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2013. CA Cancer J. Clin. 2013, 63, 11–30. [Google Scholar] [CrossRef] [PubMed]
- Punturieri, A.; Szabo, E.; Croxton, T.L.; Shapiro, S.D.; Dubinett, S.M. Lung cancer and chronic obstructive pulmonary disease: Needs and opportunities for integrated research. J. Natl. Cancer Inst. 2009, 101, 554–559. [Google Scholar] [CrossRef] [PubMed]
- Skillrud, D.M.; Offord, K.P.; Miller, R.D. Higher risk of lung cancer in chronic obstructive pulmonary disease. A prospective, matched, controlled study. Ann. Intern. Med. 1986, 105, 503–507. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Jinbo, M.; Li, T.S.; Yagi, T.; Suga, K.; Hamano, K. Computed tomography-diagnosed emphysema, not airway obstruction, is associated with the prognostic outcome of early-stage lung cancer. Clin. Cancer Res. 2006, 12, 6730–6736. [Google Scholar] [CrossRef] [PubMed]
- Mannino, D.M.; Aguayo, S.M.; Petty, T.L.; Redd, S.C. Low lung function and incident lung cancer in the United States: Data from the first national health and nutrition examination survey follow-up. Arch. Intern. Med. 2003, 163, 1475–1480. [Google Scholar] [CrossRef] [PubMed]
- Snider, G.L. Chronic obstructive pulmonary disease—A continuing challenge. Am. Rev. Respir. Dis. 1986, 133, 942–944. [Google Scholar] [PubMed]
- Tockman, M.S.; Anthonisen, N.R.; Wright, E.C.; Donithan, M.G. Airways obstruction and the risk for lung cancer. Ann. Intern. Med. 1987, 106, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Vermaelen, K.; Brusselle, G. Exposing a deadly alliance: Novel insights into the biological links between COPD and lung cancer. Pulm. Pharmacol. Ther. 2013, 26, 544–554. [Google Scholar] [CrossRef]
- Church, D.F.; Pryor, W.A. Free-radical chemistry of cigarette smoke and its toxicological implications. Environ. Health Perspect. 1985, 64, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Tetley, T.D. New perspectives on basic mechanisms in lung disease. 6. Proteinase imbalance: Its role in lung disease. Thorax 1993, 48, 560–565. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J.; Shapiro, S.D.; Pauwels, R.A. Chronic obstructive pulmonary disease: Molecular and cellular mechanisms. Eur. Respir. J. 2003, 22, 672–688. [Google Scholar] [CrossRef] [PubMed]
- Shiomi, T.; Lemaitre, V.; D’Armiento, J.; Okada, Y. Matrix metalloproteinases, a disintegrin and metalloproteinases, and a disintegrin and metalloproteinases with thrombospondin motifs in non-neoplastic diseases. Pathol. Int. 2010, 60, 477–496. [Google Scholar] [CrossRef] [PubMed]
- Stamenkovic, I. Extracellular matrix remodelling: The role of matrix metalloproteinases. J. Pathol. 2003, 200, 448–464. [Google Scholar] [CrossRef] [PubMed]
- Ala-aho, R.; Kahari, V.M. Collagenases in cancer. Biochimie 2005, 87, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y. Proteinases and matrix degradation. In Kelley’s Textbook of Rheumatology, 9th ed.; Firestein, G.S., Budd, R.C., Gabriel, S.E., Mcinnes, I.B., O’dell, J.R., Eds.; Elsevier Health Sciences: Philadelhia, PA, USA, 2012; Volume 1, pp. 97–115. [Google Scholar]
- Imai, K.; Dalal, S.S.; Chen, E.S.; Downey, R.; Schulman, L.L.; Ginsburg, M.; D’Armiento, J. Human collagenase (matrix metalloproteinase-1) expression in the lungs of patients with emphysema. Am. J. Respir. Crit. Care Med. 2001, 163, 786–791. [Google Scholar] [PubMed]
- Mercer, B.A.; Kolesnikova, N.; Sonett, J.; D’Armiento, J. Extracellular regulated kinase/mitogen activated protein kinase is up-regulated in pulmonary emphysema and mediates matrix metalloproteinase-1 induction by cigarette smoke. J. Biol. Chem. 2004, 279, 17690–17696. [Google Scholar] [CrossRef] [PubMed]
- Mercer, B.A.; Wallace, A.M.; Brinckerhoff, C.E.; D’Armiento, J.M. Identification of a cigarette smoke-responsive region in the distal MMP-1 promoter. Am. J. Respir. Cell Mol. Biol. 2009, 40, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Geraghty, P.; Dabo, A.J.; D’Armiento, J. TLR4 protein contributes to cigarette smoke-induced matrix metalloproteinase-1 (MMP-1) expression in chronic obstructive pulmonary disease. J. Biol. Chem. 2011, 286, 30211–30218. [Google Scholar] [CrossRef] [PubMed]
- Chang, Z.L. Important aspects of toll-like receptors, ligands and their signaling pathways. Inflamm. Res. 2010, 59, 791–808. [Google Scholar] [CrossRef] [PubMed]
- An, C.H.; Wang, X.M.; Lam, H.C.; Ifedigbo, E.; Washko, G.R.; Ryter, S.W.; Choi, A.M. TLR4 deficiency promotes autophagy during cigarette smoke-induced pulmonary emphysema. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L748–L757. [Google Scholar] [CrossRef] [PubMed]
- D’Armiento, J.; Dalal, S.S.; Okada, Y.; Berg, R.A.; Chada, K. Collagenase expression in the lungs of transgenic mice causes pulmonary emphysema. Cell 1992, 71, 955–961. [Google Scholar] [CrossRef]
- Foronjy, R.F.; Okada, Y.; Cole, R.; D’Armiento, J. Progressive adult-onset emphysema in transgenic mice expressing human MMP-1 in the lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 284, L727–L737. [Google Scholar] [PubMed]
- Mercer, B.; Brinckerhoff, C.; D’Armiento, J. Activation of the MMP-1 promoter by cigarette smoke in human small airway epithelial cells requires ERK MAP kinase signaling: Differential response of the 1 G and 2 G promoter sequences. Proc. Am. Thorac. Soc. 2006, 3, 477. [Google Scholar] [CrossRef] [PubMed]
- D’Armiento, J.; di Colandrea, T.; Dalal, S.S.; Okada, Y.; Huang, M.T.; Conney, A.H.; Chada, K. Collagenase expression in transgenic mouse skin causes hyperkeratosis and acanthosis and increases susceptibility to tumorigenesis. Mol. Cell. Biol. 1995, 15, 5732–5739. [Google Scholar] [PubMed]
- Shiomi, T.; Okada, Y.; Foronjy, R.; Schiltz, J.; Jaenish, R.; Krane, S.; D’Armiento, J. Emphysematous changes are caused by degradation of type III collagen in transgenic mice expressing MMP-1. Exp. Lung Res. 2003, 29, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; In, K.H.; Kim, J.H.; Lee, S.Y.; Shin, C.; Shim, J.J.; Kang, K.H.; Yoo, S.H.; Kim, C.H.; Kim, H.K.; et al. Proteomic analysis in lung tissue of smokers and COPD patients. Chest 2009, 135, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.P.; Shen, G.H.; Ko, J.L. Matrix metalloproteinase-13 expression is associated with bone marrow microinvolvement and prognosis in non-small cell lung cancer. Lung Cancer 2006, 52, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.L.; Tai, H.; Wang, R.; Wang, X.; Churg, A. Cigarette smoke upregulates pulmonary vascular matrix metalloproteinases via TNF-alpha signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L125–L133. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.R.; Chida, A.S.; Bauter, M.R.; Shafiq, N.; Seweryniak, K.; Maggirwar, S.B.; Kilty, I.; Rahman, I. Cigarette smoke induces proinflammatory cytokine release by activation of NF-kappaB and posttranslational modifications of histone deacetylase in macrophages. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L46–L57. [Google Scholar] [CrossRef] [PubMed]
- Knauper, V.; Smith, B.; Lopez-Otin, C.; Murphy, G. Activation of progelatinase B (proMMP-9) by active collagenase-3 (MMP-13). Eur. J. Biochem. 1997, 248, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Takaishi, H.; Kimura, T.; Dalal, S.; Okada, Y.; D’Armiento, J. Joint diseases and matrix metalloproteinases: A role for MMP-13. Curr. Pharm. Biotechnol. 2008, 9, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Visse, R.; Nagase, H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: Structure, function, and biochemistry. Circ. Res. 2003, 92, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Itoh, T.; Tanioka, M.; Matsuda, H.; Nishimoto, H.; Yoshioka, T.; Suzuki, R.; Uehira, M. Experimental metastasis is suppressed in MMP-9-deficient mice. Clin. Exp. Metastasis 1999, 17, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Leinonen, T.; Pirinen, R.; Bohm, J.; Johansson, R.; Ropponen, K.; Kosma, V.M. Expression of matrix metalloproteinases 7 and 9 in non-small cell lung cancer. Relation to clinicopathological factors, beta-catenin and prognosis. Lung Cancer 2006, 51, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, A.; LeVine, W.F.; Gardner, H.A. Low plasma levels of matrix metalloproteinase 9 permit increased tumor angiogenesis. Oncogene 2002, 21, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Zhou, W.; Park, S.; Wain, J.C.; Lynch, T.J.; Liu, G.; Christiani, D.C. Matrix metalloproteinase-1 promoter polymorphism and lung cancer risk. Cancer Epidemiol. Biomark. Prev. 2005, 14, 567–570. [Google Scholar] [CrossRef]
- Van Kempen, L.C.; Coussens, L.M. MMP9 potentiates pulmonary metastasis formation. Cancer Cell 2002, 2, 251–252. [Google Scholar] [CrossRef] [PubMed]
- Foronjy, R.; Nkyimbeng, T.; Wallace, A.; Thankachen, J.; Okada, Y.; Lemaitre, V.; D’Armiento, J. Transgenic expression of matrix metalloproteinase-9 causes adult-onset emphysema in mice associated with the loss of alveolar elastin. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L1149–L1157. [Google Scholar] [CrossRef] [PubMed]
- Vernooy, J.H.; Lindeman, J.H.; Jacobs, J.A.; Hanemaaijer, R.; Wouters, E.F. Increased activity of matrix metalloproteinase-8 and matrix metalloproteinase-9 in induced sputum from patients with COPD. Chest 2004, 126, 1802–1810. [Google Scholar] [CrossRef] [PubMed]
- Segura-Valdez, L.; Pardo, A.; Gaxiola, M.; Uhal, B.D.; Becerril, C.; Selman, M. Upregulation of gelatinases A and B, collagenases 1 and 2, and increased parenchymal cell death in COPD. Chest 2000, 117, 684–694. [Google Scholar] [CrossRef] [PubMed]
- Stamenkovic, I. Matrix metalloproteinases in tumor invasion and metastasis. Semin. Cancer Biol. 2000, 10, 415–433. [Google Scholar] [CrossRef] [PubMed]
- Biswas, C.; Zhang, Y.; de Castro, R.; Guo, H.; Nakamura, T.; Kataoka, H.; Nabeshima, K. The human tumor cell-derived collagenase stimulatory factor (renamed EMMPRIN) is a member of the immunoglobulin superfamily. Cancer Res. 1995, 55, 434–439. [Google Scholar] [PubMed]
- Pahwa, S.; Stawikowski, M.J.; Fields, G.B. Monitoring and inhibiting MT1-MMP during cancer initiation and progression. Cancers 2014, 6, 416–435. [Google Scholar] [CrossRef] [PubMed]
- Bergers, G.; Brekken, R.; McMahon, G.; Vu, T.H.; Itoh, T.; Tamaki, K.; Tanzawa, K.; Thorpe, P.; Itohara, S.; Werb, Z.; et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat. Cell Biol. 2000, 2, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Hiratsuka, S.; Nakamura, K.; Iwai, S.; Murakami, M.; Itoh, T.; Kijima, H.; Shipley, J.M.; Senior, R.M.; Shibuya, M. MMP9 induction by vascular endothelial growth factor receptor-1 is involved in lung-specific metastasis. Cancer Cell 2002, 2, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Li, R.; Zucker, S.; Toole, B.P. EMMPRIN (CD147), an inducer of matrix metalloproteinase synthesis, also binds interstitial collagenase to the tumor cell surface. Cancer Res. 2000, 60, 888–891. [Google Scholar] [PubMed]
- Pendas, A.M.; Balbin, M.; Llano, E.; Jimenez, M.G.; Lopez-Otin, C. Structural analysis and promoter characterization of the human collagenase-3 gene (MMP13). Genomics 1997, 40, 222–233. [Google Scholar] [CrossRef]
- Pendas, A.M.; Uria, J.A.; Jimenez, M.G.; Balbin, M.; Freije, J.P.; Lopez-Otin, C. An overview of collagenase-3 expression in malignant tumors and analysis of its potential value as a target in antitumor therapies. Clin. Chim. Acta 2000, 291, 137–155. [Google Scholar] [CrossRef] [PubMed]
- Hattori, N.; Mochizuki, S.; Kishi, K.; Nakajima, T.; Takaishi, H.; D’Armiento, J.; Okada, Y. MMP-13 plays a role in keratinocyte migration, angiogenesis, and contraction in mouse skin wound healing. Am. J. Pathol. 2009, 175, 533–546. [Google Scholar] [CrossRef] [PubMed]
- Uria, J.A.; Stahle-Backdahl, M.; Seiki, M.; Fueyo, A.; Lopez-Otin, C. Regulation of collagenase-3 expression in human breast carcinomas is mediated by stromal-epithelial cell interactions. Cancer Res. 1997, 57, 4882–4888. [Google Scholar] [PubMed]
- Morrison, C.; Mancini, S.; Cipollone, J.; Kappelhoff, R.; Roskelley, C.; Overall, C. Microarray and proteomic analysis of breast cancer cell and osteoblast co-cultures: Role of osteoblast matrix metalloproteinase (MMP)-13 in bone metastasis. J. Biol. Chem. 2011, 286, 34271–34285. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.P.; Shai, S.E.; Hsia, J.Y.; Chen, C.Y. Clinical significance of bone marrow microinvolvement in nonsmall cell lung carcinoma. Cancer 2004, 100, 794–800. [Google Scholar] [CrossRef] [PubMed]
- Cierna, Z.; Mego, M.; Janega, P.; Karaba, M.; Minarik, G.; Benca, J.; Sedlackova, T.; Cingelova, S.; Gronesova, P.; Manasova, D.; et al. Matrix metalloproteinase 1 and circulating tumor cells in early breast cancer. BMC Cancer 2014, 14, 472. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.A.; Kraut, N.; Beug, H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr. Opin. Cell Biol. 2005, 17, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Araya, J.; Cambier, S.; Markovics, J.A.; Wolters, P.; Jablons, D.; Hill, A.; Finkbeiner, W.; Jones, K.; Broaddus, V.C.; Sheppard, D.; et al. Squamous metaplasia amplifies pathologic epithelial-mesenchymal interactions in COPD patients. J. Clin. Investig. 2007, 117, 3551–3562. [Google Scholar] [CrossRef] [PubMed]
- Denholm, R.; Schuz, J.; Straif, K.; Stucker, I.; Jockel, K.H.; Brenner, D.R.; de Matteis, S.; Boffetta, P.; Guida, F.; Bruske, I.; et al. Is previous respiratory disease a risk factor for lung cancer? Am. J. Respir. Crit. Care Med. 2014, 190, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Borish, E.T.; Pryor, W.A.; Venugopal, S.; Deutsch, W.A. DNA synthesis is blocked by cigarette tar-induced DNA single-strand breaks. Carcinogenesis 1987, 8, 1517–1520. [Google Scholar] [CrossRef] [PubMed]
- Pryor, W.A. Cigarette smoke radicals and the role of free radicals in chemical carcinogenicity. Environ. Health Perspect. 1997, 105, 875–882. [Google Scholar] [CrossRef] [PubMed]
- Joos, L.; He, J.Q.; Shepherdson, M.B.; Connett, J.E.; Anthonisen, N.R.; Pare, P.D.; Sandford, A.J. The role of matrix metalloproteinase polymorphisms in the rate of decline in lung function. Hum. Mol. Genet. 2002, 11, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Kanamori, Y.; Matsushima, M.; Minaguchi, T.; Kobayashi, K.; Sagae, S.; Kudo, R.; Terakawa, N.; Nakamura, Y. Correlation between expression of the matrix metalloproteinase-1 gene in ovarian cancers and an insertion/deletion polymorphism in its promoter region. Cancer Res. 1999, 59, 4225–4227. [Google Scholar] [PubMed]
- Nishioka, Y.; Kobayashi, K.; Sagae, S.; Ishioka, S.; Nishikawa, A.; Matsushima, M.; Kanamori, Y.; Minaguchi, T.; Nakamura, Y.; Tokino, T.; et al. A single nucleotide polymorphism in the matrix metalloproteinase-1 promoter in endometrial carcinomas. Jpn. J. Cancer Res. 2000, 91, 612–615. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.; Dhillon, S.; Turner, S.J.; Bateman, A.C.; Theaker, J.M.; Pickering, R.M.; Day, I.; Howell, W.M. Invasiveness of cutaneous malignant melanoma is influenced by matrix metalloproteinase 1 gene polymorphism. Cancer Res. 2001, 61, 1296–1298. [Google Scholar] [PubMed]
- Finlay, G.A.; O’Driscoll, L.R.; Russell, K.J.; D’Arcy, E.M.; Masterson, J.B.; FitzGerald, M.X.; O’Connor, C.M. Matrix metalloproteinase expression and production by alveolar macrophages in emphysema. Am. J. Respir. Crit. Care Med. 1997, 156, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Shaykhiev, R.; Sackrowitz, R.; Fukui, T.; Zuo, W.L.; Chao, I.W.; Strulovici-Barel, Y.; Downey, R.J.; Crystal, R.G. Smoking-induced CXCl14 expression in the human airway epithelium links chronic obstructive pulmonary disease to lung cancer. Am. J. Respir. Cell Mol. Biol. 2013, 49, 418–425. [Google Scholar] [CrossRef]
- Cavallaro, U.; Christofori, G. Cell adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat. Rev. Cancer 2004, 4, 118–132. [Google Scholar] [CrossRef] [PubMed]
- Henriet, P.; Rousseau, G.G.; Eeckhout, Y. Cloning and sequencing of mouse collagenase cDNA. Divergence of mouse and rat collagenases from the other mammalian collagenases. FEBS Lett. 1992, 310, 175–178. [Google Scholar] [CrossRef]
- Balbin, M.; Fueyo, A.; Knauper, V.; Pendas, A.M.; Lopez, J.M.; Jimenez, M.G.; Murphy, G.; Lopez-Otin, C. Collagenase 2 (MMP-8) expression in murine tissue-remodeling processes. Analysis of its potential role in postpartum involution of the uterus. J. Biol. Chem. 1998, 273, 23959–23968. [Google Scholar] [CrossRef] [PubMed]
- Balbin, M.; Fueyo, A.; Knauper, V.; Lopez, J.M.; Alvarez, J.; Sanchez, L.M.; Quesada, V.; Bordallo, J.; Murphy, G.; Lopez-Otin, C. Identification and enzymatic characterization of two diverging murine counterparts of human interstitial collagenase (MMP-1) expressed at sites of embryo implantation. J. Biol. Chem. 2001, 276, 10253–10262. [Google Scholar] [CrossRef] [PubMed]
- Carver, P.I.; Anguiano, V.; D’Armiento, J.M.; Shiomi, T. Mmp1a and MMP1B are not functional orthologs to human MMP1 in cigarette smoke induced lung disease. Exp. Toxicol. Pathol. 2015, 67, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Pei, D. Matrix metalloproteinases target protease-activated receptors on the tumor cell surface. Cancer Cell 2005, 7, 207–208. [Google Scholar] [PubMed]
- Goerge, T.; Barg, A.; Schnaeker, E.M.; Poppelmann, B.; Shpacovitch, V.; Rattenholl, A.; Maaser, C.; Luger, T.A.; Steinhoff, M.; Schneider, S.W. Tumor-derived matrix metalloproteinase-1 targets endothelial proteinase-activated receptor 1 promoting endothelial cell activation. Cancer Res. 2006, 66, 7766–7774. [Google Scholar] [CrossRef] [PubMed]
- Tauro, M.; McGuire, J.; Lynch, C.C. New approaches to selectively target cancer-associated matrix metalloproteinase activity. Cancer Metastasis Rev. 2014, 33, 1043–1057. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Woode, D.; Shiomi, T.; D'Armiento, J. Collagenolytic Matrix Metalloproteinases in Chronic Obstructive Lung Disease and Cancer. Cancers 2015, 7, 329-341. https://doi.org/10.3390/cancers7010329
Woode D, Shiomi T, D'Armiento J. Collagenolytic Matrix Metalloproteinases in Chronic Obstructive Lung Disease and Cancer. Cancers. 2015; 7(1):329-341. https://doi.org/10.3390/cancers7010329
Chicago/Turabian StyleWoode, Denzel, Takayuki Shiomi, and Jeanine D'Armiento. 2015. "Collagenolytic Matrix Metalloproteinases in Chronic Obstructive Lung Disease and Cancer" Cancers 7, no. 1: 329-341. https://doi.org/10.3390/cancers7010329
APA StyleWoode, D., Shiomi, T., & D'Armiento, J. (2015). Collagenolytic Matrix Metalloproteinases in Chronic Obstructive Lung Disease and Cancer. Cancers, 7(1), 329-341. https://doi.org/10.3390/cancers7010329