
The Role of Intracellular Calcium for the Development and Treatment of Neuroblastoma



Abstract
:
1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stage | Description |
|---|---|
| L1 | Localized tumour without any detectable image-defined risk factors |
| L2 | Localized tumour with one or more image defined risk factors |
| M | Metastatic disease |
| MS | Metastatic disease with metastases confined to skin, liver, and/or bone marrow (confined to children ≤ 18 month) |
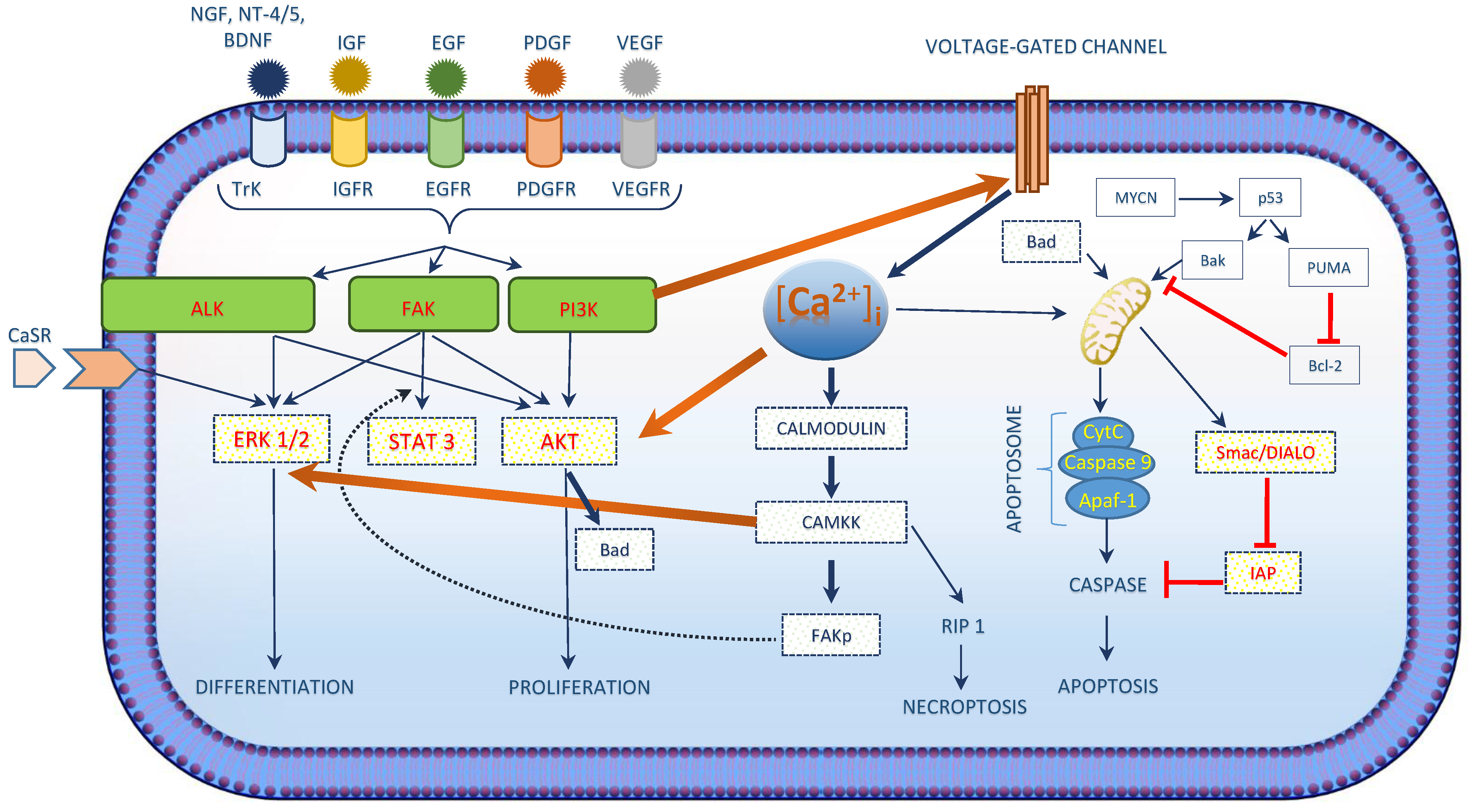
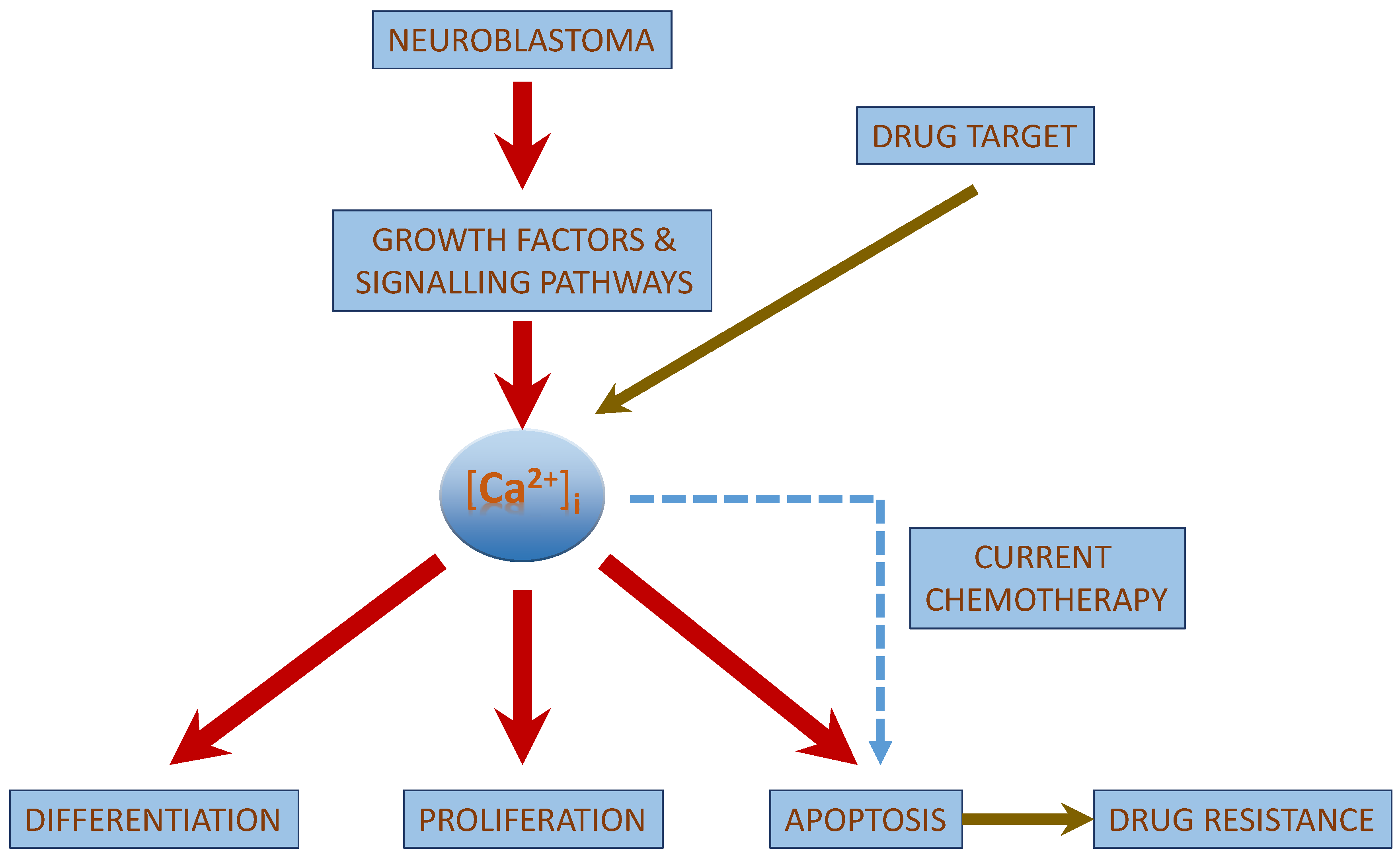
2. Role of [Ca2+]i in Neuroblastoma

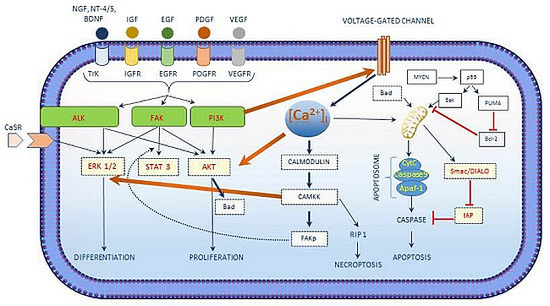
| [Ca2+]i in Neuroblastoma |
|---|
| [Ca2+]i interacts with the growth factor signaling cascade in neuroblastoma. |
| Three main kinases involved in cell survival signaling in neuroblastoma include PI3K/AKT, ALK and FAK. |
| [Ca2+]i activated CAM kinases activates ERK1/2 exerts its role in neuroblastoma differentiation. |
| [Ca2+]i regulated apoptosis in neuroblastoma involves the intrinsic pathway and the activation of CaSR. |
| Chemotherapeutic drug treatment shows an increase in [Ca2+]i concentrations. |
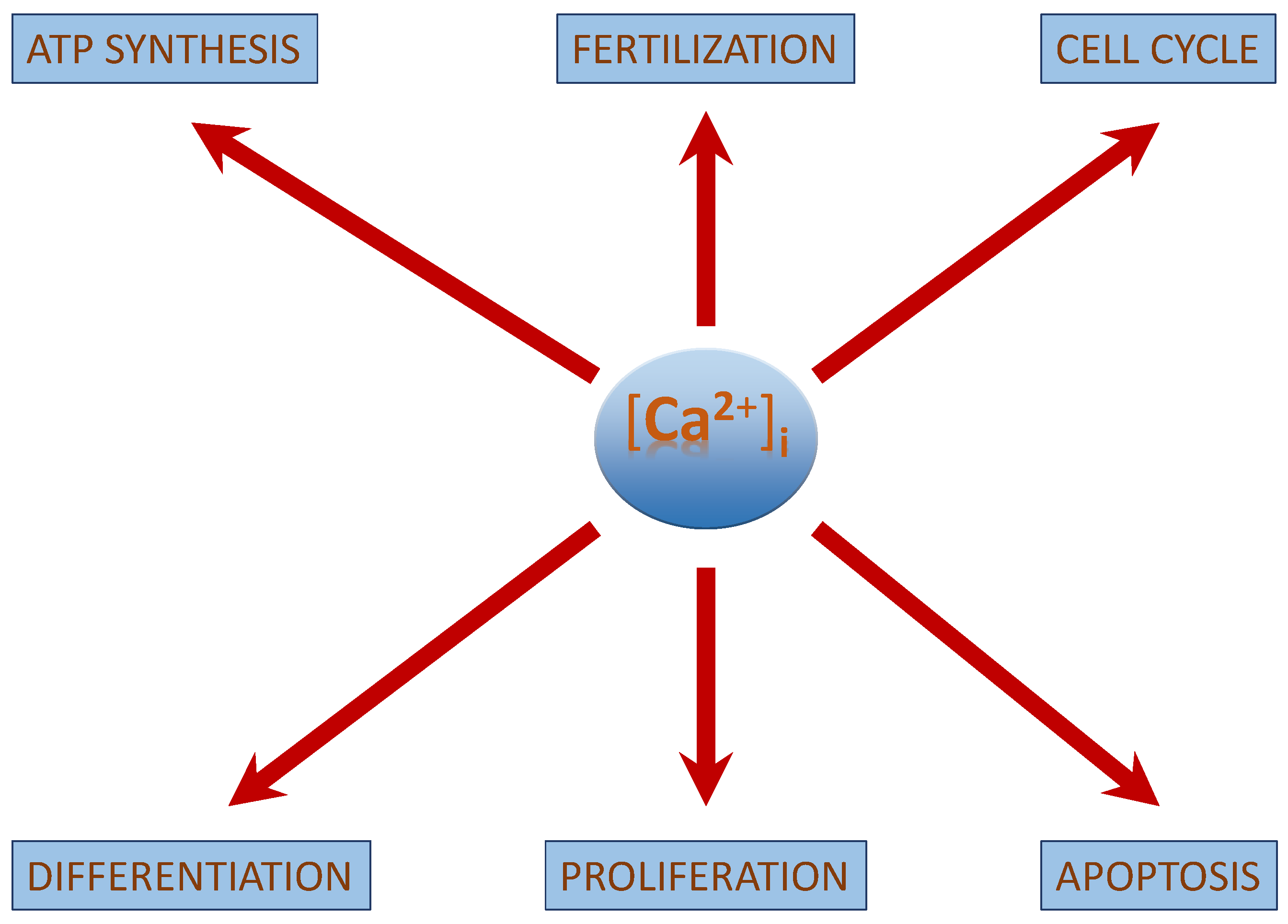
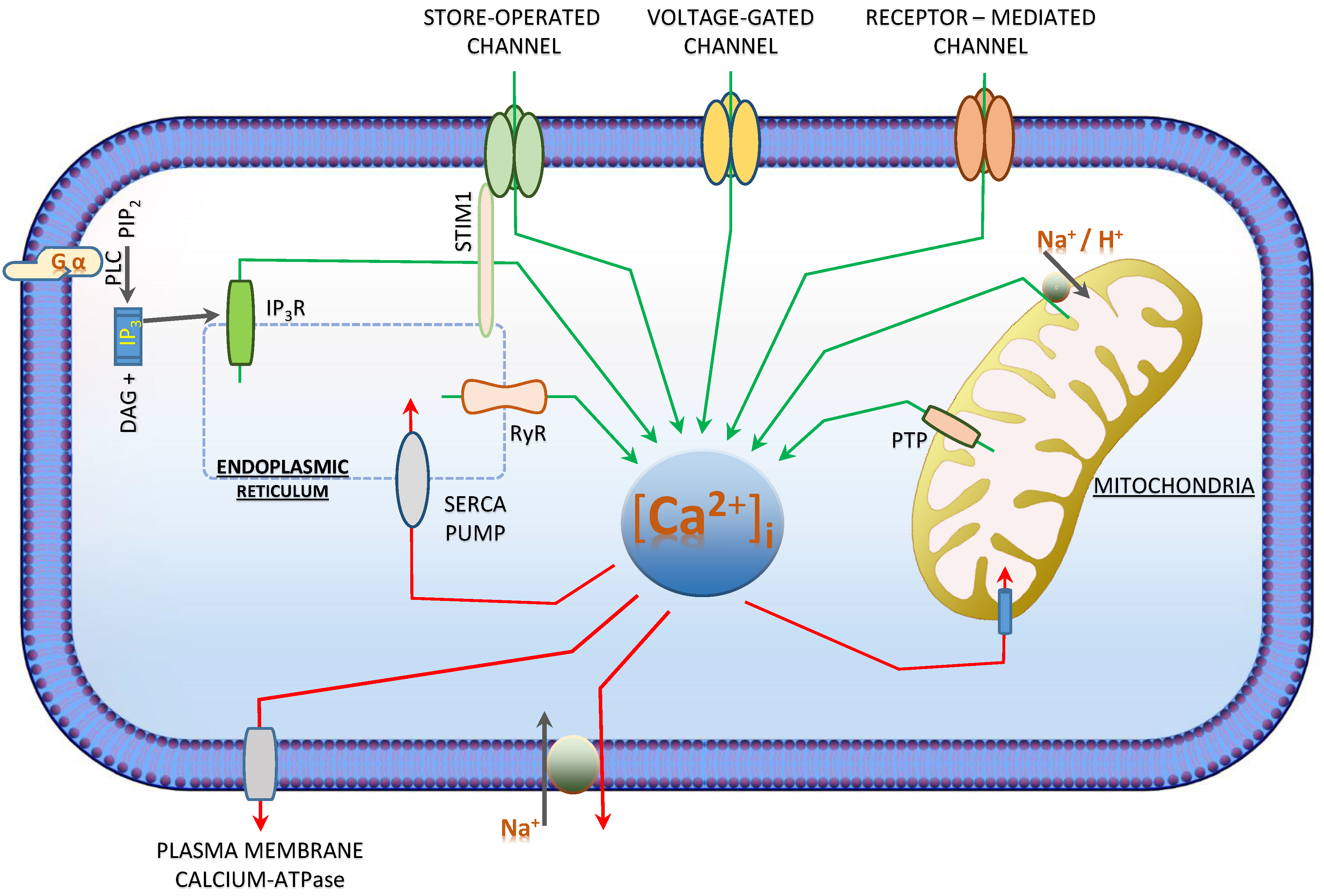
3. [Ca2+]i—Regulation and Signalling

4. Signalling Pathways in Neuroblastoma and Their Dependence on [Ca2+]i

4.1. [Ca2+]i as a Key Factor which Determines the Fate of Neuroblastoma Cells
4.2. [Ca2+]i Induces Differentiation and Proliferation in Neuroblastoma
4.3. [Ca2+]i Induces Apoptosis in Neuroblastoma
| Sl.No | Cell Lines | Orgin | Treatment | Receptor | Concentration | Ca2+ Release | Basal [Ca2+]i | Increased [Ca2+]i | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1 | IMR-32 | H | Orexin-A (GPCR) | Orexin Type 1 Receptor (GPCR) | 3 nM | Store Release (IP3R) | 50 nM | 4 fold | [24] |
| 2 | SH-SY5Y | H | Retinoic Acid | Retinoid X receptor (Nuclear Receptors) | 10 µM | Store Release | 98 nM | No increase | [25] |
| 3 | SH-SY5Y | H | Retinoic Acid | Retinoid X receptor (Nuclear Receptors) | 1 µM | Store Operated calcium Channel | 10 nM | 4 fold | [26] |
| 4 | SH-SY5Y | H | Oxotremorine-M | Muscarinic Receptor (GPCR) | 10 µM | Store Release (IP3R) | 50 nM | 2 fold | [27] |
| 5 | SH-SY5Y | H | Methacholine | Muscarinic Receptor (GPCR) | 1 mM | Store Release (IP3R) | 98 nM | 2 fold | [25] |
| 6 | SH-SY5Y | H | Carbachol | Muscarinic Receptor (GPCR) | 1 mM | Store Release | - | 3.5 fold | [28] |
| 7 | SK-N-SH | H | Carbachol | Muscarinic Receptor (GPCR) | 100 µM | Store Release | 59 nM | 2 fold | [29] |
| 8 | SH-SY5Y | H | Bradykinin | Bradykinin Receptor (GPCR) | 10 µM | Store Release (IP3R) | 98 nM | 1 fold | [25] |
| 9 | SH-SY5Y | H | Bradykinin | Bradykinin Receptor (GPCR) | 10 µM | Store Release | - | 2 fold | [28] |
| 10 | SH-SY5Y | H | Arsenic Trioxide | - | 1 µM | Store Release (IP3R and RyR) | 75 nM | 2 fold | [31] |
| 11 | SH-SY5Y | H | Arsenic Trioxide | - | 1 µM | Store Release (IP3R and RyR) | 70 nM | 3 fold | [32] |
| 12 | SH-SY5Y | H | Trimethyltin Chloride | - | 0.1 µM | Store Release | - | 2 fold | [33] |
| 13 | SH-SY5Y | H | Cisplatin | - | 1 µM | Extracellular Space | 75 nM | 2 fold | [31] |
| 14 | SK-N-SH | H | CB-64D | Sigma 2 receptor | 100 µM | Thapsigargin insensitive calcium store | - | 4 fold | [30] |
| 15 | SK-N-SH | H | JL-II-147 | Sigma 2 receptor | 100 µM | Thapsigargin insensitive calcium store | - | 2 fold | [30] |
| 16 | SK-N-SH | H | BD737 | Sigma 2 receptor | 100 µM | Thapsigargin insensitive calcium store | - | 1 fold | [30] |
| 17 | SK-N-SH | H | LR172 | Sigma 2 receptor | 100 µM | Thapsigargin insensitive calcium store | - | 1 fold | [30] |
| 18 | SK-N-SH | H | BD1008 | Sigma 2 receptor | 100 µM | Thapsigargin insensitive calcium store | - | 1 fold | [30] |
| 19 | SK-N-SH | H | Haloperidol | Sigma 2 receptor | 100 µM | Thapsigargin insensitive calcium store | - | 1 fold | [30] |
| 20 | SK-N-SH | H | Ibogaine | Sigma 2 receptor | 100 µM | Thapsigargin insensitive calcium store | - | 1 fold | [30] |

4.4. Tumour Suppressor Functions of the Calcium-Sensing Receptor (CaSR) in Neuroblastoma
5. [Ca2+]i Modulations with Chemotherapeutic Treatment of Neuroblastoma
| Stage of Neuroblastoma | Tumour Characteristics | Treatment Protocol |
|---|---|---|
| Stage 1 (Low) | Single site specific | Surgery |
| Stage 2A (Low) | Single site specific and could not be removed completely by surgery. | Surgery and Chemotherapy |
| Stage 2B (Low) | Single site specific and could be removed completely by surgery. Cancer development could be present at lymph nodes around the tumour. | Surgery |
| Stage 3 (Intermediate Risk) | Cancer could be present in one or both sides of the body and lymph nodes. | Chemotherapy |
| Stage 4 (High Risk) | Cancer spread to distant body parts (bone, liver, skin, bone marrow and other organs) and distant lymph nodes. | Surgery, Chemotherapy, Radiotherapy, Immunotherapy and Retinoid Therapy |
| Stage 4S (High Risk) | Child is younger than 12 months with cancer spread on one side of the body. Lymph nodes on the same side of the body also affected. | Surgery, Chemotherapy and Radiotherapy |
| Relapsed/Recurrent | - | Chemotherapy, Immunotherapy, Retinoid Therapy, Tyrosine kinase and Aurora kinase inhibitors and targeted delivery of radionuclides. |
6. Drug Resistance in Neuroblastoma
7. Future Studies with [Ca2+]i in Neuroblastoma
8. Conclusions
Abbreviations
| INRGSS | International Neuroblastoma Risk Group Staging System |
| Ca2+ | Calcium Ions |
| [Ca2+]i | Intracellular Calcium |
| CaSR | Cancer-Sensing Receptors |
| ATP | Adenosine Triphosphate |
| ER | Endoplasmic Reticulum |
| IP3 | Ionsitol Triphosphate |
| InsP3R | Inositol-1,4,5 Triphosphate Receptor |
| RyR | Ryanodine Receptor |
| NAADP | Nicotinic Acid Dinucleotide Phosphate |
| S1P | Sphingosine-1 Phosphate |
| cADPR | Cyclic ADP Ribose |
| NGF | Nerve Growth Factor |
| IGF | Insulin-like Growth Factor |
| EGF | Epidermal Growth Factor |
| PDGF | Platelet-derived Growth Factor |
| VEGF | Vascular Endothelial Growth Factor |
| ALK | Anaplastic Lymphoma Kinase |
| BDNF | Brain-Derived Neurotrophic Factor |
| NT-4/5 | Neurotrophin-4/5 |
| MCU | Mitochondria Calcium Uniporter |
| Δψm | Mitochondrial Membrane Potential |
| MOMP | Mitochondrial Outer Membrane Permeabilization |
| As2O3 | Arsenic Trioxide |
| TMT | Trimethyltin Chloride |
| MRP1 | Multi-Drug Resistant-Associated Protein |
| VCR | Vincristine |
| DOX | Doxorubicin |
| NCAM | Neural Cell Adhesion Molecule |
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Orbach, D.; Sarnacki, S.; Brisse, H.J.; Gauthier-Villars, M.; Jarreau, P.H.; Tsatsaris, V.; Baruchel, A.; Zerah, M.; Seigneur, E.; Peuchmaur, M.; et al. Neonatal cancer. Lancet Oncol. 2013, 14, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Maris, J.M. Recent advances in neuroblastoma. N. Engl. J. Med. 2010, 362, 2202–2211. [Google Scholar] [CrossRef] [PubMed]
- Colon, N.C.; Chung, D.H. Neuroblastoma. Adv. Pediatr. 2011, 58, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Domingo-Fernandez, R.; Watters, K.; Piskareva, O.; Stallings, R.L.; Bray, I. The role of genetic and epigenetic alterations in neuroblastoma disease pathogenesis. Pediatr. Surg. Int. 2013, 29, 101–119. [Google Scholar] [CrossRef] [PubMed]
- Kamijo, T.; Nakagawara, A. Molecular and genetic bases of neuroblastoma. Int. J. Clin. Oncol. 2012, 17, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Ward, E.; DeSantis, C.; Robbins, A.; Kohler, B.; Jemal, A. Childhood and adolescent cancer statistics, 2014. CA Cancer J. Clin. 2014, 64, 83–103. [Google Scholar] [CrossRef] [PubMed]
- Fisher, J.P.; Tweddle, D.A. Neonatal neuroblastoma. Semin Fetal Neonatal Med. 2012, 17, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Stanke, J.; Lahti, J.M. The connections between neural crest development and neuroblastoma. Curr. Top Dev. Biol. 2011, 94, 77–127. [Google Scholar] [PubMed]
- Casala, C.; Gil-Guinon, E.; Ordonez, J.L.; Miguel-Queralt, S.; Rodriguez, E.; Galvan, P.; Lavarino, C.; Munell, F.; de Alava, E.; Mora, J.; et al. The calcium-sensing receptor is silenced by genetic and epigenetic mechanisms in unfavorable neuroblastomas and its reactivation induces ERK1/2-dependent apoptosis. Carcinogenesis 2013, 34, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Maris, J.M.; Weiss, M.J.; Mosse, Y.; Hii, G.; Guo, C.; White, P.S.; Hogarty, M.D.; Mirensky, T.; Brodeur, G.M.; Rebbeck, T.R.; et al. Evidence for a hereditary neuroblastoma predisposition locus at chromosome 16p12–13. Cancer Res. 2002, 62, 6651–6658. [Google Scholar] [PubMed]
- Monclair, T.; Brodeur, G.M.; Ambros, P.F.; Brisse, H.J.; Cecchetto, G.; Holmes, K.; Kaneko, M.; London, W.B.; Matthay, K.K.; Nuchtern, J.G.; et al. The international neuroblastoma risk group (INRG) staging system: An inrg task force report. J. Clin. Oncol. 2009, 27, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Brisse, H.J.; McCarville, M.B.; Granata, C.; Krug, K.B.; Wootton-Gorges, S.L.; Kanegawa, K.; Giammarile, F.; Schmidt, M.; Shulkin, B.L.; Matthay, K.K.; et al. Guidelines for imaging and staging of neuroblastic tumors: Consensus report from the international neuroblastoma risk group project. Radiology 2011, 261, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Cohn, S.L.; Pearson, A.D.; London, W.B.; Monclair, T.; Ambros, P.F.; Brodeur, G.M.; Faldum, A.; Hero, B.; Iehara, T.; Machin, D.; et al. The international neuroblastoma risk group (INRG) classification system: An INRG task force report. J. Clin. Oncol. 2009, 27, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R. Ca2+ signaling, intracellular pH and cell volume in cell proliferation. J. Membr. Biol. 2005, 205, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Capiod, T.; Shuba, Y.; Skryma, R.; Prevarskaya, N. Calcium signalling and cancer cell growth. Subcell. Biochem. 2007, 45, 405–427. [Google Scholar] [PubMed]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell. Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Al-Taweel, N.; Varghese, E.; Florea, A.M.; Busselberg, D. Cisplatin (CDDP) triggers cell death of MCF-7 cells following disruption of intracellular calcium ([Ca2+]i) homeostasis. J. Toxicol. Sci. 2014, 39, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Varghese, E.; Busselberg, D. Auranofin, an anti-rheumatic gold compound, modulates apoptosis by elevating the intracellular calcium concentration ([Ca2+]i) in MCF-7 breast cancer cells. Cancers (Basel) 2014, 6, 2243–2258. [Google Scholar] [CrossRef]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Targeting Ca2+ transport in cancer: Close reality or long perspective? Expert Opin. Ther. Targets 2013, 17, 225–241. [Google Scholar] [CrossRef] [PubMed]
- Florea, A.M.; Busselberg, D. Anti-cancer drugs interfere with intracellular calcium signaling. Neurotoxicology 2009, 30, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Masvidal, L.; Iniesta, R.; Casala, C.; Galvan, P.; Rodriguez, E.; Lavarino, C.; Mora, J.; de Torres, C. Polymorphisms in the calcium-sensing receptor gene are associated with clinical outcome of neuroblastoma. PLoS ONE 2013, 8, e59762. [Google Scholar] [CrossRef] [PubMed]
- Nasman, J.; Bart, G.; Larsson, K.; Louhivuori, L.; Peltonen, H.; Akerman, K.E. The orexin OX1 receptor regulates Ca2+ entry via diacylglycerol-activated channels in differentiated neuroblastoma cells. J. Neurosci. 2006, 26, 10658–10666. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.K.; Nahorski, S.R.; Willars, G.B. Complex relationship between Ins(1,4,5)P3 accumulation and Ca2+-signalling in a human neuroblastoma revealed by cellular differentiation. Br. J. Pharmacol. 1999, 126, 1559–1566. [Google Scholar] [CrossRef] [PubMed]
- Bell, N.; Hann, V.; Redfern, C.P.; Cheek, T.R. Store-operated Ca2+ entry in proliferating and retinoic acid-differentiated N- and S-type neuroblastoma cells. Biochim. Biophys. Acta 2013, 1833, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Grudt, T.J.; Usowicz, M.M.; Henderson, G. Ca2+ entry following store depletion in SH-SY5Y neuroblastoma cells. Brain Res. Mol. Brain Res. 1996, 36, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Marini, P.; Moriello, A.S.; Cristino, L.; Palmery, M.; De Petrocellis, L.; di Marzo, V. Cannabinoid CB1 receptor elevation of intracellular calcium in neuroblastoma SH-SY5Y cells: Interactions with muscarinic and delta-opioid receptors. Biochim. Biophys. Acta 2009, 1793, 1289–1303. [Google Scholar] [CrossRef] [PubMed]
- McGinnis, K.M.; Shariat-Madar, Z.; Gnegy, M.E. Cytosolic calmodulin is increased in SK-N-SH human neuroblastoma cells due to release of calcium from intracellular stores. J. Neurochem. 1998, 70, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Vilner, B.J.; Bowen, W.D. Modulation of cellular calcium by sigma-2 receptors: Release from intracellular stores in human SK-N-SH neuroblastoma cells. J. Pharmacol. Exp. Ther. 2000, 292, 900–911. [Google Scholar] [PubMed]
- Gunes, D.A.; Florea, A.M.; Splettstoesser, F.; Busselberg, D. Co-application of arsenic trioxide (As2O3) and cisplatin (CDDP) on human SY-5Y neuroblastoma cells has differential effects on the intracellular calcium concentration ([Ca2+]i) and cytotoxicity. Neurotoxicology 2009, 30, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Florea, A.M.; Splettstoesser, F.; Busselberg, D. Arsenic trioxide (As2O3) induced calcium signals and cytotoxicity in two human cell lines: SY-5Y neuroblastoma and 293 embryonic kidney (HEK). Toxicol. Appl. Pharmacol. 2007, 220, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Florea, A.M.; Splettstoesser, F.; Dopp, E.; Rettenmeier, A.W.; Busselberg, D. Modulation of intracellular calcium homeostasis by trimethyltin chloride in human tumour cells: Neuroblastoma SY5Y and cervix adenocarcinoma HeLa S3. Toxicology 2005, 216, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Megison, M.L.; Gillory, L.A.; Beierle, E.A. Cell survival signaling in neuroblastoma. Anticancer Agents Med. Chem. 2013, 13, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, J.I.; Segerstrom, L.; Orrego, A.; Elfman, L.; Henriksson, M.; Kagedal, B.; Eksborg, S.; Sveinbjornsson, B.; Kogner, P. Inhibitors of mammalian target of rapamycin downregulate MYCN protein expression and inhibit neuroblastoma growth in vitro and in vivo. Oncogene 2008, 27, 2910–2922. [Google Scholar] [CrossRef] [PubMed]
- Abid, M.R.; Guo, S.; Minami, T.; Spokes, K.C.; Ueki, K.; Skurk, C.; Walsh, K.; Aird, W.C. Vascular endothelial growth factor activates PI3K/Akt/forkhead signaling in endothelial cells. Arterioscler Thromb Vasc Biol. 2004, 24, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Opel, D.; Poremba, C.; Simon, T.; Debatin, K.M.; Fulda, S. Activation of Akt predicts poor outcome in neuroblastoma. Cancer Res. 2007, 67, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.H.; Hong, S.I.; Kim, J.A.; Jung, Y.H.; Kim, S.Y.; Kim, H.C.; Lee, S.Y.; Jang, C.G. The neuroprotective effects of Lonicera japonica Thunb. Against hydrogen peroxide-induced apoptosis via phosphorylation of MAPKs and PI3K/Akt in SH-SY5Y cells. Food Chem. Toxicol. 2011, 49, 1011–1019. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yang, H.J.; Xia, Y.Y.; Feng, Z.W. Insulin-like growth factor 1 protects human neuroblastoma cells SH-EP1 against MPP+-induced apoptosis by AKT/GSK-3beta/JNK signaling. Apoptosis 2010, 15, 1470–1479. [Google Scholar] [CrossRef] [PubMed]
- Ho, R.; Minturn, J.E.; Hishiki, T.; Zhao, H.; Wang, Q.; Cnaan, A.; Maris, J.; Evans, A.E.; Brodeur, G.M. Proliferation of human neuroblastomas mediated by the epidermal growth factor receptor. Cancer Res. 2005, 65, 9868–9875. [Google Scholar] [CrossRef] [PubMed]
- Brodeur, G.M.; Seeger, R.C.; Schwab, M.; Varmus, H.E.; Bishop, J.M. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science 1984, 224, 1121–1124. [Google Scholar] [CrossRef] [PubMed]
- Brodeur, G.M. Neuroblastoma: Biological insights into a clinical enigma. Nat. Rev. Cancer 2003, 3, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.W.; Kirstein, M.N.; Valentine, M.B.; Dittmer, K.G.; Shapiro, D.N.; Saltman, D.L.; Look, A.T. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 1994, 263, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Palmer, R.H.; Vernersson, E.; Grabbe, C.; Hallberg, B. Anaplastic lymphoma kinase: Signalling in development and disease. Biochem J. 2009, 420, 345–361. [Google Scholar] [CrossRef] [PubMed]
- Lamant, L.; Pulford, K.; Bischof, D.; Morris, S.W.; Mason, D.Y.; Delsol, G.; Mariame, B. Expression of the ALK tyrosine kinase gene in neuroblastoma. Am. J. Pathol. 2000, 156, 1711–1721. [Google Scholar] [CrossRef] [PubMed]
- Passoni, L.; Longo, L.; Collini, P.; Coluccia, A.M.; Bozzi, F.; Podda, M.; Gregorio, A.; Gambini, C.; Garaventa, A.; Pistoia, V.; et al. Mutation-independent anaplastic lymphoma kinase overexpression in poor prognosis neuroblastoma patients. Cancer Res. 2009, 69, 7338–7346. [Google Scholar] [CrossRef] [PubMed]
- Del Grosso, F.; De Mariano, M.; Passoni, L.; Luksch, R.; Tonini, G.P.; Longo, L. Inhibition of N-linked glycosylation impairs ALK phosphorylation and disrupts pro-survival signaling in neuroblastoma cell lines. BMC Cancer 2011, 11. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Soellner, D.; Nunez, J.; Wang, H. The basal level of intracellular calcium gates the activation of phosphoinositide 3-kinase-Akt signaling by brain-derived neurotrophic factor in cortical neurons. J. Neurochem. 2008, 106, 1259–1274. [Google Scholar] [CrossRef] [PubMed]
- Chun-Jen Lin, C.; Summerville, J.B.; Howlett, E.; Stern, M. The metabotropic glutamate receptor activates the lipid kinase PI3K in drosophila motor neurons through the calcium/calmodulin-dependent protein kinase II and the nonreceptor tyrosine protein kinase DFaK. Genetics 2011, 188, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Nakagawara, A. Apoptotic cell death in neuroblastoma. Cells 2013, 2, 432–459. [Google Scholar] [CrossRef] [PubMed]
- Eggert, A.; Ikegaki, N.; Liu, X.G.; Brodeur, G.M. Prognostic and biological role of neurotrophin-receptor TrkA and TrkB in neuroblastoma. Klin. Padiatr. 2000, 212, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Brodeur, G.M.; Minturn, J.E.; Ho, R.; Simpson, A.M.; Iyer, R.; Varela, C.R.; Light, J.E.; Kolla, V.; Evans, A.E. Trk receptor expression and inhibition in neuroblastomas. Clin. Cancer Res. 2009, 15, 3244–3250. [Google Scholar] [CrossRef] [PubMed]
- De Bernardi, M.A.; Rabins, S.J.; Colangelo, A.M.; Brooker, G.; Mocchetti, I. TrkA mediates the nerve growth factor-induced intracellular calcium accumulation. J. Biol. Chem. 1996, 271, 6092–6098. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Ulme, D.S.; Dickens, G.; Chabuk, A.; Lavarreda, M.; Lazarovici, P.; Guroff, G. Both p140(trk) and p75(NGFR) nerve growth factor receptors mediate nerve growth factor-stimulated calcium uptake. J. Biol. Chem. 1997, 272, 6835–6837. [Google Scholar] [CrossRef] [PubMed]
- Nikodijevic, B.; Guroff, G. Nerve growth factor-induced increase in calcium uptake by PC12 cells. J. Neurosci. Res. 1991, 28, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Viard, P.; Butcher, A.J.; Halet, G.; Davies, A.; Nurnberg, B.; Heblich, F.; Dolphin, A.C. PI3K promotes voltage-dependent calcium channel trafficking to the plasma membrane. Nat. Neurosci. 2004, 7, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Seger, R. The extracellular signal-regulated kinase: Multiple substrates regulate diverse cellular functions. Growth Factors 2006, 24, 21–44. [Google Scholar] [CrossRef] [PubMed]
- Raman, M.; Chen, W.; Cobb, M.H. Differential regulation and properties of mapks. Oncogene 2007, 26, 3100–3112. [Google Scholar] [CrossRef] [PubMed]
- Avruch, J. Map kinase pathways: The first twenty years. Biochim. Biophys. Acta 2007, 1773, 1150–1160. [Google Scholar] [CrossRef] [PubMed]
- Chuderland, D.; Seger, R. Calcium regulates ERK signaling by modulating its protein-protein interactions. Commun. Integr. Biol. 2008, 1, 4–5. [Google Scholar] [CrossRef] [PubMed]
- Rottingen, J.; Iversen, J.G. Ruled by waves? Intracellular and intercellular calcium signalling. Acta Physiol. Scand. 2000, 169, 203–219. [Google Scholar] [CrossRef] [PubMed]
- Munaron, L. Calcium signalling and control of cell proliferation by tyrosine kinase receptors (review). Int. J. Mol. Med. 2002, 10, 671–676. [Google Scholar] [PubMed]
- Chuderland, D.; Marmor, G.; Shainskaya, A.; Seger, R. Calcium-mediated interactions regulate the subcellular localization of extracellular signal-regulated kinases. J. Biol. Chem. 2008, 283, 11176–11188. [Google Scholar] [CrossRef] [PubMed]
- Agell, N.; Bachs, O.; Rocamora, N.; Villalonga, P. Modulation of the Ras/Raf/MEK/ERK pathway by Ca(2+), and calmodulin. Cell. Signal. 2002, 14, 649–654. [Google Scholar] [CrossRef] [PubMed]
- Cullen, P.J.; Lockyer, P.J. Integration of calcium and Ras signalling. Nat. Rev. Mol. Cell. Biol. 2002, 3, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Tebar, F.; Villalonga, P.; Sorkina, T.; Agell, N.; Sorkin, A.; Enrich, C. Calmodulin regulates intracellular trafficking of epidermal growth factor receptor and the MAPK signaling pathway. Mol. Biol. Cell. 2002, 13, 2057–2068. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I.; Tokiwa, G.; Lev, S.; Courtneidge, S.A.; Schlessinger, J. A role for Pyk2 and Src in linking G-protein-coupled receptors with MAP kinase activation. Nature 1996, 383, 547–550. [Google Scholar] [CrossRef] [PubMed]
- Farnsworth, C.L.; Freshney, N.W.; Rosen, L.B.; Ghosh, A.; Greenberg, M.E.; Feig, L.A. Calcium activation of Ras mediated by neuronal exchange factor Ras-GRF. Nature 1995, 376, 524–527. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.J.; Rojas-Soto, M.; Oguni, A.; Kennedy, M.B. A synaptic Ras-GTPase activating protein (p135 SynGAP) inhibited by cam kinase II. Neuron 1998, 20, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Walker, S.A.; Gao, D.; Taylor, J.A.; Dai, Y.F.; Arkell, R.S.; Bootman, M.D.; Roderick, H.L.; Cullen, P.J.; Lockyer, P.J. CAPRI and RASAL impose different modes of information processing on Ras due to contrasting temporal filtering of Ca2+. J. Cell. Biol. 2005, 170, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Egea, J.; Espinet, C.; Soler, R.M.; Peiro, S.; Rocamora, N.; Comella, J.X. Nerve growth factor activation of the extracellular signal-regulated kinase pathway is modulated by Ca(2+) and calmodulin. Mol. Cell. Biol. 2000, 20, 1931–1946. [Google Scholar] [CrossRef] [PubMed]
- Yano, S.; Tokumitsu, H.; Soderling, T.R. Calcium promotes cell survival through CaM-K kinase activation of the protein-kinase-B pathway. Nature 1998, 396, 584–587. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V.M.; Kweh, F.A.; Cance, W.G. Focal adhesion kinase and cancer. Histol. Histopathol. 2009, 24, 503–510. [Google Scholar] [PubMed]
- Thiele, C.J.; Reynolds, C.P.; Israel, M.A. Decreased expression of N-MYC precedes retinoic acid-induced morphological differentiation of human neuroblastoma. Nature 1985, 313, 404–406. [Google Scholar] [CrossRef] [PubMed]
- Sidell, N.; Altman, A.; Haussler, M.R.; Seeger, R.C. Effects of retinoic acid (RA) on the growth and phenotypic expression of several human neuroblastoma cell lines. Exp. Cell. Res. 1983, 148, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Matthay, K.K.; Reynolds, C.P.; Seeger, R.C.; Shimada, H.; Adkins, E.S.; Haas-Kogan, D.; Gerbing, R.B.; London, W.B.; Villablanca, J.G. Long-term results for children with high-risk neuroblastoma treated on a randomized trial of myeloablative therapy followed by 13-cis-retinoic acid: A children’s oncology group study. J. Clin. Oncol. 2009, 27, 1007–1013. [Google Scholar] [CrossRef] [PubMed]
- Walton, J.D.; Kattan, D.R.; Thomas, S.K.; Spengler, B.A.; Guo, H.F.; Biedler, J.L.; Cheung, N.K.; Ross, R.A. Characteristics of stem cells from human neuroblastoma cell lines and in tumors. Neoplasia 2004, 6, 838–845. [Google Scholar] [CrossRef] [PubMed]
- Ross, R.A.; Spengler, B.A.; Domenech, C.; Porubcin, M.; Rettig, W.J.; Biedler, J.L. Human neuroblastoma I-type cells are malignant neural crest stem cells. Cell. Growth Differ. 1995, 6, 449–456. [Google Scholar] [PubMed]
- Ross, R.A.; Biedler, J.L.; Spengler, B.A. A role for distinct cell types in determining malignancy in human neuroblastoma cell lines and tumors. Cancer Lett. 2003, 197, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Ross, R.A.; Spengler, B.A. Human neuroblastoma stem cells. Semin. Cancer. Biol. 2007, 17, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Ciccarone, V.; Spengler, B.A.; Meyers, M.B.; Biedler, J.L.; Ross, R.A. Phenotypic diversification in human neuroblastoma cells: Expression of distinct neural crest lineages. Cancer Res. 1989, 49, 219–225. [Google Scholar] [PubMed]
- Ross, R.A.; Spengler, B.A.; Biedler, J.L. Coordinate morphological and biochemical interconversion of human neuroblastoma cells. J. Natl. Cancer Inst. 1983, 71, 741–747. [Google Scholar] [PubMed]
- Spengler, B.A.; Lazarova, D.L.; Ross, R.A.; Biedler, J.L. Cell lineage and differentiation state are primary determinants of MYCN gene expression and malignant potential in human neuroblastoma cells. Oncol. Res. 1997, 9, 467–476. [Google Scholar] [PubMed]
- Piacentini, M.; Piredda, L.; Starace, D.T.; Annicchiarico-Petruzzelli, M.; Mattei, M.; Oliverio, S.; Farrace, M.G.; Melino, G. Differential growth of N- and S-type human neuroblastoma cells xenografted into scid mice. Correlation with apoptosis. J. Pathol. 1996, 180, 415–422. [Google Scholar] [CrossRef] [PubMed]
- McDonald, R.L.; Kaye, D.F.; Reeve, H.L.; Ball, S.G.; Peers, C.; Vaughan, P.F. Bradykinin-evoked release of [3h]noradrenaline from the human neuroblastoma SH-SY5Y. Biochem. Pharmacol. 1994, 48, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Willars, G.B.; Nahorski, S.R. Quantitative comparisons of muscarinic and bradykinin receptor-mediated ins (1,4,5)p3 accumulation and Ca2+ signalling in human neuroblastoma cells. Br. J. Pharmacol. 1995, 114, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.S. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. 2011, 30. [Google Scholar] [CrossRef] [PubMed]
- Assuncao Guimaraes, C.; Linden, R. Programmed cell deaths. Apoptosis and alternative deathstyles. Eur. J. Biochem. 2004, 271, 1638–1650. [Google Scholar] [CrossRef] [PubMed]
- Henriquez, M.; Armisen, R.; Stutzin, A.; Quest, A.F. Cell death by necrosis, a regulated way to go. Curr. Mol. Med. 2008, 8, 187–206. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.S.; Strasser, A.; McDunn, J.E.; Swanson, P.E. Cell death. N. Engl. J. Med. 2009, 361, 1570–1583. [Google Scholar] [CrossRef] [PubMed]
- Krieger, C.; Duchen, M.R. Mitochondria, Ca2+ and neurodegenerative disease. Eur. J. Pharmacol. 2002, 447, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Hajnoczky, G.; Csordas, G.; Das, S.; Garcia-Perez, C.; Saotome, M.; Sinha Roy, S.; Yi, M. Mitochondrial calcium signalling and cell death: Approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis. Cell. Calcium 2006, 40, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.; Distelhorst, C.W. Bcl-2 protein family members: Versatile regulators of calcium signaling in cell survival and apoptosis. Annu. Rev. Physiol. 2008, 70, 73–91. [Google Scholar] [CrossRef] [PubMed]
- Kirichok, Y.; Krapivinsky, G.; Clapham, D.E. The mitochondrial calcium uniporter is a highly selective ion channel. Nature 2004, 427, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W.; et al. Loss of cyclophilin d reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005, 434, 658–662. [Google Scholar] [CrossRef] [PubMed]
- Basso, E.; Fante, L.; Fowlkes, J.; Petronilli, V.; Forte, M.A.; Bernardi, P. Properties of the permeability transition pore in mitochondria devoid of cyclophilin D. J. Biol. Chem. 2005, 280, 18558–18561. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, V.; Soriano, M.E.; Basso, E.; Bisetto, E.; Lippe, G.; Forte, M.A.; Bernardi, P. Cyclophilin D in mitochondrial pathophysiology. Biochim. Biophys. Acta. 2010, 1797, 1113–1118. [Google Scholar] [CrossRef] [PubMed]
- Scorrano, L.; Penzo, D.; Petronilli, V.; Pagano, F.; Bernardi, P. Arachidonic acid causes cell death through the mitochondrial permeability transition. Implications for tumor necrosis factor-alpha aopototic signaling. J. Biol. Chem. 2001, 276, 12035–12040. [Google Scholar] [CrossRef] [PubMed]
- Starkov, A.A.; Chinopoulos, C.; Fiskum, G. Mitochondrial calcium and oxidative stress as mediators of ischemic brain injury. Cell. Calcium 2004, 36, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Kroemer, G. The pathophysiology of mitochondrial cell death. Science 2004, 305, 626–629. [Google Scholar] [CrossRef] [PubMed]
- Parsons, M.J.; Green, D.R. Mitochondria in cell death. Essays Biochem. 2010, 47, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Nakagawara, A.; Arima-Nakagawara, M.; Scavarda, N.J.; Azar, C.G.; Cantor, A.B.; Brodeur, G.M. Association between high levels of expression of the TRK gene and favorable outcome in human neuroblastoma. N. Engl. J. Med. 1993, 328, 847–854. [Google Scholar] [CrossRef] [PubMed]
- Wright, K.M.; Vaughn, A.E.; Deshmukh, M. Apoptosome dependent caspase-3 activation pathway is non-redundant and necessary for apoptosis in sympathetic neurons. Cell. Death Differ. 2007, 14, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Nakagawara, A.; Azar, C.G.; Scavarda, N.J.; Brodeur, G.M. Expression and function of TRK-B and BDNF in human neuroblastomas. Mol. Cell. Biol. 1994, 14, 759–767. [Google Scholar] [PubMed]
- Caren, H.; Kryh, H.; Nethander, M.; Sjoberg, R.M.; Trager, C.; Nilsson, S.; Abrahamsson, J.; Kogner, P.; Martinsson, T. High-risk neuroblastoma tumors with 11q-deletion display a poor prognostic, chromosome instability phenotype with later onset. Proc. Natl. Acad. Sci. USA 2010, 107, 4323–4328. [Google Scholar] [CrossRef] [PubMed]
- Westermark, U.K.; Wilhelm, M.; Frenzel, A.; Henriksson, M.A. The MYCN oncogene and differentiation in neuroblastoma. Semin. Cancer Biol. 2011, 21, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Eilers, M.; Eisenman, R.N. Myc’s broad reach. Genes Dev. 2008, 22, 2755–2766. [Google Scholar] [CrossRef] [PubMed]
- Bell, E.; Premkumar, R.; Carr, J.; Lu, X.; Lovat, P.E.; Kees, U.R.; Lunec, J.; Tweddle, D.A. The role of MYCN in the failure of MYCN amplified neuroblastoma cell lines to G1 arrest after DNA damage. Cell Cycle 2006, 5, 2639–2647. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.H.; Rychahou, P.G.; Ishola, T.A.; Qiao, J.; Evers, B.M.; Chung, D.H. MYCN silencing induces differentiation and apoptosis in human neuroblastoma cells. Biochem. Biophys. Res. Commun. 2006, 351, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Nara, K.; Kusafuka, T.; Yoneda, A.; Oue, T.; Sangkhathat, S.; Fukuzawa, M. Silencing of MYCN by RNA interference induces growth inhibition, apoptotic activity and cell differentiation in a neuroblastoma cell line with MYCN amplification. Int. J. Oncol. 2007, 30, 1189–1196. [Google Scholar] [PubMed]
- Tauszig-Delamasure, S.; Yu, L.Y.; Cabrera, J.R.; Bouzas-Rodriguez, J.; Mermet-Bouvier, C.; Guix, C.; Bordeaux, M.C.; Arumae, U.; Mehlen, P. The TrkC receptor induces apoptosis when the dependence receptor notion meets the neurotrophin paradigm. Proc. Natl. Acad. Sci. USA 2007, 104, 13361–13366. [Google Scholar] [CrossRef] [PubMed]
- Bouzas-Rodriguez, J.; Cabrera, J.R.; Delloye-Bourgeois, C.; Ichim, G.; Delcros, J.G.; Raquin, M.A.; Rousseau, R.; Combaret, V.; Benard, J.; Tauszig-Delamasure, S.; et al. Neurotrophin-3 production promotes human neuroblastoma cell survival by inhibiting trkc-induced apoptosis. J. Clin. Investig. 2010, 120, 850–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, D.R.; Miller, F.D. Neurotrophin signal transduction in the nervous system. Curr. Opin. Neurobiol. 2000, 10, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Iraci, N.; Gherardi, S.; Gamble, L.D.; Wood, K.M.; Perini, G.; Lunec, J.; Tweddle, D.A. P53 is a direct transcriptional target of mycn in neuroblastoma. Cancer Res. 2010, 70, 1377–1388. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Vousden, K.H. Puma, a novel proapoptotic gene, is induced by p53. Mol. Cell. 2001, 7, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Seoane, J.; Le, H.V.; Massague, J. Myc suppression of the p21(Cip1) Cdk inhibitor influences the outcome of the p53 response to DNA damage. Nature 2002, 419, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Lutz, W.; Schwab, M.; Debatin, K.M. MYCN sensitizes neuroblastoma cells for drug-induced apoptosis. Oncogene 1999, 18, 1479–1486. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Li, T.; Ding, H.F. Linking of N-MYC to death receptor machinery in neuroblastoma cells. J. Biol. Chem. 2005, 280, 9474–9481. [Google Scholar] [CrossRef] [PubMed]
- Berninger, B.; Garcia, D.E.; Inagaki, N.; Hahnel, C.; Lindholm, D. BDNF and NT-3 induce intracellular Ca2+ elevation in hippocampal neurones. Neuroreport 1993, 4, 1303–1306. [Google Scholar] [CrossRef] [PubMed]
- Levine, E.S.; Dreyfus, C.F.; Black, I.B.; Plummer, M.R. Differential effects of NGF and BDNF on voltage-gated calcium currents in embryonic basal forebrain neurons. J. Neurosci. 1995, 15, 3084–3091. [Google Scholar] [PubMed]
- Bonnington, J.K.; McNaughton, P.A. Signalling pathways involved in the sensitisation of mouse nociceptive neurones by nerve growth factor. J. Physiol. 2003, 551, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Baldelli, P.; Forni, P.E.; Carbone, E. BDNF, NT-3 and NGF induce distinct new Ca2+ channel synthesis in developing hippocampal neurons. Eur. J. Neurosci. 2000, 12, 4017–4032. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Romagnoli, A.; Pinton, P.; Rizzuto, R. Ca2+ signaling, mitochondria and cell death. Curr. Mol. Med. 2008, 8, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A. Calcium and cell death. Subcell Biochem. 2007, 45, 465–480. [Google Scholar] [PubMed]
- Nomura, M.; Ueno, A.; Saga, K.; Fukuzawa, M.; Kaneda, Y. Accumulation of cytosolic calcium induces necroptotic cell death in human neuroblastoma. Cancer Res. 2014, 74, 1056–1066. [Google Scholar] [CrossRef] [PubMed]
- Rodland, K.D. The role of the calcium-sensing receptor in cancer. Cell. Calcium 2004, 35, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Saidak, Z.; Mentaverri, R.; Brown, E.M. The role of the calcium-sensing receptor in the development and progression of cancer. Endocr. Rev. 2009, 30, 178–195. [Google Scholar] [CrossRef] [PubMed]
- de Torres, C.; Beleta, H.; Diaz, R.; Toran, N.; Rodriguez, E.; Lavarino, C.; Garcia, I.; Acosta, S.; Sunol, M.; Mora, J. The calcium-sensing receptor and parathyroid hormone-related protein are expressed in differentiated, favorable neuroblastic tumors. Cancer 2009, 115, 2792–2803. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarty, S.; Wang, H.; Canaff, L.; Hendy, G.N.; Appelman, H.; Varani, J. Calcium sensing receptor in human colon carcinoma: Interaction with Ca(2+) and 1,25-dihydroxyvitamin d(3). Cancer Res. 2005, 65, 493–498. [Google Scholar] [PubMed]
- Singh, N.; Promkan, M.; Liu, G.; Varani, J.; Chakrabarty, S. Role of calcium sensing receptor (CaSR) in tumorigenesis. Best Pract. Res. Clin. Endocrinol. Metab. 2013, 27, 455–463. [Google Scholar] [CrossRef]
- Maris, J.M.; Mosse, Y.P.; Bradfield, J.P.; Hou, C.; Monni, S.; Scott, R.H.; Asgharzadeh, S.; Attiyeh, E.F.; Diskin, S.J.; Laudenslager, M.; et al. Chromosome 6p22 locus associated with clinically aggressive neuroblastoma. N. Engl. J. Med. 2008, 358, 2585–2593. [Google Scholar] [CrossRef] [PubMed]
- Nguyen le, B.; Diskin, S.J.; Capasso, M.; Wang, K.; Diamond, M.A.; Glessner, J.; Kim, C.; Attiyeh, E.F.; Mosse, Y.P.; Cole, K.; et al. Phenotype restricted genome-wide association study using a gene-centric approach identifies three low-risk neuroblastoma susceptibility loci. PLoS Genet. 2011, 7, e1002026. [Google Scholar] [CrossRef] [PubMed]
- Capasso, M.; Devoto, M.; Hou, C.; Asgharzadeh, S.; Glessner, J.T.; Attiyeh, E.F.; Mosse, Y.P.; Kim, C.; Diskin, S.J.; Cole, K.A.; et al. Common variations in bard1 influence susceptibility to high-risk neuroblastoma. Nat. Genet. 2009, 41, 718–723. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Diskin, S.J.; Zhang, H.; Attiyeh, E.F.; Winter, C.; Hou, C.; Schnepp, R.W.; Diamond, M.; Bosse, K.; Mayes, P.A.; et al. Integrative genomics identifies LMO1 as a neuroblastoma oncogene. Nature 2011, 469, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Capasso, M.; Diskin, S.J.; Totaro, F.; Longo, L.; De Mariano, M.; Russo, R.; Cimmino, F.; Hakonarson, H.; Tonini, G.P.; Devoto, M.; et al. Replication of GWAS-identified neuroblastoma risk loci strengthens the role of BARD1 and affirms the cumulative effect of genetic variations on disease susceptibility. Carcinogenesis 2013, 34, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Diskin, S.J.; Capasso, M.; Schnepp, R.W.; Cole, K.A.; Attiyeh, E.F.; Hou, C.; Diamond, M.; Carpenter, E.L.; Winter, C.; Lee, H.; et al. Common variation at 6q16 within HACE1 and LIN28B influences susceptibility to neuroblastoma. Nat. Genet. 2012, 44, 1126–1130. [Google Scholar] [CrossRef] [PubMed]
- Bhatnagar, S.N.; Sarin, Y.K. Neuroblastoma: A review of management and outcome. Indian J. Pediatr. 2012, 79, 787–792. [Google Scholar] [CrossRef] [PubMed]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef] [PubMed]
- Florea, A.M.; Busselberg, D. Cisplatin as an anti-tumor drug: Cellular mechanisms of activity, drug resistance and induced side effects. Cancers (Basel) 2011, 3, 1351–1371. [Google Scholar] [CrossRef]
- Splettstoesser, F.; Florea, A.M.; Busselberg, D. IP(3) receptor antagonist, 2-APB, attenuates cisplatin induced Ca2+-influx in HeLa-S3 cells and prevents activation of calpain and induction of apoptosis. Br. J. Pharmacol. 2007, 151, 1176–1186. [Google Scholar] [CrossRef] [PubMed]
- Florea, A.M.; Yamoah, E.N.; Dopp, E. Intracellular calcium disturbances induced by arsenic and its methylated derivatives in relation to genomic damage and apoptosis induction. Environ. Health Perspect. 2005, 113, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Keshelava, N.; Seeger, R.C.; Groshen, S.; Reynolds, C.P. Drug resistance patterns of human neuroblastoma cell lines derived from patients at different phases of therapy. Cancer Res. 1998, 58, 5396–5405. [Google Scholar] [PubMed]
- Peaston, A.E.; Gardaneh, M.; Franco, A.V.; Hocker, J.E.; Murphy, K.M.; Farnsworth, M.L.; Catchpoole, D.R.; Haber, M.; Norris, M.D.; Lock, R.B.; et al. MRP1 gene expression level regulates the death and differentiation response of neuroblastoma cells. Br. J. Cancer 2001, 85, 1564–1571. [Google Scholar] [CrossRef] [PubMed]
- Keshelava, N.; Zuo, J.J.; Chen, P.; Waidyaratne, S.N.; Luna, M.C.; Gomer, C.J.; Triche, T.J.; Reynolds, C.P. Loss of p53 function confers high-level multidrug resistance in neuroblastoma cell lines. Cancer Res. 2001, 61, 6185–6193. [Google Scholar] [PubMed]
- Kotchetkov, R.; Cinatl, J.; Blaheta, R.; Vogel, J.U.; Karaskova, J.; Squire, J.; Hernaiz Driever, P.; Klingebiel, T.; Cinatl, J., Jr. Development of resistance to vincristine and doxorubicin in neuroblastoma alters malignant properties and induces additional karyotype changes: A preclinical model. Int. J. Cancer 2003, 104, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Blaheta, R.A.; Michaelis, M.; Natsheh, I.; Hasenberg, C.; Weich, E.; Relja, B.; Jonas, D.; Doerr, H.W.; Cinatl, J., Jr. Valproic acid inhibits adhesion of vincristine- and cisplatin-resistant neuroblastoma tumour cells to endothelium. Br. J. Cancer 2007, 96, 1699–1706. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, J.; Edsjo, A.; Pahlman, S.; Pettersson, H.M. Multidrug-resistant neuroblastoma cells are responsive to arsenic trioxide at both normoxia and hypoxia. Mol. Cancer Ther. 2005, 4, 1128–1135. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Satheesh, N.J.; Büsselberg, D. The Role of Intracellular Calcium for the Development and Treatment of Neuroblastoma. Cancers 2015, 7, 823-848. https://doi.org/10.3390/cancers7020811
Satheesh NJ, Büsselberg D. The Role of Intracellular Calcium for the Development and Treatment of Neuroblastoma. Cancers. 2015; 7(2):823-848. https://doi.org/10.3390/cancers7020811
Chicago/Turabian StyleSatheesh, Noothan Jyothi, and Dietrich Büsselberg. 2015. "The Role of Intracellular Calcium for the Development and Treatment of Neuroblastoma" Cancers 7, no. 2: 823-848. https://doi.org/10.3390/cancers7020811
APA StyleSatheesh, N. J., & Büsselberg, D. (2015). The Role of Intracellular Calcium for the Development and Treatment of Neuroblastoma. Cancers, 7(2), 823-848. https://doi.org/10.3390/cancers7020811