
Tumor and Stromal-Based Contributions to Head and Neck Squamous Cell Carcinoma Invasion

{kind=link}
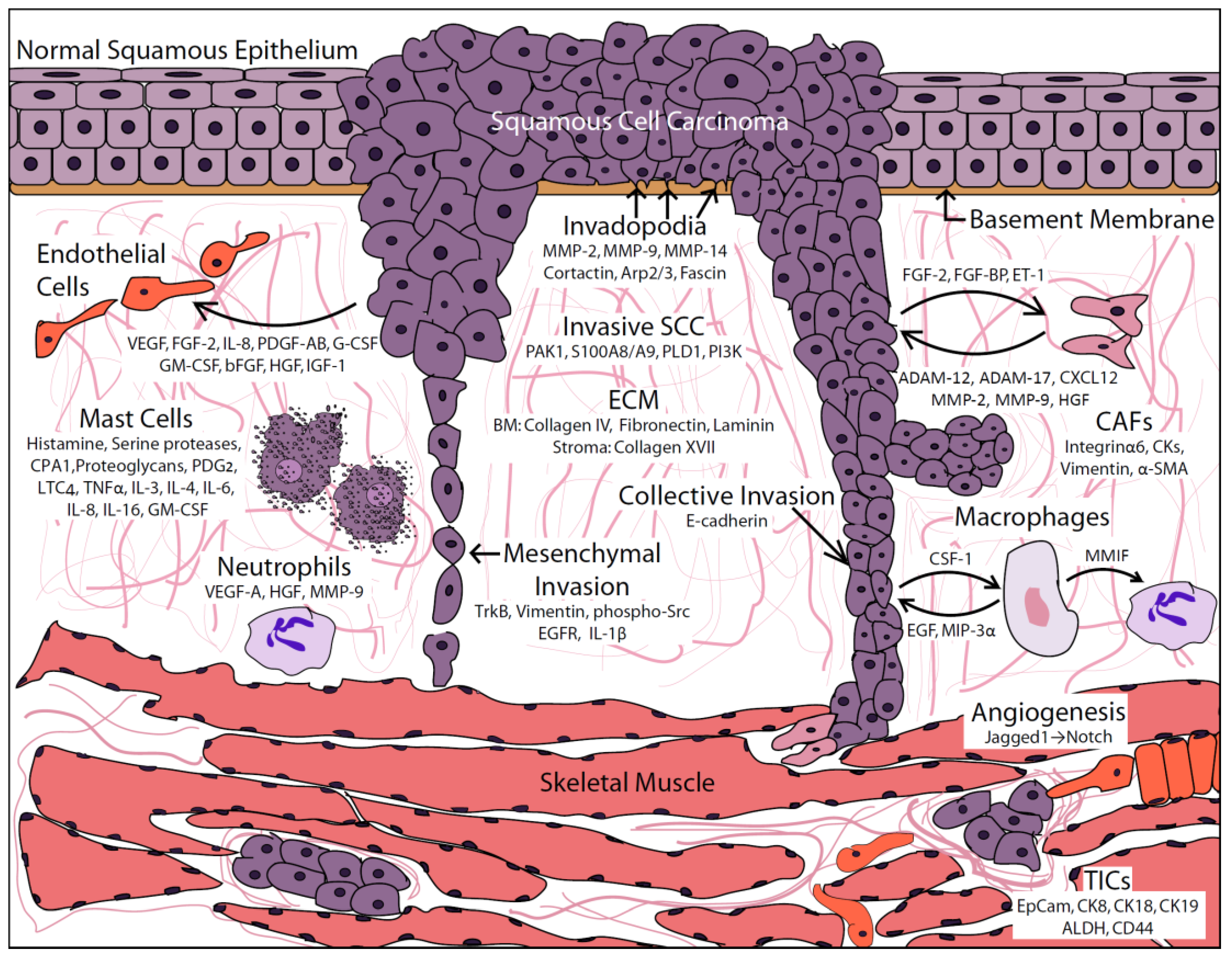
Abstract
:1. Introduction
2. Tumor Cell Contributions
2.1. Cell-ECM Interactions
2.2. Cell-Cell Interactions
2.3. Angiogenesis and Neo-Vascularization
2.4. Metastasis to Distant Sites
3. Stromal Cell Contributions
3.1. Mast Cells
3.2. Neutrophils
3.3. Macrophages
3.4. Endothelial Cells
3.5. Fibroblasts
4. Anti-Metastatic Therapeutic Approaches
5. Conclusions

Acknowledgments
Author Contributions
Abbreviations
| ALDH | aldehyde dehydrogenase |
| ADAM-12 | a disintegrin and metalloprotease 12 |
| ADAM-17 | a disintegrin and metalloprotease 17 |
| bFGF | basic fibroblast growth factor |
| CAF | cancer associated fibroblast |
| CK | cytokeratin |
| CK8 | cytokeratin 8 |
| CK18 | cytokeratin 18 |
| CK19 | cytokeratin 19 |
| CSC | cancer stem cell |
| CTC | circulating tumor cell |
| CXCL12 | chemokine C-X-C motif ligand 12 |
| CXCR4 | chemokine C-X-C motif receptor type 4 |
| E-cadherin | epithelial-cadherin |
| ECM | extracellular matrix |
| EGF | epidermal growth factor |
| EGFR | epidermal growth factor receptor |
| EMT | epithelial to mesenchymal transition |
| EpCAM | epidermal cell adhesion molecule |
| ET-1 | endothelin-1 |
| F-actin | filamentous-actin |
| FAK | focal adhesion kinase |
| FGF-2 | fibroblast growth factor 2 |
| FGF-3 | fibroblast growth factor 3 |
| FGF-4 | fibroblast growth factor 4 |
| FGF-19 | fibroblast growth factor 19 |
| FGF-BP | fibroblast growth factor binding protein |
| G-CSF | granulocyte colony stimulating factor |
| GM-CSF | granulocyte macrophage colony stimulating factor |
| HER3 | human epidermal growth factor receptor 3 |
| HGF | hepatocyte growth factor |
| HNSCC | head and neck squamous cell carcinoma |
| IGF-1 | insulin-like growth factor 1 |
| IGF-1R | insulin-like growth factor receptor 1 |
| IL-1β | interleukin 1β |
| IL-3 | interleukin 3 |
| IL-4 | interleukin 4 |
| IL-5 | interleukin 5 |
| IL-6 | interleukin 6 |
| IL-8 | interleukin 8 |
| IL-10 | interleukin 10 |
| IL-13 | interleukin 13 |
| IL-16 | interleukin 16 |
| LIMK | LIM kinase |
| LTC4 | leukotriene C4 |
| MAPK | mitogen activated protein kinase |
| MIP-3α | macrophage inhibitor protein 3α |
| MLC | myosin light chain |
| MLCK | myosin light chain kinase |
| MMIF | macrophage migration inhibitory factor |
| MMP | matrix metalloproteinase |
| MMP-1 | matrix metalloproteinase 1 |
| MMP-2 | matrix metalloproteinase 2 |
| MMP-3 | matrix metalloproteinase 3 |
| MMP-9 | matrix metalloproteinase 9 |
| MMP-14 | matrix metalloproteinase 14 |
| MT1-MMP | membrane type 1-matrix metalloproteinase |
| PAK1 | p21 protein (Cdc42/Rac)-activated kinase 1 |
| PDGF | platelet-derived growth factor |
| PDGF-AB | platelet-derived growth factor AB |
| PDGFR | platelet-derived growth factor receptor |
| PGD2 | prostaglandin D2 |
| PI3K | phosphoinsositide-3-kinase |
| TAM | tumor associated macrophage |
| TGF-β | transforming growth factor β |
| TIC | tumor initiating cell |
| TNF-α | tumor necrosis factor α |
| TrkB | tyrosine receptor kinase B |
| VEGF | vascular endothelial growth factor |
| VEGF-A | vascular endothelial growth factor A |
| VEGFR | vascular endothelial growth factor receptor |
Conflicts of Interest
References
- Probert, J.C.; Thompson, R.W.; Bagshaw, M.A. Patterns of spread of distant metastases in head and neck cancer. Cancer 1974, 33, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Onken, M.D.; Winkler, A.E.; Kanchi, K.-L.; Chalivendra, V.; Law, J.H.; Rickert, C.G.; Kallogjeri, D.; Judd, N.P.; Dunn, G.P.; Piccirillo, J.F.; et al. A surprising cross-species conservation in the genomic landscape of mouse and human oral cancer identifies a transcriptional signature predicting metastatic disease. Clin. Cancer Res. 2014, 20, 2873–2884. [Google Scholar] [CrossRef] [PubMed]
- Leemans, C.R.; Braakhuis, B.J.M.; Brakenhoff, R.H. The molecular biology of head and neck cancer. Nat. Rev. Cancer 2011, 11, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Koontongkaew, S. The tumor microenvironment contribution to development, growth, invasion and metastasis of head and neck squamous cell carcinomas. J. Cancer 2013, 4, 66–83. [Google Scholar] [CrossRef] [PubMed]
- Bowden, E.T.; Barth, M.; Thomas, D.; Glazer, R.I.; Mueller, S.C. An invasion-related complex of cortactin, paxillin and PKCmu associates with invadopodia at sites of extracellular matrix degradation. Oncogene 1999, 18, 4440–4449. [Google Scholar] [CrossRef] [PubMed]
- Branch, K.M.; Hoshino, D.; Weaver, A.M. Adhesion rings surround invadopodia and promote maturation. Biol. Open 2012, 1, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Wyckoff, J.; Condeelis, J. Cell migration in tumors. Curr. Opin. Cell Biol. 2005, 17, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Baldassarre, M.; Pompeo, A.; Beznoussenko, G.; Castaldi, C.; Cortellino, S.; McNiven, M.A.; Luini, A.; Buccione, R. Dynamin participates in focal extracellular matrix degradation by invasive cells. Mol. Biol. Cell 2003, 14, 1074–1084. [Google Scholar] [CrossRef] [PubMed]
- Leong, H.S.; Robertson, A.E.; Stoletov, K.; Leith, S.J.; Chin, C.A.; Chien, A.E.; Hague, M.N.; Ablack, A.; Carmine-Simmen, K.; McPherson, V.A.; et al. Invadopodia are required for cancer cell extravasation and are a therapeutic target for metastasis. Cell Rep. 2014, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lohmer, L.L.; Kelley, L.C.; Hagedorn, E.J.; Sherwood, D.R. Invadopodia and basement membrane invasion in vivo. Cell Adh. Migr. 2014, 8, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Paz, H.; Pathak, N.; Yang, J. Invading one step at a time: The role of invadopodia in tumor metastasis. Oncogene 2014, 33, 4193–4202. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, E.; Matrisian, L.M. Matrix metalloproteases in head and neck cancer. Head Neck 2006, 28, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Clark, E.S.; Whigham, A.S.; Yarbrough, W.G.; Weaver, A.M. Cortactin is an essential regulator of matrix metalloproteinase secretion and extracellular matrix degradation in invadopodia. Cancer Res. 2007, 67, 4227–4235. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Parsons, J.T. Cortactin, an 80/85-Kilodalton pp60Src substrate, is a Filamentous Actin-binding Protein Enriched in the Cell Cortex. J. Cell Biol. 1993, 120, 1417–1426. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Reynolds, A.B.; Kanner, S.B.; Vines, R.R.; Parsons, J.T. Identification and characterization of a novel cytoskeleton-associated pp60src substrate. Mol. Cell. Biol. 1991, 11, 5113–5124. [Google Scholar] [PubMed]
- Weed, S.A.; Du, Y.; Parsons, J.T. Translocation of cortactin to the cell periphery is mediated by the small GTPase Rac1. J. Cell Sci. 1998, 111, 2433–2443. [Google Scholar] [PubMed]
- Siton, O.; Ideses, Y.; Albeck, S.; Unger, T.; Bershadsky, A.D.; Gov, N.S.; Bernheim-Groswasser, A. Cortactin releases the brakes in actin- based motility by enhancing WASP-VCA detachment from Arp2/3 branches. Curr. Biol. 2011, 21, 2092–2097. [Google Scholar] [CrossRef] [PubMed]
- Schuuring, E.; Verhoeven, E.; Litvinov, S.; Michalides, R.J.A.M. The product of the EMS1 gene, amplified and overexpressed in human carcinomas, is homologous to a v-src substrate and is located in cell-substratum contact sites. Mol. Cell. Biol. 1993, 13, 2891–2898. [Google Scholar] [PubMed]
- Schuuring, E.; Verhoeven, E.; Mooi, W.J.; Michalides, R.J.A.M. Identification and cloning of two overexpressed genes, U21B31/PRAD1 and EMS1, within the amplified chromosome 11q13 region in human carcinomas. Oncogene 1992, 7, 355–361. [Google Scholar] [PubMed]
- Clark, E.S.; Brown, B.; Whigham, A.S.; Kochaishvili, A.; Yarbrough, W.G.; Weaver, A.M. Aggressiveness of HNSCC tumors depends on expression levels of cortactin, a gene in the 11q13 amplicon. Oncogene 2009, 28, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Liu, J.; Haudenschild, C.C.; Zhan, X. The Role of Tyrosine Phosphorylation of Cortactin in the Locomotion of Endothelial Cells. J. Biol. Chem. 1998, 273, 25770–25776. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.S.; Schechter, G.L.; Wasilenko, W.J.; Somers, K.D. Overexpression of EMS1/cortactin in NIH3T3 fibroblasts causes increased cell motility and invasion in vitro. Oncogene 1998, 16, 3227–3232. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, J.R.; Egile, C.; Gil, S.; Snapper, S.B.; Li, R.; Thomas, S.M. Cortactin regulates cell migration through activation of N-WASP. J. Cell Sci. 2005, 118, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Rodrigo, J.P.; Garcı, L.A.; Ramos, S.; Lazo, P.S.; Sua, C. EMS1 gene amplification correlates with poor prognosis in squamous cell carcinomas of the head and neck. Clin. Cancer Res. 2000, 6, 3177–3182. [Google Scholar] [PubMed]
- Rothschild, B.L.; Shim, A.H.; Ammer, A.G.; Kelley, L.C.; Irby, K.B.; Head, J.A.; Chen, L.; Varella-Garcia, M.; Sacks, P.G.; Frederick, B.; et al. Cortactin overexpression regulates actin-related protein 2/3 complex activity, motility, and invasion in carcinomas with chromosome 11q13 amplification. Cancer Res. 2006, 66, 8017–8025. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.C.; Hayes, K.E.; Ammer, A.G.; Martin, K.H.; Weed, S.A. Cortactin phosphorylated by ERK1/2 localizes to sites of dynamic actin regulation and is required for carcinoma lamellipodia persistence. PLoS One 2010, 5, e13847. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.C.; Ammer, A.G.; Hayes, K.E.; Martin, K.H.; Machida, K.; Jia, L.; Mayer, B.J.; Weed, S.A. Oncogenic Src requires a wild-type counterpart to regulate invadopodia maturation. J. Cell Sci. 2010, 123, 3923–3932. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.-L.; Shen, X.-M.; Zhang, Y.; Wei, F.; Xu, X.; Cai, Y.; Zhang, X.; Sun, Y.-T.; Zhan, Q.-M.; Wu, M.; et al. Amplification and overexpression of CTTN (EMS1) contribute to the metastasis of esophageal squamous cell carcinoma by promoting cell migration and anoikis resistance. Cancer Res. 2006, 66, 11690–11699. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tondravi, M.; Liu, J.; Smith, E.; Haudenschild, C.C.; Kaczmarek, M.; Zhan, X. Cortactin potentiates bone metastasis of breast cancer cells. Cancer Res. 2001, 61, 6906–6911. [Google Scholar] [PubMed]
- Mader, C.C.; Oser, M.; Magalhaes, M.A.O.; Bravo-Cordero, J.J.; Condeelis, J.; Koleske, A.J.; Gil-Henn, H. An EGFR-Src-Arg-Cortactin pathway mediates functional maturation of invadopodia and breast cancer cell invasion. Cancer Res. 2011, 71, 1730–1741. [Google Scholar] [CrossRef] [PubMed]
- Bowden, E.T.; Onikoyi, E.; Slack, R.; Myoui, A.; Yoneda, T.; Yamada, K.M.; Mueller, S.C. Co-localization of cortactin and phosphotyrosine identifies active invadopodia in human breast cancer. Exp. Cell Res. 2006, 321, 1240–1253. [Google Scholar] [CrossRef]
- Clark, E.S.; Weaver, A.M. A new role for cortactin in invadopodia: Regulation of protease secretion. Eur. J. Cell Biol. 2008, 87, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.P.; Shah, S.V.; Shukla, S.N.; Shah, P.M.; Patel, P.S. Clinical significance of MMP-2 and MMP-9 in patients with oral cancer. Head Neck 2007, 29, 564–572. [Google Scholar] [CrossRef] [PubMed]
- Koontongkaew, S.; Amornphimoltham, P.; Monthanpisut, P.; Saensuk, T.; Leelakriangsak, M. Fibroblasts and extracellular matrix differently modulate MMP activation by primary and metastatic head and neck cancer cells. Med. Oncol. 2012, 29, 690–703. [Google Scholar] [CrossRef] [PubMed]
- Papaspyrou, K.; Brochhausen, C.; Schmidtmann, I.; Fruth, K.; Gouveris, H.; Kirckpatrick, J.; Mann, W.; Brieger, J. Fascin upregulation in primary head and neck squamous cell carcinoma is associated with lymphatic metastasis. Oncol. Lett. 2014, 7, 2041–2046. [Google Scholar] [PubMed]
- Karasavvidou, F.; Barbanis, S.; Pappa, D.; Moutzouris, G.; Tzortzis, V.; Melekos, M.D.; Koukoulis, G. Fascin determination in urothelial carcinomas of the urinary bladder: A marker of invasiveness. Arch. Pathol. Lab. Med. 2008, 132, 1912–1915. [Google Scholar] [PubMed]
- Adams, J.C. Roles of fascin in cell adhesion and motility. Curr. Opin. Cell Biol. 2004, 16, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Jawhari, A.U.; Buda, A.; Jenkins, M.; Shehzad, K.; Sarraf, C.; Noda, M.; Farthing, M.J.G.; Pignatelli, M.; Adams, J.C. Fascin, an actin-bundling protein, modulates colonic epithelial cell invasiveness and differentiation in vitro. Am. J. Pathol. 2003, 162, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Kim, J.-M.; Park, J.K.; Huang, S.; Kwak, S.Y.; Ryu, K.A.; Kong, G.; Park, J.; Koo, B.S. Association of p21-activated kinase-1 activity with aggressive tumor behavior and poor prognosis of head and neck cancer. Head Neck 2014. [Google Scholar] [CrossRef]
- McCarty, S.K.; Saji, M.; Zhang, X.; Jarjoura, D.; Fusco, A.; Vasko, V.V.; Ringel, M.D. Group I p21-activated kinases regulate thyroid cancer cell migration and are overexpressed and activated in thyroid cancer invasion. Endocr. Relat. Cancer 2010, 17, 989–999. [Google Scholar] [CrossRef] [PubMed]
- King, H.; Nicholas, N.S.; Wells, C.M. Role of p-21-activated kinases in cancer progression. Int. Rev. Cell Mol. Biol. 2014, 309, 347–387. [Google Scholar] [PubMed]
- Dharmawardhane, S.; Brownson, D.; Lennartz, M.; Bokoch, G.M. Localization of p21-activated kinase 1 (PAK1) to pseudopodia, membrane ruffles, and phagocytic cups in activated human neutrophils. J. Leukoc. Biol. 1999, 66, 521–527. [Google Scholar] [PubMed]
- Dharmawardhane, S.; Sanders, L.C.; Martin, S.S.; Daniels, R.H.; Bokoch, G.M. Localization of p21-activated kinase 1 (PAK1) to pinocytic vesicles and cortical actin structures in stimulated cells. J. Cell Biol. 1997, 138, 1265–1278. [Google Scholar] [CrossRef] [PubMed]
- Delorme-Walker, V.D.; Peterson, J.R.; Chernoff, J.; Waterman, C.M.; Danuser, G.; DerMardirossian, C.; Bokoch, G.M. Pak1 regulates focal adhesion strength, myosin IIA distribution, and actin dynamics to optimize cell migration. J. Cell Biol. 2011, 193, 1289–1303. [Google Scholar] [CrossRef] [PubMed]
- Sanders, L.C. Inhibition of myosin light chain kinase by p21-activated kinase. Science 1999, 283, 2083–2085. [Google Scholar] [CrossRef] [PubMed]
- Stockton, R.A.; Schaefer, E.; Schwartz, M.A. p21-Activated kinase regulates endothelial permeability through modulation of contractility. J. Biol. Chem. 2004, 279, 46621–46630. [Google Scholar] [CrossRef] [PubMed]
- Arber, S.; Barbayannis, F.A.; Hanser, H.; Schneider, C.; Stanyon, C.A.; Bernard, O.; Caroni, P. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature 1998, 393, 805–809. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Higuchi, O.; Ohashi, K.; Nagata, K.; Wada, A.; Kangawa, K.; Nishida, E.; Mizuno, K. Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature 1998, 393, 809–812. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.C.; Shahab, S.; Weed, S.A. Actin cytoskeletal mediators of motility and invasion amplified and overexpressed in head and neck cancer. Clin. Exp. Metastasis 2008, 25, 289–304. [Google Scholar] [CrossRef] [PubMed]
- Vadlamudi, R.K.; Li, F.; Barnes, C.J.; Bagheri-Yarmand, R.; Kumar, R. P41-Arc subunit of human Arp2/3 complex is a p21-activated kinase-1-interacting substrate. EMBO Rep. 2004, 5, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.A.; Zhou, S.; Eves, R.; Shen, L.; Jia, L.; Mak, A.S. Phosphorylation of cortactin by p21-activated kinase. Arch. Biochem. Biophys. 2006, 456, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Elloul, S.; Vaksman, O.; Stavnes, H.T.; Trope, C.G.; Davidson, B.; Reich, R. Mesenchymal-to-epithelial transition determinants as characteristics of ovarian carcinoma effusions. Clin. Exp. Metastasis 2010, 27, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Goc, A.; Al-Azayzih, A.; Abdalla, M.; Al-Husein, B.; Kavuri, S.; Lee, J.; Moses, K.; Somanath, P.R. P21 activated kinase-1 (Pak1) promotes prostate tumor growth and microinvasion via inhibition of transforming growth factor β expression and enhanced matrix metalloproteinase 9 secretion. J. Biol. Chem. 2013, 288, 3025–3035. [Google Scholar] [CrossRef] [PubMed]
- Rider, L.; Oladimeji, P.; Diakonova, M. PAK1 regulates breast cancer cell invasion through secretion of matrix metalloproteinases in response to prolactin and three-dimensional collagen IV. Mol. Endocrinol. 2013, 27, 1048–1064. [Google Scholar] [CrossRef] [PubMed]
- Srikrishna, G. S100A8 and S100A9: New insights into their roles in malignancy. J. Innate Immun. 2012, 4, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Donato, R. S100: A multigenic family of calcium-modulated proteins of the EF-hand type with intracellular and extracellular functional roles. Int. J. Biochem. Cell Biol. 2001, 33, 637–668. [Google Scholar] [CrossRef] [PubMed]
- Heizmann, C.W.; Fritz, G.; Schäfer, B.W. S100 proteins: Structure, functions and pathology. Front. Biosci. 2002, 7, d1356–d1368. [Google Scholar] [CrossRef] [PubMed]
- Leukert, N.; Vogl, T.; Strupat, K.; Reichelt, R.; Sorg, C.; Roth, J. Calcium-dependent tetramer formation of S100A8 and S100A9 is essential for biological activity. J. Mol. Biol. 2006, 359, 961–972. [Google Scholar] [CrossRef] [PubMed]
- Silva, E.J.; Argyris, P.; Zou, X.; Ross, K.F.; Herzberg, M.C. S100A8/A9 regulates MMP-2 expression and invasion and migration by carcinoma cells. Int. J. Biochem. Cell Biol. 2014, 55, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Fung, L.F.; Lo, A.K.F.; Yuen, P.W.; Wang, X.H.; Tsao, S.W. Differential gene expression in nasopharyngeal carcinoma cells. Life Sci. 2000, 67, 923–936. [Google Scholar] [CrossRef] [PubMed]
- Ghavami, S.; Chitayat, S.; Hashemi, M.; Eshraghi, M.; Chazin, W.J.; Halayko, A.J.; Kerkhoff, C. S100A8/A9: A Janus-faced molecule in cancer therapy and tumorgenesis. Eur. J. Pharmacol. 2009, 625, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Fogh, B.S.; Multhaupt, H.A.B.; Couchman, J.R. Protein kinase C, focal adhesions and the regulation of cell migration. J. Histochem. Cytochem. 2014, 62, 172–184. [Google Scholar] [CrossRef] [PubMed]
- Zaidel-Bar, R.; Itzkovitz, S.; Ma’ayan, A.; Iyengar, R.; Geiger, B. Functional atlas of the integrin adhesome. Nat. Cell Biol. 2007, 9, 858–867. [Google Scholar] [CrossRef] [PubMed]
- Zaidel-Bar, R.; Cohen, M.; Addadi, L.; Geiger, B. Hierarchical assembly of cell-matrix adhesion complexes. Biochem. Soc. Trans. 2004, 32, 416–420. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Li, A.; Bi, J.; Veltman, D.M.; Zech, T.; Spence, H.J.; Yu, X.; Timpson, P.; Insall, R.H.; Frame, M.C.; et al. Loss of Scar/WAVE complex promotes N-WASP- and FAK-dependent invasion. Curr. Biol. 2013, 23, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Schlaepfer, D.D.; Mitra, S.K. Multiple connections link FAK to cell motility and invasion. Curr. Opin. Genet. Dev. 2004, 14, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Canel, M.; Secades, P.; Rodrigo, J.-P.; Cabanillas, R.; Herrero, A.; Suarez, C.; Chiara, M.-D. Overexpression of focal adhesion kinase in head and neck squamous cell carcinoma is independent of fak gene copy number. Clin. Cancer Res. 2006, 12, 3272–3279. [Google Scholar] [CrossRef] [PubMed]
- Gabarra-Niecko, V.; Schaller, M.D.; Dunty, J.M. FAK regulates biological processes important for the pathogenesis of cancer. Cancer Metastasis Rev. 2003, 22, 359–374. [Google Scholar] [CrossRef] [PubMed]
- Hsia, D.A.; Mitra, S.K.; Hauck, C.R.; Streblow, D.N.; Nelson, J.A.; Ilic, D.; Huang, S.; Li, E.; Nemerow, G.R.; Leng, J.; et al. Differential regulation of cell motility and invasion by FAK. J. Cell Biol. 2003, 160, 753–767. [Google Scholar] [CrossRef] [PubMed]
- McLean, G.W.; Carragher, N.O.; Avizienyte, E.; Evans, J.; Brunton, V.G.; Frame, M.C. The role of focal-adhesion kinase in cancer—A new therapeutic opportunity. Nat. Rev. Cancer 2005, 5, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Schaller, M.D.; Borgman, C.A.; Cobb, B.S.; Vines, R.R.; Reynolds, A.B.; Parsons, J.T. pp125FAK a structurally distinctive protein-tyrosine kinase associated with focal adhesions. Proc. Natl. Acad. Sci. USA 1992, 89, 5192–5196. [Google Scholar] [CrossRef] [PubMed]
- Shanthi, E.; Krishna, M.H.; Arunesh, G.M.; Venkateswara Reddy, K.; Sooriya Kumar, J.; Viswanadhan, V.N. Focal adhesion kinase inhibitors in the treatment of metastatic cancer: A patent review. Expert Opin. Ther. Pat. 2014, 24, 1077–1100. [Google Scholar] [CrossRef] [PubMed]
- Dao, P.; Smith, N.; Tomkiewicz-Raulet, C.; Yen-Pon, E.; Camacho-Artacho, M.; Lietha, D.; Herbeuval, J.-P.; Coumoul, X.; Garbay, C.; Chen, H. Design, synthesis, and evaluation of novel imidazo(1,2-a)(1,3,5)triazines and their derivatives as focal adhesion kinase inhibitors with antitumor activity. J. Med. Chem. 2014, 58, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-J.; Su, J.-H.; Tsai, C.-C.; Chen, Y.-J.; Liao, M.-H.; Wu, Y.-J. 11-epi-Sinulariolide acetate reduces cell migration and invasion of human hepatocellular carcinoma by reducing the activation of ERK1/2, p38MAPK and FAK/PI3K/AKT/mTOR signaling pathways. Mar. Drugs 2014, 12, 4783–4798. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Su, J.; Zhang, T.; Wang, K.; Li, X. Fangchinoline as a kinase inhibitor targets FAK and suppresses FAK-mediated signaling pathway in A549. J. Drug Target. 2014, 24, 1–9. [Google Scholar]
- Steed, P.M.; Chow, A.H. Intracellular signaling by phospholipase D as a therapeutic target. Curr. Pharm. Biotechnol. 2001, 2, 241–256. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, G.M.; Frohman, M.A. Phospholipase D: A lipid centric review. Cell. Mol. Life Sci. 2005, 62, 2305–2316. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, H.; Jeon, H.; Suh, P.-G.; Ryu, S.H. Phospholipase D1 regulates cell migration in a lipase activity-independent manner. J. Biol. Chem. 2006, 281, 15747–15756. [Google Scholar] [CrossRef] [PubMed]
- Ahn, B.-H.; Kim, S.Y.; Kim, E.H.; Choi, K.S.; Kwon, T.K.; Lee, Y.H.; Chang, J.-S.; Kim, M.-S.; Jo, Y.-H.; Min, D.S. Transmodulation between phospholipase D and c-Src enhances cell proliferation. Mol. Cell. Biol. 2003, 23, 3103–3115. [Google Scholar] [CrossRef] [PubMed]
- Martinez-quiles, N.; Ho, H.H.; Kirschner, M.W.; Ramesh, N.; Geha, R.S. Erk/Src phosphorylation of cortactin acts as a switch on-switch off mechanism that controls its ability to activate N-WASP. Mol. Cell. Biol. 2004, 24, 5269–5280. [Google Scholar] [CrossRef] [PubMed]
- Noh, D.Y.; Ahn, S.J.; Lee, R.A.; Park, I.A.; Kim, J.H.; Suh, P.G.; Ryu, S.H.; Lee, K.H.; Han, J.S. Overexpression of phospholipase D1 in human breast cancer tissues. Cancer Lett. 2000, 161, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.C.; Hayes, K.E.; Ammer, A.G.; Martin, K.H.; Weed, S.A. Revisiting the ERK/Src cortactin switch. Commun. Integr. Biol. 2011, 4, 205–207. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, M.C.; Scott, S.A.; Brown, K.A.; Oguin, T.H.; Thomas, P.G.; Daniels, J.S.; Morrison, R.; Brown, H.A.; Lindsley, C.W. Development of dual PLD1/2 and PLD2 selective inhibitors from a common 1,3,8-Triazaspiro(4.5)decane Core: Discovery of Ml298 and Ml299 that decrease invasive migration in U87-MG glioblastoma cells. J. Med. Chem. 2013, 56, 2695–2699. [Google Scholar] [CrossRef] [PubMed]
- Park, M.H.; Min, D.S. Quercetin-induced downregulation of phospholipase D1 inhibits proliferation and invasion in U87 glioma cells. Biochem. Biophys. Res. Commun. 2011, 412, 710–715. [Google Scholar] [CrossRef] [PubMed]
- Simpson, D.R.; Mell, L.K.; Cohen, E.E.W. Targeting the PI3K/AKT/mTOR pathway in squamous cell carcinoma of the head and neck. Oral Oncol. 2014, in press. [Google Scholar]
- Lui, V.W.Y.; Hedberg, M.L.; Li, H.; Vangara, B.S.; Pendleton, K.; Zeng, Y.; Lu, Y.; Zhang, Q.; Du, Y.; Gilbert, B.R.; et al. Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer Discov. 2013, 3, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Volinia, S.; Hiles, I.; Ormondroyd, E.; Nizetic, D.; Antonacci, R.; Rocchi, M.; Waterfield, M.D. Molecular cloning, cDNA sequence, and chromosomal localization of the human phosphatidylinositol 3-kinase p110 alpha (PIK3CA) gene. Genomics 1994, 24, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Roymans, D.; Slegers, H. Phosphatidylinositol 3-kinases in tumor progression. Eur. J. Biochem. 2001, 268, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Estilo, C.L.; O-Charoenrat, P.; Ngai, I.; Patel, S.G.; Reddy, P.G.; Dao, S.; Shaha, A.R.; Kraus, D.H.; Boyle, J.O.; Wong, R.J.; et al. The role of novel oncogenes squamous cell carcinoma-related oncogene and phosphatidylinositol 3-kinase p110alpha in squamous cell carcinoma of the oral tongue. Clin. Cancer Res. 2003, 9, 2300–2306. [Google Scholar] [PubMed]
- Katoh, M. WNT and FGF gene clusters. Int. J. Oncol. 2002, 21, 1269–1273. [Google Scholar] [PubMed]
- Marshall, M.E.; Hinz, T.K.; Kono, S.A.; Singleton, K.R.; Bichon, B.; Ware, K.E.; Marek, L.; Frederick, B.A.; Raben, D.; Heasley, L.E. Fibroblast growth factor receptors are components of autocrine signaling networks in head and neck squamous cell carcinoma cells. Clin. Cancer Res. 2011, 17, 5016–5025. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, C.; Juhn, S.K.; Ondrey, F.G.; Lin, J. Expression of fibroblast growth factor binding protein in head and neck cancer. Arch. Otolaryngol. Head Neck Surg. 2009, 135, 896–901. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Locker, J.; Sahai, E.; Segall, J.E. Classifying collective cancer cell invasion. Nat. Cell Biol. 2012, 14, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Alexander, S. Cancer invasion and the microenvironment: Plasticity and reciprocity. Cell 2011, 147, 992–1009. [Google Scholar] [CrossRef] [PubMed]
- Mandal, M.; Myers, J.N.; Lippman, S.M.; Johnson, F.M.; Williams, M.D.; Rayala, S.; Ohshiro, K.; Rosenthal, D.I.; Weber, R.S.; Gallick, G.E.; et al. Epithelial to mesenchymal transition in head and neck squamous carcinoma: Association of Src activation with E-cadherin down-regulation, vimentin expression, and aggressive tumor features. Cancer 2008, 112, 2088–2100. [Google Scholar] [CrossRef] [PubMed]
- Sok, J.C.; Coppelli, F.M.; Thomas, S.M.; Lango, M.N.; Xi, S.; Hunt, J.L.; Freilino, M.L.; Graner, M.W.; Wikstrand, C.J.; Bigner, D.D.; et al. Mutant epidermal growth factor receptor (EGFRvIII) contributes to head and neck cancer growth and resistance to EGFR targeting. Clin. Cancer Res. 2006, 12, 5064–5073. [Google Scholar] [CrossRef] [PubMed]
- Xi, S.; Zhang, Q.; Dyer, K.F.; Lerner, E.C.; Smithgall, T.E.; Gooding, W.E.; Kamens, J.; Grandis, J.R. Src kinases mediate STAT growth pathways in squamous cell carcinoma of the head and neck. J. Biol. Chem. 2003, 278, 31574–31583. [Google Scholar] [CrossRef] [PubMed]
- Bryne, M.; Koppang, H.S.; Lilleng, R.; Kjaerheim, A. Malignancy grading of the deep invasive margins of oral squamous cell carcinomas has high prognostic value. J. Pathol. 1992, 166, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Basu, D.; Bewley, A.F.; Sperry, S.M.; Montone, K.T.; Gimotty, P.A.; Rasanen, K.; Facompre, N.D.; Weinstein, G.S.; Nakagawa, H.; Diehl, J.A.; et al. EGFR inhibition promotes an aggressive invasion pattern mediated by mesenchymal-like tumor cells within squamous cell carcinomas. Mol. Cancer Ther. 2013, 12, 2176–2186. [Google Scholar] [CrossRef] [PubMed]
- Basu, D.; Montone, K.T.; Wang, L.-P.; Gimotty, P.A.; Hammond, R.; Diehl, J.A.; Rustgi, A.K.; Lee, J.T.; Rasanen, K.; Weinstein, G.S.; et al. Detecting and targeting mesenchymal-like subpopulations within squamous cell carcinomas. Cell Cycle 2011, 10, 2008–2016. [Google Scholar] [CrossRef] [PubMed]
- Basu, D.; Nguyen, T.-T.K.; Montone, K.T.; Zhang, G.; Wang, L.-P.; Diehl, J.A.; Rustgi, A.K.; Lee, J.T.; Weinstein, G.S.; Herlyn, M. Evidence for mesenchymal-like sub-populations within squamous cell carcinomas possessing chemoresistance and phenotypic plasticity. Oncogene 2010, 29, 4170–4182. [Google Scholar] [CrossRef] [PubMed]
- Kupferman, M.E.; Jiffar, T.; El-Naggar, A.; Yilmaz, T.; Zhou, G.; Xie, T.; Feng, L.; Wang, J.; Holsinger, F.C.; Yu, D.; et al. TrkB induces EMT and has a key role in invasion of head and neck squamous cell carcinoma. Oncogene 2010, 29, 2049–2059. [Google Scholar] [CrossRef]
- Sawatsubashi, M.; Yamada, T.; Fukushima, N.; Mizokami, H.; Tokunaga, O.; Shin, T. Association of vascular endothelial growth factor and mast cells with angiogenesis in laryngeal squamous cell carcinoma. Virchows Arch. 2000, 436, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Ninck, S.; Reisser, C.; Dyckhoff, G.; Helmke, B.; Bauer, H.; Herold-Mende, C. Expression profiles of angiogenic growth factors in squamous cell carcinomas of the head and neck. Int. J. Cancer 2003, 106, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Liss, C.; Fekete, M.J.; Hasina, R.; Lam, C.D.; Lingen, M.W. Paracrine angiogenic loop between head-and-neck squamous-cell carcinomas and macrophages. Int. J. Cancer 2001, 93, 781–785. [Google Scholar] [CrossRef] [PubMed]
- Bran, B.; Bran, G.; Hormann, K.; Riedel, F. The platelet-derived growth factor receptor as a target for vascular endothelial growth factor-mediated anti-angiogeneic therapy in head and neck cancer. Int. J. Oncol. 2009, 34, 255–261. [Google Scholar] [PubMed]
- Ali, M.A. Lymphatic microvessel density and the expression of lymphangiogenic factors in oral squamous cell carcinoma. Med. Princ. Pract. 2008, 17, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Shintani, S.; Terakada, N.; Klosek, S.K.; Ishikawa, T.; Nakashiro, K.; Hamakawa, H. Microvessel density and expression of vascular endothelial growth factor, basic fibroblast growth factor, and platelet-derived endothelial growth factor in oral squamous cell carcinomas. Int. J. Oral Maxillofac. Surg. 2005, 34, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Sedivy, R.; Beck-Mannagetta, J.; Haverkampf, C.; Battistutti, W.; Honigschnabl, S. Expression of vascular endothelial growth factor-C correlates with the lymphatic microvessel density and the nodal status in oral squamous cell cancer. J. Oral Pathol. Med. 2003, 32, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Beasley, N.J.P.; Prevo, R.; Banerji, S.; Leek, R.D.; Moore, J.; van Trappen, P.; Cox, G.; Harris, A.L.; Jackson, D.G. Intratumoral lymphangiogenesis and lymph node metastasis in head and neck cancer. Cancer Res. 2002, 62, 1315–1320. [Google Scholar] [PubMed]
- Jatana, K.R.; Balasubramanian, P.; Lang, J.C.; Yang, L.; Jatana, C.; White, E.; Agrawal, A.; Ozer, E.; Schuller, D.E.; Teknos, T.N.; et al. Significance of cirulating tumors cells in patients with squamous cell carcinoma of the head and neck. Arch. Otolaryngol. Head Neck Surg. 2010, 136, 1274–1279. [Google Scholar] [CrossRef] [PubMed]
- Buglione, M.; Grisanti, S.; Almici, C.; Mangoni, M.; Polli, C.; Consoli, F.; Verardi, R.; Costa, L.; Paiar, F.; Pasinetti, N.; et al. Circulating tumor cells in locally advanced cancer: Preliminary report about their possible role in predicting response to non-surgical treatment and survival. Eur. J. Cancer 2012, 48, 3019–3026. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, S.; Warner, K.A.; Dong, Z.; Imai, A.; Nör, C.; Ward, B.B.; Helman, J.I.; Taichman, R.S.; Bellile, E.L.; McCauley, L.K.; et al. Endothelial interleukin-6 defines the tumorigenic potential of primary human cancer stem cells. Stem Cells 2014, 32, 2845–2857. [Google Scholar] [CrossRef] [PubMed]
- Nör, C.; Zhang, Z.; Warner, K.A.; Bernardi, L.; Visioli, F.; Helman, J.I.; Roesler, R.; Nör, J.E. Cisplatin induces Bmi-1 and enhances the stem cell fraction in head and neck cancer. Neoplasia 2014, 16, 137–146. [Google Scholar] [PubMed]
- Zuo, J.-H.; Zhu, W.; Li, M.-Y.; Li, X.-H.; Yi, H.; Zeng, G.-Q.; Wan, X.-X.; He, Q.-Y.; Li, J.-H.; Qu, J.-Q.; et al. Activation of EGFR promotes squamous carcinoma SCC10A cell migration and invasion via inducing EMT-like phenotype change and MMP-9-mediated degradation of E-cadherin. J. Cell. Biochem. 2011, 112, 2508–2517. [Google Scholar] [CrossRef] [PubMed]
- St. John, M.A.; Dohadwala, M.; Luo, J.; Wang, G.; Lee, G.; Shih, H.; Heinrich, E.; Krysan, K.; Walser, T.; Hazra, S.; et al. Proinflammatory mediators upregulate snail in head and neck squamous cell carcinoma. Clin. Cancer Res. 2009, 15, 6018–6027. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.-L.; Xu, R.-J.; Zheng, Y.-Y.; Liu, J.; Teng, Y.-S.; Li, Y.; Zhu, J. Expression of type IV collagen, metalloproteinase-2, metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 in laryngeal squamous cell carcinomas. Asian Pac. J. Cancer Prev. 2011, 12, 3245–3249. [Google Scholar] [PubMed]
- Nielsen, J.D.; Moeslund, M.; Wandall, H.H.; Dabelsteen, S. Influences of tumor stroma on the malignant phenotype. J. Oral Pathol. Med. 2008, 37, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Parikka, M.; Nissienen, L.; Kainulainen, T.; Bruckner-Tuderman, L.; Salo, T.; Heino, J.; Tasanen, K. Collagen XVII promotes integrin-mediated squamous cell carcinoma transmigration—A novel role for αIIb integrin and tirofiban. Exp. Cell Res. 2006, 312, 1431–1438. [Google Scholar] [CrossRef] [PubMed]
- Prussin, C.; Metcalfe, D.D. IgE, mast cells, basophils, and eosinophils. J. Allergy Clin. Imunol. 2003, 111, S486–S494. [Google Scholar] [CrossRef]
- Barth, P.J.; Schweinsberg zu Schweinsberg, T.; Ramaswamy, A.; Moll, R. CD34+ fibrocytes, alpha-smooth muscle antigen-positive myofibroblasts, and CD117 expression in the stroma of invasive squamous cell carcinomas of the oral cavity, pharynx, and larynx. Virchows Arch. 2004, 444, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Iamaroon, A.; Pongsiriwet, S.; Jittidecharaks, S.; Pattanaporn, K.; Prapayasatok, S.; Wanachantararak, S. Increase of mast cells and tumor angiogenesis in oral squamous cell carcinoma. J. Oral Pathol. Med. 2003, 32, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Beckhove, P.; Helmke, B.M.; Ziouta, Y.; Bucur, M.; Borner, W.; Mogler, C.; Dyckhoff, G.; Herold-Mende, C. Heparanase expression at the invasion front of human head and neck cancers and correlation with poor prognosis. Clin. Cancer Res. 2005, 11, 2899–2906. [Google Scholar] [CrossRef] [PubMed]
- Friedmann, Y.; Vlodavsky, I.; Aingorn, H.; Aviv, A.; Peretz, T.; Pecker, I.; Pappo, O. Expression of heparanase in normal, dysplastic and neoplastic human colonic mucosa and stroma. Am. J. Pathol. 2000, 157, 1167–1175. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C. Neutrophils and immunity: Challenges and opportunities. Nat. Rev. Immunol. 2006, 6, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Waugh, D.J.J.; Wilson, C. The interleukin-8 pathway in cancer. Clin. Cancer Res. 2008, 14, 6735–6741. [Google Scholar] [CrossRef] [PubMed]
- Scapini, P.; Bazzoni, F.; Cassatella, M.A. Regulation of B-cell-activating factor (BAFF)/B lymphocyte stimulator (BLyS) expression in human neutrophils. Immunol. Lett. 2008, 116, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Scapini, P.; Morini, M.; Tecchio, C.; Minghelli, S.; di Carlo, E.; Tanghetti, E.; Albini, A.; Lowell, C.; Berton, G.; Noonan, D.M.; et al. CXCL1/Macrophage inflammatory protein-2-induced angiogenesis in vivo is mediated by neutrophil-derived vascular endothelial growth factor-A. J. Immunol. 2004, 172, 5034–5040. [Google Scholar] [CrossRef] [PubMed]
- Benelli, R.; Morini, M.; Carrozzino, F.; Ferrari, N.; Minghelli, S.; Santi, L.; Cassatella, M.; Noonan, D.M.; Albini, A. Neutrophils as a key cellular target for angiostatin: Implications for regulation of angiogenesis and inflammation. FASEB J. 2002, 2, 267–269. [Google Scholar]
- Eming, S.A.; Krieg, T.; Davidson, J.M. Inflammation in wound repair: Molecular and cellular mechanisms. J. Investig. Dermatol. 2007, 127, 514–525. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Shintani, S.; Terakada, N.; Nakashiro, K.; Hamakawa, H. Infiltration of tumor-associated macrophages in human oral squamous cell carcinoma. Oncol. Rep. 2002, 9, 1219–1223. [Google Scholar] [PubMed]
- Marcus, B.; Arenberg, D.; Lee, J.; Kleer, C.; Chepeha, D.B.; Schmalbach, C.E.; Islam, M.; Paul, S.; Pan, Q.; Hanash, S.; et al. Prognostic factors in oral cavity and oropharyngeal squamous cell carcinoma. Cancer 2004, 101, 2779–2787. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.-Y.; Chang, L.-C.; Pan, L.-F.; Hung, Y.-J.; Lee, C.-H.; Shieh, Y.-S. Clinicopathologic significance of tumor cell-lined vessel and microenvironment in oral squamous cell carcinoma. Oral Oncol. 2008, 44, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Pixley, F.; Condeelis, J. Invadopodia and podosomes in tumor invasion. Eur. J. Cell Biol. 2006, 85, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Wyckoff, J.; Wang, W.; Lin, E.Y.; Wang, Y.; Pixley, F.; Stanley, E.R.; Graf, T.; Pollard, J.W.; Segall, J.; Condeelis, J. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res. 2004, 64, 7022–7029. [Google Scholar] [CrossRef] [PubMed]
- Goswami, S.; Sahai, E.; Wyckoff, J.; Cammer, M.; Cox, D.; Pixley, F.J.; Stanley, E.R.; Segall, J.E.; Condeelis, J.S. Macrophages promote the invasion of breast carcinoma cells via a colony-stimulating factor-1/epidermal growth factor paracrine loop. Cancer Res. 2005, 65, 5278–5283. [Google Scholar] [CrossRef] [PubMed]
- Dumitru, C.A.; Gholaman, H.; Trellakis, S.; Bruderek, K.; Dominas, N.; Gu, X.; Bankfalvi, A.; Whiteside, T.L.; Lang, S.; Brandau, S. Tumor-derived macrophage migration inhibitory factor modulates the biology of head and neck cancer cells via neutrophil activation. Int. J. Cancer 2011, 129, 859–869. [Google Scholar] [CrossRef] [PubMed]
- Linder, S.; Nelson, D.; Weiss, M.; Aepfelbacher, M. Wiskott-Aldrich syndrome protein regulates podosomes in primary human macrophages. Proc. Natl. Acad. Sci. USA 1999, 96, 9648–9653. [Google Scholar] [CrossRef] [PubMed]
- Schachtner, H.; Calaminus, S.D.J.; Thomas, S.G.; Machesky, L.M. Podosomes in adhesion, migration, mechanosensing and matrix remodeling. Cytoskeleton 2013, 70, 572–589. [Google Scholar] [CrossRef] [PubMed]
- Varon, C.; Tatin, F.; Moreau, V.; van Obberghen-Schilling, E.; Fernandez-Sauze, S.; Reuzeau, E.; Kramer, I.; Genot, E. Transforming growth factor beta induces rosettes of podosomes in primary aortic endothelial cells. Mol. Cell. Biol. 2006, 26, 3582–3594. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.-P.; Kao, H.-K.; Yen, T.-C.; Chang, Y.-L.; Liang, Y.; Liu, S.-C.; Lee, L.-Y.; Chang, Y.-L.; Kang, C.-J.; Chen, I.-H.; et al. Overexpression of macrophage inflammatory protein-3alpha in oral cavity squamous cell carcinoma is associated with nodal metastasis. Oral Oncol. 2011, 47, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Osiak, A.-E.; Zenner, G.; Linder, S. Subconfluent endothelial cells form podosomes downstream of cytokine and RhoGTPase signaling. Exp. Cell Res. 2005, 307, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Lorenz, M.; Kempiak, S.; Sarmieto, C.; Coniglio, S.; Symons, M.; Segall, J.; Eddy, R.; Miki, H.; Takenawa, T.; et al. Molecular mechanisms of invadopodium formation: The role of the N-WASP-Arp2/3 complex pathway and cofilin. J. Cell Biol. 2005, 168, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, K.; Miki, H.; He, H.; Maruta, H.; Takenawa, T. Essential role of neural Wiskot-Aldrich syndrome protein in podosome formation and degradation of extracellular matrix in src-transformed fibroblasts. Cancer Res. 2002, 62, 669–674. [Google Scholar] [PubMed]
- Spinardi, L.; Rietdorf, J.; Nitsch, L.; Bono, M.; Tacchetti, C.; Way, M.; Marchisio, P.C. A dynamic podosome-like structure of endothelial cells. Exp. Cell Res. 2004, 295, 360–374. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Li, S.; Chepeha, D.B.; Giordano, T.J.; Li, J.; Zhang, H.; Polverini, P.J.; Nor, J.; Kitajewski, J.; Wang, C.-Y. Crosstalk between tumor and endothelial cells promotes tumor angiogenesis by MAPK activation of Notch signaling. Cancer Cell 2005, 8, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Goetz, J.G.; Minguet, S.; Navarro-Lerida, I.; Lazcano, J.J.; Samaniego, R.; Calvo, E.; Tello, M.; Osteso-Ibanez, T.; Pellinen, T.; Echarri, A.; et al. Biomechanical remodeling of the microenvironment by stromal caveolin-1 favors tumor invasion and metastasis. Cell 2011, 146, 148–163. [Google Scholar] [CrossRef] [PubMed]
- Gaggioli, C.; Hooper, S.; Hidalgo-Carcedo, C.; Grosse, R.; Marshall, J.F.; Harrington, K.; Sahai, E. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat. Cell Biol. 2007, 9, 1392–1400. [Google Scholar] [CrossRef] [PubMed]
- Kelly, T.; Mueller, S.C.; Yeh, Y.; Chen, W.T. Invadopodia promote proteolysis of a wide variety of extracellular matrix proteins. J. Cell. Physiol. 1994, 158, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.P.; Cirillo, N.; Hassona, Y.; Wei, W.; Thurlow, J.K.; Cheong, S.C.; Pitiyage, G.; Parkinson, E.K.; Prime, S.S. Fibroblast gene expression profile reflects the stage of tumour progression in oral squamous cell carcinoma. J. Pathol. 2011, 223, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Abram, C.L.; Seals, D.F.; Pass, I.; Salinsky, D.; Maurer, L.; Roth, T.M.; Courtneidge, S.A. The adaptor protein fish associates with members of the ADAMs family and localizes to podosomes of Src-transformed cells. J. Biol. Chem. 2003, 278, 16844–16851. [Google Scholar] [CrossRef] [PubMed]
- Seals, D.F.; Azucena, E.F.J.; Pass, I.; Tesfay, L.; Gordon, R.; Woodrow, M.; Resau, J.H.; Courtneidge, S.A. The adaptor protein Tks5/Fish is required for podosome formation and function, and for the protease-driven invasion of cancer cells. Cancer Cell 2005, 7, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Hinsley, E.E.; Hunt, S.; Hunter, K.D.; Whawell, S.A.; Lambert, D.W. Endothelin-1 stimulates motility of head and neck squamous carcinoma cells by promoting stromal-epithelial interactions. Int. J. Cancer 2012, 130, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Nakashiro, K.; Klosek, S.K.; Goda, H.; Hara, S.; Uchida, D.; Hamakawa, H. Hypoxia enhances CXCR4 expression by activating HIF-1 in oral squamous cell carcinoma. Oncol. Rep. 2009, 21, 707–712. [Google Scholar] [PubMed]
- Uchida, D.; Begum, N.M.; Almofti, A.; Nakashiro, K.; Kawamata, H.; Tateishi, Y.; Hamakawa, H.; Yoshida, H.; Sato, M. Possible role of stromal-cell-derived factor-1/CXCR4 signaling on lymph node metastasis of oral squamous cell carcinoma. Exp. Cell Res. 2003, 290, 289–302. [Google Scholar] [CrossRef] [PubMed]
- Chuma, M.; Sakamoto, M.; Yasuda, J.; Fujii, G.; Nakanishi, K.; Tsuchiya, A.; Asaka, M.; Hirohashi, S. Overexpression of cortactin is involved in motility and metastasis of hepatocellular carcinoma. J. Hepatol. 2004, 41, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hu, T.; Shen, J.; Li, S.F.; Lin, J.W.; Zheng, X.H.; Gao, Q.H.; Zhou, H.M. Separation, cultivation and biological characteristics of oral carcinoma-associated fibroblasts. Oral Dis. 2006, 12, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Lewis, M.P.; Lygoe, K.A.; Nystrom, M.L.; Anderson, W.P.; Speight, P.M.; Marshall, J.F.; Thomas, G.J. Tumor-derived TGF-β1 modulates myofibroblast differentiation and promotes HGF/SF-dependent invasion of squamous carcinoma cells. Br. J. Cancer 2004, 90, 822–832. [Google Scholar] [CrossRef] [PubMed]
- Marsh, D.; Suchak, K.; Moutasim, K.A.; Vallath, S.; Hopper, C.; Jerjes, W.; Upile, T.; Kalavrezos, N.; Violette, S.M.; Weinreb, P.H.; et al. Stromal features are predictive of disease mortality in oral cancer patients. J. Pathol. 2011, 223, 470–481. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, S.E.; Shi, H.; Lin, F.; Dasari, S.; Bednash, J.; Thorne, S.; Watkins, S.; Joshi, R.; Thomas, S.M. Enhancement of head and neck squamous cell carcinoma proliferation, invasion, and metastasis by tumor-associated fibroblasts in preclinical models. Head Neck 2014, 36, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Taddei, M.L.; Cavallini, L.; Comito, G.; Giannoni, E.; Folini, M.; Marini, A.; Gandellini, P.; Morandi, A.; Pintus, G.; Raspollini, M.R.; et al. Senescent stroma promotes prostate cancer progression: The role of miR-210. Mol. Oncol. 2014, 8, 1729–1746. [Google Scholar] [CrossRef] [PubMed]
- Hassona, Y.; Cirillo, N.; Heesom, K.; Parkinson, E.K.; Prime, S.S. Senescent cancer-associated fibroblasts secrete active MMP-2 that promotes keratinocyte dis-cohesion and invasion. Br. J. Cancer 2014, 111, 1230–1237. [Google Scholar] [CrossRef] [PubMed]
- Hassona, Y.; Cirillo, N.; Lim, K.P.; Herman, A.; Mellone, M.; Thomas, G.J.; Pitiyage, G.N.; Parkinson, E.K.; Prime, S.S. Progression of genotype-specific oral cancer leads to senescence of cancer-associated fibroblasts and is mediated by oxidative stress and TGF-β. Carcinogenesis 2013, 34, 1286–1295. [Google Scholar] [CrossRef] [PubMed]
- Ren, C.; Cheng, X.; Lu, B.; Yang, G. Activation of interleukin-6/signal transducer and activator of transcription 3 by human papillomavirus early proteins 6 induces fibroblast senescence to promote cervical tumourigenesis through autocrine and paracrine pathways in tumour microenvironment. Eur. J. Cancer 2013, 49, 3889–3899. [Google Scholar] [CrossRef] [PubMed]
- Capparelli, C.; Chiavarina, B.; Whitaker-Menezes, D.; Pestell, T.G.; Pestell, R.G.; Hulit, J.; Andò, S.; Howell, A.; Martinez-Outschoorn, U.E.; Sotgia, F.; et al. CDK inhibitors (p16/p19/p21) induce senescence and autophagy in cancer-associated fibroblasts, “fueling” tumor growth via paracrine interactions, without an increase in neo-angiogenesis. Cell Cycle 2012, 11, 3599–3610. [Google Scholar] [CrossRef] [PubMed]
- Capparelli, C.; Whitaker-Menezes, D.; Guido, C.; Balliet, R.; Pestell, T.G.; Howell, A.; Sneddon, S.; Pestell, R.G.; Martinez-Outschoorn, U.; Lisanti, M.P.; et al. CTGF drives autophagy, glycolysis and senescence in cancer-associated fibroblasts via HIF1 activation, metabolically promoting tumor growth. Cell Cycle 2012, 11, 2272–2284. [Google Scholar] [CrossRef] [PubMed]
- Ammer, A.G.; Kelley, L.C.; Hayes, K.E.; Evans, J.V.; Lopez-skinner, A.; Martin, K.H.; Frederick, B.; Rothschild, B.L.; Elvin, P.; Green, T.P.; et al. Saracatinib impairs head and neck squamous cell carcinoma invasion by disruption invadopodia function. J. Cancer Sci. Ther. 2009, 1, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Nozawa, H.; Howell, G.; Suzuki, S.; Zhang, Q.; Qi, Y.; Klein-Seetharaman, J.; Wells, A.; Grandis, J.R.; Thomas, S.M. Combined inhibition of PLCγ-1 and c-Src abrogates epidermal growth factor receptor-mediated head and neck squamous cell carcinoma invasion. Clin. Cancer Res. 2008, 14, 4336–4344. [Google Scholar] [CrossRef] [PubMed]
- Koppikar, P.; Choi, S.-H.; Egloff, A.M.; Cai, Q.; Suzuki, S.; Freilino, M.; Nozawa, H.; Thomas, S.M.; Gooding, W.E.; Siegfried, J.M.; et al. Combined inhibition of c-Src and epidermal growth factor receptor abrogates growth and invasion of head and neck squamous cell carcinoma. Clin. Cancer Res. 2008, 14, 4284–4291. [Google Scholar] [CrossRef] [PubMed]
- Fury, M.G.; Baxi, S.; Shen, R.; Kelly, K.W.; Lipson, B.L.; Carlson, D.; Stambuk, H.; Haque, S.; Pfister, D.G. Phase II study of saracatinib (AZD0530) for patients with recurrent or metastatic head and neck squamous cell carcinoma (HNSCC). Anticancer Res. 2011, 31, 249–253. [Google Scholar] [PubMed]
- Johnson, F.M.; Saigal, B.; Talpaz, M.; Donato, N.J. Dasatinib (BMS-354825) tyrosine kinase inhibitor suppresses invasion and induces cell cycle arrest and apoptosis of head and neck squamous cell carcinoma and non-small cell lung cancer cells. Clin. Cancer Res. 2005, 11, 6924–6932. [Google Scholar] [CrossRef] [PubMed]
- Brooks, H.D.; Glisson, B.S.; Bekele, B.N.; Johnson, F.M.; Ginsberg, L.E.; El-Naggar, A.; Culotta, K.S.; Takebe, N.; Wright, J.; Tran, H.T.; et al. Phase 2 study of dasatinib in the treatment of head and neck squamous cell carcinoma. Cancer 2011, 117, 2112–2119. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, D.; Plattner, R. Activation of Abl tyrosine kinases promotes invasion of aggressive breast cancer cells. Cancer Res. 2006, 66, 5648–5655. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Sun, T.; Ji, L.; Deng, W.; Roth, J.; Minna, J.; Arlinghaus, R. Oncogenic activation of c-Abl in non-small cell lung cancer cells lacking FUS1 expression: Inhibition of c-Abl by the tumor suppressor gene product Fus1. Oncogene 2007, 26, 6989–6996. [Google Scholar] [CrossRef] [PubMed]
- Furlan, A.; Stagni, V.; Hussain, A.; Richelme, S.; Conti, F.; Prodosmo, A.; Destro, A.; Roncalli, M.; Barilà, D.; Maina, F. Abl interconnects oncogenic Met and p53 core pathways in cancer cells. Cell Death Differ. 2011, 18, 1608–1616. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, S.S.; Fiore, L.S.; Sims, J.T.; Friend, J.W.; Srinivasan, D.; Thacker, M.A.; Cibull, M.L.; Wang, C.; Novak, M.; Kaetzel, D.M.; et al. C-Abl and Arg are activated in human primary melanomas, promote melanoma cell invasion via distinct pathways, and drive metastatic progression. Oncogene 2012, 31, 1804–1816. [Google Scholar] [CrossRef] [PubMed]
- Smith-Pearson, P.S.; Greuber, E.K.; Yogalingam, G.; Pendergast, A.M. Abl kinases are required for invadopodia formation and chemokine-induced invasion. J. Biol. Chem. 2010, 285, 40201–40211. [Google Scholar] [CrossRef] [PubMed]
- Hayes, K.E.; Walk, E.L.; Ammer, A.G.; Kelley, L.C.; Martin, K.H.; Weed, S.A. Ableson kinases negatively regulate invadopodia function and invasion in head and neck squamous cell carcinoma by inhibiting an HB-EGF autocrine loop. Oncogene 2013, 32, 4766–4777. [Google Scholar] [CrossRef] [PubMed]
- Tsao, A.S.; Liu, S.; Fujimoto, J.; Wistuba, I.I.; Lee, J.J.; Marom, E.M.; Charnsangavej, C.; Fossella, F.V.; Tran, H.T.; Blumenschein, G.R.; et al. Phase II trials of imatinib mesylate and docetaxel in patients with metastatic non-small cell lung cancer and head and neck squamous cell carcinoma. J. Thorac. Oncol. 2011, 6, 2104–2111. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Kim, E.S.; Harari, P.M. IMC-C225, an anti-epidermal growth factor receptor monoclonal antibody, for treatment of head and neck cancer. Expert Opin. Biol. Ther. 2001, 1, 719–732. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.M.; Bock, J.M.; Harari, P.M. Epidermal growth factor receptor blockade with C225 modulates proliferation, apoptosis, and radiosensitivity in squamous cell carcinomas of the head and neck. Cancer Res. 1999, 59, 1935–1940. [Google Scholar] [PubMed]
- Huang, S.M.; Harari, P.M. Modulation of radiation response after epidermal growth factor receptor blockade in squamous cell carcinomas: Inhibition of damage repair, cell cycle kinetics, and tumor angiogenesis. Clin. Cancer Res. 2000, 6, 2166–2174. [Google Scholar] [PubMed]
- Fury, M.G.; Sherman, E.; Lisa, D.; Agarwal, N.; Algazy, K.; Brockstein, B.; Langer, C.; Lim, D.; Mehra, R.; Rajan, S.K.; et al. A randomized phase II study of cetuximab every 2 weeks at either 500 or 750 mg/m2 for patients with recurrent or metastatic head and neck squamous cell cancer. J. Natl. Compr. Canc. Netw. 2012, 10, 1391–1398. [Google Scholar] [PubMed]
- Merlano, M.; Russi, E.; Benasso, M.; Corvò, R.; Colantonio, I.; Vigna-Taglianti, R.; Vigo, V.; Bacigalupo, A.; Numico, G.; Crosetto, N.; et al. Cisplatin-based chemoradiation plus cetuximab in locally advanced head and neck cancer: A phase II clinical study. Ann. Oncol. 2011, 22, 712–717. [Google Scholar] [CrossRef] [PubMed]
- Licitra, L.; Störkel, S.; Kerr, K.M.; van Cutsem, E.; Pirker, R.; Hirsch, F.R.; Vermorken, J.B.; von Heydebreck, A.; Esser, R.; Celik, I.; et al. Predictive value of epidermal growth factor receptor expression for first-line chemotherapy plus cetuximab in patients with head and neck and colorectal cancer: Analysis of data from the EXTREME and CRYSTAL studies. Eur. J. Cancer 2013, 49, 1161–1168. [Google Scholar] [CrossRef] [PubMed]
- Licitra, L.; Mesia, R.; Rivera, F.; Remenár, E.; Hitt, R.; Erfán, J.; Rottey, S.; Kawecki, A.; Zabolotnyy, D.; Benasso, M.; et al. Evaluation of EGFR gene copy number as a predictive biomarker for the efficacy of cetuximab in combination with chemotherapy in the first-line treatment of recurrent and/or metastatic squamous cell carcinoma of the head and neck: EXTREME study. Ann. Oncol. 2011, 22, 1078–1087. [Google Scholar] [CrossRef] [PubMed]
- Rivera, F.; García-Castaño, A.; Vega, N.; Vega-Villegas, M.E.; Gutiérrez-Sanz, L. Cetuximab in metastatic or recurrent head and neck cancer: The EXTREME trial. Expert Rev. Anticancer Ther. 2009, 9, 1421–1428. [Google Scholar] [CrossRef] [PubMed]
- William, W.N.; Kim, E.S.; Herbst, R.S. Cetuximab therapy for patients with advanced squamous cell carcinomas of the head and neck. Nat. Clin. Pract. Oncol. 2009, 6, 132–133. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, D.; Heinold, A.; Opelz, G.; Daniel, V.; Naujokat, C. Salinomycin induces apoptosis and overcomes apoptosis resistance in human cancer cells. Biochem. Biophys. Res. Commun. 2009, 390, 743–749. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Markwell, S.M.; Weed, S.A. Tumor and Stromal-Based Contributions to Head and Neck Squamous Cell Carcinoma Invasion. Cancers 2015, 7, 382-406. https://doi.org/10.3390/cancers7010382
Markwell SM, Weed SA. Tumor and Stromal-Based Contributions to Head and Neck Squamous Cell Carcinoma Invasion. Cancers. 2015; 7(1):382-406. https://doi.org/10.3390/cancers7010382
Chicago/Turabian StyleMarkwell, Steven M., and Scott A. Weed. 2015. "Tumor and Stromal-Based Contributions to Head and Neck Squamous Cell Carcinoma Invasion" Cancers 7, no. 1: 382-406. https://doi.org/10.3390/cancers7010382
APA StyleMarkwell, S. M., & Weed, S. A. (2015). Tumor and Stromal-Based Contributions to Head and Neck Squamous Cell Carcinoma Invasion. Cancers, 7(1), 382-406. https://doi.org/10.3390/cancers7010382