
Hepatocyte Growth Factor from a Clinical Perspective: A Pancreatic Cancer Challenge


Abstract
:1. Introduction
2. HGF-the Good, the Bad and the Ugly

3. HGF and Desmoplasia
4. HGF and Hypoxia
5. HGF and Acidic Environment
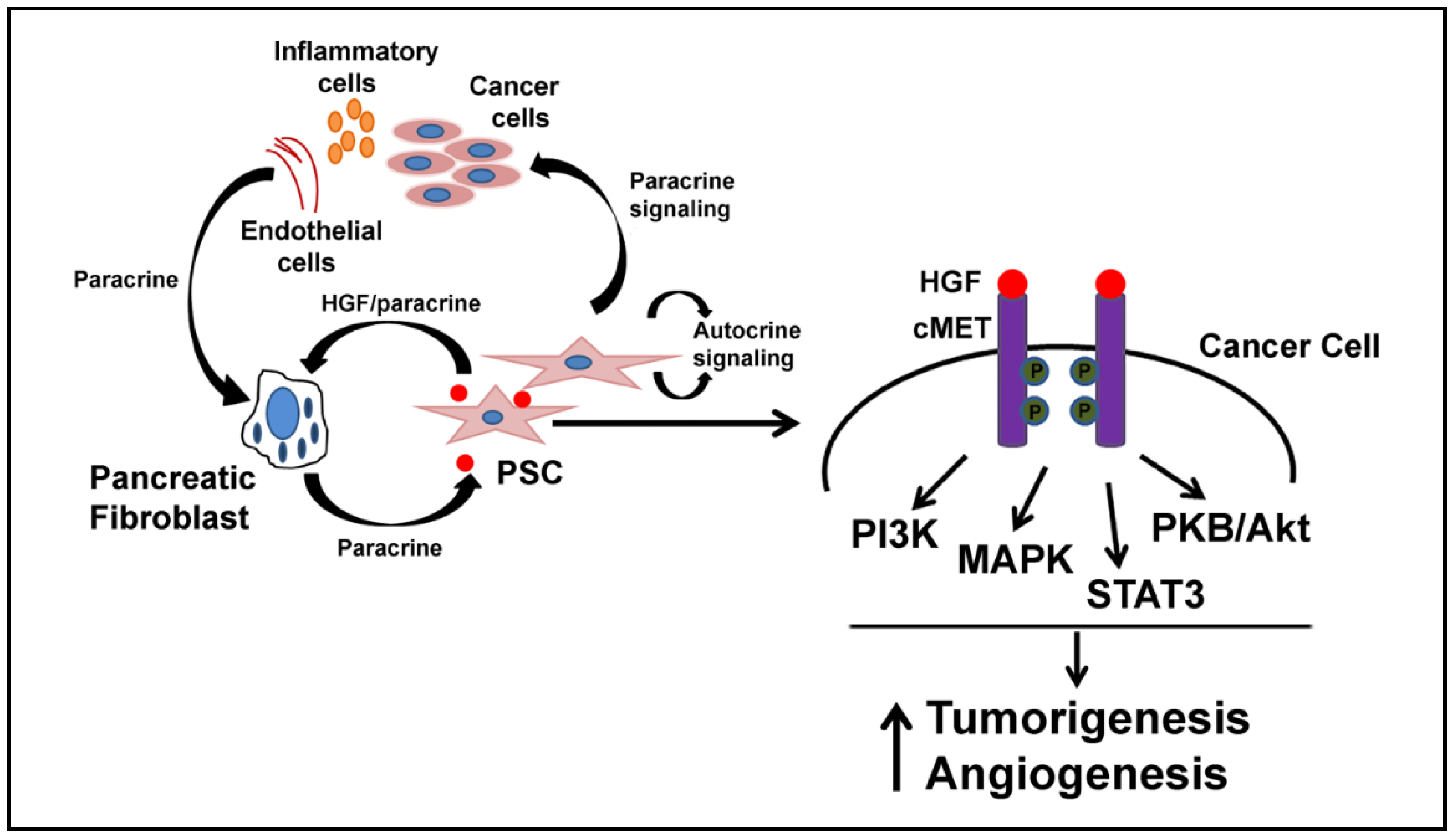
6. HGF and Inflammation
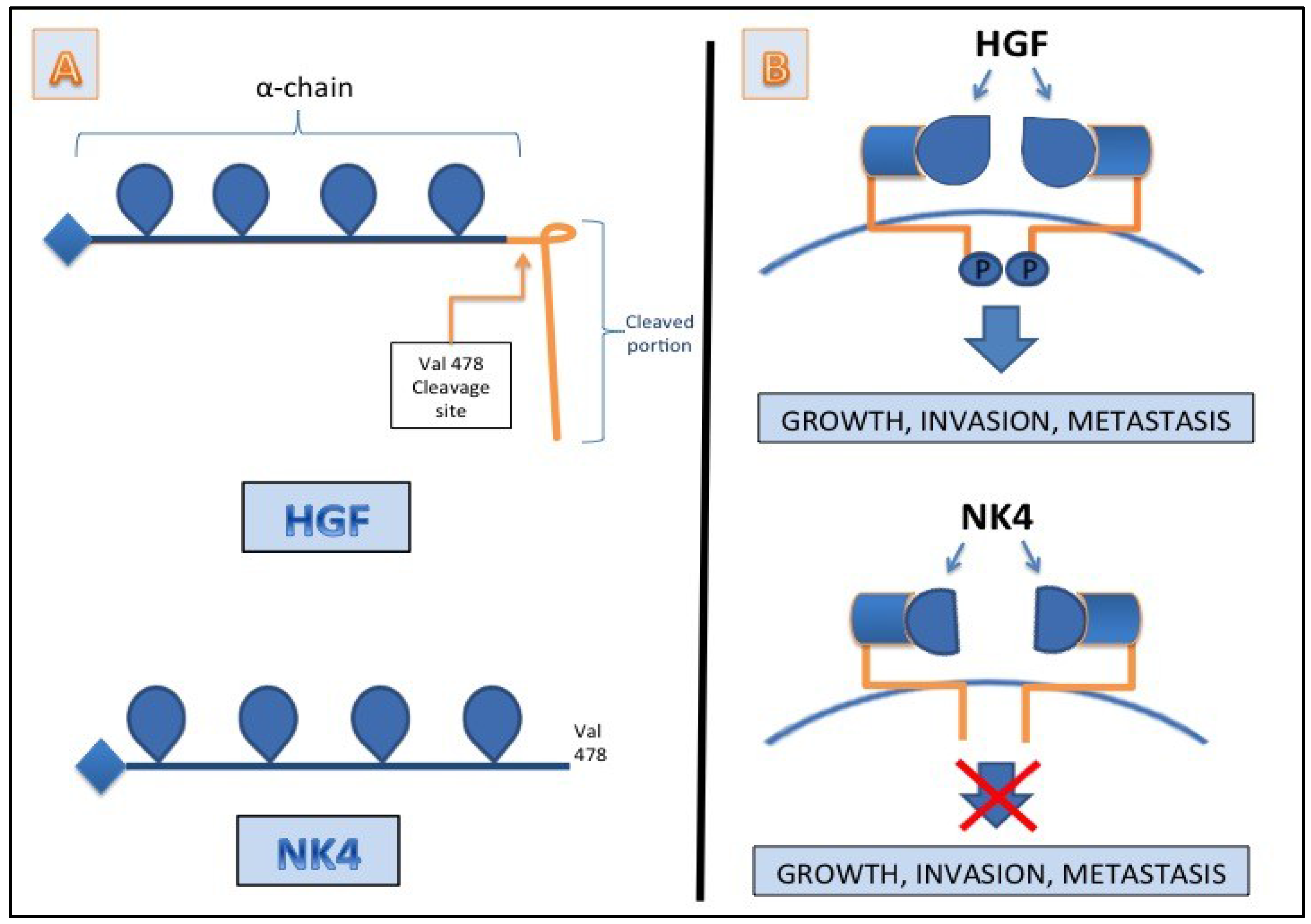
7. HGF/c-MET as a Target

{kind=link}
{kind=link}
| Category | Drug Name | Trial Phase | Target Neoplasm | Median Progression Free Survival | Side Effects | Conclusions | Source |
|---|---|---|---|---|---|---|---|
| HGF/SF Mab | AMG102 (Rilotumumab) +Bevacizumab | II | Renal cell carcinoma | 3.7 months at 10 mg/kg and 2 months at 20 mg/kg | Edema (45.9%) | AMG102 is tolerated, but not definitively growth inhibitory | Schoffski et al. [104] |
| Fatigue (37.7%) | |||||||
| Nausea(27.9%) | |||||||
| HGF/SF Mab | AMG 102 (Rilotumumab) vs. AMG 102 after previous Bevacizumab Therapy | II | Recurrent Glioblastoma | [AMG102 only] 4.1 weeks vs. [previous Bevacizumab] 4.3 weeks | Fatigue (38%), | AMG 102 monotherapy not associated with statistically significant anti-tumor activity | Wen et al. [105] |
| Headache (33%) | |||||||
| Peripheral Edema (23%). | |||||||
| HGF/SF Mab | AMG 102 (Rilotumumab) plus mitoxantrone and prednisone | II | Castration Resistant Prostate Cancer | 3.0 months [AMG 102] vs. 2.9 months [control] | Pulmonary Embolism (6%) | Addition of AMG 102 showed no efficacy improvements | Ryan et al. [106] |
| Fatigue (3%) | |||||||
| Met Kinase Inhibitor | Tivantinib plus Erlotinib versus Placebo plus Erlotinib | III | Nonsquamous, Non-Small-Cell Lung Cancer | 3.8 months [Erlotinib + Tivantinib] vs. 2.3 months for Erlotinib+Placebo | Rash | Addition of Tivantinib showed a significant delay in metastasis when compared to Erlotinib alone | Scagliotti et al. [107] |
| Diarrhea | |||||||
| Fatigue | |||||||
| Vomiting | |||||||
| Dyspnea | |||||||
| Met Kinase Inhibitor | Tivantinib vs. Placebo | II | Hepatocellular Carcinoma | 1–6 months [Titantivib] vs. 1–4 months [Placebo] | Neutropenia (14%) | Beneficial second line treatment for c-MET-high advanced HCC. | Santoro et al. [108] |
| Anemia (11%) | |||||||
| Met Kinase Inhibitor | Tivantinib | II | Microphthalmia transcription factor (MITF)-associated (MiT) tumors | 3.6 months [overall] vs. 5.5 months [ASPS] vs. 1.9 months [CCS and tRCC] | Anemia (4%) | Safe and tolerable at doses of 360mg BID, with moderate antitumor response | Wagner et al. [109] |
| Neutropenia (4%). | |||||||
| Thrombocytopenia | |||||||
| Deep vein thrombosis (6.4%) | |||||||
| Met Kinase Inhibitor | PF-02341066 (Crizotinib) vs. Pemetrexed or Docetaxel | III | ALK+ Non-Small Cell Lung Cancer | 7.7 months [crizotinib] vs. 3.0 months [control] | Visual disorder | Crizotinib is superior to standard chemotherapy in terms of progression free survival, symptomology, and quality of life | Shaw et al. [110] |
| GI SE | |||||||
| Elevated liver aminotransferase levels | |||||||
| Met Kinase Inhibitor | Cabozantinib | III | Medullary Thyroid Carcinoma | 11.2 months [cabozantinib] vs. 4.0 months [placebo] | Diarrhea | Cabozantinib resulted in statistically significant increased progression free survival length of time. | Elisei et al. [111] |
| Palmar-plantar erythrodysesthesia Decreased weight and appetite Nausea | |||||||
| Fatigue | |||||||
| Met Kinase Inhibitor | Foretinib | II | Papillary Renal Cell Carcinoma | 9.3 months [Foretinib] vs. 1.3 months [Sunitinib] | Fatigue, Hypertension, Gastrointestinal toxicities | Foretinib demonstrated a high response rate in cancers with known germline MET mutations | Choueiri et al. [112] |
| Pulmonary Emboli. | |||||||
| Met Kinase Inhibitor | Foretinib | II | Gastric Cancer | 1.7 months vs. [no comparison] | Hypertension (35%) | Foretinib is an insufficient monotherapy in the treatment of gastric cancer | Shah et al. [113] |
| Elevated Aspartate Aminotransferase (23%) |

8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Howe, H.L.; Wu, X.; Ries, L.A.; Cokkinides, V.; Ahmed, F.; Jemal, A.; Miller, B.; Williams, M.; Ward, E.; Wingo, P.A.; et al. Annual report to the nation on the status of cancer, 1975–2003, featuring cancer among U.S. Hispanic/Latino populations. Cancer 2006, 107, 1711–1742. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Nawa, K.; Ichihara, A. Partial purification and characterization of hepatocyte growth factor from serum of hepatectomized rats. Biochem. Biophys. Res. Commun. 1984, 122, 1450–1459. [Google Scholar] [CrossRef]
- Nakamura, T.; Nishizawa, T.; Hagiya, M.; Seki, T.; Shimonishi, M.; Sugimura, A.; Tashiro, K.; Shimizu, S. Molecular cloning and expression of human hepatocyte growth factor. Nature 1989, 342, 440–443. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T. Structure and function of hepatocyte growth factor. Prog. Growth Factor. Res. 1991, 3, 67–85. [Google Scholar] [CrossRef]
- Gherardi, E.; Birchmeier, W.; Birchmeier, C.; vande Woude, G. Targeting MET in cancer: rationale and progress. Nat. Rev. Cancer 2012, 12, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Mizuno, S. The discovery of hepatocyte growth factor (HGF) and its significance for cell biology, life sciences and clinical medicine. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2010, 86, 588–610. [Google Scholar] [CrossRef] [PubMed]
- Ohmichi, H.; Koshimizu, U.; Matsumoto, K.; Nakamura, T. Hepatocyte growth factor (HGF) acts as a mesenchyme-derived morphogenic factor during fetal lung development. Development 1998, 125, 1315–1324. [Google Scholar] [PubMed]
- Noji, S.; Tashiro, K.; Koyama, E.; Nohno, T.; Ohyama, K.; Taniguchi, S.; Nakamura, T. Expression of hepatocyte growth factor gene in endothelial and Kupffer cells of damaged rat livers, as revealed by in situ hybridization. Biochem. Biophys. Res. Commun. 1990, 173, 42–47. [Google Scholar] [CrossRef]
- Yoshida, S.; Yamaguchi, Y.; Itami, S.; Yoshikawa, K.; Tabata, Y.; Matsumoto, K.; Nakamura, T. Neutralization of hepatocyte growth factor leads to retarded cutaneous wound healing associated with decreased neovascularization and granulation tissue formation. J. Invest. Dermatol. 2003, 120, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Hisanaga, M.; Nakajima, Y.; Mizuno, S.; Matsumoto, K.; Nakamura, T.; Nakano, H. Enhanced expression of hepatocyte growth factor by pulmonary ischemia-reperfusion injury in the rat. Am. J. Respir. Crit. Care Med. 2000, 162, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Morishita, R.; Hayashi, S.; Matsushita, H.; Nakagami, H.; Moriguchi, A.; Matsumoto, K.; Nakamura, T.; Kaneda, Y.; Ogihara, T. Contribution of Bcl-2, but not Bcl-xL and Bax, to antiapoptotic actions of hepatocyte growth factor in hypoxia-conditioned human endothelial cells. Hypertension 2001, 37, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Okunishi, K.; Dohi, M.; Nakagome, K.; Tanaka, R.; Mizuno, S.; Matsumoto, K.; Miyazaki, J.; Nakamura, T.; Yamamoto, K. A novel role of hepatocyte growth factor as an immune regulator through suppressing dendritic cell function. J. Immunol. 2005, 175, 4745–4753. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.M.; Delous, M.; Bosch, J.A.; Ye, L.; Robertson, M.A.; Hesselson, D.; Stainier, D.Y. Hepatocyte growth factor signaling in intrapancreatic ductal cells drives pancreatic morphogenesis. PLoS Genet. 2013, 9, e1003650. [Google Scholar] [CrossRef] [PubMed]
- Delitto, D.; Vertes-George, E.; Hughes, S.J.; Behrns, K.E.; Trevino, J.G. C-Met signaling in the development of tumorigenesis and chemoresistance: potential applications in pancreatic cancer. World J. Gastroenterol. 2014, 20, 8458–8470. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, A.; Gotze, T.; Korc, M. Hepatocyte growth factor-mediated cell invasion in pancreatic cancer cells is dependent on neuropilin-1. Cancer Res. 2007, 67, 10309–10316. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.W.; Mizumoto, K.; Maehara, N.; Ohuchida, K.; Inadome, N.; Saimura, M.; Nagai, E.; Matsumoto, K.; Nakamura, T.; Tanaka, M. Co-cultivation of pancreatic cancer cells with orthotopic tumor-derived fibroblasts: fibroblasts stimulate tumor cell invasion via HGF secretion whereas cancer cells exert a minor regulative effect on fibroblasts HGF production. Cancer Lett. 2003, 190, 105–112. [Google Scholar] [CrossRef]
- Watanabe, S.; Kishimoto, T.; Yokosuka, O. Hepatocyte growth factor inhibits anoikis of pancreatic carcinoma cells through phosphatidylinositol 3-kinase pathway. Pancreas 2011, 40, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wientjes, M.G.; Au, J.L. Pancreatic cancer: pathobiology, treatment options, and drug delivery. AAPS J. 2010, 12, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Neesse, A.; Michl, P.; Frese, K.K.; Feig, C.; Cook, N.; Jacobetz, M.A.; Lolkema, M.P.; Buchholz, M.; Olive, K.P.; Gress, T.M.; et al. Stromal biology and therapy in pancreatic cancer. Gut 2011, 60, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Michl, P.; Gress, T.M. Current concepts and novel targets in advanced pancreatic cancer. Gut 2013, 62, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Farrow, B.; Evers, B.M. Inflammation and the development of pancreatic cancer. Surg. Oncol. 2002, 10, 153–169. [Google Scholar] [CrossRef]
- Luttges, J.; Kloppel, G. Pancreatic ductal adenocarcinoma and its precursors. Der Pathologe 2005, 26, 12–17. [Google Scholar] [PubMed]
- Yen, T.W.; Aardal, N.P.; Bronner, M.P.; Thorning, D.R.; Savard, C.E.; Lee, S.P.; Bell, R.H., Jr. Myofibroblasts are responsible for the desmoplastic reaction surrounding human pancreatic carcinomas. Surgery 2002, 131, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Whatcott, C.J.; Posner, R.G.; von Hoff, D.D.; Han, H. Desmoplasia and chemoresistance in pancreatic cancer. In Pancreatic Cancer and Tumor Microenvironment; Grippo, P.J., Munshi, H.G., Eds.; Transworld Research Network: Trivandrum, India, 2012. [Google Scholar]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Mueller, M.M.; Fusenig, N.E. Friends or foes—bipolar effects of the tumour stroma in cancer. Nat. Rev. Cancer 2004, 4, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349–363. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. Stromal fibroblasts in cancer initiation and progression. Nature 2004, 432, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Lin, J.E.; Chervoneva, I.; Schulz, S.; Waldman, S.A.; Pitari, G.M. Homeostatic control of the crypt-villus axis by the bacterial enterotoxin receptor guanylyl cyclase C restricts the proliferating compartment in intestine. Am. J. Pathol. 2007, 171, 1847–1858. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.E.; Li, P.; Snook, A.E.; Schulz, S.; Dasgupta, A.; Hyslop, T.M.; Gibbons, A.V.; Marszlowicz, G.; Pitari, G.M.; Waldman, S.A. The hormone receptor GUCY2C suppresses intestinal tumor formation by inhibiting AKT signaling. Gastroenterology 2010, 138, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Pitari, G.M.; di Guglielmo, M.D.; Park, J.; Schulz, S.; Waldman, S.A. Guanylyl cyclase C agonists regulate progression through the cell cycle of human colon carcinoma cells. Proc. Natl. Acad. Sci. USA 2001, 98, 7846–7851. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, A.V.; Lin, J.E.; Kim, G.W.; Marszalowicz, G.P.; Li, P.; Stoecker, B.A.; Blomain, E.S.; Rattan, S.; Snook, A.E.; Schulz, S.; et al. Intestinal GUCY2C prevents TGF-beta secretion coordinating desmoplasia and hyperproliferation in colorectal cancer. Cancer Res. 2013, 73, 6654–6666. [Google Scholar] [CrossRef] [PubMed]
- Oyanagi, J.; Kojima, N.; Sato, H.; Higashi, S.; Kikuchi, K.; Sakai, K.; Matsumoto, K.; Miyazaki, K. Inhibition of transforming growth factor-beta signaling potentiates tumor cell invasion into collagen matrix induced by fibroblast-derived hepatocyte growth factor. Exp. Cell Res. 2014, 326, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Ohba, N.; Funatomi, H.; Seki, T.; Makino, R.; Mitamura, K. Hepatocyte growth factor stimulates cell growth and enhances the expression of transforming growth factor alpha mRNA in AsPC-1 human pancreatic cancer cells. J. Gastroenterol. 1999, 34, 498–504. [Google Scholar] [CrossRef] [PubMed]
- Ueda, T.; Takeyama, Y.; Hori, Y.; Nishikawa, J.; Yamamoto, M.; Saitoh, Y. Hepatocyte growth factor in assessment of acute pancreatitis: comparison with C-reactive protein and interleukin-6. J. Gastroenterol. 1997, 32, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Kemik, O.; Purisa, S.; Kemik, A.S.; Tuzun, S. Increase in the circulating level of hepatocyte growth factor in pancreatic cancer patients. Bratisl. Lek. Listy 2009, 110, 627–629. [Google Scholar] [PubMed]
- Shek, F.W.; Benyon, R.C.; Walker, F.M.; McCrudden, P.R.; Pender, S.L.; Williams, E.J.; Johnson, P.A.; Johnson, C.D.; Bateman, A.C.; Fine, D.R.; et al. Expression of transforming growth factor-beta 1 by pancreatic stellate cells and its implications for matrix secretion and turnover in chronic pancreatitis. Am. J. Pathol. 2002, 160, 1787–1798. [Google Scholar] [CrossRef]
- Mahadevan, D.; von Hoff, D.D. Tumor-stroma interactions in pancreatic ductal adenocarcinoma. Mol. Cancer Ther. 2007, 6, 1186–1197. [Google Scholar] [CrossRef] [PubMed]
- Mews, P.; Phillips, P.; Fahmy, R.; Korsten, M.; Pirola, R.; Wilson, J.; Apte, M. Pancreatic stellate cells respond to inflammatory cytokines: potential role in chronic pancreatitis. Gut 2002, 50, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Matsuo, Y.; Ma, J.; Koide, S.; Ochi, N.; Yasuda, A.; Funahashi, H.; Okada, Y.; Takeyama, H. Cancer cell-derived IL-1alpha promotes HGF secretion by stromal cells and enhances metastatic potential in pancreatic cancer cells. J. Surg. Oncol. 2010, 102, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Wang, Y.; Ye, Z.; Xie, H.; Zhou, L.; Zheng, S. Mechanism of TNF-alpha-induced migration and hepatocyte growth factor production in human mesenchymal stem cells. J. Cell Biochem. 2010, 111, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Weng, J.; Mohan, R.R.; Li, Q.; Wilson, S.E. IL-1 upregulates keratinocyte growth factor and hepatocyte growth factor mRNA and protein production by cultured stromal fibroblast cells: interleukin-1 beta expression in the cornea. Cornea 1997, 16, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Unger, B.L.; McGee, D.W. Hepatocyte growth factor and keratinocyte growth factor enhance IL-1-induced IL-8 secretion through different mechanisms in Caco-2 epithelial cells. In Vitro Cell Dev. Biol. Anim. 2011, 47, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Trevino, J.G.; Summy, J.M.; Gray, M.J.; Nilsson, M.B.; Lesslie, D.P.; Baker, C.H.; Gallick, G.E. Expression and activity of SRC regulate interleukin-8 expression in pancreatic adenocarcinoma cells: implications for angiogenesis. Cancer Res. 2005, 65, 7214–7222. [Google Scholar] [CrossRef] [PubMed]
- Summy, J.M.; Trevino, J.G.; Lesslie, D.P.; Baker, C.H.; Shakespeare, W.C.; Wang, Y.; Sundaramoorthi, R.; Metcalf, C.A., III; Keats, J.A.; Sawyer, T.K.; et al. AP23846, a novel and highly potent Src family kinase inhibitor, reduces vascular endothelial growth factor and interleukin-8 expression in human solid tumor cell lines and abrogates downstream angiogenic processes. Mol. Cancer Ther. 2005, 4, 1900–1911. [Google Scholar] [CrossRef] [PubMed]
- Trevino, J.G.; Summy, J.M.; Lesslie, D.P.; Parikh, N.U.; Hong, D.S.; Lee, F.Y.; Donato, N.J.; Abbruzzese, J.L.; Baker, C.H.; Gallick, G.E. Inhibition of SRC expression and activity inhibits tumor progression and metastasis of human pancreatic adenocarcinoma cells in an orthotopic nude mouse model. Am. J. Pathol. 2006, 168, 962–972. [Google Scholar] [CrossRef] [PubMed]
- Trevino, J.G.; Gray, M.J.; Nawrocki, S.T.; Summy, J.M.; Lesslie, D.P.; Evans, D.B.; Sawyer, T.K.; Shakespeare, W.C.; Watowich, S.S.; Chiao, P.J.; et al. Src activation of Stat3 is an independent requirement from NF-kappaB activation for constitutive IL-8 expression in human pancreatic adenocarcinoma cells. Angiogenesis 2006, 9, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Pennacchietti, S.; Michieli, P.; Galluzzo, M.; Mazzone, M.; Giordano, S.; Comoglio, P.M. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell 2003, 3, 347–361. [Google Scholar] [CrossRef]
- Kitajima, Y.; Ide, T.; Ohtsuka, T.; Miyazaki, K. Induction of hepatocyte growth factor activator gene expression under hypoxia activates the hepatocyte growth factor/c-Met system via hypoxia inducible factor-1 in pancreatic cancer. Cancer Sci. 2008, 99, 1341–1347. [Google Scholar] [CrossRef] [PubMed]
- Spivak-Kroizman, T.R.; Hostetter, G.; Posner, R.; Aziz, M.; Hu, C.; Demeure, M.J.; von Hoff, D.; Hingorani, S.R.; Palculict, T.B.; Izzo, J.; et al. Hypoxia triggers hedgehog-mediated tumor-stromal interactions in pancreatic cancer. Cancer Res. 2013, 73, 3235–3247. [Google Scholar] [CrossRef] [PubMed]
- Bussolino, F.; di Renzo, M.F.; Ziche, M.; Bocchietto, E.; Olivero, M.; Naldini, L.; Gaudino, G.; Tamagnone, L.; Coffer, A.; Comoglio, P.M. Hepatocyte growth factor is a potent angiogenic factor which stimulates endothelial cell motility and growth. J. Cell. Biol. 1992, 119, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Morishita, R.; Higaki, J.; Kida, I.; Aoki, M.; Moriguchi, A.; Yamada, K.; Hayashi, S.; Yo, Y.; Nakano, H.; et al. Hepatocyte growth factor is a novel member of the endothelium-specific growth factors: Additive stimulatory effect of hepatocyte growth factor with basic fibroblast growth factor but not with vascular endothelial growth factor. J. Hypertens. 1996, 14, 1067–1072. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.B.; Pothula, S.P.; Xu, Z.; Lee, A.K.; Goldstein, D.; Pirola, R.C.; Apte, M.V.; Wilson, J.S. The role of the hepatocyte growth factor/c-MET pathway in pancreatic stellate cell-endothelial cell interactions: antiangiogenic implications in pancreatic cancer. Carcinogenesis 2014, 35, 1891–1900. [Google Scholar] [CrossRef] [PubMed]
- Laterra, J.; Nam, M.; Rosen, E.; Rao, J.S.; Lamszus, K.; Goldberg, I.D.; Johnston, P. Scatter factor/hepatocyte growth factor gene transfer enhances glioma growth and angiogenesis in vivo. Lab. Invest. 1997, 76, 565–577. [Google Scholar] [PubMed]
- Kubota, T.; Taiyoh, H.; Matsumura, A.; Murayama, Y.; Ichikawa, D.; Okamoto, K.; Fujiwara, H.; Ikoma, H.; Nakanishi, M.; Kikuchi, S.; et al. NK4, an HGF antagonist, prevents hematogenous pulmonary metastasis by inhibiting adhesion of CT26 cells to endothelial cells. Clin. Exp. Metastasis 2009, 26, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.G.; Martin, T.A.; Matsumoto, K.; Nakamura, T.; Mansel, R.E. Hepatocyte growth factor/scatter factor decreases the expression of occludin and transendothelial resistance (TER) and increases paracellular permeability in human vascular endothelial cells. J. Cell. Physiol. 1999, 181, 319–329. [Google Scholar] [CrossRef]
- Li, H.; Shimura, H.; Aoki, Y.; Date, K.; Matsumoto, K.; Nakamura, T.; Tanaka, M. Hepatocyte growth factor stimulates the invasion of gallbladder carcinoma cell lines in vitro. Clin. Exp. Metastasis 1998, 16, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Nagakawa, O.; Murakami, K.; Yamaura, T.; Fujiuchi, Y.; Murata, J.; Fuse, H.; Saiki, I. Expression of membrane-type 1 matrix metalloproteinase (MT1-MMP) on prostate cancer cell lines. Cancer Lett. 2000, 155, 173–179. [Google Scholar] [CrossRef]
- Jiang, Y.; Xu, W.; Lu, J.; He, F.; Yang, X. Invasiveness of hepatocellular carcinoma cell lines: contribution of hepatocyte growth factor, c-met, and transcription factor Ets-1. Biochem. Biophys. Res. Commun. 2001, 286, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Ide, T.; Kitajima, Y.; Miyoshi, A.; Ohtsuka, T.; Mitsuno, M.; Ohtaka, K.; Koga, Y.; Miyazaki, K. Tumor-stromal cell interaction under hypoxia increases the invasiveness of pancreatic cancer cells through the hepatocyte growth factor/c-Met pathway. Int. J. Cancer 2006, 119, 2750–2759. [Google Scholar] [CrossRef] [PubMed]
- Itakura, J.; Ishiwata, T.; Friess, H.; Fujii, H.; Matsumoto, Y.; Buchler, M.W.; Korc, M. Enhanced expression of vascular endothelial growth factor in human pancreatic cancer correlates with local disease progression. Clin. Cancer Res. 1997, 3, 1309–1316. [Google Scholar] [PubMed]
- Seo, Y.; Baba, H.; Fukuda, T.; Takashima, M.; Sugimachi, K. High expression of vascular endothelial growth factor is associated with liver metastasis and a poor prognosis for patients with ductal pancreatic adenocarcinoma. Cancer 2000, 88, 2239–2245. [Google Scholar] [CrossRef]
- Korc, M. Role of growth factors in pancreatic cancer. Surg. Oncol. Clin. N. Am. 1998, 7, 25–41. [Google Scholar] [PubMed]
- Balaz, P.; Friess, H.; Buchler, M.W. Growth factors in pancreatic health and disease. Pancreatology 2001, 1, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, A.; Kubota, T.; Taiyoh, H.; Fujiwara, H.; Okamoto, K.; Ichikawa, D.; Shiozaki, A.; Komatsu, S.; Nakanishi, M.; Kuriu, Y.; et al. HGF regulates VEGF expression via the c-Met receptor downstream pathways, PI3K/Akt, MAPK and STAT3, in CT26 murine cells. Int. J. Oncol. 2013, 42, 535–542. [Google Scholar] [PubMed]
- Gatenby, R.A.; Gillies, R.J. A microenvironmental model of carcinogenesis. Nat Rev Cancer 2008, 8, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Riva, C.; Chauvin, C.; Pison, C.; Leverve, X. Cellular physiology and molecular events in hypoxia-induced apoptosis. Anticancer Res. 1998, 18, 4729–4736. [Google Scholar] [PubMed]
- Williams, A.C.; Collard, T.J.; Paraskeva, C. An acidic environment leads to p53 dependent induction of apoptosis in human adenoma and carcinoma cell lines: implications for clonal selection during colorectal carcinogenesis. Oncogene 1999, 18, 3199–3204. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Moellering, R.E.; Black, K.C.; Krishnamurty, C.; Baggett, B.K.; Stafford, P.; Rain, M.; Gatenby, R.A.; Gillies, R.J. Acid treatment of melanoma cells selects for invasive phenotypes. Clin. Exp. Metastasis 2008, 25, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Alfarouk, K.O.; Shayoub, M.E.; Muddathir, A.K.; Elhassan, G.O.; Bashir, A.H. Evolution of tumor metabolism might reflect carcinogenesis as a reverse evolution process (dismantling of multicellularity). Cancers 2011, 3, 3002–3017. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [PubMed]
- Zhou, W.; Capello, M.; Fredolini, C.; Racanicchi, L.; Piemonti, L.; Liotta, L.A.; Novelli, F.; Petricoin, E.F. Proteomic analysis reveals Warburg effect and anomalous metabolism of glutamine in pancreatic cancer cells. J. Proteome Res. 2012, 11, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Grinstein, S.; Rotin, D.; Mason, M.J. Na+/H+ exchange and growth factor-induced cytosolic pH changes. Role in cellular proliferation. Biochim. Biophys. Acta 1989, 988, 73–97. [Google Scholar] [CrossRef]
- Harguindey, S.; Orive, G.; Luis Pedraz, J.; Paradiso, A.; Reshkin, S.J. The role of pH dynamics and the Na+/H+ antiporter in the etiopathogenesis and treatment of cancer. Two faces of the same coin—one single nature. Biochim. Biophys. Acta 2005, 1756, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Putney, L.K.; Denker, S.P.; Barber, D.L. The changing face of the Na+/H+ exchanger, NHE1: Structure, regulation, and cellular actions. Annu. Rev. Pharmacol. Toxicol. 2002, 42, 527–552. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, D.L.; Boron, W.F. Long-term expression of c-H-ras stimulates Na-H and Na(+)-dependent Cl-HCO3 exchange in NIH-3T3 fibroblasts. J. Biol. Chem. 1994, 269, 4116–4124. [Google Scholar] [PubMed]
- Martinez-Zaguilan, R.; Seftor, E.A.; Seftor, R.E.; Chu, Y.W.; Gillies, R.J.; Hendrix, M.J. Acidic pH enhances the invasive behavior of human melanoma cells. Clin. Exp. Metastasis 1996, 14, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Paradiso, A.; Cardone, R.A.; Bellizzi, A.; Bagorda, A.; Guerra, L.; Tommasino, M.; Casavola, V.; Reshkin, S.J. The Na+-H+ exchanger-1 induces cytoskeletal changes involving reciprocal RhoA and Rac1 signaling, resulting in motility and invasion in MDA-MB-435 cells. Breast Cancer Res. 2004, 6, R616–R628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneko, A.; Hayashi, N.; Tanaka, Y.; Horimoto, M.; Ito, T.; Sasaki, Y.; Fusamoto, H.; Kamada, T. Activation of Na+/H+ exchanger by hepatocyte growth factor in hepatocytes. Hepatology 1995, 22, 629–636. [Google Scholar] [PubMed]
- Steffan, J.J.; Williams, B.C.; Welbourne, T.; Cardelli, J.A. HGF-induced invasion by prostate tumor cells requires anterograde lysosome trafficking and activity of Na+-H+ exchangers. J. Cell Sci. 2010, 123, 1151–1159. [Google Scholar] [CrossRef] [PubMed]
- Lowenfels, A.B.; Maisonneuve, P.; Cavallini, G.; Ammann, R.W.; Lankisch, P.G.; Andersen, J.R.; Dimagno, E.P.; Andren-Sandberg, A.; Domellof, L. Pancreatitis and the risk of pancreatic cancer. International Pancreatitis Study Group. N. Eng. J. Med. 1993, 328, 1433–1437. [Google Scholar] [CrossRef] [PubMed]
- Clark, C.E.; Hingorani, S.R.; Mick, R.; Combs, C.; Tuveson, D.A.; Vonderheide, R.H. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007, 67, 9518–9527. [Google Scholar] [CrossRef] [PubMed]
- De Monte, L.; Reni, M.; Tassi, E.; Clavenna, D.; Papa, I.; Recalde, H.; Braga, M.; di Carlo, V.; Doglioni, C.; Protti, M.P. Intratumor T helper type 2 cell infiltrate correlates with cancer-associated fibroblast thymic stromal lymphopoietin production and reduced survival in pancreatic cancer. J. Exp. Med. 2011, 208, 469–478. [Google Scholar]
- Ebrahimi, B.; Tucker, S.L.; Li, D.; Abbruzzese, J.L.; Kurzrock, R. Cytokines in pancreatic carcinoma: Correlation with phenotypic characteristics and prognosis. Cancer 2004, 101, 2727–2736. [Google Scholar] [CrossRef] [PubMed]
- Scholz, A.; Heinze, S.; Detjen, K.M.; Peters, M.; Welzel, M.; Hauff, P.; Schirner, M.; Wiedenmann, B.; Rosewicz, S. Activated signal transducer and activator of transcription 3 (STAT3) supports the malignant phenotype of human pancreatic cancer. Gastroenterology 2003, 125, 891–905. [Google Scholar] [CrossRef]
- Lee, L.S.; Banks, P.A.; Bellizzi, A.M.; Sainani, N.I.; Kadiyala, V.; Suleiman, S.; Conwell, D.L.; Paulo, J.A. Inflammatory protein profiling of pancreatic cyst fluid using EUS-FNA in tandem with cytokine microarray differentiates between branch duct IPMN and inflammatory cysts. J. Immunol. Methods 2012, 382, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Jacobetz, M.A.; Chan, D.S.; Neesse, A.; Bapiro, T.E.; Cook, N.; Frese, K.K.; Feig, C.; Nakagawa, T.; Caldwell, M.E.; Zecchini, H.I.; et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2013, 62, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yao, W.; Newton, R.C.; Scherle, P.A. Targeting the c-MET signaling pathway for cancer therapy. Expert Opin. Investig. Drugs 2008, 17, 997–1011. [Google Scholar] [CrossRef] [PubMed]
- Birchmeier, C.; Birchmeier, W.; Gherardi, E.; vande Woude, G.F. Met, metastasis, motility and more. Nat. Rev. Mol. Cell Biol. 2003, 4, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Di Renzo, M.F.; Olivero, M.; Martone, T.; Maffe, A.; Maggiora, P.; Stefani, A.D.; Valente, G.; Giordano, S.; Cortesina, G.; Comoglio, P.M. Somatic mutations of the MET oncogene are selected during metastatic spread of human HNSC carcinomas. Oncogene 2000, 19, 1547–1555. [Google Scholar]
- Ferracini, R.; Di Renzo, M.F.; Scotlandi, K.; Baldini, N.; Olivero, M.; Lollini, P.; Cremona, O.; Campanacci, M.; Comoglio, P.M. The Met/HGF receptor is over-expressed in human osteosarcomas and is activated by either a paracrine or an autocrine circuit. Oncogene 1995, 10, 739–749. [Google Scholar] [PubMed]
- Bean, J.; Brennan, C.; Shih, J.Y.; Riely, G.; Viale, A.; Wang, L.; Chitale, D.; Motoi, N.; Szoke, J.; Broderick, S.; et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc. Natl. Acad. Sci. USA 2007, 104, 20932–20937. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, L.; Duh, F.M.; Chen, F.; Kishida, T.; Glenn, G.; Choyke, P.; Scherer, S.W.; Zhuang, Z.; Lubensky, I.; Dean, M.; et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat. Genet. 1997, 16, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Houldsworth, J.; Cordon-Cardo, C.; Ladanyi, M.; Kelsen, D.P.; Chaganti, R.S. Gene amplification in gastric and esophageal adenocarcinomas. Cancer Res. 1990, 50, 6417–6422. [Google Scholar] [PubMed]
- Clinical Trials Involving HGF/SF-Met Inhibitor. Available online: www.vai.org/metclinicaltrials (accessed on 21 July 2015).
- Sharma, N.; Adjei, A.A. In the clinic: ongoing clinical trials evaluating c-MET-inhibiting drugs. Ther. Adv. Med. Oncol. 2011, 3, S37–S50. [Google Scholar] [CrossRef] [PubMed]
- A Randomized Phase 2 Study of ARQ 197 Versus Gemcitabine in Treatment-Naïve Patients With Unresectable Locally Advanced or Metastatic Pancreatic Adenocarcinoma. Available online: https://clinicaltrials.gov/ct2/show/NCT00558207 (accessed on 21 July 2015).
- Brandes, F.; Schmidt, K.; Wagner, C.; Redekopf, J.; Schlitt, H.J.; Geissler, E.K.; Lang, S.A. Targeting cMET with INC280 impairs tumour growth and improves efficacy of gemcitabine in a pancreatic cancer model. BMC Cancer 2015, 15, 71. [Google Scholar] [CrossRef] [PubMed]
- Schoffski, P.; Garcia, J.A.; Stadler, W.M.; Gil, T.; Jonasch, E.; Tagawa, S.T.; Smitt, M.; Yang, X.; Oliner, K.S.; Anderson, A.; et al. A phase II study of the efficacy and safety of AMG 102 in patients with metastatic renal cell carcinoma. BJU Int. 2011, 108, 679–686. [Google Scholar] [PubMed]
- Wen, P.Y.; Schiff, D.; Cloughesy, T.F.; Raizer, J.J.; Laterra, J.; Smitt, M.; Wolf, M.; Oliner, K.S.; Anderson, A.; Zhu, M.; et al. A phase II study evaluating the efficacy and safety of AMG 102 (rilotumumab) in patients with recurrent glioblastoma. Neuro Oncol. 2011, 13, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Ryan, C.J.; Rosenthal, M.; Ng, S.; Alumkal, J.; Picus, J.; Gravis, G.; Fizazi, K.; Forget, F.; Machiels, J.P.; Srinivas, S.; et al. Targeted MET inhibition in castration-resistant prostate cancer: A randomized phase II study and biomarker analysis with rilotumumab plus mitoxantrone and prednisone. Clin. Cancer Res. 2013, 19, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Scagliotti, G.V.; Novello, S.; Schiller, J.H.; Hirsh, V.; Sequist, L.V.; Soria, J.C.; von Pawel, J.; Schwartz, B.; von Roemeling, R.; Sandler, A.B. Rationale and design of MARQUEE: A phase III, randomized, double-blind study of tivantinib plus erlotinib versus placebo plus erlotinib in previously treated patients with locally advanced or metastatic, nonsquamous, non-small-cell lung cancer. Clin. Lung Cancer 2012, 13, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Santoro, A.; Rimassa, L.; Borbath, I.; Daniele, B.; Salvagni, S.; Van Laethem, J.L.; van Vlierberghe, H.; Trojan, J.; Kolligs, F.T.; Weiss, A.; et al. Tivantinib for second-line treatment of advanced hepatocellular carcinoma: A randomised, placebo-controlled phase 2 study. Lancet Oncol. 2013, 14, 55–63. [Google Scholar] [CrossRef]
- Wagner, A.J.; Goldberg, J.M.; Dubois, S.G.; Choy, E.; Rosen, L.; Pappo, A.; Geller, J.; Judson, I.; Hogg, D.; Senzer, N.; et al. Tivantinib (ARQ 197), a selective inhibitor of MET, in patients with microphthalmia transcription factor-associated tumors: results of a multicenter phase 2 trial. Cancer 2012, 118, 5894–5902. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Kim, D.W.; Nakagawa, K.; Seto, T.; Crino, L.; Ahn, M.J.; De Pas, T.; Besse, B.; Solomon, B.J.; Blackhall, F.; et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N. Eng. J. Med. 2013, 368, 2385–2394. [Google Scholar] [CrossRef] [PubMed]
- Elisei, R.; Schlumberger, M.J.; Muller, S.P.; Schoffski, P.; Brose, M.S.; Shah, M.H.; Licitra, L.; Jarzab, B.; Medvedev, V.; Kreissl, M.C.; et al. Cabozantinib in progressive medullary thyroid cancer. J. Clin. Oncol. 2013, 31, 3639–3646. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Vaishampayan, U.; Rosenberg, J.E.; Logan, T.F.; Harzstark, A.L.; Bukowski, R.M.; Rini, B.I.; Srinivas, S.; Stein, M.N.; Adams, L.M.; et al. Phase II and biomarker study of the dual MET/VEGFR2 inhibitor foretinib in patients with papillary renal cell carcinoma. J. Clin. Oncol. 2013, 31, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.A.; Wainberg, Z.A.; Catenacci, D.V.; Hochster, H.S.; Ford, J.; Kunz, P.; Lee, F.C.; Kallender, H.; Cecchi, F.; Rabe, D.C.; et al. Phase II study evaluating 2 dosing schedules of oral foretinib (GSK1363089), cMET/VEGFR2 inhibitor, in patients with metastatic gastric cancer. PLoS ONE 2013, 8, e54014. [Google Scholar] [CrossRef] [PubMed]
- Michieli, P.; Basilico, C.; Pennacchietti, S.; Maffe, A.; Tamagnone, L.; Giordano, S.; Bardelli, A.; Comoglio, P.M. Mutant Met-mediated transformation is ligand-dependent and can be inhibited by HGF antagonists. Oncogene 1999, 18, 5221–5231. [Google Scholar] [CrossRef] [PubMed]
- Tomioka, D.; Maehara, N.; Kuba, K.; Mizumoto, K.; Tanaka, M.; Matsumoto, K.; Nakamura, T. Inhibition of growth, invasion, and metastasis of human pancreatic carcinoma cells by NK4 in an orthotopic mouse model. Cancer Res. 2001, 61, 7518–7524. [Google Scholar] [PubMed]
- Qian, L.W.; Mizumoto, K.; Inadome, N.; Nagai, E.; Sato, N.; Matsumoto, K.; Nakamura, T.; Tanaka, M. Radiation stimulates HGF receptor/c-Met expression that leads to amplifying cellular response to HGF stimulation via upregulated receptor tyrosine phosphorylation and MAP kinase activity in pancreatic cancer cells. Int. J. Cancer 2003, 104, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Date, K.; Matsumoto, K.; Shimura, H.; Tanaka, M.; Nakamura, T. HGF/NK4 is a specific antagonist for pleiotrophic actions of hepatocyte growth factor. FEBS Lett. 1997, 420, 1–6. [Google Scholar] [CrossRef]
- Date, K.; Matsumoto, K.; Kuba, K.; Shimura, H.; Tanaka, M.; Nakamura, T. Inhibition of tumor growth and invasion by a four-kringle antagonist (HGF/NK4) for hepatocyte growth factor. Oncogene 1998, 17, 3045–3054. [Google Scholar] [CrossRef] [PubMed]
- Gherardi, E.; Sandin, S.; Petoukhov, M.V.; Finch, J.; Youles, M.E.; Ofverstedt, L.G.; Miguel, R.N.; Blundell, T.L.; Vande Woude, G.F.; Skoglund, U.; et al. Structural basis of hepatocyte growth factor/scatter factor and MET signalling. Proc. Natl. Acad. Sci. USA 2006, 103, 4046–4051. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, S.; Nakamura, T. HGF-MET cascade, a key target for inhibiting cancer metastasis: The impact of NK4 discovery on cancer biology and therapeutics. Int. J. Mol. Sci. 2013, 14, 888–919. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rizwani, W.; Allen, A.E.; Trevino, J.G. Hepatocyte Growth Factor from a Clinical Perspective: A Pancreatic Cancer Challenge. Cancers 2015, 7, 1785-1805. https://doi.org/10.3390/cancers7030861
Rizwani W, Allen AE, Trevino JG. Hepatocyte Growth Factor from a Clinical Perspective: A Pancreatic Cancer Challenge. Cancers. 2015; 7(3):1785-1805. https://doi.org/10.3390/cancers7030861
Chicago/Turabian StyleRizwani, Wasia, Amanda E. Allen, and Jose G. Trevino. 2015. "Hepatocyte Growth Factor from a Clinical Perspective: A Pancreatic Cancer Challenge" Cancers 7, no. 3: 1785-1805. https://doi.org/10.3390/cancers7030861
APA StyleRizwani, W., Allen, A. E., & Trevino, J. G. (2015). Hepatocyte Growth Factor from a Clinical Perspective: A Pancreatic Cancer Challenge. Cancers, 7(3), 1785-1805. https://doi.org/10.3390/cancers7030861