
Differences in Redox Regulatory Systems in Human Lung and Liver Tumors Suggest Different Avenues for Therapy

Abstract
:1. Introduction
2. Results
2.1. Lung and Liver Tumor and Normal Tissues

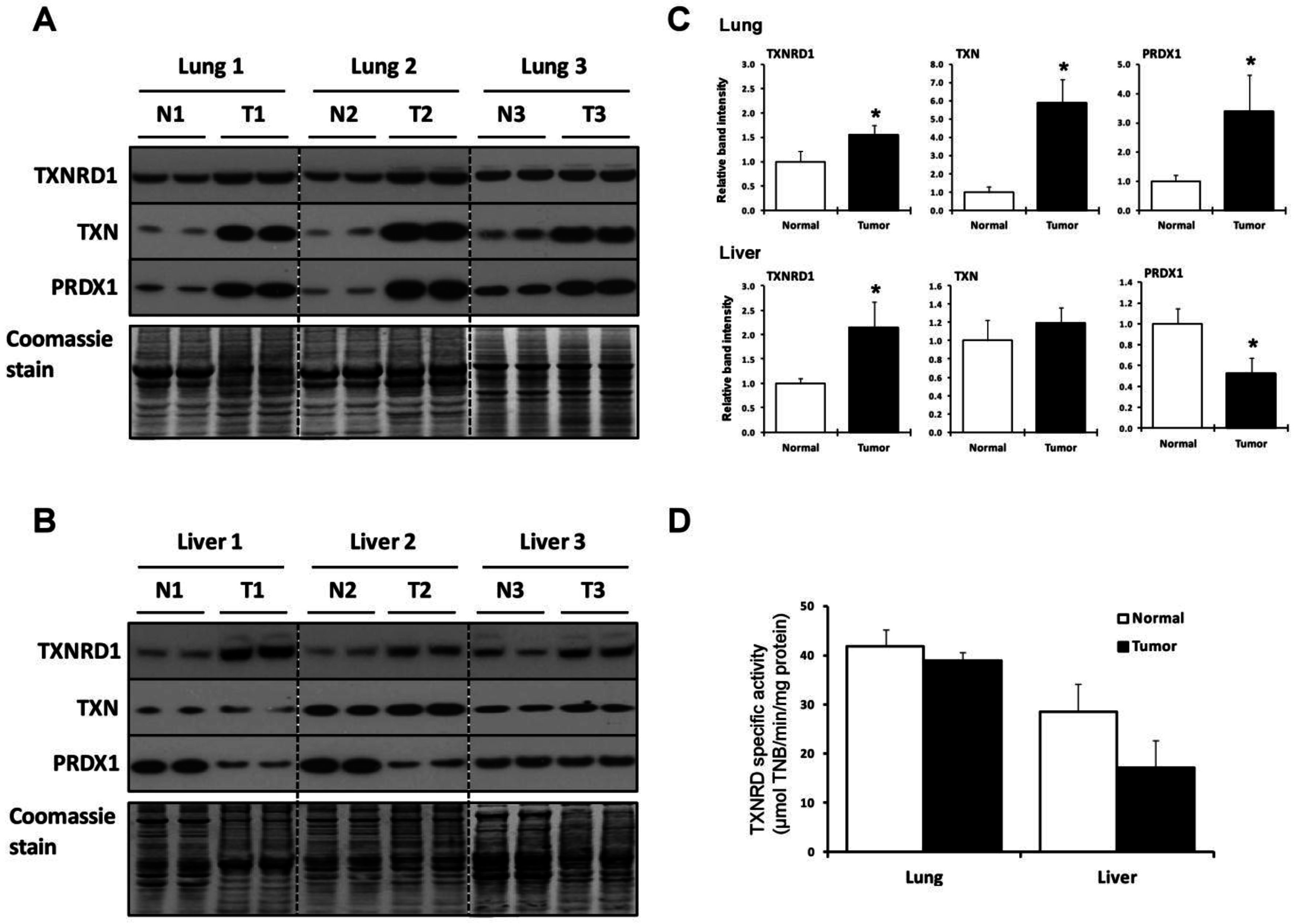{kind=link}
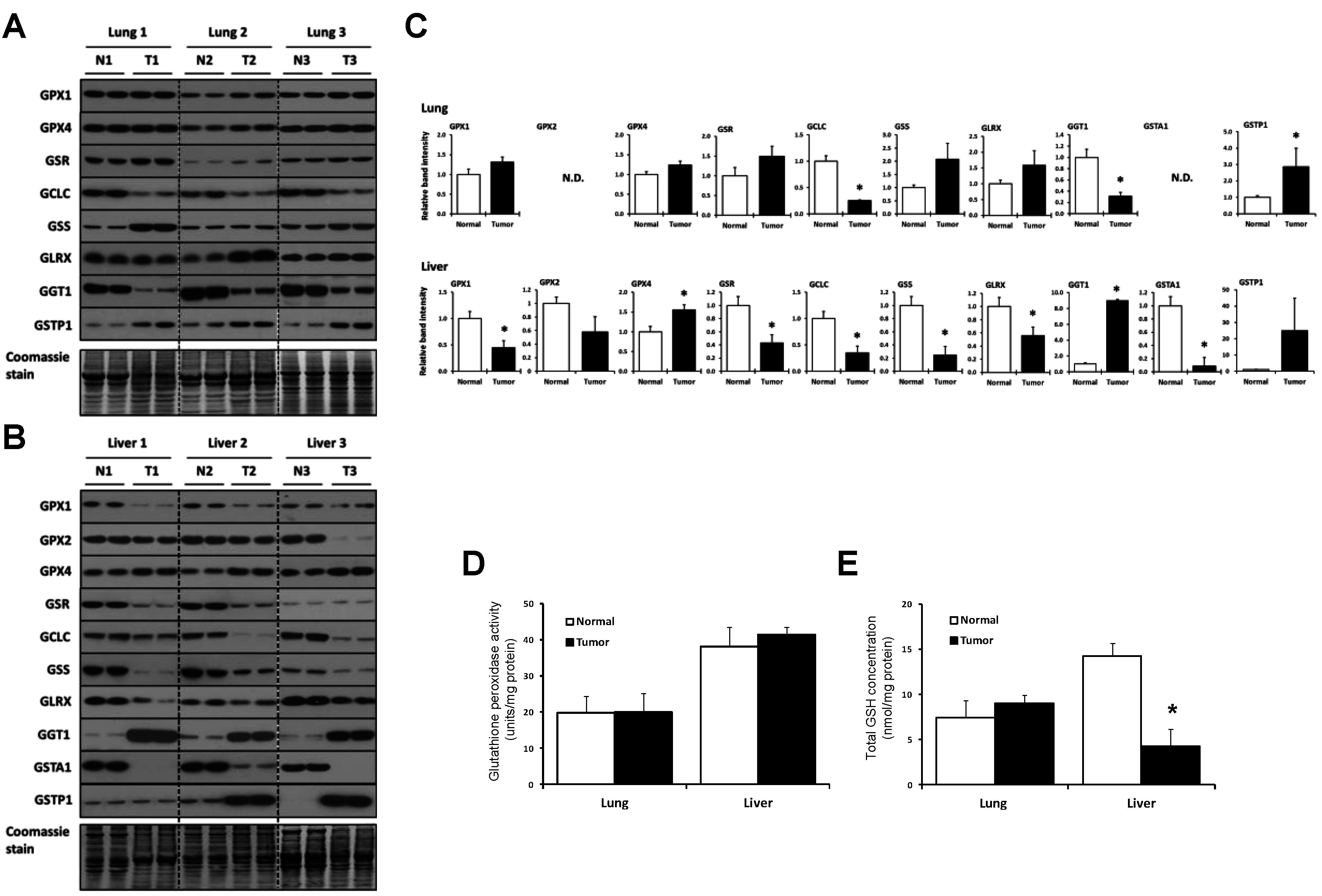{kind=link}
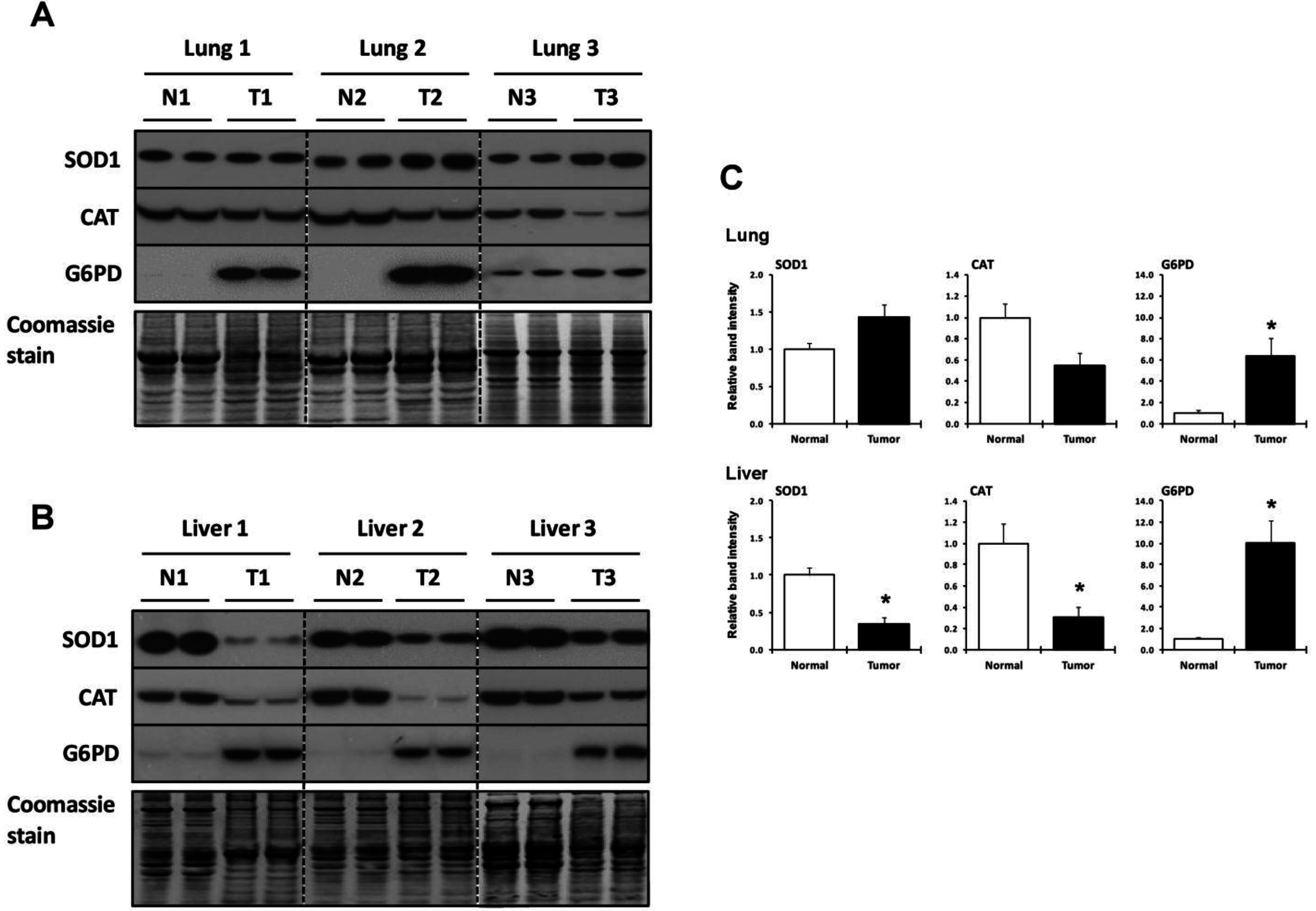{kind=link}
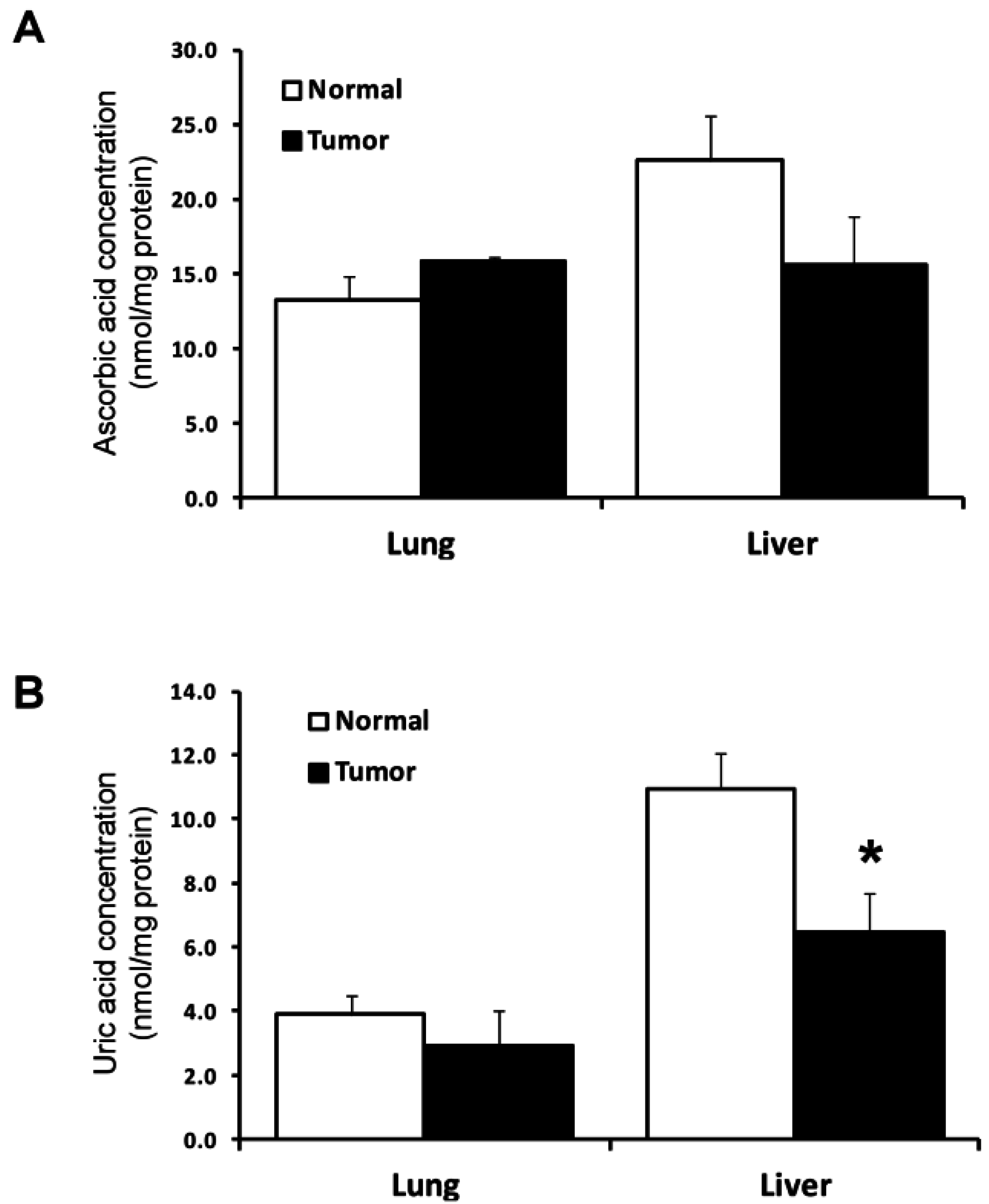{kind=link}
| Sample No. | Name | Pathological Diagnosis 2 | Cancer Cell % | Grade | Stage | Sex | Age |
|---|---|---|---|---|---|---|---|
| 1 | N1 | Normal Adjacent Tissue | - | - | - | M | 48 |
| T1 | Adenocarcinoma | 70% | 2 | IIB | |||
| 2 | N2 | Normal Adjacent Tissue | - | - | - | M | 56 |
| T2 | Adenocarcinoma | 70% | 2 | IIB | |||
| 3 | N3 | Normal Adjacent Tissue | - | - | - | M | 59 |
| T3 | Adenocarcinoma | 70% | 2 | IIB |
| Sample No. | Name | Pathological Diagnosis | Cancer Cell % | Grade | Stage | Sex | Age |
|---|---|---|---|---|---|---|---|
| 1 | N1 | Normal Adjacent Tissue | - | - | - | M | 64 |
| T1 | Hepatocellular Carcinoma | 90% | 2 | II | |||
| 2 | N2 | Normal Adjacent Tissue | - | - | - | M | 50 |
| T2 | Hepatocellular Carcinoma | 90% | 2 | II | |||
| 3 | N3 | Normal Adjacent Tissue | - | - | - | M | 63 |
| T3 | Hepatocellular Carcinoma | 90% | 2 | II |
2.2. Lung and Liver Tumors Utilize the TXN System Differently

2.3. Lung and Liver Tumors Utilize the GSH System Differently


2.4. Utilization of Other Antioxidant Proteins and Compounds in Lung and Liver Tumor and Normal Tissues

3. Discussion
| Antioxidant | Lung 2 | Liver 2 |
|---|---|---|
| TXNRD1 | ↑ | ↑ |
| TXN | ↑ | NS |
| PRDX1 | ↑ | ↓ |
| GPX1 | NS | ↓ |
| GPX2 | ND | NS |
| GPX4 | NS | ↑ |
| GSR | NS | ↓ |
| GCLC | ↓ | ↓ |
| GSS | NS | ↓ |
| GLRX | NS | ↓ |
| GGT1 | ↓ | ↑ |
| GSTA1 | ND | ↓ |
| GSTP1 | ↑ | NS |
| SOD1 | NS | ↓ |
| CAT | NS | ↓ |
| G6PD | ↑ | ↑ |
| Ascorbic acid | NS | NS |
| Uric acid | NS | ↓ |
4. Experimental Section
4.1. Tissues and Other Materials
4.2. Tissue Preparation, Protein Concentration, and Western Blotting
4.3. Enzyme Activity Assay
4.4. Determination of Total Glutathione, Ascorbic Acid, and Uric Acid Concentrations
4.5. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; Tuveson, D.A. The promise and perils of antioxidants for cancer patients. N. Engl. J. Med. 2014, 371, 177–178. [Google Scholar] [PubMed]
- Glasauer, A.; Chandel, N.S. Targeting antioxidants for cancer therapy. Biochem. Pharmacol. 2014, 92, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.C.; Hevia, D.; Patchva, S.; Park, B.; Koh, W.; Aggarwal, B.B. Upsides and downsides of reactive oxygen species for cancer: The roles of reactive oxygen species in tumorigenesis, prevention, and therapy. Antioxid. Redox Signal. 2012, 16, 1295–1322. [Google Scholar] [CrossRef] [PubMed]
- Kardeh, S.; Ashkani-Esfahani, S.; Alizadeh, A.M. Paradoxical action of reactive oxygen species in creation and therapy of cancer. Eur. J. Pharmacol. 2014, 735, 150–168. [Google Scholar] [CrossRef] [PubMed]
- Pietraforte, D.; Malorni, W. Focusing at the double-edged sword of redox imbalance: Signals for cell survival or for cell death? Antioxid. Redox Signal. 2014, 21, 52–55. [Google Scholar] [CrossRef] [PubMed]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ros in cancer: Initiators, amplifiers or an achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Saeidnia, S.; Abdollahi, M. Antioxidants: Friends or foe in prevention or treatment of cancer: The debate of the century. Toxicol. Appl. Pharmacol. 2013, 271, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Dammeyer, P.; Arner, E.S. Human protein atlas of redox systems—What can be learnt? Biochim. Biophys. Acta 2011, 1810, 111–138. [Google Scholar] [CrossRef] [PubMed]
- Godoy, J.R.; Funke, M.; Ackermann, W.; Haunhorst, P.; Oesteritz, S.; Capani, F.; Elsasser, H.P.; Lillig, C.H. Redox atlas of the mouse. Immunohistochemical detection of glutaredoxin-, peroxiredoxin-, and thioredoxin-family proteins in various tissues of the laboratory mouse. Biochim. Biophys. Acta 2011, 1810, 2–92. [Google Scholar] [CrossRef] [PubMed]
- Harris, I.S.; Treloar, A.E.; Inoue, S.; Sasaki, M.; Gorrini, C.; Lee, K.C.; Yung, K.Y.; Brenner, D.; Knobbe-Thomsen, C.B.; Cox, M.A.; et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 2015, 27, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Hellfritsch, J.; Kirsch, J.; Schneider, M.; Fluege, T.; Wortmann, M.; Frijhoff, J.; Dagnell, M.; Fey, T.; Esposito, I.; Kolle, P.; et al. Knockout of mitochondrial thioredoxin reductase stabilizes prolyl hydroxylase 2 and inhibits tumor growth and tumor-derived angiogenesis. Antioxid. Redox Signal. 2015, 22, 938–950. [Google Scholar] [CrossRef] [PubMed]
- Kiebala, M.; Skalska, J.; Casulo, C.; Brookes, P.S.; Peterson, D.R.; Hilchey, S.P.; Dai, Y.; Grant, S.; Maggirwar, S.B.; Bernstein, S.H. Dual targeting of the thioredoxin and glutathione antioxidant systems in malignant b cells: A novel synergistic therapeutic approach. Exp. Hematol. 2015, 43, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Kwee, J.K. A paradoxical chemoresistance and tumor suppressive role of antioxidant in solid cancer cells: A strange case of Dr. Jekyll and Mr. Hyde. Biomed. Res. Int. 2014, 2014, 209845. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, D.F.; Abderrazak, A.; el Hadri, K.; Simmet, T.; Rouis, M. The thioredoxin system as a therapeutic target in human health and disease. Antioxid. Redox Signal. 2013, 19, 1266–1303. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.K.; Schneider, M.; Kolle, P.; Kuhlencordt, P.; Forster, H.; Beck, H.; Bornkamm, G.W.; Conrad, M. Loss of thioredoxin reductase 1 renders tumors highly susceptible to pharmacologic glutathione deprivation. Cancer Res. 2010, 70, 9505–9514. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Xu, J.; Arner, E.S. Thiophosphate and selenite conversely modulate cell death induced by glutathione depletion or cisplatin: Effects related to activity and sec contents of thioredoxin reductase. Biochem. J. 2012, 447, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Traverso, N.; Ricciarelli, R.; Nitti, M.; Marengo, B.; Furfaro, A.L.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. Role of glutathione in cancer progression and chemoresistance. Oxid. Med. Cell. Longev. 2013, 2013, 972913. [Google Scholar] [CrossRef] [PubMed]
- Sayin, V.I.; Ibrahim, M.X.; Larsson, E.; Nilsson, J.A.; Lindahl, P.; Bergo, M.O. Antioxidants accelerate lung cancer progression in mice. Sci. Transl. Med. 2014, 6, 221ra215. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Yan, J.; Deng, S.; Lan, L.; He, F.; Kuang, B.; Zeng, H. A thioredoxin reductase inhibitor induces growth inhibition and apoptosis in five cultured human carcinoma cell lines. Cancer Lett. 2006, 236, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Tang, J.; Xu, J.; Cao, L.; Jia, G.; Long, D.; Liu, G.; Chen, X.; Wang, K. Selenoprotein genes exhibit differential expression patterns between hepatoma HepG2 and normal hepatocytes LO2 cell lines. Biol. Trace Elem. Res. 2015, 167, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Novoselov, S.V.; Calvisi, D.F.; Labunskyy, V.M.; Factor, V.M.; Carlson, B.A.; Fomenko, D.E.; Moustafa, M.E.; Hatfield, D.L.; Gladyshev, V.N. Selenoprotein deficiency and high levels of selenium compounds can effectively inhibit hepatocarcinogenesis in transgenic mice. Oncogene 2005, 24, 8003–8011. [Google Scholar] [CrossRef] [PubMed]
- Kasaikina, M.V.; Turanov, A.A.; Avanesov, A.; Schweizer, U.; Seeher, S.; Bronson, R.T.; Novoselov, S.N.; Carlson, B.A.; Hatfield, D.L.; Gladyshev, V.N. Contrasting roles of dietary selenium and selenoproteins in chemically induced hepatocarcinogenesis. Carcinogenesis 2013, 34, 1089–1095. [Google Scholar] [CrossRef] [PubMed]
- Brozmanova, J.; Manikova, D.; Vlckova, V.; Chovanec, M. Selenium: A double-edged sword for defense and offence in cancer. Arch. Toxicol. 2010, 84, 919–938. [Google Scholar] [CrossRef] [PubMed]
- Hatfield, D.L.; Tsuji, P.A.; Carlson, B.A.; Gladyshev, V.N. Selenium and selenocysteine: Roles in cancer, health, and development. Trends Biochem. Sci. 2014, 39, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Steinbrenner, H.; Speckmann, B.; Sies, H. Toward understanding success and failures in the use of selenium for cancer prevention. Antioxid. Redox Signal. 2013, 19, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Vinceti, M.; Crespi, C.M.; Malagoli, C.; del Giovane, C.; Krogh, V. Friend or foe? The current epidemiologic evidence on selenium and human cancer risk. J. Environ. Sci. Health C Environ. Carcinog. Ecotoxicol. Rev. 2013, 31, 305–341. [Google Scholar] [CrossRef] [PubMed]
- Carlson, B.A.; Yoo, M.H.; Tobe, R.; Mueller, C.; Naranjo-Suarez, S.; Hoffmann, V.J.; Gladyshev, V.N.; Hatfield, D.L. Thioredoxin reductase 1 protects against chemically induced hepatocarcinogenesis via control of cellular redox homeostasis. Carcinogenesis 2012, 33, 1806–1813. [Google Scholar] [CrossRef] [PubMed]
- Cebula, M.; Schmidt, E.E.; Arner, E.S. TrxR1 as a potent regulator of the nrf2-keap1 response system. Antioxid. Redox Signal. 2015, 23, 823–853. [Google Scholar] [CrossRef] [PubMed]
- Dai, B.; Yoo, S.Y.; Bartholomeusz, G.; Graham, R.A.; Majidi, M.; Yan, S.; Meng, J.; Ji, L.; Coombes, K.; Minna, J.D.; et al. Keap1-dependent synthetic lethality induced by akt and txnrd1 inhibitors in lung cancer. Cancer Res. 2013, 73, 5532–5543. [Google Scholar] [CrossRef] [PubMed]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonen, A.L. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; Villeneuve, N.F.; Sun, Z.; Wong, P.K.; Zhang, D.D. Dual roles of Nrf2 in cancer. Pharmacol. Res. 2008, 58, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Satoh, H.; Moriguchi, T.; Takai, J.; Ebina, M.; Yamamoto, M. Nrf2 prevents initiation but accelerates progression through the Kras signaling pathway during lung carcinogenesis. Cancer Res. 2013, 73, 4158–4168. [Google Scholar] [CrossRef] [PubMed]
- Yoo, M.H.; Xu, X.M.; Carlson, B.A.; Gladyshev, V.N.; Hatfield, D.L. Thioredoxin reductase 1 deficiency reverses tumor phenotype and tumorigenicity of lung carcinoma cells. J. Biol. Chem. 2006, 281, 13005–13008. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, S.E.; Prast-Nielsen, S.; Flaberg, E.; Szekely, L.; Arner, E.S. High levels of thioredoxin reductase 1 modulate drug-specific cytotoxic efficacy. Free Radic. Biol. Med. 2009, 47, 1661–1671. [Google Scholar] [CrossRef] [PubMed]
- Suadicani, P.; Hein, H.O.; Gyntelberg, F. Serum selenium level and risk of lung cancer mortality: A 16-year follow-up of the copenhagen male study. Eur. Respir. J. 2012, 39, 1443–1448. [Google Scholar] [CrossRef] [PubMed]
- Fritz, H.; Kennedy, D.; Fergusson, D.; Fernandes, R.; Cooley, K.; Seely, A.; Sagar, S.; Wong, R.; Seely, D. Selenium and lung cancer: A systematic review and meta analysis. PLoS ONE 2011, 6, e26259. [Google Scholar] [CrossRef] [PubMed]
- Jaworska, K.; Gupta, S.; Durda, K.; Muszynska, M.; Sukiennicki, G.; Jaworowska, E.; Grodzki, T.; Sulikowski, M.; Waloszczyk, P.; Wojcik, J.; et al. A low selenium level is associated with lung and laryngeal cancers. PLoS ONE 2013, 8, e59051. [Google Scholar] [CrossRef] [PubMed]
- Karp, D.D.; Lee, S.J.; Keller, S.M.; Wright, G.S.; Aisner, S.; Belinsky, S.A.; Johnson, D.H.; Johnston, M.R.; Goodman, G.; Clamon, G.; et al. Randomized, double-blind, placebo-controlled, phase III chemoprevention trial of selenium supplementation in patients with resected stage I non-small-cell lung cancer: ECOG 5597. J. Clin. Oncol. 2013, 31, 4179–4187. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.X.; Wu, F.X.; Luo, M.; Ma, L.; Gao, K.F.; Li, J.; Wu, W.J.; Huang, S.; Yang, Q.; Liu, K.; et al. The glutathione s-transferase p1 341c>t polymorphism and cancer risk: A meta-analysis of 28 case-control studies. PLoS ONE 2013, 8, e56722. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C. Regulation of hepatic glutathione synthesis: Current concepts and controversies. FASEB J. 1999, 13, 1169–1183. [Google Scholar] [CrossRef] [PubMed]
- Schnekenburger, M.; Karius, T.; Diederich, M. Regulation of epigenetic traits of the glutathione s-transferase p1 gene: From detoxification toward cancer prevention and diagnosis. Front. Pharmacol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Singh, S. Cytoprotective and regulatory functions of glutathione s-transferases in cancer cell proliferation and cell death. Cancer Chemother. Pharmacol. 2015, 75. [Google Scholar] [CrossRef] [PubMed]
- Balendiran, G.K.; Dabur, R.; Fraser, D. The role of glutathione in cancer. Cell Biochem. Funct. 2004, 22, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Arner, E.S.; Holmgren, A. Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 2000, 267, 6102–6109. [Google Scholar] [CrossRef] [PubMed]
- Arner, E.S.J. Focus on mammalian thioredoxin reductases—Important selenoproteins with versatile functions. Biochim. Biophys. Acta Gen. Subj. 2009, 1790, 495–526. [Google Scholar] [CrossRef] [PubMed]
- Rundlof, A.K.; Arner, E.S. Regulation of the mammalian selenoprotein thioredoxin reductase 1 in relation to cellular phenotype, growth, and signaling events. Antioxid. Redox Signal. 2004, 6, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Rundlof, A.K.; Janard, M.; Miranda-Vizuete, A.; Arner, E.S. Evidence for intriguingly complex transcription of human thioredoxin reductase 1. Free Radic. Biol. Med. 2004, 36, 641–656. [Google Scholar] [CrossRef] [PubMed]
- Arner, E.S.J.; Holmgren, A. The thioredoxin system in cancer. Semin. Cancer Biol. 2006, 16, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Biaglow, J.E.; Miller, R.A. The thioredoxin reductase/thioredoxin system: Novel redox targets for cancer therapy. Cancer Biol. Ther. 2005, 4, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Carlson, B.A. Dual functions of selenoproteins in cancer: Thioredoxin reductase. In Diversity of Selenium Functions in Health and Disease; Brigelius-Flohe, R., Sies, H., Eds.; CRC Press: Boca Raton, FL, USA, 2015; p. 430. [Google Scholar]
- Gromer, S.; Urig, S.; Becker, K. The thioredoxin system—From science to clinic. Med. Res. Rev. 2004, 24, 40–89. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.; Awwad, R.T.; Smart, D.D.; Spitz, D.R.; Gius, D. Thioredoxin reductase as a novel molecular target for cancer therapy. Cancer Lett. 2006, 236, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Mimura, J.; Yamamoto, M. Discovery of the negative regulator of Nrf2, Keap1: A historical overview. Antioxid. Redox Signal. 2010, 13, 1665–1678. [Google Scholar] [CrossRef] [PubMed]
- Kensler, T.W.; Wakabayashi, N. Nrf2: Friend or foe for chemoprevention? Carcinogenesis 2010, 31, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Moon, E.J.; Giaccia, A. Dual roles of Nrf2 in tumor prevention and progression: Possible implications in cancer treatment. Free Radic. Biol. Med. 2015, 79, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Suvorova, E.S.; Lucas, O.; Weisend, C.M.; Rollins, M.F.; Merrill, G.F.; Capecchi, M.R.; Schmidt, E.E. Cytoprotective Nrf2 pathway is induced in chronically txnrd 1-deficient hepatocytes. PLoS ONE 2009, 4, e6158. [Google Scholar] [CrossRef] [PubMed]
- Murakami, S.; Motohashi, H. Roles of Nrf2 in cell proliferation and differentiation. Free Radic. Biol. Med. 2015. [Google Scholar] [CrossRef] [PubMed]
- Osburn, W.O.; Karim, B.; Dolan, P.M.; Liu, G.; Yamamoto, M.; Huso, D.L.; Kensler, T.W. Increased colonic inflammatory injury and formation of aberrant crypt foci in Nrf2-deficient mice upon dextran sulfate treatment. Int. J. Cancer 2007, 121, 1883–1891. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Huang, M.T.; Shen, G.; Yuan, X.; Lin, W.; Khor, T.O.; Conney, A.H.; Kong, A.N. Inhibition of 7,12-dimethylbenz(a)anthracene-induced skin tumorigenesis in c57bl/6 mice by sulforaphane is mediated by nuclear factor e2-related factor 2. Cancer Res. 2006, 66, 8293–8296. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhang, Z.; Zhu, Y.; Qin, S. Glucose-6-phosphate dehydrogenase: A biomarker and potential therapeutic target for cancer. Anticancer Agents Med. Chem. 2014, 14, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Du, W.; Wu, M. Regulation of the pentose phosphate pathway in cancer. Protein Cell 2014, 5, 592–602. [Google Scholar] [CrossRef] [PubMed]
- Glorieux, C.; Zamocky, M.; Sandoval, J.M.; Verrax, J.; Calderon, P.B. Regulation of catalase expression in healthy and cancerous cells. Free Radic. Biol. Med. 2015, 87, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Papa, L.; Manfredi, G.; Germain, D. Sod1, an unexpected novel target for cancer therapy. Genes Cancer 2014, 5, 15–21. [Google Scholar] [PubMed]
- Elchuri, S.; Oberley, T.D.; Qi, W.; Eisenstein, R.S.; Jackson Roberts, L.; van Remmen, H.; Epstein, C.J.; Huang, T.T. Cuznsod deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene 2005, 24, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, P.A.; Carlson, B.A.; Yoo, M.H.; Naranjo-Suarez, S.; Xu, X.M.; He, Y.; Asaki, E.; Seifried, H.E.; Reinhold, W.C.; Davis, C.D.; et al. The 15kda selenoprotein and thioredoxin reductase 1 promote colon cancer by different pathways. PLoS ONE 2015, 10, e0124487. [Google Scholar] [CrossRef] [PubMed]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by Gpx4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.I.; Combs, G.F.J. Selenium as a cancer preventive agent. In Selenium: Its Molecular Biology and Role in Human Health, 3rd ed.; Hatfield, D.L., Berry, M.J., Gladyshev, V.N., Eds.; Springer Science + Business Media, LLC: New York, NY, USA, 2012. [Google Scholar]
- Aldridge, G.M.; Podrebarac, D.M.; Greenough, W.T.; Weiler, I.J. The use of total protein stains as loading controls: An alternative to high-abundance single-protein controls in semi-quantitative immunoblotting. J. Neurosci. Methods 2008, 172, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.A.; An, J.; Peller, D.; Bowser, R. Total protein is an effective loading control for cerebrospinal fluid western blots. J. Neurosci. Methods 2015, 251, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Eaton, S.L.; Roche, S.L.; Llavero Hurtado, M.; Oldknow, K.J.; Farquharson, C.; Gillingwater, T.H.; Wishart, T.M. Total protein analysis as a reliable loading control for quantitative fluorescent western blotting. PLoS ONE 2013, 8, e72457. [Google Scholar] [CrossRef] [PubMed]
- Klein, D.; Kern, R.M.; Sokol, R.Z. A method for quantification and correction of proteins after transfer to immobilization membranes. Biochem. Mol. Biol. Int. 1995, 36, 59–66. [Google Scholar] [PubMed]
- Welinder, C.; Ekblad, L. Coomassie staining as loading control in western blot analysis. J. Proteome Res. 2011, 10, 1416–1419. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tobe, R.; Carlson, B.A.; Tsuji, P.A.; Lee, B.J.; Gladyshev, V.N.; Hatfield, D.L. Differences in Redox Regulatory Systems in Human Lung and Liver Tumors Suggest Different Avenues for Therapy. Cancers 2015, 7, 2262-2276. https://doi.org/10.3390/cancers7040889
Tobe R, Carlson BA, Tsuji PA, Lee BJ, Gladyshev VN, Hatfield DL. Differences in Redox Regulatory Systems in Human Lung and Liver Tumors Suggest Different Avenues for Therapy. Cancers. 2015; 7(4):2262-2276. https://doi.org/10.3390/cancers7040889
Chicago/Turabian StyleTobe, Ryuta, Bradley A. Carlson, Petra A. Tsuji, Byeong Jae Lee, Vadim N. Gladyshev, and Dolph L. Hatfield. 2015. "Differences in Redox Regulatory Systems in Human Lung and Liver Tumors Suggest Different Avenues for Therapy" Cancers 7, no. 4: 2262-2276. https://doi.org/10.3390/cancers7040889
APA StyleTobe, R., Carlson, B. A., Tsuji, P. A., Lee, B. J., Gladyshev, V. N., & Hatfield, D. L. (2015). Differences in Redox Regulatory Systems in Human Lung and Liver Tumors Suggest Different Avenues for Therapy. Cancers, 7(4), 2262-2276. https://doi.org/10.3390/cancers7040889