
The Tip of the Iceberg: Clinical Implications of Genomic Sequencing Projects in Head and Neck Cancer

Abstract
:1. Background
2. Further Need for Genetic Analysis and Follow-Up Genomic Studies

{kind=link}
{kind=link}
{kind=link}
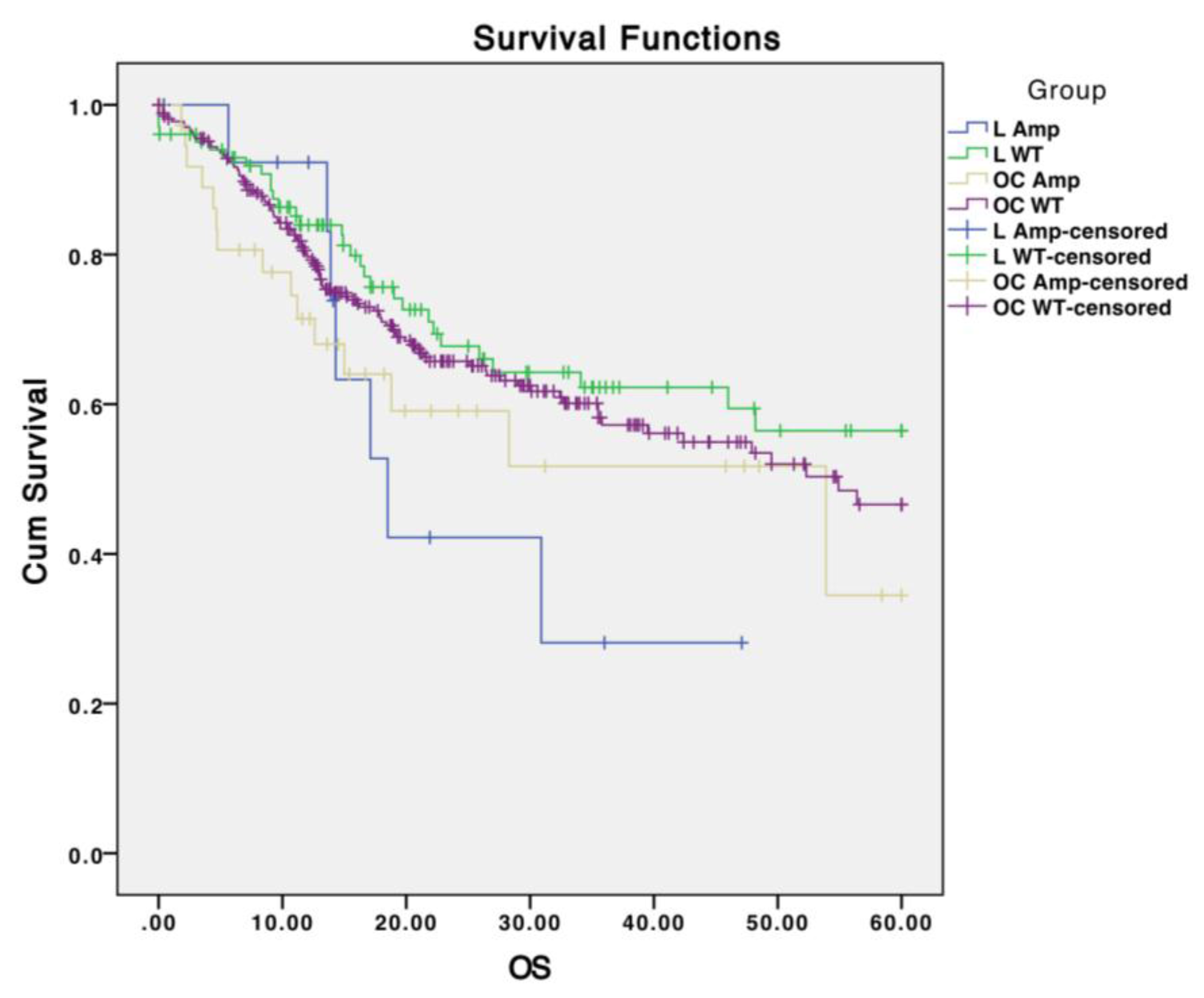{kind=link}
| Gene | Stransky et al. [3] n = 74 | Agrawal et al. [4] n = 32 | TCGA [2] n = 279 | Pickering et al. [5] n = 44 | Total n = 429 |
|---|---|---|---|---|---|
| TP53 | 46 (62%) | 22 (69%) | 204 (73%) | 31 (70%) | 303 (71%) |
| FAT1 | 9 (12%) | 0 (0%) | 64 (23%) | 8 (18%) | 81 (19%) |
| CDKN2A | 9 (12%) | 0 (0%) | 63 (23%) | 2 (5%) | 74 (17%) |
| PIK3CA | 6 (8%) | 3 (9%) | 58 (21%) | 3 (7%) | 70 (16%) |
| NOTCH1 | 9 (12%) | 4 (13%) | 52 (19%) | 9 (20%) | 74 (17%) |
| CASP8 | 6 (8%) | 1 (3%) | 24 (9%) | 4 (9%) | 35 (8%) |
2.1. Limited Data on Early Stage, HPV+, Non-Caucasian Patients
2.2. Subsequent Sequencing Studies
2.3. Genetic Biomarkers
| Altered Median OS (mo) | WT Median OS (mo) | Log-Rank p-Value | |
|---|---|---|---|
| TP53 | 19.2 | 65.8 | 0.04 |
| FAT1 | 18.8 | 26.4 | 0.80 |
| CDKN2A | 17.2 | 52.3 | 0.24 |
| PIK3CA | 28.3 | 21.9 | 0.41 |
| NOTCH1 | 18.0 | 28.3 | 0.49 |
| CASP8 | 18.8 | 26.4 | 0.31 |
| EGFR | 17.2 | 30.0 | 0.03 |


2.4. Insight across Multiple TCGA Cohorts
| HNSCC (n = 279) | Cervical (n = 171) | Lung (n = 178) | Esophageal (n = 88) | Nasopharyngeal (n = 56) | |
|---|---|---|---|---|---|
| TP53 | 73.1% | 2.9% | 93.3% | 83.0% | 12.5% |
| FAT1 | 22.9% | 4.1% | 14.6% | 4.5% | 3.6% |
| CDKN2A | 22.6% | 1.2% | 18.0% | 4.5% | 0.0% |
| PIK3CA | 20.8% | 26.3% | 15.7% | 4.5% | 1.8% |
| NOTCH1 | 18.6% | 6.4% | 8.4% | 9.1% | 3.6% |
| CASP8 | 8.6% | 4.7% | 1.7% | 0.0% | 1.8% |
| HNSCC (n = 522) | Cervical (n = 295) | Lung (n = 501) | |
|---|---|---|---|
| 9p21 (CDKN2A, CDKN2B) | 27.7%–31.3% | 0.3% | 23.0%–27.6% |
| Deletion | Deletion | Deletion | |
| 11q13 (FGF3/4/19,CCND1) | 23.4%–24.2% | 2.9%–10.4% | 14.1%–14.3% |
| Amplification | Amplification | Amplification | |
| 3q28 (TP63, EIF4A2, FGF12) | 18.7%–21.5% | 19.2%–21.4% | 38.9%–41.9% |
| Amplification | Amplification | Amplification | |
| 3q26 (SOX2, PIK3CA) | 18.7%%–20.8% | 19.5%–21.1% | 43.5%–48.0% |
| Amplification | Amplification | Amplification | |
| 8q24 (MYC, PLEC, EPPK1) | 8.3%–12.3% | 2.6%–4.9% | 6.9%–10.5% |
| Amplification | Amplification | Amplification |
3. Further Need for Tumor Stratification
3.1. Driver vs. Passenger Mutations
3.2. Early vs. Late-Stage Mutations
3.3. Tumor Temporal and Spatial Heterogeneity
3.4. Cancer Stem Cells
3.5. Molecular Heterogeneity

3.6. Tissue-Specific Mutational Profiles
| OC (n = 172) | OP (n = 33) | L (n = 72) | |
|---|---|---|---|
| HPV+ | 7.0% | 66.7% | 1.4% |
| TP53 | 75.6% | 27.3% | 88.9% |
| FAT1 | 26.2% | 12.1% | 19.4% |
| CDKN2A | 25.6% | 6.1% | 23.6% |
| PIK3CA | 17.4% | 30.3% | 25.0% |
| NOTCH1 | 21.5% | 6.1% | 18.1% |

| OC (n = 172) | OP (n = 33) | L (n = 72) | |
|---|---|---|---|
| 9p21 (CDKN2A, CDKN2B) | 26.2%–28.5% | 18.2% | 30.6%–31.9% |
| Deletion | Deletion | Deletion | |
| 11q13 (FGF3/4/19, CCND1) | 25.0% | 24.2% | 36.1% |
| Amplification | Amplification | Amplification | |
| 3q28 (TP63, EIF4A2, FGF12) | 12.2%–12.8% | 27.3%–30.3% | 30.6%–34.7% |
| Amplification | Amplification | Amplification | |
| 3q26 (SOX2, PIK3CA) | 12.8%–13.4% | 27.3%–30.3% | 34.7%–37.5% |
| Amplification | Amplification | Amplification | |
| 8q24 (MYC, PLEC, EPPK1) | 9.3%–11.0% | 9.1%–12.1% | 12.5%–16.7% |
| Amplification | Amplification | Amplification |
4. Challenges to Targeted Therapy
4.1. What is Actionable?
4.2. Creating Targeted Therapeutics
4.3. Development of Resistance to Monotherapy
4.4. Organ-Specific Response to Therapy
4.5. Targeting Drivers of Cancer Stem Cells
5. Translation to Clinical Targeted Therapy Paradigms
5.1. When to Use Targeted Therapy
5.2. Incorporating Targeted Therapy with Other Treatment Paradigms (Immunotherapy, Surgery, Chemoradiation)
5.3. Employing Agents Approved in Other Cancers
5.4. Precision Medicine Clinical Trials
5.5. Precision Medicine Tumor Boards
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Pulte, D.; Brenner, H. Changes in survival in head and neck cancers in the late 20th and early 21st century: A period analysis. Oncologist 2006, 15, 994–1001. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar]
- Stransky, N.; Egloff, A.M.; Tward, A.D.; Kostic, A.D.; Cibulskis, K.; Sivachenko, A.; Kryukov, G.V.; Lawrence, M.S.; Sougnez, C.; McKenna, A.; et al. The mutational landscape of head and neck squamous cell carcinoma. Science 2011, 333, 1157–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agrawal, N.; Frederick, M.J.; Pickering, C.R.; Bettegowda, C.; Chang, K.; Li, R.J.; Fakhry, C.; Xie, T.X.; Zhang, J.; Wang, J.; et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science 2011, 333, 1154–1157. [Google Scholar] [CrossRef] [PubMed]
- Pickering, C.R.; Zhang, J.; Neskey, D.M.; Zhao, M.; Jasser, S.A.; Wang, J.; Tsai, C.J.; Ortega Alves, M.V.; Zhou, J.H.; Drummond, J.; et al. Squamous cell carcinoma of the oral tongue in young non-smokers is genomically similar to tumors in older smokers. Clin. Cancer Res. 2014, 20, 3842–3848. [Google Scholar] [CrossRef] [PubMed]
- Liu, V.W.; Hedberg, M.L.; Li, H.; Vangara, B.S.; Pendleton, K.; Zeng, Y.; Lu, Y.; Zhang, Q.; Du, Y.; Gilbert, B.R.; et al. Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer Discov. 2013, 3, 761–769. [Google Scholar]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parfenov, M.; Pedamallu, C.S.; Gehlenborg, N.; Freeman, S.S.; Danilova, L.; Bristow, C.A.; Lee, S.; Hadjipanayis, A.G.; Ivanova, E.V.; Wilkerson, M.D.; et al. Characterization of HPV and host genome interactions in primary head and neck cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 15544–15549. [Google Scholar] [PubMed]
- Seiwert, T.Y.; Zuo, Z.; Keck, M.K.; Khattri, A.; Pedamallu, C.S.; Stricker, T.; Brown, C.; Pugh, T.J.; Stojanov, P.; Cho, J.; et al. Integrative and comparative genomic analysis of HPV-positive and HPV-negative head and neck squamous cell carcinomas. Clin. Cancer Res. 2015, 21, 632–641. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.H.; Guthrie, V.B.; Masica, D.L.; Tokheim, C.; Kang, H.; Richmon, J.; Agrawal, N.; Fakhry, C.; Quon, H.; Subramaniam, R.M.; et al. Genomic alterations in head and neck squamous cell carcinoma determined by cancer gene-targeted sequencing. Ann. Oncol. 2015, 26, 1216–1223. [Google Scholar] [CrossRef] [PubMed]
- Akagi, K.; Li, J.; Broutian, T.R.; Padilla-Nash, H.; Xiao, W.; Jiang, B.; Rocco, J.W.; Teknos, T.N.; Kumar, B.; Wangsa, D.; et al. Genome-wide analysis of HPV integration in human cancers reveals recurrent, focal genomic instability. Genome Res. 2014, 24, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Bradford, C.R.; Kumar, B.; Bellile, E.; Lee, J.; Taylor, J.; D’Silva, N.; Cordell, K.; Kleer, C.; Kupfer, R.; Kumar, P.; et al. Biomarkers in advanced larynx cancer. Laryngoscope 2014, 124, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Ang, K.K.; Berkey, B.A.; Tu, X.; Zhang, H.Z.; Katz, R.; Hammond, E.H.; Fu, K.K.; Milas, L. Impact of epidermal growth factor receptor expression on survival and pattern of relapse in patients with advanced head and neck carcinoma. Cancer Res. 2002, 62, 7350–7536. [Google Scholar] [PubMed]
- Temam, S.; Kawaguchi, H.; El-Naggar, A.K.; Jelinek, J.; Tang, H.; Liu, D.D.; Lang, W.; Issa, J.P.; Lee, J.J.; Mao, L. Epidermal growth factor receptor copy number alterations correlate with poor clinical outcome in patients with head and neck squamous cancer. J. Clin. Oncol. 2007, 25, 2164–2170. [Google Scholar] [CrossRef] [PubMed]
- Poeta, M.L.; Manola, J.; Goldwasser, M.A.; Forastiere, A.; Benoit, N.; Califano, J.A.; Ridge, J.A.; Goodwin, J.; Kenady, D.; Saunders, J.; et al. TP53 mutations and survival in squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2007, 357, 2552–2561. [Google Scholar] [CrossRef] [PubMed]
- Ojesina, A.I.; Lichtenstein, L.; Freeman, S.S.; Pedamallu, C.S.; Imaz-Rosshandler, I.; Pugh, T.J.; Cherniack, A.D.; Ambrogio, L.; Cibulskis, K.; Bertelsen, B.; et al. Landscape of genomic alterations in cervical carcinomas. Nature 2014, 506, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [Google Scholar]
- Song, Y.; Li, L.; Ou, Y.; Gao, Z.; Li, E.; Li, X.; Zhang, W.; Wang, J.; Xu, L.; Zhou, Y.; et al. Identification of genomic alterations in oesophageal squamous cell cancer. Nature 2014, 509, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.C.; Meng, X.; Hazawa, M.; Nagata, Y.; Varela, A.M.; Xu, L.; Sato, Y.; Liu, L.Z.; Ding, L.W.; Sharma, A.; et al. The genomic landscape of nasopharyngeal carcinoma. Nat. Genet. 2014, 46, 866–871. [Google Scholar] [CrossRef] [PubMed]
- Schwaederle, M.; Elkin, S.K.; Tomson, B.N.; Carter, J.L.; Kurzrock, R. Squamousness: Next-generation sequencing reveals shared molecular features across squamous tumor types. Cell Cycle 2015, 14, 2355–2361. [Google Scholar] [CrossRef] [PubMed]
- Ciriello, G.; Miller, M.; Aksoy, B.A.; Senbabaoglu, Y.; Schultz, N.; Sander, C. Emerging landscape of oncogenic signatures across human cancers. Nat. Genet. 2013, 45, 1127–1133. [Google Scholar] [CrossRef] [PubMed]
- Haber, D.A.; Settleman, J. Cancer: Drivers and passengers. Nature 2007, 446, 145–146. [Google Scholar] [CrossRef] [PubMed]
- Neskey, D.M.; Osman, A.A.; Ow, T.J.; Katsonis, P.; McDonald, T.; Hicks, S.C.; Hsu, T.K.; Pickering, C.R.; Ward, A.; Patel, A.; et al. Evolutionary action score of TP53 identifies high-risk mutations associated with decreased survival and increased distant metastases in head and neck cancer. Cancer Res. 2015, 75, 1527–1536. [Google Scholar] [CrossRef] [PubMed]
- Waclaw, B.; Bozic, I.; Pittman, M.E.; Hruban, R.H.; Vogelstein, B.; Nowak, M.A. A spatial model predicts that dispersal and cell turnover limit intratumour heterogeneity. Nature 2015, 525, 261–264. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Swanton, C. Intratumor heterogeneity: Evolution through space and time. Cancer Res. 2012, 72, 4975–4882. [Google Scholar] [CrossRef] [PubMed]
- Tillman, B.N.; Yanik, M.; Birkeland, A.C.; Liu, C.; Hovelson, D.H.; Cani, A.K.; Palanisamy, N.; Carskadon, S.; Carey, T.E.; Bradford, C.R.; et al. Targeted sequencing of an epidemiologically low risk patient defines Fibroblast Growth Factor family aberrations as a putative driver of head and neck squamous cell carcinoma. Head Neck 2015, in press. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Li, S.C.; Tachiki, L.M.; Kabeer, M.H.; Dethlefs, B.A.; Anthony, M.J.; Loudon, W.G. Cancer genomic research at the crossroads: Realizing the changing genetic landscape as intratumoral spatial and temporal heterogeneity becomes a confounding factor. Cancer Cell Int. 2014, 14, 115. [Google Scholar] [CrossRef]
- Mroz, E.A.; Tward, A.D.; Hammon, R.J.; Ren, Y.; Rocco, J.W. Intra-tumor genetic heterogeneity and mortality in head and neck cancer: Analysis of data from the Cancer Genome Atlas. PLoS Med. 2015, 12, e1001786. [Google Scholar] [CrossRef] [PubMed]
- Rocco, J.W. Mutant allele tumor heterogeneity (MATH) and head and neck squamous cell carcinoma. Head Neck Pathol. 2015, 9, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Navin, N.; Kendall, J.; Troge, J.; Andrews, P.; Rodgers, L.; McIndoo, J.; Cook, K.; Stepansky, A.; Levy, D.; Esposito, D.; et al. Tumour evolution inferred by single-cell sequencing. Nature 2011, 472, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Prince, M.E.; Sivanandan, R.; Kaczorowski, A.; Wolf, G.T.; Kaplan, M.J.; Dalerba, P.; Weissman, I.L.; Clarke, M.F.; Ailles, L.E. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 2007, 104, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Chinn, S.B.; Darr, O.A.; Owen, J.H.; Bellile, E.; McHugh, J.B.; Spector, M.E.; Papagerakis, S.M.; Chepeha, D.B.; Bradford, C.R.; Carey, T.E.; et al. Cancer stem cells: Mediators of tumorigenesis and metastasis in head and neck squamous cell carcinoma. Head Neck 2015, 37, 317–236. [Google Scholar] [CrossRef] [PubMed]
- Joshua, B.; Kaplan, M.J.; Doweck, I.; Pai, R.; Weissman, I.L.; Prince, M.E.; Ailles, L.E. Frequency of cells expressing CD44, a head and neck cancer stem cell marker: Correlation with tumor aggressiveness. Head Neck 2012, 34, 42–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yap, L.; Lee, D.; Khairuddin, A.; Pairan, M.; Puspita, B.; Siar, C.; Paterson, I. The opposing roles of NOTCH signaling in head and neck cancer: A mini review. Oral. Dis. 2015. [Google Scholar] [CrossRef] [PubMed]
- Brenner, C.; Birkeland, A.C. Personalizing medicine in head and neck squamous cell carcinoma: The rationale for combination therapies. Med. Res. Arch. 2015, 3. [Google Scholar] [CrossRef]
- Bonner, J.A.; Harari, P.M.; Giralt, J.; Azarnia, N.; Shin, D.M.; Cohen, R.B.; Jones, C.U.; Sur, R.; Raben, D.; Jassem, D.; et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2006, 354, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Vermorken, J.B.; Mesia, R.; Rivera, F.; Remenar, E.; Kawecki, A.; Rottey, S.; Erfan, J.; Zabolotnyy, D.; Kienzer, H.R.; Cupissol, D.; et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N. Engl. J. Med. 2008, 359, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Nazarian, R.; Shi, H.; Wang, Q.; Kong, X.; Koya, R.C.; Lee, H.; Chen, Z.; Lee, M.K.; Attar, N.; Sazegar, H.; et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010, 468, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Ware, K.E.; Hinz, T.K.; Kleczko, E.; Singleton, K.R.; Marek, L.A.; Helfrich, B.A.; Cummings, C.T.; Graham, D.K.; Astling, D.; Tan, A.C.; Heasley, L.E. A mechanism of resistance to gefitinib mediated by cellular reprogramming and the acquisition of an FGF2-FGFR1 autocrine growth loop. Oncogenesis 2013, 2, e39. [Google Scholar] [CrossRef] [PubMed]
- Tassone, P.; Old, M.; Teknos, T.N.; Pan, Q. p53-based therapeutics for head and neck squamous cell carcinoma. Oral. Oncol. 2013, 49, 733–737. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Pan, S.; Hsieh, M.H.; Ng, N.; Sun, F.; Wang, T.; Kasibhatla, S.; Schuller, A.G.; Li, A.G.; Cheng, D.; et al. Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc. Natl. Acad. Sci. USA 2013, 110, 20224–20229. [Google Scholar] [CrossRef] [PubMed]
- NCI. NCI prepares to launch MATCH trial. Cancer Discov. 2015, 5, 685. [Google Scholar]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoms with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef] [PubMed]
- Bookman, M.A.; Darcy, K.M.; Clarke-Pearson, D.; Boothby, R.A.; Horowitz, I.R. Evaluation of monoclonal humanized anti-HER2 antibody, trastuzumab, in patients with recurrent or refractory ovarian or primary peritoneal carcinoma with overexpression of HER2: A phase II trial of the Gynecologic Oncology Group. J. Clin. Oncol. 2003, 21, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Urba, S.; Wolf, G.; Eisbruch, A.; Worden, F.; Lee, J.; Bradford, C.; Teknos, T.; Chepeha, D.; Prince, M.; Hogikyan, N.; et al. Single-cycle induction chemotherapy selects patients with advanced laryngeal cancer for combined chemoradiation: A new treatment paradigm. J. Clin. Oncol. 2006, 24, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Seiwart, T.Y.; Haddad, R.I.; Gupta, S.; Mehra, R.; Tahara, M.; Berger, R.; Lee, S.; Burtness, B.; Le, D.T.; Heath, K.; et al. Antitumor activity and safety of pembrolizumab in patients (pts) with advanced squamous cell carcinoma of the head and neck (SCCHN): Preliminary results from KEYNOTE-012 expansion cohort. In Proceedings of the American Society of Clinical Oncology Annual Meeting, Chicago, IL, USA, 29 May–2 June 2015.
- Birkeland, A.C.; Yanik, M.L.; Tillman, B.N.; Scott, M.V.; Foltin, S.K.; Mann, J.E.; Michmerhuizen, N.L.; Ludwig, M.L.; Sandelski, M.M.; Komarck, C.M.; et al. Identification of targetable HER2 amplifications in head and neck squamous cell carcinoma. 2015. submitted for publication. [Google Scholar]
- Breuskin, I.; Even, C.; Ileana, E.; Massard, C.; Lezghed, N.; Lacroix, L.; Hollebecque, A.; Bahleda, R.; Ngo-Camus, M.; Loriot, L.; et al. Molecular screening for cancer treatment optimization in head and neck cancer (MOSCATO 01): A prospective molecular triage trial; interim analysis of 78 patients with recurrent or metastatic head and neck cancers. In Proceedings of the American Head and Neck Society Translational Meeting, Boston, MA, USA, 21–22 April 2015; p. S021.
- Beltran, H.; Eng, K.; Mosquera, J.; Sigaras, A.; Romanel, A.; Rennert, H.; Kossai, M.; Pauli, C.; Faltas, B.; Fontugne, J.; et al. Whole-exome sequencing of metastatic cancer and biomarkers of treatment response. JAMA Oncol. 2015, 1, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Roychowdhury, S.; Iver, M.K.; Robinson, D.R.; Lonigro, R.J.; Wu, Y.M.; Cao, X.; Kalyana-Sundaram, S.; Sam, L.; Balbin, O.A.; Quist, M.J.; et al. Personalized oncology through integrative high-throughput sequencing: A pilot study. Sci. Transl. Med. 2011, 3, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Birkeland, A.C.; Uhlmann, W.R.; Brenner, J.C.; Shuman, A.G. Getting personal: Head and neck cancer management in the era of genomic medicine. Head Neck 2015. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Birkeland, A.C.; Ludwig, M.L.; Meraj, T.S.; Brenner, J.C.; Prince, M.E. The Tip of the Iceberg: Clinical Implications of Genomic Sequencing Projects in Head and Neck Cancer. Cancers 2015, 7, 2094-2109. https://doi.org/10.3390/cancers7040879
Birkeland AC, Ludwig ML, Meraj TS, Brenner JC, Prince ME. The Tip of the Iceberg: Clinical Implications of Genomic Sequencing Projects in Head and Neck Cancer. Cancers. 2015; 7(4):2094-2109. https://doi.org/10.3390/cancers7040879
Chicago/Turabian StyleBirkeland, Andrew C., Megan L. Ludwig, Taha S. Meraj, J. Chad Brenner, and Mark E. Prince. 2015. "The Tip of the Iceberg: Clinical Implications of Genomic Sequencing Projects in Head and Neck Cancer" Cancers 7, no. 4: 2094-2109. https://doi.org/10.3390/cancers7040879
APA StyleBirkeland, A. C., Ludwig, M. L., Meraj, T. S., Brenner, J. C., & Prince, M. E. (2015). The Tip of the Iceberg: Clinical Implications of Genomic Sequencing Projects in Head and Neck Cancer. Cancers, 7(4), 2094-2109. https://doi.org/10.3390/cancers7040879