
Dichotomy in the Epigenetic Mark Lysine Acetylation is Critical for the Proliferation of Prostate Cancer Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
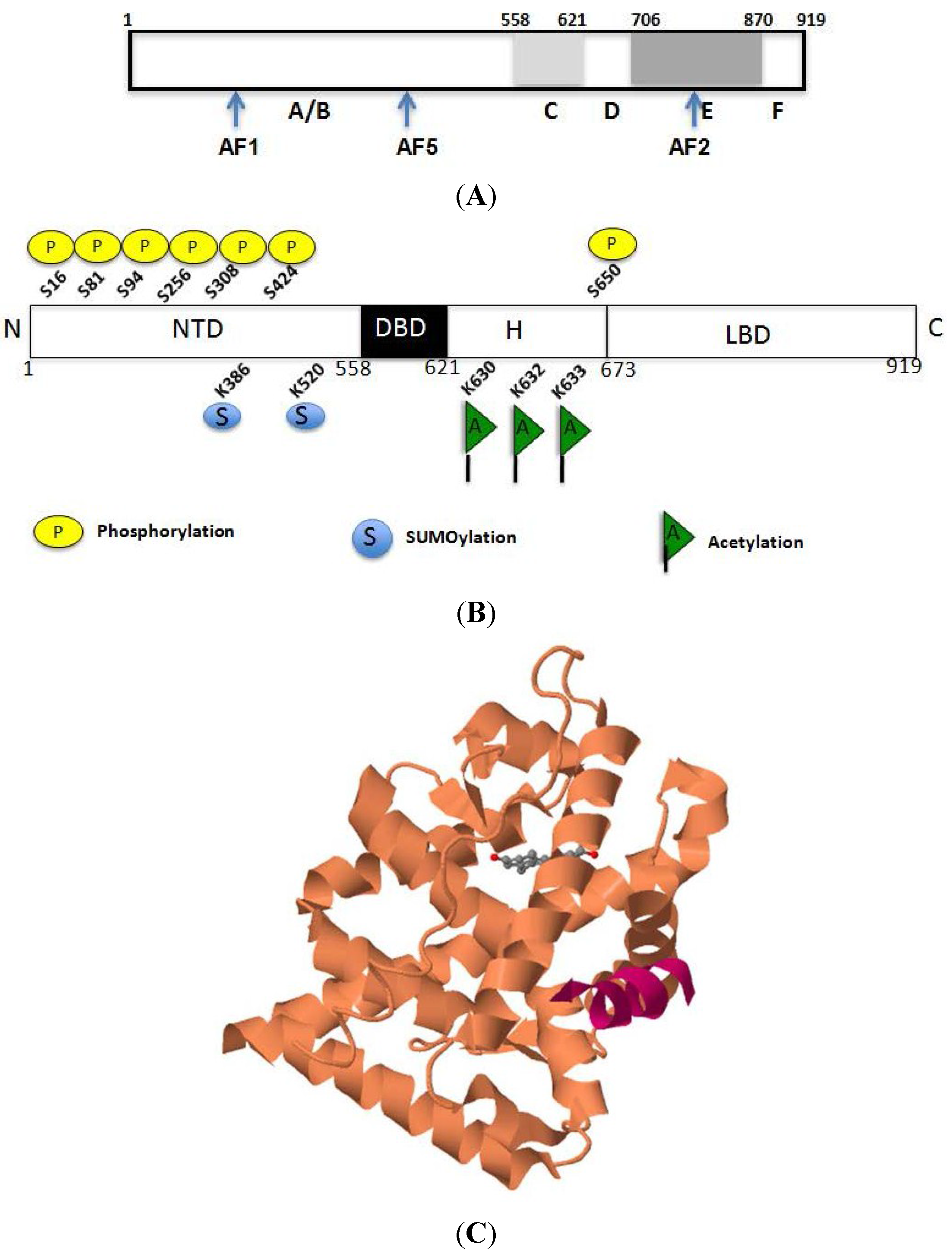
2.1. Structural Basis for the Transcriptional Functions of AR

2.2. Acetylation-Mediated Molecular Interactions of AR


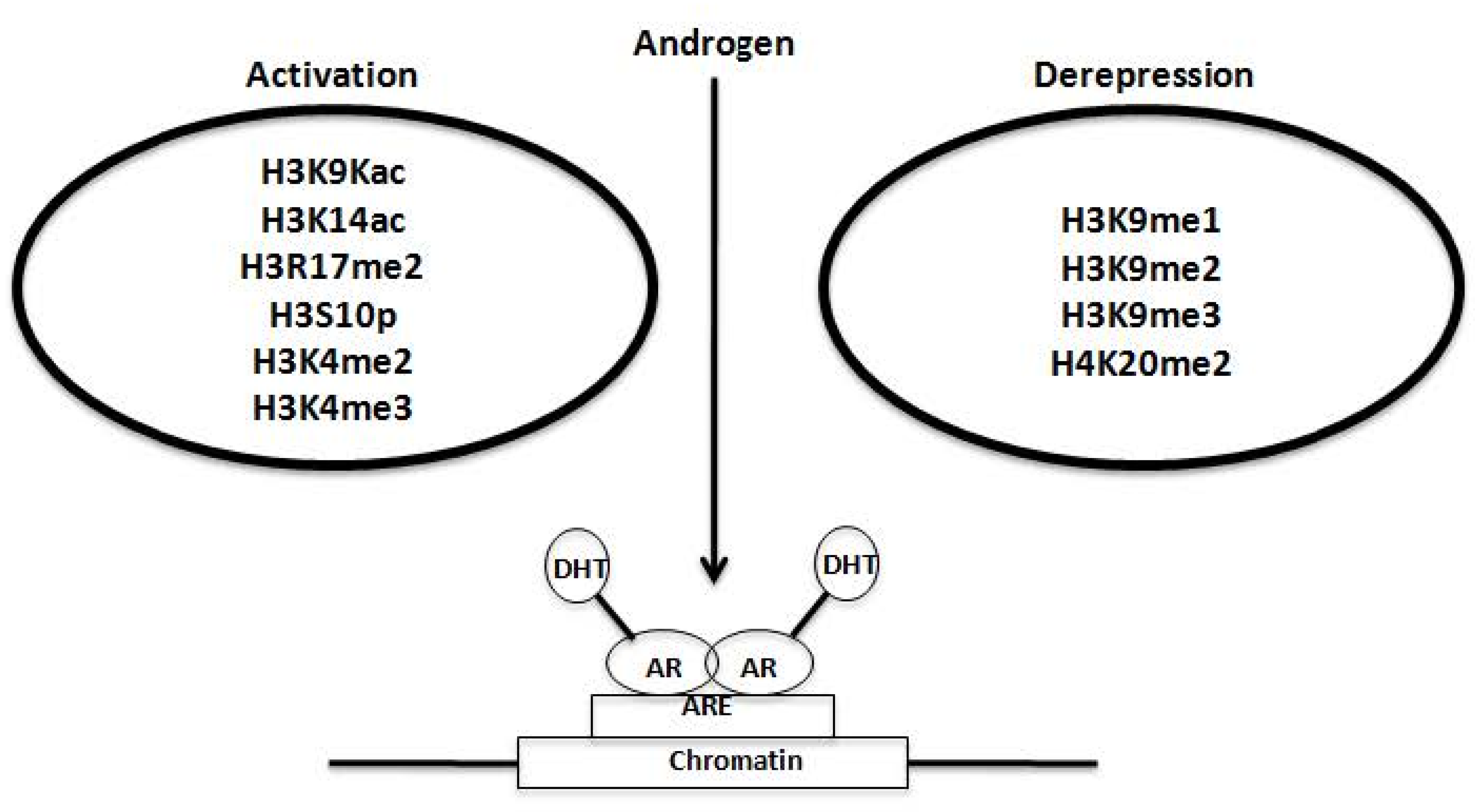
2.3. Regulation of AR Transcriptional Functions by Acetylation and Deacetylation

2.4. Synergy of AR with Key Molecules to Promote Metastasis and Growth of PCa
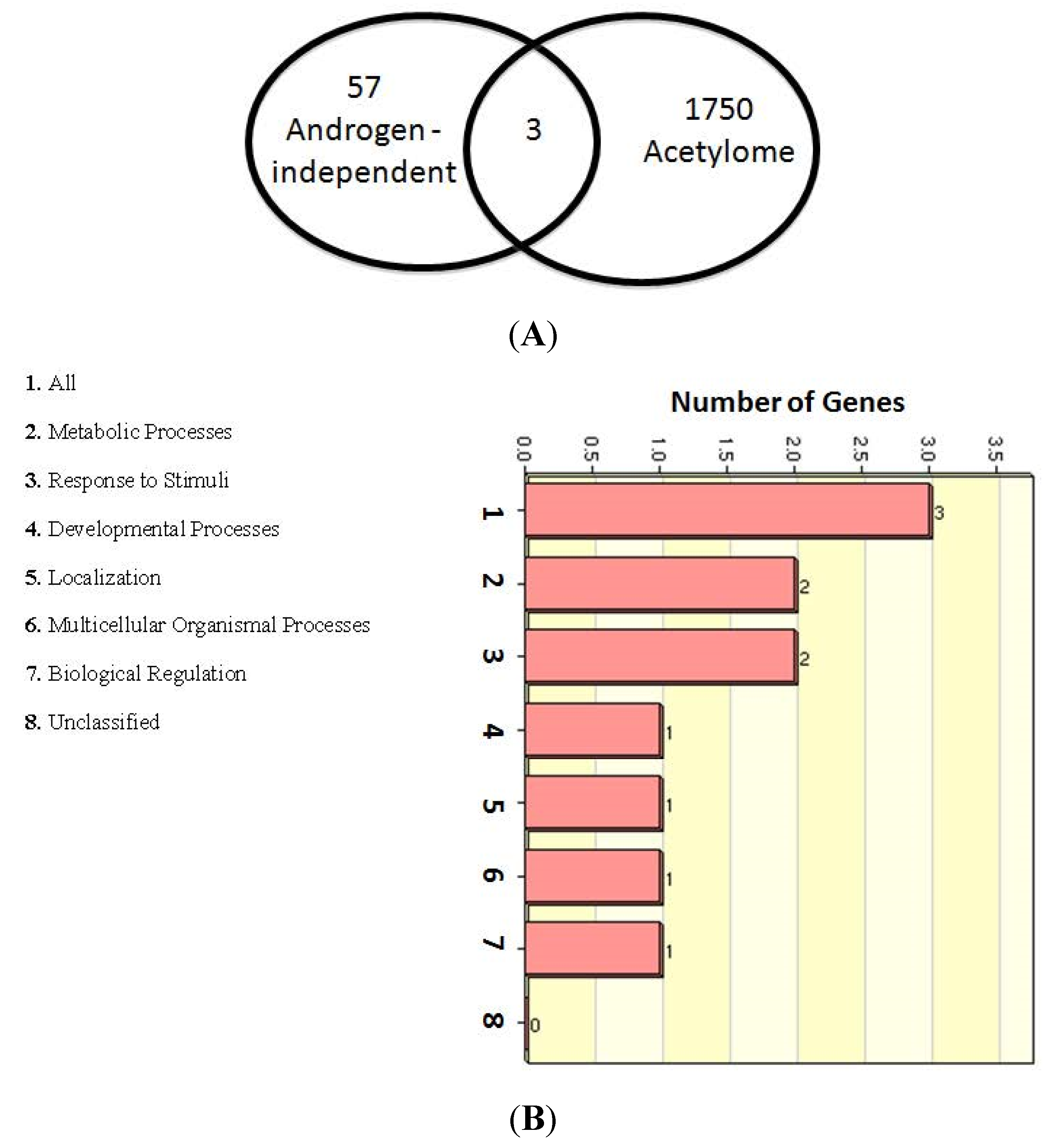
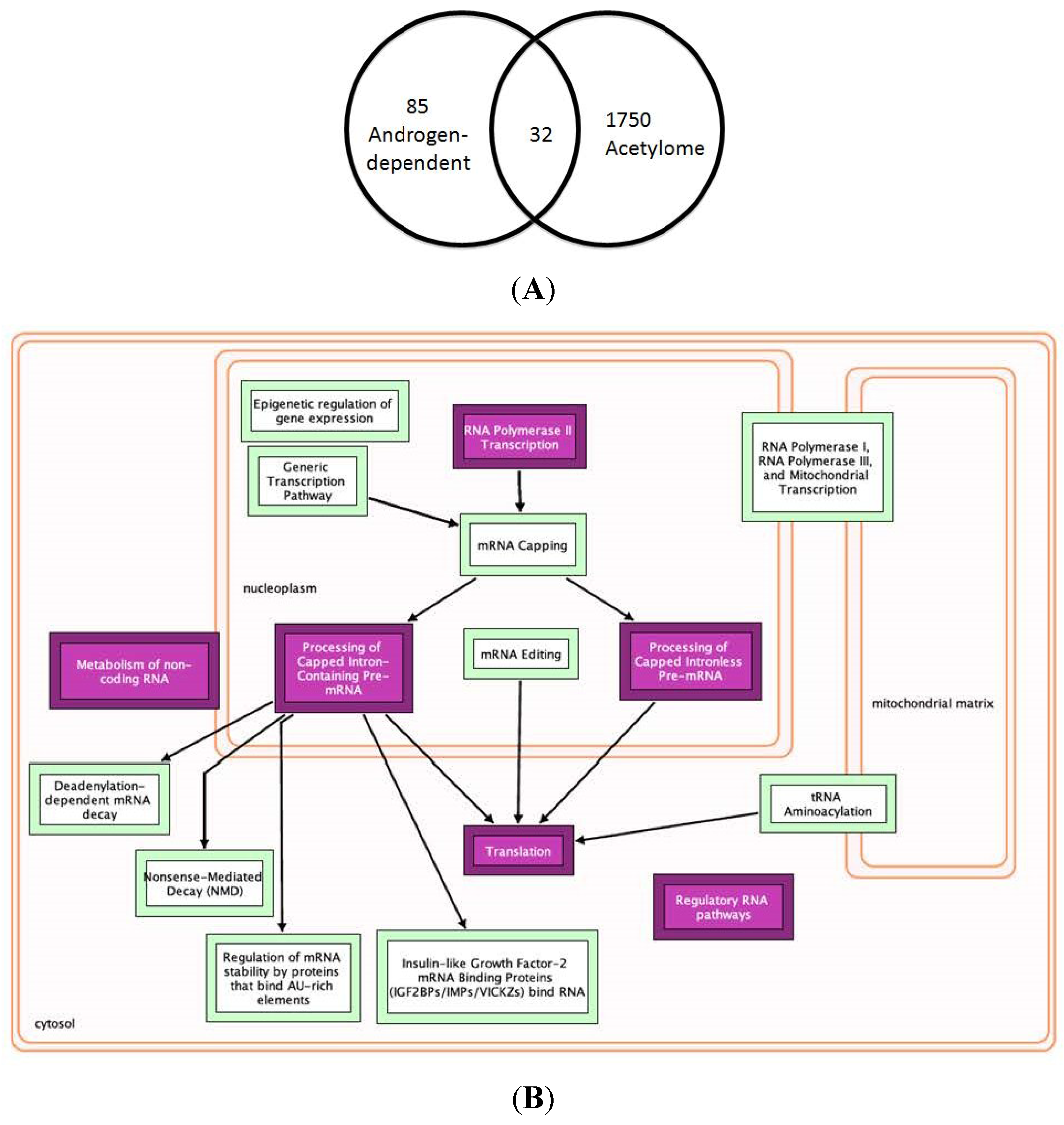
2.5. Role of Lysine-acetylated Proteins in AR-independent Progression of PCa


2.6. Multiple Treatment Strategies Target AR-Mediated Signaling
3. Discussion
4. Methods of Analysis
4.1. Construction of AR Interactome
4.2. Network Visualizations and Reactome Analysis
4.3. Functional Enrichment Analysis
Supplementary Files
Supplementary File 1Acknowledgements
Author Contributions
Conflicts of Interest
Abbreviations
| PCa | Prostate Cancer |
| AR | Androgen receptor mainly localized to prostate cells that binds to androgen |
| HATs | Histone acetyltransferases that was identified first to enzymatically acetylate histone proteins also acetylate, cellular proteins including transcription factors and metabolic enzymes |
| HDACs | Histone deacetylases enzymatically removes the acetyl marks imprinted by HATs |
| PSA | Prostate Specific Antigen is a target gene directly regulated by the transcription factor AR. The presence of PSA beyond normal level in the blood is an indicator for altered prostate functions |
References
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2013. A Cancer J. Clin. 2013, 63, 11–30. [Google Scholar] [CrossRef] [PubMed]
- Klein, E. Prostate cancer. Urologic Oncol. 2008, 26, 494. [Google Scholar] [CrossRef] [PubMed]
- Balk, S.P.; Knudsen, K.E. AR, the cell cycle, and prostate cancer. Nucl. Recept. Signal. 2008, 6, e001. [Google Scholar] [CrossRef] [PubMed]
- Feldman, B.J.; Feldman, D. The development of androgen-independent prostate cancer. Nat. Rev. Cancer 2001, 1, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Hemberger, M.; Dean, W. Epigenetic arbitration of cell fate decisions: Tipping the bias. Dev. Cell 2007, 12, 176–178. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, I.; Otte, A.P.; Allis, C.D.; Reinberg, D.; Heard, E. Epigenetic dynamics of imprinted X inactivation during early mouse development. Science 2004, 303, 644–649. [Google Scholar] [CrossRef] [PubMed]
- Sims, R.J., 3rd; Reinberg, D. Is there a code embedded in proteins that is based on post-translational modifications? Nat. Rev. Mol. Cell Biol. 2008, 9, 815–820. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Shiina, H.; Kawano, H.; Sato, T.; Kato, S. Androgen receptor functions in male and female physiology. J. Steroid Biochem. Mol. Biol. 2008, 109, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, M.E.; Huang, H.; Tindall, D.J. Androgen receptor signaling in androgen-refractory prostate cancer. J. Natl. Cancer Inst. 2001, 93, 1687–1697. [Google Scholar] [CrossRef] [PubMed]
- Roth, S.Y.; Denu, J.M.; Allis, C.D. Histone acetyltransferases. Annu. Rev. Biochem. 2001, 70, 81–120. [Google Scholar] [CrossRef] [PubMed]
- Debes, J.D.; Comuzzi, B.; Schmidt, LJ.; Dehm, S.M.; Culig, Z.; Tindall, D.J. p300 regulates androgen receptor-independent expression of prostate-specific antigen in prostate cancer cells treated chronically with interleukin-6. Cancer Res. 2005, 65, 5965–5973. [Google Scholar] [CrossRef] [PubMed]
- Debes, J.D.; Schmidt, L.J.; Huang, H.; Tindall, D.J. p300 mediates androgen-independent transactivation of the androgen receptor by interleukin 6. Cancer Res. 2002, 62, 5632–5636. [Google Scholar] [PubMed]
- Jaganathan, A.; Chaurasia, P.; Xiao, G.Q.; Philizaire, M.; Lv, X.; Yao, S.; Burnstein, K.L.; Liu, D.P.; Levine, A.C.; Mujtaba, S. Coactivator MYST1 regulates nuclear factor-kappaB and androgen receptor functions during proliferation of prostate cancer cells. Mol. Endocrinol. 2014, 28, 872–885. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.; Pathak, R.R.; Mujtaba, S. The biology of lysine acetylation integrates transcriptional programming and metabolism. Nutr. Metab. (Lond) 2011, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Roeder, R.G. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 1997, 90, 595–606. [Google Scholar] [CrossRef]
- Fu, M.; Rao, M.; Wang, C.; Sakamaki, T.; Wang, J.; Di Vizio, D.; Zhang, X.; Albanese, C.; Balk, S.; Chang, C.; et al. Acetylation of androgen receptor enhances coactivator binding and promotes prostate cancer cell growth. Mol. Cell Biol. 2003, 23, 8563–8575. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Wang, M.; Liu, X.; Luo, L.; Li, K.; Zhang, S.; Wang, Y.; Yang, Y.; Ding, F.; Gu, X. PCAF improves glucose homeostasis by suppressing the gluconeogenic activity of PGC-1alpha. Cell Rep. 2014, 9, 2250–2262. [Google Scholar] [CrossRef] [PubMed]
- Lantier, L.; Williams, A.S.; Williams, I.M.; Yang, K.K.; Bracy, D.P.; Goelzer, M.; James, F.D.; Gius, D.; Wasserman, D.H. SIRT3 is crucial for maintaining skeletal muscle insulin action and protects against severe insulin resistance in high fat fed mice. Diabetes 2015. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.W.; Gatza, E.; Hou, G.; Sun, Y.; Whitfield, J.; Song, Y.; Oravecz-Wilson, K.; Tawara, I.; Dinarello, C.A.; Reddy, P. Histone deacetylase inhibition regulates inflammation and enhances Tregs after allogeneic hematopoietic cell transplantation in humans. Blood 2015, 125, 815–819. [Google Scholar] [CrossRef] [PubMed]
- Brady, M.E.; Ozanne, D.M.; Gaughan, L.; Waite, I.; Cook, S.; Neal, D.E.; Robson, C.N. Tip60 is a nuclear hormone receptor coactivator. J. Biol. Chem. 1999, 274, 17599–17604. [Google Scholar] [CrossRef] [PubMed]
- Gaughan, L.; Logan, I.R.; Cook, S.; Neal, D.E.; Robson, C.N. Tip60 and histone deacetylase 1 regulate androgen receptor activity through changes to the acetylation status of the receptor. J. Biol. Chem. 2002, 277, 25904–25913. [Google Scholar] [CrossRef] [PubMed]
- Halkidou, K.; Gnanapragasam, V.J.; Mehta, P.B.; Logan, I.R.; Brady, M.E.; Cook, S.; Leung, H.Y.; Neal, D.E.; Robson, C.N. Expression of Tip60, an androgen receptor coactivator, and its role in prostate cancer development. Oncogene 2003, 22, 2466–2477. [Google Scholar] [CrossRef] [PubMed]
- Shiota, M.; Yokomizo, A.; Masubuchi, D.; Tada, Y.; Inokuchi, J.; Eto, M.; Uchiumi, T.; Fujimoto, N.; Naito, S. Tip60 promotes prostate cancer cell proliferation by translocation of androgen receptor into the nucleus. Prostate 2010, 70, 540–554. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Wang, C.; Zhang, X.; Pestell, R.G. Acetylation of nuclear receptors in cellular growth and apoptosis. Biochem. Pharmacol. 2004, 68, 1199–1208. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Liu, M.; Sauve, A.A.; Jiao, X.; Zhang, X.; Wu, X.; Powell, M.J.; Yang, T.; Gu, W.; Avantaggiati, M.L.; et al. Hormonal control of androgen receptor function through SIRT1. Mol. Cell. Biol. 2006, 26, 8122–8135. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Rao, M.; Wu, K.; Wang, C.; Zhang, X.; Hessien, M.; Yeung, Y.G.; Gioeli, D.; Weber, M.J.; Pestell, R.G. The androgen receptor acetylation site regulates cAMP and AKT but not ERK-induced activity. J. Biol. Chem. 2004, 279, 29436–29449. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Wang, C.; Reutens, A.T.; Wang, J.; Angeletti, R.H.; Siconolfi-Baez, L.; Ogryzko, V.; Avantaggiati, M.L.; Pestell, R.G. p300 and p300/cAMP-response element-binding protein-associated factor acetylate the androgen receptor at sites governing hormone-dependent transactivation. J. Biol. Chem. 2000, 275, 20853–20860. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Tindall, D.J. Androgen action in prostate cancer. Horm Cancer 2010, 1, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Andriole, G.; Bruchovsky, N.; Chung, L.W.; Matsumoto, A.M.; Rittmaster, R.; Roehrborn, C.; Russell, D.; Tindall, D. Dihydrotestosterone and the prostate: The scientific rationale for 5alpha-reductase inhibitors in the treatment of benign prostatic hyperplasia. J. Urol. 2004, 172, 1399–1403. [Google Scholar] [CrossRef] [PubMed]
- Culig, Z.; Bartsch, G. Androgen axis in prostate cancer. J. Cell Biochem. 2006, 99, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Dehm, S.M.; Tindall, D.J. Regulation of androgen receptor signaling in prostate cancer. Expert. Rev. Anticancer Ther. 2005, 5, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Chmelar, R.; Buchanan, G.; Need, E.F.; Tilley, W.; Greenberg, N.M. Androgen receptor coregulators and their involvement in the development and progression of prostate cancer. Int. J. Cancer 2007, 120, 719–733. [Google Scholar] [CrossRef] [PubMed]
- Mader, S.; Chambon, P.; White, J.H. Defining a minimal estrogen receptor DNA binding domain. Nucleic Acids Res. 1993, 21, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Schoenmakers, E.; Alen, P.; Verrijdt, G.; Peeters, B.; Verhoeven, G.; Rombauts, W.; Claessens, F. Differential DNA binding by the androgen and glucocorticoid receptors involves the second Zn-finger and a C-terminal extension of the DNA-binding domains. Biochem. J. 1999, 341, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Hard, T.; Kellenbach, E.; Boelens, R.; Maler, B.A.; Dahlman, K.; Freedman, L.P.; Carlstedt-Duke, J.; Yamamoto, K.R.; Gustafsson, J.A.; Kaptein, R. Solution structure of the glucocorticoid receptor DNA-binding domain. Science 1990, 249, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Gewirth, D.T.; Sigler, P.B. The basis for half-site specificity explored through a non-cognate steroid receptor-DNA complex. Nat. Struct. Biol. 1995, 2, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Dahlman-Wright, K.; Grandien, K.; Nilsson, S.; Gustafsson, J.A.; Carlstedt-Duke, J. Protein-protein interactions between the DNA-binding domains of nuclear receptors: Influence on DNA-binding. J. Steroid. Biochem. Mol. Biol. 1993, 45, 239–250. [Google Scholar] [CrossRef]
- Tanner, T.; Claessens, F.; Haelens, A. The hinge region of the androgen receptor plays a role in proteasome-mediated transcriptional activation. Ann. N. Y. Acad. Sci. 2004, 1030, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Saporita, A.J.; Zhang, Q.; Navai, N.; Dincer, Z.; Hahn, J.; Cai, X.; Wang, Z. Identification and characterization of a ligand-regulated nuclear export signal in androgen receptor. J. Biol. Chem. 2003, 278, 41998–42005. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.X.; Kemppainen, J.A.; Wilson, E.M. Identification of three proline-directed phosphorylation sites in the human androgen receptor. Mol. Endocrinol. 1995, 9, 605–615. [Google Scholar] [PubMed]
- Gioeli, D.; Ficarro, S.B.; Kwiek, J.J.; Aaronson, D.; Hancock, M.; Catling, A.D.; White, F.M.; Christian, R.E.; Settlage, R.E.; Shabanowitz, J.; et al. Androgen receptor phosphorylation. regulation and identification of the phosphorylation sites. J. Biol. Chem. 2002, 277, 29304–29314. [Google Scholar] [CrossRef] [PubMed]
- Matias, P.M.; Ficarro, S.B.; Kwiek, J.J.; Aaronson, D.; Hancock, M.; Catling, A.D.; White, F.M.; Christian, R.E.; Settlage, R.E.; Shabanowitz, J.; et al. Structural evidence for ligand specificity in the binding domain of the human androgen receptor. Implications for pathogenic gene mutations. J. Biol. Chem. 2000, 275, 26164–26171. [Google Scholar] [CrossRef] [PubMed]
- Sack, J.S.; Kish, K.F.; Wang, C.; Attar, R.M.; Kiefer, S.E.; An, Y.; Wu, G.Y.; Scheffler, J.E.; Salvati, M.E.; Krystek, S.R., Jr.; et al. Crystallographic structures of the ligand-binding domains of the androgen receptor and its T877A mutant complexed with the natural agonist dihydrotestosterone. Proc. Natl. Acad. Sci. USA 2001, 98, 4904–4909. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Gampe, R.T., Jr.; Kole, A.J.; Hnat, A.T.; Stanley, T.B.; An, G.; Stewart, E.L.; Kalman, R.I.; Minges, J.T.; Wilson, E.M. Structural basis for androgen receptor interdomain and coactivator interactions suggests a transition in nuclear receptor activation function dominance. Mol. Cell 2004, 16, 425–438. [Google Scholar] [CrossRef] [PubMed]
- Moreland, J.L.; Gramada, A.; Buzko, O.V.; Zhang, Q.; Bourne, P.E. The Molecular Biology Toolkit (MBT): A modular platform for developing molecular visualization applications. BMC Bioinform. 2005, 6, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tora, L.; White, J.; Brou, C.; Tasset, D.; Webster, N.; Scheer, E.; Plerre Chambon, P. The human estrogen receptor has two independent nonacidic transcriptional activation functions. Cell 1989, 59, 477–487. [Google Scholar] [CrossRef]
- Bevan, C.L.; Hoare, S.; Claessens, F.; Heery, D.M.; Parker, M.G. The AF1 and AF2 domains of the androgen receptor interact with distinct regions of SRC1. Mol. Cell. Biol. 1999, 19, 8383–8392. [Google Scholar] [PubMed]
- Dehm, S.M.; Tindall, D.J. Ligand-independent androgen receptor activity is activation function-2-independent and resistant to antiandrogens in androgen refractory prostate cancer cells. J. Biol. Chem. 2006, 281, 27882–27893. [Google Scholar] [CrossRef] [PubMed]
- Steers, W.D. 5alpha-reductase activity in the prostate. Urology 2001, 58, 17–24. [Google Scholar] [CrossRef]
- Navarro, D.; Luzardo, O.P.; Fernandez, L.; Chesa, N.; Diaz-Chico, B.N. Transition to androgen-independence in prostate cancer. J. Steroid Biochem. Mol. Biol. 2002, 81, 191–201. [Google Scholar] [CrossRef]
- Heinlein, C.A.; Chang, C. Androgen receptor in prostate cancer. Endocr. Rev. 2004, 25, 276–308. [Google Scholar] [CrossRef] [PubMed]
- Claessens, F.; Verrijdta, G.; Schoenmakersa, E.; Haelensa, A.; Peetersa, B.; Verhoevenb, G.; Rombautsa, W. Selective DNA binding by the androgen receptor as a mechanism for hormone-specific gene regulation. J. Steroid Biochem. Mol. Biol. 2001, 76, 23–30. [Google Scholar] [CrossRef]
- Khorasanizadeh, S.; Rastinejad, F. Nuclear-receptor interactions on DNA-response elements. Trends Biochem. Sci. 2001, 26, 384–390. [Google Scholar] [CrossRef]
- Glass, C.K.; Rosenfeld, M.G. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 2000, 14, 121–141. [Google Scholar] [PubMed]
- Rosenfeld, M.G.; Lunyak, V.V.; Glass, C.K. Sensors and signals: A coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev. 2006, 20, 1405–1428. [Google Scholar] [CrossRef] [PubMed]
- Van Royen, M.E.; van Cappellen, W.A.; de Vos, C.; Houtsmuller, A.B.; Trapman, J. Stepwise androgen receptor dimerization. J. Cell Sci. 2012, 125, 1970–1979. [Google Scholar] [CrossRef] [PubMed]
- Abate-Shen, C. Deregulated homeobox gene expression in cancer: Cause or consequence? Nat. Rev. Cancer 2002, 2, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Bouras, T.; Fu, M.; Sauve, A.A.; Wang, F.; Quong, A.A.; Perkins, N.D.; Hay, R.T.; Gu, W.; Pestell, R.G. SIRT1 deacetylation and repression of p300 involves lysine residues 1020/1024 within the cell cycle regulatory domain 1. J. Biol. Chem. 2005, 280, 10264–10276. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Rao, M.; Bouras, T.; Wang, C.; Wu, K.; Zhang, X.; Li, Z.; Yao, T.P.; Pestell, R.G. Cyclin D1 inhibits peroxisome proliferator-activated receptor gamma-mediated adipogenesis through histone deacetylase recruitment. J. Biol. Chem. 2005, 280, 16934–16941. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Wang, C.; Rao, M.; Wu, X.; Bouras, T.; Zhang, X.; Li, Z.; Jiao, X.; Yang, J.; Li, A.; et al. Cyclin D1 represses p300 transactivation through a cyclin-dependent kinase-independent mechanism. J. Biol. Chem. 2005, 280, 29728–29742. [Google Scholar] [CrossRef] [PubMed]
- Fulco, M.; Schiltz, R.L.; Iezzi, S.; King, M.T.; Zhao, P.; Kashiwaya, Y.; Hoffman, E.; Veech, R.L.; Sartorelli, V. Sir2 regulates skeletal muscle differentiation as a potential sensor of the redox state. Mol. Cell 2003, 12, 51–62. [Google Scholar] [CrossRef]
- Henshall, S.M.; Quinn, D.I.; Lee, C.S.; Head, D.R.; Golovsky, D.; Brenner, P.C.; Delprado, W.; Stricker, P.D.; Grygiel, J.J.; Sutherland, R.L. Altered expression of androgen receptor in the malignant epithelium and adjacent stroma is associated with early relapse in prostate cancer. Cancer Res. 2001, 61, 423–427. [Google Scholar] [PubMed]
- Chodak, G.W.; Kranc, D.M.; Puy, L.A.; Takeda, H.; Johnson, K.; Chang, C. Nuclear localization of androgen receptor in heterogeneous samples of normal, hyperplastic and neoplastic human prostate. J. Urol. 1992, 147, 798–803. [Google Scholar] [PubMed]
- Ruizeveld de Winter, J.A.; Janssen, P.J.; Sleddens, H.M.; Verleun-Mooijman, M.C.; Trapman, J.; Brinkmann, A.O.; Santerse, A.B.; Schröder, F.H.; van der Kwast, T.H. Androgen receptor status in localized and locally progressive hormone refractory human prostate cancer. Am. J. Pathol. 1994, 144, 735–746. [Google Scholar] [PubMed]
- Sadi, M.V.; Barrack, E.R. Androgen receptors and growth fraction in metastatic prostate cancer as predictors of time to tumour progression after hormonal therapy. Cancer Surv. 1991, 11, 195–215. [Google Scholar] [PubMed]
- Van der Kwast, T.H.; Schalken, J.; Ruizeveld de Winter, J.A.; van Vroonhoven, C.C.; Mulder, E.; Boersma, W.; Trapman, J. Androgen receptors in endocrine-therapy-resistant human prostate cancer. Int. J. Cancer 1991, 48, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Trapman, J.; Cleutjens, K.B. Androgen-regulated gene expression in prostate cancer. Semin. Cancer Biol. 1997, 8, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Cleutjens, K.B.; van Eekelen, C.C.; van der Korput, H.A.; Brinkmann, A.O.; Trapman, J. Two androgen response regions cooperate in steroid hormone regulated activity of the prostate-specific antigen promoter. J. Biol. Chem. 1996, 271, 6379–6388. [Google Scholar] [PubMed]
- Luk, S.U.; Xue, H.; Cheng, H.; Lin, D.; Gout, P.W.; Fazli, L.; Collins, C.C.; Gleave, M.E.; Wang, Y. The BIRC6 gene as a novel target for therapy of prostate cancer: Dual targeting of inhibitors of apoptosis. Oncotarget 2014, 5, 6896–6908. [Google Scholar] [PubMed]
- Paule, B.; Herelle, M.O.; Rage, E.; Ducreux, M.; Adam, R.; Guettier, C.; Bralet, M.P. Cetuximab plus gemcitabine-oxaliplatin (GEMOX) in patients with refractory advanced intrahepatic cholangiocarcinomas. Oncology 2007, 72, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Paule, B.; Terry, S.; Kheuang, L.; Soyeux, P.; Vacherot, F.; de la Taille, A. The NF-kappaB/IL-6 pathway in metastatic androgen-independent prostate cancer: New therapeutic approaches? World J. Urol. 2007, 25, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.C.; Hobisch, A.; Lin, D.L.; Culig, Z.; Keller, E.T. Interleukin-6 and prostate cancer progression. Cytokine Growth Factor Rev. 2001, 12, 33–40. [Google Scholar] [CrossRef]
- Smith, P.C.; Keller, E.T. Anti-interleukin-6 monoclonal antibody induces regression of human prostate cancer xenografts in nude mice. Prostate 2001, 48, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Mellado, B.; Codony, J.; Ribal, M.J.; Visa, L.; Gascon, P. Molecular biology of androgen-independent prostate cancer: The role of the androgen receptor pathway. Clin. Transl. Oncol. 2009, 11, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Roy-Burman, P.; Tindall, D.J.; Robins, D.M.; Greenberg, N.M.; Hendrix, M.J.; Mohla, S.; Getzenberg, R.H.; Isaacs, J.T.; Pienta, K.J. Androgens and prostate cancer: Are the descriptors valid? Cancer Biol. Ther. 2005, 4, 4–5. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, G.; Irvine, R.A.; Coetzee, G.A.; Tilley, W.D. Contribution of the androgen receptor to prostate cancer predisposition and progression. Cancer Metastasis Rev. 2001, 20, 207–223. [Google Scholar] [CrossRef] [PubMed]
- Eder, I.E.; Culig, Z.; Ramoner, R.; Thurnher, M.; Putz, T.; Nessler-Menardi, C.; Tiefenthaler, M.; Bartsch, G.; Klocker, H. Inhibition of LncaP prostate cancer cells by means of androgen receptor antisense oligonucleotides. Cancer Gene Ther. 2000, 7, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Haag, P.; Bektic, J.; Bartsch, G.; Klocker, H.; Eder, I.E. Androgen receptor down regulation by small interference RNA induces cell growth inhibition in androgen sensitive as well as in androgen independent prostate cancer cells. J. Steroid Biochem. Mol. Biol. 2005, 96, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Tang, S.; Thrasher, J.B.; Griebling, T.L.; Li, B. Small-interfering RNA-induced androgen receptor silencing leads to apoptotic cell death in prostate cancer. Mol. Cancer Ther. 2005, 4, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Zegarra-Moro, O.L.; Schmidt, L.J.; Huang, H.; Tindall, D.J. Disruption of androgen receptor function inhibits proliferation of androgen-refractory prostate cancer cells. Cancer Res. 2002, 62, 1008–1013. [Google Scholar] [PubMed]
- Zhu, B.; Kyprianou, N. Transforming growth factor beta and prostate cancer. Cancer Treat Res. 2005, 126, 157–173. [Google Scholar] [PubMed]
- Memarzadeh, S.; Xin, L.; Mulholland, D.J.; Mansukhani, A.; Wu, H.; Teitell, M.A.; Witte, O.N. Enhanced paracrine FGF10 expression promotes formation of multifocal prostate adenocarcinoma and an increase in epithelial androgen receptor. Cancer Cell 2007, 12, 572–585. [Google Scholar] [CrossRef] [PubMed]
- Rosini, P.; Bonaccorsi, L.; Baldi, E.; Chiasserini, C.; Forti, G.; de Chiara, G.; Lucibello, M.; Mongiat, M.; Iozzo, R.V.; Garaci, E.; et al. Androgen receptor expression induces FGF2, FGF-binding protein production, and FGF2 release in prostate carcinoma cells: Role of FGF2 in growth, survival, and androgen receptor down-modulation. Prostate 2002, 53, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Boddy, J.L.; Fox, S.B.; Han, C.; Campo, L.; Turley, H.; Kanga, S.; Malone, P.R.; Harris, A.L. The androgen receptor is significantly associated with vascular endothelial growth factor and hypoxia sensing via hypoxia-inducible factors HIF-1a, HIF-2a, and the prolyl hydroxylases in human prostate cancer. Clin. Cancer Res. 2005, 11, 7658–7663. [Google Scholar] [CrossRef] [PubMed]
- Vellaichamy, A.; Dezső, Z.; JeBailey, L.; Chinnaiyan, A.M.; Sreekumar, A.; Nesvizhskii, A.I.; Omenn, G.S.; Bugrim, A. “Topological significance” analysis of gene expression and proteomic profiles from prostate cancer cells reveals key mechanisms of androgen response. PLoS ONE 2010, 5, e10936. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Duncan, D.; Shi, Z.; Zhang, B. WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): Update 2013. Nucleic Acids Res. 2013, 41, W77–W83. [Google Scholar] [CrossRef] [PubMed]
- Saraon, P.; Musrap, N.; Cretu, D.; Karagiannis, G.S.; Batruch, I.; Smith, C.; Drabovich, A.P.; Trudel, D.; van der Kwast, T.; Morrissey, C.; et al. Proteomic profiling of androgen-independent prostate cancer cell lines reveals a role for protein S during the development of high grade and castration-resistant prostate cancer. J. Biol. Chem. 2012, 287, 34019–34031. [Google Scholar] [CrossRef] [PubMed]
- Djavan, B.; Nasu, Y. Prostate cancer gene therapy-what have we learned and where are we going? Rev. Urol. 2001, 3, 179–186. [Google Scholar] [PubMed]
- Harris, K.A.; Reese, D.M. Treatment options in hormone-refractory prostate cancer: Current and future approaches. Drugs 2001, 61, 2177–2192. [Google Scholar] [CrossRef] [PubMed]
- Wright, M.E.; Tsai, M.J.; Aebersold, R. Androgen receptor represses the neuroendocrine transdifferentiation process in prostate cancer cells. Mol. Endocrinol. 2003, 17, 1726–1737. [Google Scholar] [CrossRef] [PubMed]
- Murillo, H.; Huang, H.; Schmidt, L.J.; Smith, D.I.; Tindall, D.J. Role of PI3K signaling in survival and progression of LNCaP prostate cancer cells to the androgen refractory state. Endocrinology 2001, 142, 4795–4805. [Google Scholar] [CrossRef] [PubMed]
- Graff, J.R.; Konicek, B.W.; McNulty, A.M.; Wang, Z.; Houck, K.; Allen, S.; Paul, J.D.; Hbaiu, A.; Goode, R.G.; Sandusky, G.E.; et al. Increased AKT activity contributes to prostate cancer progression by dramatically accelerating prostate tumor growth and diminishing p27Kip1 expression. J. Biol. Chem. 2000, 275, 24500–24505. [Google Scholar] [CrossRef] [PubMed]
- Colombel, M.; Symmans, F.; Gil, S.; O’Toole, K.M.; Chopin, D.; Benson, M.; Olsson, C.A.; Korsmeyer, S.; Buttyan, R. Detection of the apoptosis-suppressing oncoprotein bc1-2 in hormone-refractory human prostate cancers. Am. J. Pathol. 1993, 143, 390–400. [Google Scholar] [PubMed]
- Krajewska, M.; Krajewski, S.; Epstein, J.I.; Shabaik, A.; Sauvageot, J.; Song, K.; Kitada, S.; Reed, J.C. Immunohistochemical analysis of bcl-2, bax, bcl-X, and mcl-1 expression in prostate cancers. Am. J. Pathol. 1996, 148, 1567–1576. [Google Scholar] [PubMed]
- McDonnell, T.J.; Troncoso, P.; Brisbay, S.M.; Logothetis, C.; Chung, L.W.; Hsieh, J.T.; Tu, S.M.; Campbell, M.L. Expression of the protooncogene bcl-2 in the prostate and its association with emergence of androgen-independent prostate cancer. Cancer Res. 1992, 52, 6940–6944. [Google Scholar] [PubMed]
- Isaacs, J.T. The biology of hormone refractory prostate cancer. Why does it develop? Urol. Clin. North. Am. 1999, 26, 263–273. [Google Scholar] [CrossRef]
- Huang, H.; Cheville, J.C.; Pan, Y.; Roche, P.C.; Schmidt, L.J.; Tindall, D.J. PTEN induces chemosensitivity in PTEN-mutated prostate cancer cells by suppression of Bcl-2 expression. J. Biol. Chem. 2001, 276, 38830–38836. [Google Scholar] [CrossRef] [PubMed]
- Schwer, B.; Bunkenborg, J.; Verdin, R.O.; Andersen, J.S.; Verdin, E. Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2. Proc. Natl. Acad. Sci. USA 2006, 103, 10224–10229. [Google Scholar] [CrossRef] [PubMed]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Sykes, S.M.; Mellert, H.S.; Holbert, M.A.; Li, K.; Marmorstein, R.; Lane, W.S.; McMahon, S.B. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol. Cell 2006, 24, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, Y.; Yang, C.; Xiong, H.; Lin, Y.; Yao, J.; Li, H.; Xie, L.; Zhao, W.; Yao, Y.; et al. Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science 2010, 327, 1004–1007. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.J.; Seto, E. Lysine acetylation: Codified crosstalk with other posttranslational modifications. Mol. Cell 2008, 31, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Verdin, E.; Ott, M. 50 years of protein acetylation: From gene regulation to epigenetics, metabolism and beyond. Nat. Rev. Mol. Cell Biol. 2015, 16, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Richon, V.M.; Emiliani, S.; Verdin, E.; Webb, Y.; Breslow, R.; Rifkind, R.A.; Marks, P.A. A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases. Proc. Natl. Acad. Sci. USA 1998, 95, 3003–3007. [Google Scholar] [CrossRef] [PubMed]
- Borah, J.C.; Mujtaba, S.; Karakikes, I.; Zeng, L.; Muller, M.; Patel, J.; Moshkina, N.; Morohashi, K.; Zhang, W.; Gerona-Navarro, G.; et al. A small molecule binding to the coactivator CREB-binding protein blocks apoptosis in cardiomyocytes. Chem. Biol. 2011, 18, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Mujtaba, S.; Zhou, M.M. Anti-viral opportunities during transcriptional activation of latent HIV in the host chromatin. Methods 2010, 53, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef] [PubMed]
- Croft, D.; Mundo, A.F.; Haw, R.; Milacic, M.; Weiser, J.; Wu, G.; Caudy, M.; Garapati, P.; Gillespie, M.; Kamdar, M.R.; et al. The Reactome pathway knowledgebase. Nucleic Acids Res. 2014, 42, D472–D477. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Demchak, B.; Ideker, T. Cytoscape tools for the web age: D3.js and Cytoscape.js exporters. F1000 Res. 2014, 3, 143. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pathak, R.; Philizaire, M.; Mujtaba, S. Dichotomy in the Epigenetic Mark Lysine Acetylation is Critical for the Proliferation of Prostate Cancer Cells. Cancers 2015, 7, 1622-1642. https://doi.org/10.3390/cancers7030854
Pathak R, Philizaire M, Mujtaba S. Dichotomy in the Epigenetic Mark Lysine Acetylation is Critical for the Proliferation of Prostate Cancer Cells. Cancers. 2015; 7(3):1622-1642. https://doi.org/10.3390/cancers7030854
Chicago/Turabian StylePathak, Ravi, Marc Philizaire, and Shiraz Mujtaba. 2015. "Dichotomy in the Epigenetic Mark Lysine Acetylation is Critical for the Proliferation of Prostate Cancer Cells" Cancers 7, no. 3: 1622-1642. https://doi.org/10.3390/cancers7030854
APA StylePathak, R., Philizaire, M., & Mujtaba, S. (2015). Dichotomy in the Epigenetic Mark Lysine Acetylation is Critical for the Proliferation of Prostate Cancer Cells. Cancers, 7(3), 1622-1642. https://doi.org/10.3390/cancers7030854