
Involvement of 14-3-3 Proteins in Regulating Tumor Progression of Hepatocellular Carcinoma

Abstract
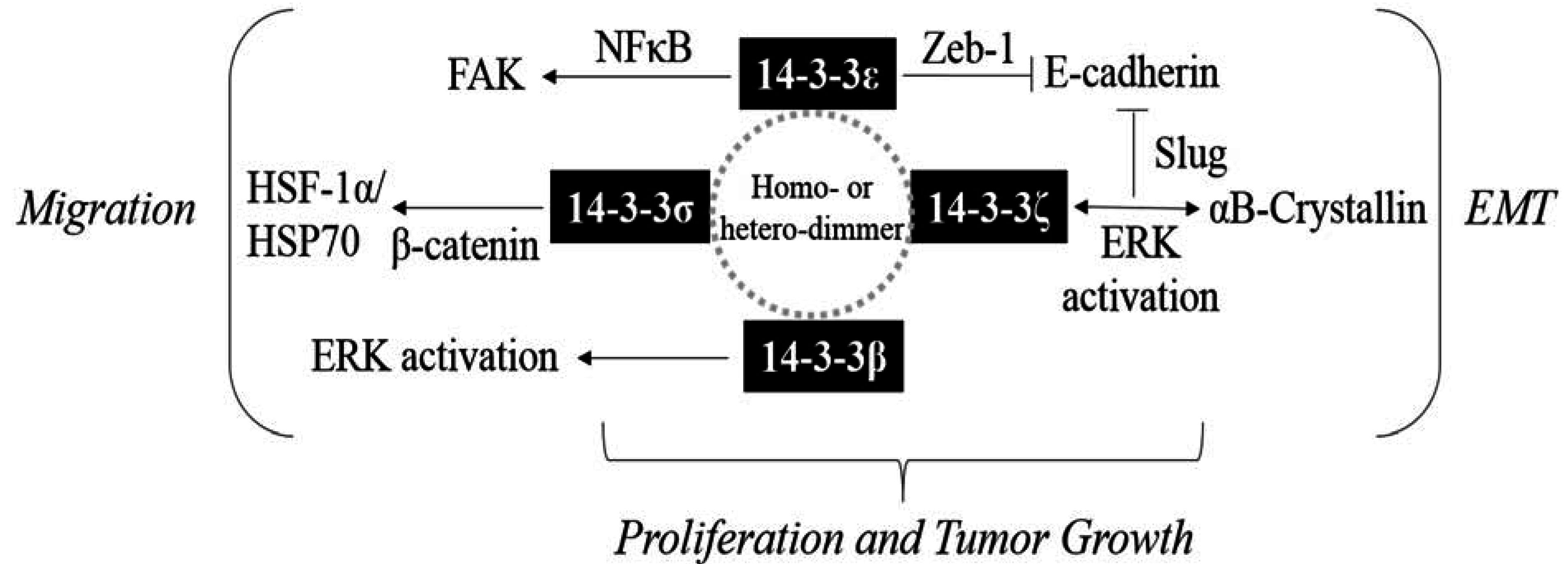
:1. Introduction
2. Expression of 14-3-3 Proteins in HCC
3. 14-3-3 Proteins Regulate HCC Cell Proliferation
4. Anti-Apoptotic Effects of 14-3-3
5. 14-3-3 Proteins Contribute to HCC EMT
6. 14-3-3 Proteins Promote HCC Cell Migration
7. Conclusions

{kind=link}
| Ref. | Year | Isoform | Result |
|---|---|---|---|
| Iwata et al. [55] | 2000 | 14-3-3σ | Promoter of 14-3-3σ is hypermethylated, and 14-3-3σ is downregulated in HCC |
| Komiya et al. [61] | 2008 | 14-3-3β | 14-3-3β promotes tumorigenicity and metastasis in rat hepatoma |
| Chen et al. [56] | 2010 | 14-3-3σ | 14-3-3σ is detected in HCC |
| Ko et al. [32] | 2011 | 14-3-3ε | 14-3-3ε is overexpressed and associates with extrahepatic metastasis and survival of HCC |
| Ko et al. [33] | 2011 | 14-3-3γ | 14-3-3γ is overexpressed and associates with extrahepatic metastasis and survival of HCC |
| Liu et al. [34] | 2011 | 14-3-3β | 14-3-3β is overexpressed and associates with extrahepatic metastasis and survival of HCC; 14-3-3β promotes cancer cell migration, proliferation and tumor growth of HCC |
| Choi et al. [35] | 2011 | 14-3-3ζ | 14-3-3ζ is overexpressed in HCC. Silencing of 14-3-3ζ inhibits cell proliferation of HCC |
| Jan et al. [74] | 2013 | 14-3-3ε | 14-3-3ε expression is correlated with Par-3 in HCC |
| Liu et al. [69] | 2013 | 14-3-3ε | 14-3-3ε contributes to EMT in HCC cells; 14-3-3ε expression is reverse correlated with E-cadherin in HCC; a combination of 14-3-3ε/E-cadherin expression is associated with the prognosis of HCC |
| Huang et al. [36] | 2013 | 14-3-3ζ | 14-3-3ζ complexes with αB-crystallin to promote EMT and enhance resistance to sorafenib of HCC |
| Ko et al. [85] | 2013 | 14-3-3ε | 14-3-3ε induces FAK expression, and 14-3-3ε expression is correlated with FAK in HCC |
| Liu et al. [37] | 2014 | 14-3-3σ | 14-3-3σ is overexpressed and promotes cell migration of HCC |
| Zhang et al. [38] | 2014 | 14-3-3σ | 14-3-3σ is overexpressed in HCC |

Acknowledgments
Author Contributions
Conflicts of Interest
References
- Aitken, A. Post-translational modification of 14-3-3 isoforms and regulation of cellular function. Semin. Cell Dev. Biol. 2011, 22, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Lee, W.H.; Sobott, F.; Papagrigoriou, E.; Robinson, C.V.; Grossmann, J.G.; Sundstrom, M.; Doyle, D.A.; Elkins, J.M. Structural basis for protein-protein interactions in the 14-3-3 protein family. Proc. Natl. Acad. Sci. USA 2006, 103, 17237–17242. [Google Scholar] [CrossRef] [PubMed]
- Morrison, D.K. The 14-3-3 proteins: Integrators of diverse signaling cues that impact cell fate and cancer development. Trends. Cell Biol. 2009, 19, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Tzivion, G.; Avruch, J. 14-3-3 proteins: Active cofactors in cellular regulation by serine/threonine phosphorylation. J. Biol. Chem. 2002, 277, 3061–3064. [Google Scholar] [CrossRef] [PubMed]
- Tzivion, G.; Gupta, V.S.; Kaplun, L.; Balan, V. 14-3-3 proteins as potential oncogenes. Semin. Cancer Biol. 2006, 16, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Liou, J.Y.; Lee, S.; Ghelani, D.; Matijevic-Aleksic, N.; Wu, K.K. Protection of endothelial survival by peroxisome proliferator-activated receptor-δ mediated 14-3-3 upregulation. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1481–1487. [Google Scholar] [CrossRef] [PubMed]
- Liou, J.Y.; Ghelani, D.; Yeh, S.; Wu, K.K. Nonsteroidal anti-inflammatory drugs induce colorectal cancer cell apoptosis by suppressing 14-3-3epsilon. Cancer Res. 2007, 67, 3185–3191. [Google Scholar] [CrossRef] [PubMed]
- Liou, J.Y.; Wu, C.C.; Chen, B.R.; Yen, L.B.; Wu, K.K. Nonsteroidal anti-inflammatory drugs induced endothelial apoptosis by perturbing peroxisome proliferator-activated receptor-σ transcriptional pathway. Mol. Pharmacol. 2008, 74, 1399–1406. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhao, J.; Du, Y.; Park, H.R.; Sun, S.Y.; Bernal-Mizrachi, L.; Aitken, A.; Khuri, F.R.; Fu, H. Down-regulation of 14-3-3ζ suppresses anchorage-independent growth of lung cancer cells through anoikis activation. Proc. Natl. Acad. Sci. USA 2008, 105, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Neal, C.L.; Yao, J.; Yang, W.; Zhou, X.; Nguyen, N.T.; Lu, J.; Danes, C.G.; Guo, H.; Lan, K.H.; Ensor, J.; et al. 14-3-3ζ overexpression defines high risk for breast cancer recurrence and promotes cancer cell survival. Cancer Res. 2009, 69, 3425–3432. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Liu, X.; Qiao, D.; Martinez, J.D. Isoform-specific expression of 14-3-3 proteins in human lung cancer tissues. Int. J. Cancer 2005, 113, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Shen, G.; Liu, Q.; Xu, Y.; Zhou, L.; Xiao, S.; Xu, Z.; Gong, F.; You, C.; Wei, Y. Isoform-specific expression and characterization of 14-3-3 proteins in human glioma tissues discovered by stable isotope labeling with amino acids in cell culture-based proteomic analysis. Proteomics Clin. Appl. 2009, 3, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Cao, W.; Lin, H.; Zhang, W.; Lin, W.; Cao, L.; Zhen, H.; Huo, J.; Zhang, X. Isoform-specific expression of 14-3-3 proteins in human astrocytoma. J. Neurol. Sci. 2009, 276, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Li, D.Q.; Wang, L.; Fei, F.; Hou, Y.F.; Luo, J.M.; Zeng, R.; Wu, J.; Lu, J.S.; Di, G.H.; Ou, Z.L.; et al. Identification of breast cancer metastasis-associated proteins in an isogenic tumor metastasis model using two-dimensional gel electrophoresis and liquid chromatography-ion trap-mass spectrometry. Proteomics 2006, 6, 3352–3368. [Google Scholar] [CrossRef] [PubMed]
- Ko, B.S.; Chang, T.C.; Hsu, C.; Chen, Y.C.; Shen, T.L.; Chen, S.C.; Wang, J.; Wu, K.K.; Jan, Y.J.; Liou, J.Y. Overexpression of 14-3-3ε predicts tumour metastasis and poor survival in hepatocellular carcinoma. Histopathology 2011, 58, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Ko, B.S.; Lai, I.R.; Chang, T.C.; Liu, T.A.; Chen, S.C.; Wang, J.; Jan, Y.J.; Liou, J.Y. Involvement of 14-3-3γ overexpression in extrahepatic metastasis of hepatocellular carcinoma. Hum. Pathol. 2011, 42, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.A.; Jan, Y.J.; Ko, B.S.; Chen, S.C.; Liang, S.M.; Hung, Y.L.; Hsu, C.; Shen, T.L.; Lee, Y.M.; Chen, P.F.; et al. Increased expression of 14-3-3β promotes tumor progression and predicts extrahepatic metastasis and worse survival in hepatocellular carcinoma. Am. J. Pathol. 2011, 179, 2698–2708. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.E.; Hur, W.; Jung, C.K.; Piao, L.S.; Lyoo, K.; Hong, S.W.; Kim, S.W.; Yoon, H.Y.; Yoon, S.K. Silencing of 14-3-3ζ over-expression in hepatocellular carcinoma inhibits tumor growth and enhances chemosensitivity to cis-diammined dichloridoplatium. Cancer Lett. 2011, 303, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.Y.; Ke, A.W.; Shi, G.M.; Zhang, X.; Zhang, C.; Shi, Y.H.; Wang, X.Y.; Ding, Z.B.; Xiao, Y.S.; Yan, J.; et al. αB-crystallin complexes with 14-3-3ζ to induce epithelial-mesenchymal transition and resistance to sorafenib in hepatocellular carcinoma. Hepatology 2013, 57, 2235–2247. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Jan, Y.J.; Ko, B.S.; Wu, Y.M.; Liang, S.M.; Chen, S.C.; Lee, Y.M.; Liu, T.A.; Chang, T.C.; Wang, J.; et al. 14-3-3σ induces heat shock protein 70 expression in hepatocellular carcinoma. BMC Cancer 2014, 14, 425. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, Y.; Lin, C.; Ding, J.; Liao, G.; Tang, B. Aberrant upregulation of 14-3-3σ and ezh2 expression serves as an inferior prognostic biomarker for hepatocellular carcinoma. PLoS ONE 2014, 9, e107251. [Google Scholar] [CrossRef] [PubMed]
- Ajjappala, B.S.; Kim, Y.S.; Kim, M.S.; Lee, M.Y.; Lee, K.Y.; Ki, H.Y.; Cha, D.H.; Baek, K.H. 14-3-3γ is stimulated by il-3 and promotes cell proliferation. J. Immunol. 2009, 182, 1050–1060. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.W.; Qi, W.; Brabant, M.; Bosco, G.; Martinez, J.D. Human 14-3-3γ protein results in abnormal cell proliferation in the developing eye of drosophila melanogaster. Cell Div. 2008, 3, 2. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.N.; Chen, C.H.; Sheu, J.C.; Lee, H.S.; Huang, G.T.; Yu, C.Y.; Lu, F.J.; Chow, L.P. Identification of human hepatocellular carcinoma-related biomarkers by two-dimensional difference gel electrophoresis and mass spectrometry. J. Proteome Res. 2005, 4, 2062–2069. [Google Scholar] [CrossRef] [PubMed]
- Takihara, Y.; Matsuda, Y.; Hara, J. Role of the β isoform of 14-3-3 proteins in cellular proliferation and oncogenic transformation. Carcinogenesis 2000, 21, 2073–2077. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, A.; Miyagi, Y.; Komiya, Y.; Kurabe, N.; Kitanaka, C.; Kato, N.; Nagashima, Y.; Kuchino, Y.; Tashiro, F. Forced expression of antisense 14-3-3β RNA suppresses tumor cell growth in vitro and in vivo. Carcinogenesis 2003, 24, 1549–1559. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liu, J.Y.; Zhang, J.T. 14-3-3σ, the double-edged sword of human cancers. Am. J. Transl. Res. 2009, 1, 326–340. [Google Scholar] [PubMed]
- Lodygin, D.; Hermeking, H. The role of epigenetic inactivation of 14-3-3σ in human cancer. Cell Res. 2005, 15, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Hermeking, H. The 14-3-3 cancer connection. Nat. Rev. Cancer 2003, 3, 931–943. [Google Scholar] [CrossRef] [PubMed]
- Neupane, D.; Korc, M. 14-3-3σ modulates pancreatic cancer cell survival and invasiveness. Clin. Cancer Res. 2008, 14, 7614–7623. [Google Scholar] [CrossRef] [PubMed]
- Guweidhi, A.; Kleeff, J.; Giese, N.; El Fitori, J.; Ketterer, K.; Giese, T.; Buchler, M.W.; Korc, M.; Friess, H. Enhanced expression of 14-3-3σ in pancreatic cancer and its role in cell cycle regulation and apoptosis. Carcinogenesis 2004, 25, 1575–1585. [Google Scholar] [CrossRef] [PubMed]
- Perathoner, A.; Pirkebner, D.; Brandacher, G.; Spizzo, G.; Stadlmann, S.; Obrist, P.; Margreiter, R.; Amberger, A. 14-3-3σ expression is an independent prognostic parameter for poor survival in colorectal carcinoma patients. Clin. Cancer Res. 2005, 11, 3274–3279. [Google Scholar] [CrossRef] [PubMed]
- Ide, M.; Saito, K.; Tsutsumi, S.; Tsuboi, K.; Yamaguchi, S.; Asao, T.; Kuwano, H.; Nakajima, T. Over-expression of 14-3-3σ a in budding colorectal cancer cells modulates cell migration in the presence of tenascin-c. Oncol. Rep. 2007, 18, 1451–1456. [Google Scholar] [PubMed]
- Zhou, W.H.; Tang, F.; Xu, J.; Wu, X.; Feng, Z.Y.; Li, H.G.; Lin, D.J.; Shao, C.K.; Liu, Q. Aberrant upregulation of 14-3-3o expression serves as an inferior prognostic biomarker for gastric cancer. BMC Cancer 2011, 11, 397. [Google Scholar] [CrossRef] [PubMed]
- Shiba-Ishii, A.; Noguchi, M. Aberrant stratifin overexpression is regulated by tumor-associated cpg demethylation in lung adenocarcinoma. Am. J. Pathol. 2012, 180, 1653–1662. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, V.M.; Jensen, T.J.; Cui, H.; Futscher, B.W.; Martinez, J.D. Hypomethylation of the 14-3-3σ promoter leads to increased expression in non-small cell lung cancer. Gene. Chromosome. Canc. 2011, 50, 830–836. [Google Scholar] [CrossRef] [PubMed]
- Shiba-Ishii, A.; Kano, J.; Morishita, Y.; Sato, Y.; Minami, Y.; Noguchi, M. High expression of stratifin is a universal abnormality during the course of malignant progression of early-stage lung adenocarcinoma. Int. J. Cancer 2011, 129, 2445–2453. [Google Scholar] [CrossRef] [PubMed]
- Iwata, N.; Yamamoto, H.; Sasaki, S.; Itoh, F.; Suzuki, H.; Kikuchi, T.; Kaneto, H.; Iku, S.; Ozeki, I.; Karino, Y.; et al. Frequent hypermethylation of cpg islands and loss of expression of the 14-3-3σ gene in human hepatocellular carcinoma. Oncogene 2000, 19, 5298–5302. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.L.; Zhou, L.; Yang, J.; Shen, F.K.; Zhao, S.P.; Wang, Y.L. Hepatocellular carcinoma-associated protein markers investigated by maldi-tof ms. Mol. Med. Rep. 2010, 3, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Tzivion, G.; Luo, Z.; Avruch, J. A dimeric 14-3-3 protein is an essential cofactor for raf kinase activity. Nature 1998, 394, 88–92. [Google Scholar] [PubMed]
- Roy, S.; McPherson, R.A.; Apolloni, A.; Yan, J.; Lane, A.; Clyde-Smith, J.; Hancock, J.F. 14-3-3 facilitates ras-dependent raf-1 activation in vitro and in vivo. Mol. Cell Biol. 1998, 18, 3947–3955. [Google Scholar] [PubMed]
- Thorson, J.A.; Yu, L.W.; Hsu, A.L.; Shih, N.Y.; Graves, P.R.; Tanner, J.W.; Allen, P.M.; Piwnica-Worms, H.; Shaw, A.S. 14-3-3 proteins are required for maintenance of Raf-1 phosphorylation and kinase activity. Mol. Cell Biol. 1998, 18, 5229–5238. [Google Scholar] [PubMed]
- DeYoung, M.P.; Horak, P.; Sofer, A.; Sgroi, D.; Ellisen, L.W. Hypoxia regulates tsc1/2-mtor signaling and tumor suppression through redd1-mediated 14-3-3 shuttling. Genes Dev. 2008, 22, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Liu, X.; Chen, W.; Li, Q.; Martinez, J.D. Overexpression of 14-3-3γ causes polyploidization in h322 lung cancer cells. Mol. Carcinog. 2007, 46, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Musholt, T.J.; Brehm, C.; Hanack, J.; von Wasielewski, R.; Musholt, P.B. Identification of differentially expressed genes in papillary thyroid carcinomas with and without rearrangements of the tyrosine kinase receptors ret and/or ntrk1. J. Surg. Res. 2006, 131, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Li, Y.; Chen, R.S.; He, X.; Yang, L.; Li, W. Overexpression of xct induces up-regulation of 14-3-3β in kaposi’s sarcoma. Biosci. Rep. 2010, 30, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Komiya, Y.; Kurabe, N.; Katagiri, K.; Ogawa, M.; Sugiyama, A.; Kawasaki, Y.; Tashiro, F. A novel binding factor of 14-3-3β functions as a transcriptional repressor and promotes anchorage-independent growth, tumorigenicity, and metastasis. J. Biol. Chem. 2008, 283, 18753–18764. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.M.; Cory, S. The Bcl-2 protein family: Arbiters of cell survival. Science 1998, 281, 1322–1326. [Google Scholar] [CrossRef] [PubMed]
- Cheng, E.H.; Wei, M.C.; Weiler, S.; Flavell, R.A.; Mak, T.W.; Lindsten, T.; Korsmeyer, S.J. Bcl-2, Bcl-XL sequester BH3 domain-only molecules preventing bax- and bak-mediated mitochondrial apoptosis. Mol. Cell 2001, 8, 705–711. [Google Scholar] [CrossRef]
- Zha, J.; Harada, H.; Osipov, K.; Jockel, J.; Waksman, G.; Korsmeyer, S.J. BH3 domain of BAD is required for heterodimerization with BCL-XL and pro-apoptotic activity. J. Biol. Chem. 2000, 272, 24101–24104. [Google Scholar] [CrossRef]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. AKT phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef]
- Scheid, M.P.; Schubert, K.M.; Duronio, V. Regulation of bad phosphorylation and association with Bcl-XL by the MAPK/Erk kinase. J. Biol. Chem. 1999, 274, 31108–31113. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Ruan, H.; Demeter, M.R.; Comb, M.J. p90(RSK) blocks bad-mediated cell death via a protein kinase C-dependent pathway. J. Biol. Chem. 1999, 274, 34859–34867. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Demeter, M.R.; Ruan, H.; Comb, M.J. BAD Ser-155 phosphorylation regulates BAD/Bcl-XL interaction and cell survival. J. Biol. Chem. 2000, 275, 25865–25869. [Google Scholar] [CrossRef] [PubMed]
- Lizcano, J.M.; Morrice, N.; Cohen, P. Regulation of BAD by cAMP-dependent protein kinase is mediated via phosphorylation of a novel site, Ser155. Biochem. J. 2000, 349, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.M.; Liu, Y.; Payne, G.; Lutz, R.J.; Chittenden, T. Growth factors inactivate the cell death promoter BAD by phosphorylation of its BH3 domain on Ser155. J. Biol. Chem. 2000, 275, 25046–25051. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, R.R.; Zhang, H.; Wang, H.; Ichijo, H.; Miyashita, T.; Fu, H. Interaction of apoptosis signal-regulating kinase 1 with isoforms of 14-3-3 proteins. Exp. Cell Res. 2004, 294, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chen, J.; Fu, H. Suppression of apoptosis signal-regulating kinase 1-induced cell death by 14-3-3 proteins. Proc. Natl. Acad. Sci. USA 1999, 96, 8511–8515. [Google Scholar] [CrossRef] [PubMed]
- Alavi, A.; Hood, J.D.; Frausto, R.; Stupack, D.G.; Cheresh, D.A. Role of Raf in vascular protection from distinct apoptotic stimuli. Science 2003, 301, 94–96. [Google Scholar] [CrossRef] [PubMed]
- Shimamura, A.; Ballif, B.A.; Richards, S.A.; Blenis, J. Rsk1 mediates a MEK-MAP kinase cell survival signal. Curr. Biol. 2000, 10, 127–135. [Google Scholar] [CrossRef]
- Bonni, A.; Brunet, A.; West, A.E.; Datta, S.R.; Takasu, M.A.; Greenberg, M.E. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science 1999, 286, 1358–1362. [Google Scholar] [CrossRef] [PubMed]
- Jeanes, A.; Gottardi, C.J.; Yap, A.S. Cadherins and cancer: How does cadherin dysfunction promote tumor progression? Oncogene 2008, 27, 6920–6929. [Google Scholar] [CrossRef] [PubMed]
- Eger, A.; Stockinger, A.; Park, J.; Langkopf, E.; Mikula, M.; Gotzmann, J.; Mikulits, W.; Beug, H.; Foisner, R. β-catenin and TGFβ signalling cooperate to maintain a mesenchymal phenotype after foser-induced epithelial to mesenchymal transition. Oncogene 2004, 23, 2672–2680. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, T.; Makino, R.; Mitamura, K. Frequent down-regulation of e-cadherin by genetic and epigenetic changes in the malignant progression of hepatocellular carcinomas. Clin. Cancer Res. 2001, 7, 594–599. [Google Scholar] [PubMed]
- Kanai, Y.; Ushijima, S.; Hui, A.M.; Ochiai, A.; Tsuda, H.; Sakamoto, M.; Hirohashi, S. The e-cadherin gene is silenced by cpg methylation in human hepatocellular carcinomas. Int. J. Cancer 1997, 71, 355–359. [Google Scholar] [CrossRef]
- Yang, B.; Guo, M.; Herman, J.G.; Clark, D.P. Aberrant promoter methylation profiles of tumor suppressor genes in hepatocellular carcinoma. Am. J. Pathol. 2003, 163, 1101–1107. [Google Scholar] [CrossRef]
- Moreno-Bueno, G.; Portillo, F.; Cano, A. Transcriptional regulation of cell polarity in EMT and cancer. Oncogene 2008, 27, 6958–6969. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Neilson, E.G. Biomarkers for epithelial-mesenchymal transitions. J. Clin. Investig. 2009, 119, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.A.; Jan, Y.J.; Ko, B.S.; Liang, S.M.; Chen, S.C.; Wang, J.; Hsu, C.; Wu, Y.M.; Liou, J.Y. 14-3-3ε overexpression contributes to epithelial-mesenchymal transition of hepatocellular carcinoma. PLoS ONE 2013, 8, e57968. [Google Scholar] [CrossRef] [PubMed]
- Hou, Z.; Peng, H.; White, D.E.; Wang, P.; Lieberman, P.M.; Halazonetis, T.; Rauscher, F.J., III. 14-3-3 binding sites in the snail protein are essential for snail-mediated transcriptional repression and epithelial-mesenchymal differentiation. Cancer Res. 2010, 70, 4385–4393. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Edwards, A.S.; Fawcett, J.P.; Mbamalu, G.; Scott, J.D.; Pawson, T. A mammalian par-3-par-6 complex implicated in cdc42/rac1 and apkc signalling and cell polarity. Nat. Cell Biol. 2000, 2, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Ooshio, T.; Fujita, N.; Yamada, A.; Sato, T.; Kitagawa, Y.; Okamoto, R.; Nakata, S.; Miki, A.; Irie, K.; Takai, Y. Cooperative roles of PAR-3 and afadin in the formation of adherens and tight junctions. J. Cell Sci. 2007, 120, 2352–2365. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Macara, I.G. Par-3 controls tight junction assembly through the rac exchange factor tiam1. Nat. Cell Biol. 2005, 7, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Jan, Y.J.; Ko, B.S.; Liu, T.A.; Wu, Y.M.; Liang, S.M.; Chen, S.C.; Wang, J.; Liou, J.Y. Expression of partitioning defective 3 (PAR-3) for predicting extrahepatic metastasis and survival with hepatocellular carcinoma. Int. J. Mol. Sci. 2013, 14, 1684–1697. [Google Scholar] [CrossRef] [PubMed]
- Hurd, T.W.; Fan, S.; Liu, C.J.; Kweon, H.K.; Hakansson, K.; Margolis, B. Phosphorylation-dependent binding of 14-3-3 to the polarity protein Par3 regulates cell polarity in mammalian epithelia. Curr. Biol. 2003, 13, 2082–2090. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Guo, H.; Treekitkarnmongkol, W.; Li, P.; Zhang, J.; Shi, B.; Ling, C.; Zhou, X.; Chen, T.; Chiao, P.J.; et al. 14-3-3ζ cooperates with erbb2 to promote ductal carcinoma in situ progression to invasive breast cancer by inducing epithelial-mesenchymal transition. Cancer Cell 2009, 16, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Neal, C.L.; Xu, J.; Li, P.; Mori, S.; Yang, J.; Neal, N.N.; Zhou, X.; Wyszomierski, S.L.; Yu, D. Overexpression of 14-3-3ζ in cancer cells activates pi3k via binding the p85 regulatory subunit. Oncogene 2012, 31, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Parcellier, A.; Schmitt, E.; Brunet, M.; Hammann, A.; Solary, E.; Garrido, C. Small heat shock proteins HSP27 and αB-crystallin: Cytoprotective and oncogenic functions. Antioxid. Redox Signal. 2005, 7, 404–413. [Google Scholar] [CrossRef] [PubMed]
- McLean, G.W.; Carragher, N.O.; Avizienyte, E.; Evans, J.; Brunton, V.G.; Frame, M.C. The role of focal-adhesion kinase in cancer—a new therapeutic opportunity. Nat. Rev. Cancer 2005, 5, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Van Nimwegen, M.J.; van de Water, B. Focal adhesion kinase: A potential target in cancer therapy. Biochem. Pharmacol. 2007, 73, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, M.A.; Modzelewska, K.; Kwong, L.; Keely, P.J. Focal adhesion regulation of cell behavior. Biochim. Biophys. Acta 2004, 1692, 103–119. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, L.; Earp, H.S.; Parsons, J.T.; Schaller, M.; Juliano, R.L. Cell adhesion or integrin clustering increases phosphorylation of a focal adhesion-associated tyrosine kinase. J. Biol. Chem. 1992, 267, 23439–23442. [Google Scholar] [PubMed]
- Jan, Y.J.; Ko, B.S.; Hsu, C.; Chang, T.C.; Chen, S.C.; Wang, J.; Liou, J.Y. Overexpressed focal adhesion kinase predicts a higher incidence of extrahepatic metastasis and worse survival in hepatocellular carcinoma. Hum. Pathol. 2009, 40, 1384–1390. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V.; Kaur, A.; Cance, W. Cloning and characterization of the promoter region of human focal adhesion kinase gene: Nuclear factor kappa b and p53 binding sites. Biochim. Biophys. Acta 2004, 1678, 111–125. [Google Scholar] [CrossRef] [PubMed]
- Ko, B.S.; Chang, T.C.; Chen, C.H.; Liu, C.C.; Kuo, C.C.; Hsu, C.; Shen, Y.C.; Shen, T.L.; Golubovskaya, V.M.; Chang, C.C.; et al. Bortezomib suppresses focal adhesion kinase expression via interrupting nuclear factor-kappa b. Life Sci. 2010, 86, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Ko, B.S.; Jan, Y.J.; Chang, T.C.; Liang, S.M.; Chen, S.C.; Liu, T.A.; Wu, Y.M.; Wang, J.; Liou, J.Y. Upregulation of focal adhesion kinase by 14-3-3ε via nfkappab activation in hepatocellular carcinoma. Anticancer Agents Med. Chem. 2013, 13, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Fantl, W.J.; Muslin, A.J.; Kikuchi, A.; Martin, J.A.; MacNicol, A.M.; Gross, R.W.; Williams, L.T. Activation of raf-1 by 14-3-3 proteins. Nature 1994, 371, 612–614. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Janosch, P.; Tanji, M.; Rosenfeld, G.C.; Waymire, J.C.; Mischak, H.; Kolch, W.; Sedivy, J.M. Regulation of raf-1 kinase activity by the 14-3-3 family of proteins. EMBO J. 1995, 14, 685–696. [Google Scholar] [PubMed]
- Boudreau, A.; Tanner, K.; Wang, D.; Geyer, F.C.; Reis-Filho, J.S.; Bissell, M.J. 14-3-3σ stabilizes a complex of soluble actin and intermediate filament to enable breast tumor invasion. Proc. Natl. Acad. Sci. USA 2013, 110, E3937–E3944. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Y.-J.; Jan, Y.-J.; Ko, B.-S.; Liang, S.-M.; Liou, J.-Y. Involvement of 14-3-3 Proteins in Regulating Tumor Progression of Hepatocellular Carcinoma. Cancers 2015, 7, 1022-1036. https://doi.org/10.3390/cancers7020822
Wu Y-J, Jan Y-J, Ko B-S, Liang S-M, Liou J-Y. Involvement of 14-3-3 Proteins in Regulating Tumor Progression of Hepatocellular Carcinoma. Cancers. 2015; 7(2):1022-1036. https://doi.org/10.3390/cancers7020822
Chicago/Turabian StyleWu, Yi-Ju, Yee-Jee Jan, Bor-Sheng Ko, Shu-Man Liang, and Jun-Yang Liou. 2015. "Involvement of 14-3-3 Proteins in Regulating Tumor Progression of Hepatocellular Carcinoma" Cancers 7, no. 2: 1022-1036. https://doi.org/10.3390/cancers7020822
APA StyleWu, Y. -J., Jan, Y. -J., Ko, B. -S., Liang, S. -M., & Liou, J. -Y. (2015). Involvement of 14-3-3 Proteins in Regulating Tumor Progression of Hepatocellular Carcinoma. Cancers, 7(2), 1022-1036. https://doi.org/10.3390/cancers7020822