
Prostate Cancer Stem-like Cells Contribute to the Development of Castration-Resistant Prostate Cancer

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Persistent AR Signaling Plays a Role in Developing Resistance to Inhibition of AR Signaling
3. PCSLCs are a Critical Contributor to Resistance Acquisition to AR Inhibition
3.1. Prostate Cancer Stem-Like Cells
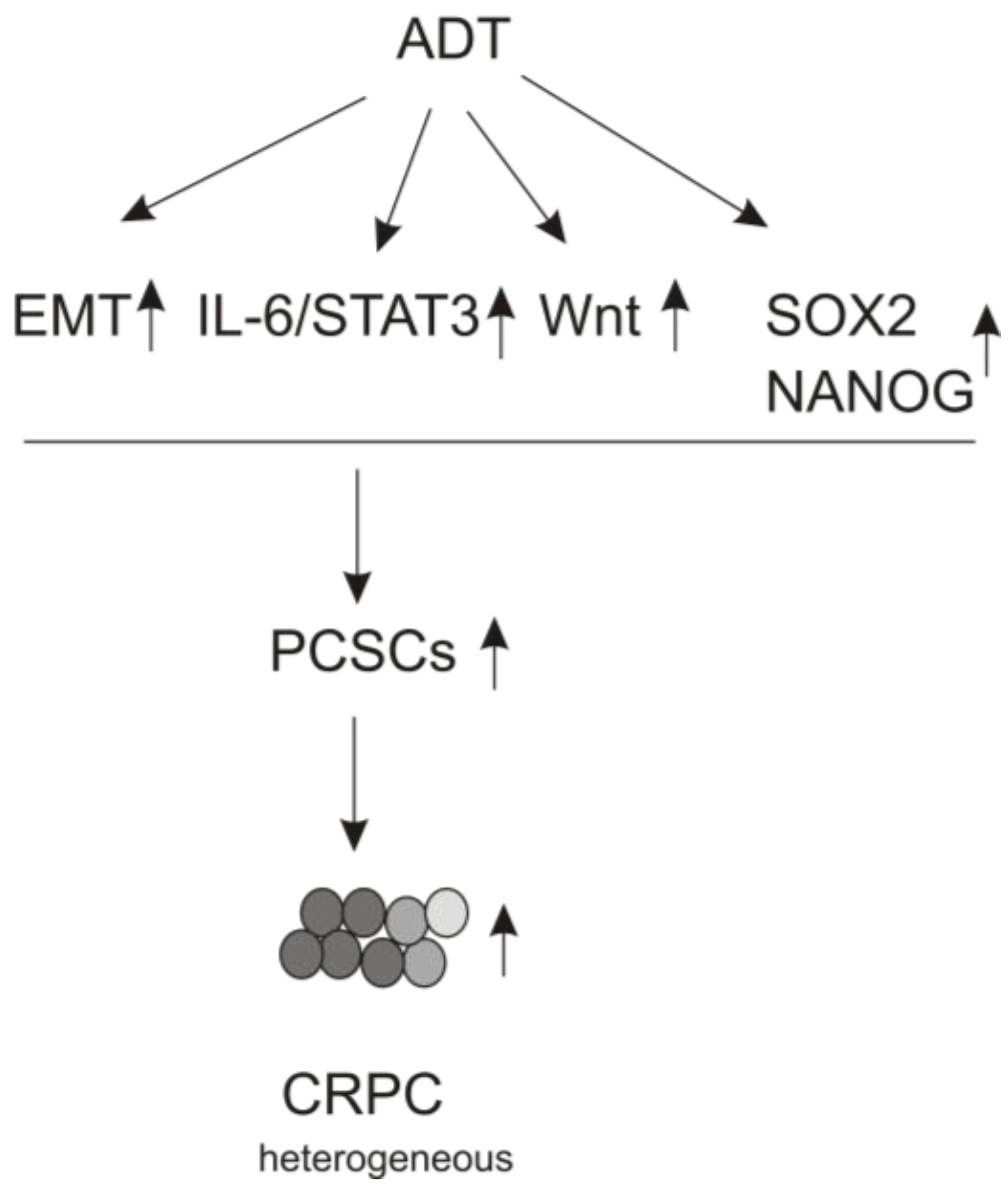
3.2. PCSLC Contributes to CRPC
3.3. ADT Leads to EMT

3.4. The IL-6/JAK/STAT3 Pathway Plays a Role in ADT-induced PCSLC Evolvement
3.5. Wnt Signaling Contributing to Resistance to ADT
3.6. ADT Stimulates PCSLC via Upregulation of Pluripotency Genes
3.6.1. SOX2
3.6.2. NANOG
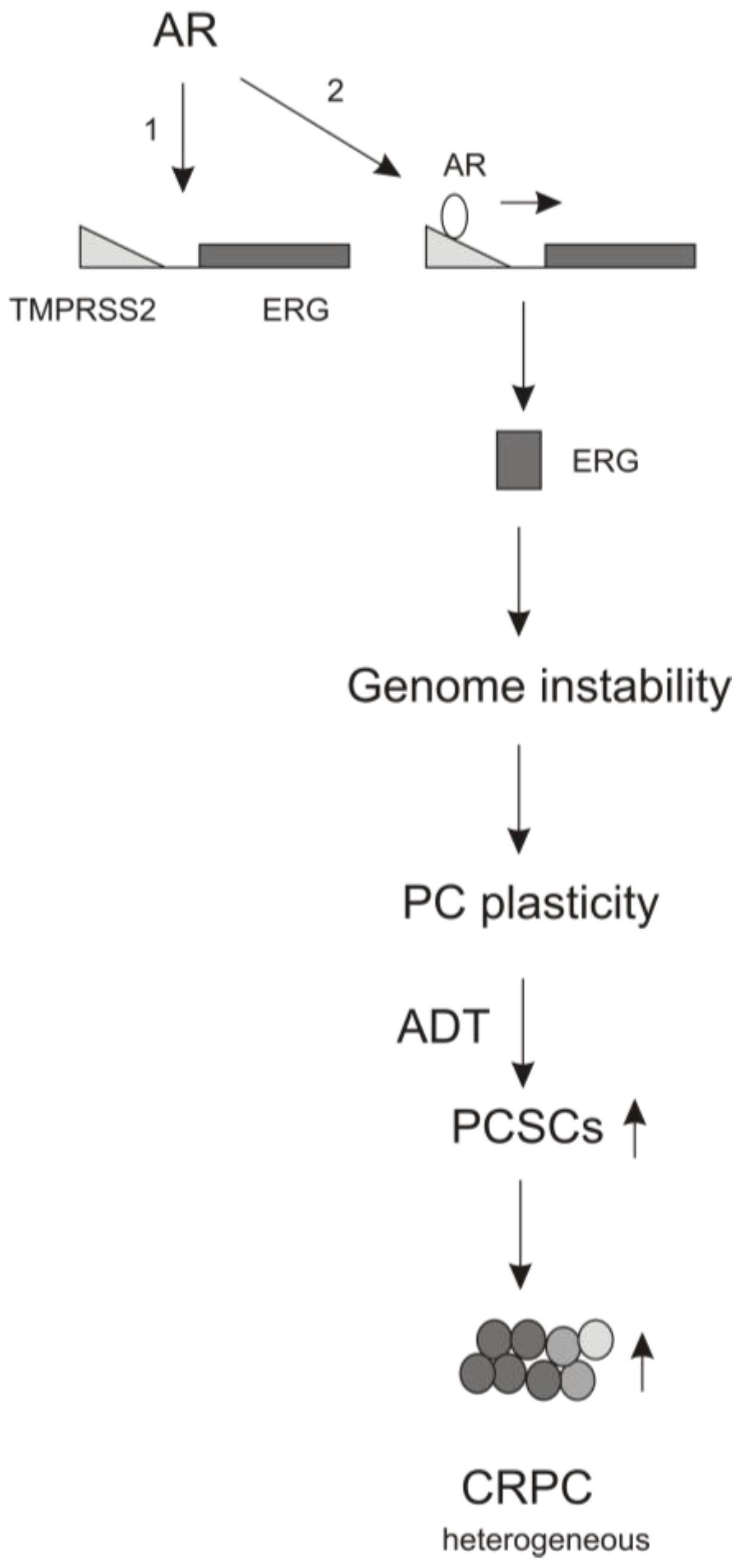
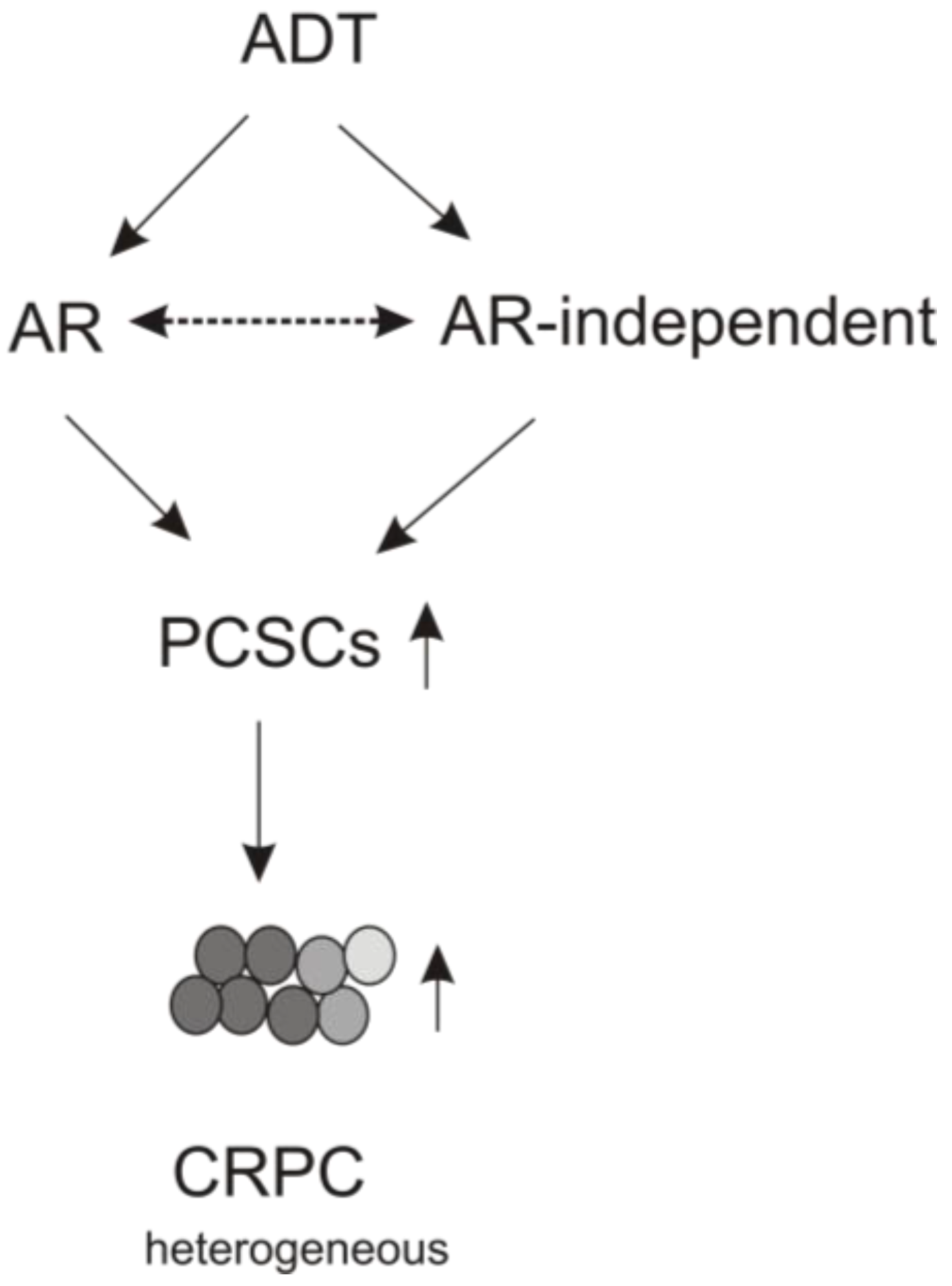
4. The Contributions of AR Signaling to ADT-induced PCSLC Expansion

5. Perspectives

Acknowledgments
Author Contributions
Conflicts of Interest
References
- Siegel, R.; Ward, E.; Brawley, O.; Jemal, A. Cancer statistics, 2011: The impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J. Clin. 2011, 61, 212–236. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.S. The androgen receptor in prostate cancer: Therapy target in search of an integrated diagnostic test. Adv. Anat. Pathol. 2007, 14, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Moon, C.; Park, J.C.; Chae, Y.K.; Yun, J.H.; Kim, S. Current status of experimental therapeutics for prostate cancer. Cancer Lett. 2008, 266, 116–134. [Google Scholar] [CrossRef] [PubMed]
- Lytton, B. Prostate cancer: A brief history and the discovery of hormonal ablation treatment. J. Urol. 2001, 165, 1859–1862. [Google Scholar] [CrossRef]
- Wong, Y.N.; Ferraldeschi, R.; Attard, G.; de Bono, J. Evolution of androgen receptor targeted therapy for advanced prostate cancer. Nat. Rev. Clin. Oncol. 2014, 11, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Heinlein, C.A.; Chang, C. Androgen receptor in prostate cancer. Endocr. Rev. 2004, 25, 276–308. [Google Scholar] [CrossRef] [PubMed]
- Tannock, I.F.; de Wit, R.; Berry, W.R.; Horti, J.; Pluzanska, A.; Chi, K.N.; Oudard, S.; Theodore, C.; James, N.D.; Turesson, I.; et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N. Engl. J. Med. 2004, 351, 1502–1512. [Google Scholar] [CrossRef] [PubMed]
- Berthold, D.R.; Pond, G.R.; Soban, F.; de Wit, R.; Eisenberger, M.; Tannock, I.F. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer: Updated survival in the tax 327 study. J. Clin. Oncol. 2008, 26, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Mitsiades, N. A road map to comprehensive androgen receptor axis targeting for castration-resistant prostate cancer. Cancer Res. 2013, 73, 4599–4605. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Smith, A.; Ong, M.; de Bono, J.S. Novel drugs targeting the androgen receptor pathway in prostate cancer. Cancer Metastasis Rev. 2014, 33, 567–579. [Google Scholar] [CrossRef] [PubMed]
- De Bono, J.S.; Logothetis, C.J.; Molina, A.; Fizazi, K.; North, S.; Chu, L.; Chi, K.N.; Jones, R.J.; Goodman, O.B., Jr.; Saad, F.; et al. Abiraterone and increased survival in metastatic prostate cancer. N. Engl. J. Med. 2011, 364, 1995–2005. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.E.; Sternberg, C.N.; Miller, K.; de Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [PubMed]
- Li, Y.; Chan, S.C.; Brand, L.J.; Hwang, T.H.; Silverstein, K.A.; Dehm, S.M. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 2013, 73, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. Ar-v7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [PubMed]
- Pienta, K.J.; Walia, G.; Simons, J.W.; Soule, H.R. Beyond the androgen receptor: New approaches to treating metastatic prostate cancer. Report of the 2013 prouts neck prostate cancer meeting. Prostate 2014, 74, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Ramadan, W.H.; Kabbara, W.K.; Al Basiouni Al Masri, H.S. Enzalutamide for patients with metastatic castration-resistant prostate cancer. OncoTargets Ther. 2015, 8, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Oudard, S. Progress in emerging therapies for advanced prostate cancer. Cancer Treat. Rev. 2013, 39, 275–289. [Google Scholar] [CrossRef] [PubMed]
- Mostaghel, E.A.; Page, S.T.; Lin, D.W.; Fazli, L.; Coleman, I.M.; True, L.D.; Knudsen, B.; Hess, D.L.; Nelson, C.C.; Matsumoto, A.M.; et al. Intraprostatic androgens and androgen-regulated gene expression persist after testosterone suppression: Therapeutic implications for castration-resistant prostate cancer. Cancer Res. 2007, 67, 5033–5041. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, R.B.; Mostaghel, E.A.; Vessella, R.; Hess, D.L.; Kalhorn, T.F.; Higano, C.S.; True, L.D.; Nelson, P.S. Maintenance of intratumoral androgens in metastatic prostate cancer: A mechanism for castration-resistant tumor growth. Cancer Res. 2008, 68, 4447–4454. [Google Scholar] [CrossRef] [PubMed]
- Mitsiades, N.; Sung, C.C.; Schultz, N.; Danila, D.C.; He, B.; Eedunuri, V.K.; Fleisher, M.; Sander, C.; Sawyers, C.L.; Scher, H.I. Distinct patterns of dysregulated expression of enzymes involved in androgen synthesis and metabolism in metastatic prostate cancer tumors. Cancer Res. 2012, 72, 6142–6152. [Google Scholar] [CrossRef] [PubMed]
- Levesque, E.; Huang, S.P.; Audet-Walsh, E.; Lacombe, L.; Bao, B.Y.; Fradet, Y.; Laverdiere, I.; Rouleau, M.; Huang, C.Y.; Yu, C.C.; et al. Molecular markers in key steroidogenic pathways, circulating steroid levels, and prostate cancer progression. Clin. Cancer Res. 2013, 19, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.; Garcia, J.A. Abiraterone acetate, a novel adrenal inhibitor in metastatic castration-resistant prostate cancer. Curr. Oncol. Rep. 2011, 13, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Vasaitis, T.S.; Bruno, R.D.; Njar, V.C. Cyp17 inhibitors for prostate cancer therapy. J. Steroid biochem. Mol. Biol. 2011, 125, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Labrie, F. Adrenal androgens and intracrinology. Semin. Reprod. Med. 2004, 22, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Scher, H.I.; Molina, A.; Logothetis, C.J.; Chi, K.N.; Jones, R.J.; Staffurth, J.N.; North, S.; Vogelzang, N.J.; Saad, F.; et al. Abiraterone acetate for treatment of metastatic castration-resistant prostate cancer: Final overall survival analysis of the COU-AA-301 randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. 2012, 13, 983–992. [Google Scholar] [CrossRef]
- Ryan, C.J.; Smith, M.R.; Fizazi, K.; Saad, F.; Mulders, P.F.; Sternberg, C.N.; Miller, K.; Logothetis, C.J.; Shore, N.D.; Small, E.J.; et al. Abiraterone acetate plus prednisone versus placebo plus prednisone in chemotherapy-naive men with metastatic castration-resistant prostate cancer (COU-AA-302): Final overall survival analysis of a randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. 2015, 16, 152–160. [Google Scholar] [CrossRef]
- Augello, M.A.; Hickey, T.E.; Knudsen, K.E. Foxa1: Master of steroid receptor function in cancer. EMBO J. 2011, 30, 3885–3894. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, W.; Zhang, Y.; Yuan, X.; Xu, K.; Yu, J.; Chen, Z.; Beroukhim, R.; Wang, H.; Lupien, M.; et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell 2009, 138, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.L.; Hickey, T.E.; Warren, A.Y.; Vowler, S.L.; Carroll, T.; Lamb, A.D.; Papoutsoglou, N.; Neal, D.E.; Tilley, W.D.; Carroll, J.S. Elevated levels of foxa1 facilitate androgen receptor chromatin binding resulting in a crpc-like phenotype. Oncogene 2014, 33, 5666–5674. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Visakorpi, T.; Hyytinen, E.; Koivisto, P.; Tanner, M.; Keinanen, R.; Palmberg, C.; Palotie, A.; Tammela, T.; Isola, J.; Kallioniemi, O.P. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat. Genet. 1995, 9, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.; Krishna, N.S.; Grigor, K.M.; Bartlett, J.M. Androgen receptor gene amplification and protein expression in hormone refractory prostate cancer. Br. J. Cancer 2003, 89, 552–556. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Dehm, S.M.; Tindall, D.J. Alternatively spliced androgen receptor variants. Endocr. Relat. Cancer 2011, 18, R183–R196. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Dunn, T.A.; Wei, S.; Isharwal, S.; Veltri, R.W.; Humphreys, E.; Han, M.; Partin, A.W.; Vessella, R.L.; Isaacs, W.B.; et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009, 69, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Yang, X.; Sun, F.; Jiang, R.; Linn, D.E.; Chen, H.; Chen, H.; Kong, X.; Melamed, J.; Tepper, C.G.; et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 2009, 69, 2305–2313. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Laitinen, S.; Khan, S.; Vihinen, M.; Kowalski, J.; Yu, G.; Chen, L.; Ewing, C.M.; Eisenberger, M.A.; Carducci, M.A.; et al. Copy number analysis indicates monoclonal origin of lethal metastatic prostate cancer. Nat. Med. 2009, 15, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, C.E.; Demichelis, F.; Rubin, M.A. Molecular genetics of prostate cancer: Emerging appreciation of genetic complexity. Histopathology 2012, 60, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.C.; Giannopoulou, E.G.; Park, K.; Mosquera, J.M.; Sboner, A.; Tewari, A.K.; Garraway, L.A.; Beltran, H.; Rubin, M.A.; Elemento, O. Epigenomic alterations in localized and advanced prostate cancer. Neoplasia 2013, 15, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Pellacani, D.; Kestoras, D.; Droop, A.P.; Frame, F.M.; Berry, P.A.; Lawrence, M.G.; Stower, M.J.; Simms, M.S.; Mann, V.M.; Collins, A.T.; et al. DNA hypermethylation in prostate cancer is a consequence of aberrant epithelial differentiation and hyperproliferation. Cell Death Differ. 2014, 21, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Logothetis, C.J.; Gallick, G.E.; Maity, S.N.; Kim, J.; Aparicio, A.; Efstathiou, E.; Lin, S.H. Molecular classification of prostate cancer progression: Foundation for marker-driven treatment of prostate cancer. Cancer Discov. 2013, 3, 849–861. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, D.; Lorente, D.; Rodriguez-Vida, A.; Omlin, A.; Pezaro, C.; Ferraldeschi, R.; Zivi, A.; Attard, G.; Chowdhury, S.; de Bono, J.S. Antitumour activity of enzalutamide (MDV3100) in patients with metastatic castration-resistant prostate cancer (CRPC) pre-treated with docetaxel and abiraterone. Eur. J. Cancer 2014, 50, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Schrader, A.J.; Boegemann, M.; Ohlmann, C.H.; Schnoeller, T.J.; Krabbe, L.M.; Hajili, T.; Jentzmik, F.; Stoeckle, M.; Schrader, M.; Herrmann, E.; et al. Enzalutamide in castration-resistant prostate cancer patients progressing after docetaxel and abiraterone. Eur. Urol. 2014, 65, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Azad, A.A.; Eigl, B.J.; Murray, R.N.; Kollmannsberger, C.; Chi, K.N. Efficacy of enzalutamide following abiraterone acetate in chemotherapy-naive metastatic castration-resistant prostate cancer patients. Eur. Urol. 2015, 67, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Noonan, K.L.; North, S.; Bitting, R.L.; Armstrong, A.J.; Ellard, S.L.; Chi, K.N. Clinical activity of abiraterone acetate in patients with metastatic castration-resistant prostate cancer progressing after enzalutamide. Ann. Oncol. 2013, 24, 1802–1807. [Google Scholar] [CrossRef] [PubMed]
- Verras, M.; Lee, J.; Xue, H.; Li, T.H.; Wang, Y.; Sun, Z. The androgen receptor negatively regulates the expression of c-Met: Implications for a novel mechanism of prostate cancer progression. Cancer Res. 2007, 67, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Carver, B.S.; Chapinski, C.; Wongvipat, J.; Hieronymus, H.; Chen, Y.; Chandarlapaty, S.; Arora, V.K.; Le, C.; Koutcher, J.; Scher, H.; et al. Reciprocal feedback regulation of PI3k and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell 2011, 19, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Mulholland, D.J.; Tran, L.M.; Li, Y.; Cai, H.; Morim, A.; Wang, S.; Plaisier, S.; Garraway, I.P.; Huang, J.; Graeber, T.G.; et al. Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell 2011, 19, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.T.; Berry, P.A.; Hyde, C.; Stower, M.J.; Maitland, N.J. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005, 65, 10946–10951. [Google Scholar] [CrossRef] [PubMed]
- Rane, J.K.; Greener, S.; Frame, F.M.; Mann, V.M.; Simms, M.S.; Collins, A.T.; Berney, D.M.; Maitland, N.J. Telomerase activity and telomere length in human benign prostatic hyperplasia stem-like cells and their progeny implies the existence of distinct basal and luminal cell lineages. European Urol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Miki, J.; Furusato, B.; Li, H.; Gu, Y.; Takahashi, H.; Egawa, S.; Sesterhenn, I.A.; McLeod, D.G.; Srivastava, S.; Rhim, J.S. Identification of putative stem cell markers, CD133 and CXCR4, in htert-immortalized primary nonmalignant and malignant tumor-derived human prostate epithelial cell lines and in prostate cancer specimens. Cancer Res. 2007, 67, 3153–3161. [Google Scholar] [CrossRef] [PubMed]
- Gu, G.; Yuan, J.; Wills, M.; Kasper, S. Prostate cancer cells with stem cell characteristics reconstitute the original human tumor in vivo. Cancer Res. 2007, 67, 4807–4815. [Google Scholar] [CrossRef] [PubMed]
- Patrawala, L.; Calhoun, T.; Schneider-Broussard, R.; Li, H.; Bhatia, B.; Tang, S.; Reilly, J.G.; Chandra, D.; Zhou, J.; Claypool, K.; et al. Highly purified CD44+ prostate cancer cells from xenograft human tumors are enriched in tumorigenic and metastatic progenitor cells. Oncogene 2006, 25, 1696–1708. [Google Scholar] [CrossRef] [PubMed]
- Huss, W.J.; Gray, D.R.; Greenberg, N.M.; Mohler, J.L.; Smith, G.J. Breast cancer resistance protein-mediated efflux of androgen in putative benign and malignant prostate stem cells. Cancer Res. 2005, 65, 6640–6650. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.A.; Mitrofanova, A.; Bergren, S.K.; Abate-Shen, C.; Cardiff, R.D.; Califano, A.; Shen, M.M. Lineage analysis of basal epithelial cells reveals their unexpected plasticity and supports a cell-of-origin model for prostate cancer heterogeneity. Nat. Cell Biol. 2013, 15, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Rycaj, K.; Liu, X.; Tang, D.G. New insights into prostate cancer stem cells. Cell Cycle 2013, 12, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhou, J.; Miki, J.; Furusato, B.; Gu, Y.; Srivastava, S.; McLeod, D.G.; Vogel, J.C.; Rhim, J.S. Telomerase-immortalized non-malignant human prostate epithelial cells retain the properties of multipotent stem cells. Exp. Cell Res. 2008, 314, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Liu, X.; Laffin, B.; Chen, X.; Choy, G.; Jeter, C.R.; Calhoun-Davis, T.; Li, H.; Palapattu, G.S.; Pang, S.; et al. The psa(-/lo) prostate cancer cell population harbors self-renewing long-term tumor-propagating cells that resist castration. Cell Stem Cell 2012, 10, 556–569. [Google Scholar] [CrossRef] [PubMed]
- Fidler, I.J.; Hart, I.R. Biological diversity in metastatic neoplasms: Origins and implications. Science 1982, 217, 998–1003. [Google Scholar] [CrossRef] [PubMed]
- Heppner, G.H.; Miller, B.E. Tumor heterogeneity: Biological implications and therapeutic consequences. Cancer Metastasis Rev. 1983, 2, 5–23. [Google Scholar] [CrossRef] [PubMed]
- Nowell, P.C. Mechanisms of tumor progression. Cancer Res. 1986, 46, 2203–2207. [Google Scholar] [PubMed]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M. Cancer stem cells as “units of selection”. Evolut. Appl. 2013, 6, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells in solid tumours: Accumulating evidence and unresolved questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Cojoc, M.; Mabert, K.; Muders, M.H.; Dubrovska, A. A role for cancer stem cells in therapy resistance: Cellular and molecular mechanisms. Semin. Cancer Biol. 2015, 31, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Vlashi, E.; Pajonk, F. Cancer stem cells, cancer cell plasticity and radiation therapy. Semin. Cancer Biol. 2015, 31, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Tang, D. Prostate cancer stem-like cells proliferate slowly and resist etoposide-induced cytotoxicity via enhancing DNA damage response. Exp. Cell Res. 2014, 328, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Frame, F.M.; Pellacani, D.; Collins, A.T.; Simms, M.S.; Mann, V.M.; Jones, G.D.; Meuth, M.; Bristow, R.G.; Maitland, N.J. HDAC inhibitor confers radiosensitivity to prostate stem-like cells. Br. J. Cancer 2013, 109, 3023–3033. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A. Epithelial plasticity: A common theme in embryonic and cancer cells. Science 2013, 342, 1234850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanpain, C.; Fuchs, E. Stem cell plasticity. Plasticity of epithelial stem cells in tissue regeneration. Science 2014, 344, 1242281. [Google Scholar] [CrossRef] [PubMed]
- Germann, M.; Wetterwald, A.; Guzman-Ramirez, N.; van der Pluijm, G.; Culig, Z.; Cecchini, M.G.; Williams, E.D.; Thalmann, G.N. Stem-like cells with luminal progenitor phenotype survive castration in human prostate cancer. Stem Cells 2012, 30, 1076–1086. [Google Scholar] [CrossRef] [PubMed]
- Colombel, M.; Eaton, C.L.; Hamdy, F.; Ricci, E.; van der Pluijm, G.; Cecchini, M.; Mege-Lechevallier, F.; Clezardin, P.; Thalmann, G. Increased expression of putative cancer stem cell markers in primary prostate cancer is associated with progression of bone metastases. Prostate 2012, 72, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Domingo-Domenech, J.; Vidal, S.J.; Rodriguez-Bravo, V.; Castillo-Martin, M.; Quinn, S.A.; Rodriguez-Barrueco, R.; Bonal, D.M.; Charytonowicz, E.; Gladoun, N.; de la Iglesia-Vicente, J.; et al. Suppression of acquired docetaxel resistance in prostate cancer through depletion of notch- and hedgehog-dependent tumor-initiating cells. Cancer Cell 2012, 22, 373–388. [Google Scholar] [CrossRef] [PubMed]
- Palapattu, G.S.; Wu, C.; Silvers, C.R.; Martin, H.B.; Williams, K.; Salamone, L.; Bushnell, T.; Huang, L.S.; Yang, Q.; Huang, J. Selective expression of CD44, a putative prostate cancer stem cell marker, in neuroendocrine tumor cells of human prostate cancer. Prostate 2009, 69, 787–798. [Google Scholar] [CrossRef] [PubMed]
- Bitting, R.L.; Schaeffer, D.; Somarelli, J.A.; Garcia-Blanco, M.A.; Armstrong, A.J. The role of epithelial plasticity in prostate cancer dissemination and treatment resistance. Cancer Metastasis Rev. 2014, 33, 441–468. [Google Scholar] [CrossRef] [PubMed]
- Burgio, S.L.; Conteduca, V.; Menna, C.; Carretta, E.; Rossi, L.; Bianchi, E.; Kopf, B.; Fabbri, F.; Amadori, D.; de Giorgi, U. Chromogranin a predicts outcome in prostate cancer patients treated with abiraterone. Endocr. Relat. Cancer 2014, 21, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Conteduca, V.; Burgio, S.L.; Menna, C.; Carretta, E.; Rossi, L.; Bianchi, E.; Masini, C.; Amadori, D.; de Giorgi, U. Chromogranin a is a potential prognostic marker in prostate cancer patients treated with enzalutamide. Prostate 2014, 74, 1691–1696. [Google Scholar] [CrossRef] [PubMed]
- Berruti, A.; Mosca, A.; Tucci, M.; Terrone, C.; Torta, M.; Tarabuzzi, R.; Russo, L.; Cracco, C.; Bollito, E.; Scarpa, R.M.; et al. Independent prognostic role of circulating chromogranin a in prostate cancer patients with hormone-refractory disease. Endocr. Relat. Cancer 2005, 12, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, D.; Singh, S.K.; Mandal, A.K.; Agarwal, M.M.; Mete, U.K.; Kumar, S.; Mavuduru, R.S.; Prasad, R. Plasma chromogranin A: Clinical implications in patients with castrate resistant prostate cancer receiving docetaxel chemotherapy. Cancer Biomark. 2010, 8, 81–87. [Google Scholar] [PubMed]
- Marin-Aguilera, M.; Codony-Servat, J.; Reig, O.; Lozano, J.J.; Fernandez, P.L.; Pereira, M.V.; Jimenez, N.; Donovan, M.; Puig, P.; Mengual, L.; et al. Epithelial-to-mesenchymal transition mediates docetaxel resistance and high risk of relapse in prostate cancer. Mol. Cancer Ther. 2014, 13, 1270–1284. [Google Scholar] [CrossRef] [PubMed]
- Puhr, M.; Hoefer, J.; Schafer, G.; Erb, H.H.; Oh, S.J.; Klocker, H.; Heidegger, I.; Neuwirt, H.; Culig, Z. Epithelial-to-mesenchymal transition leads to docetaxel resistance in prostate cancer and is mediated by reduced expression of miR-200c and miR-205. Am. J. Pathol. 2012, 181, 2188–2201. [Google Scholar] [CrossRef] [PubMed]
- Seiler, D.; Zheng, J.; Liu, G.; Wang, S.; Yamashiro, J.; Reiter, R.E.; Huang, J.; Zeng, G. Enrichment of putative prostate cancer stem cells after androgen deprivation: Upregulation of pluripotency transactivators concurs with resistance to androgen deprivation in lncap cell lines. Prostate 2013, 73, 1378–1390. [Google Scholar] [CrossRef] [PubMed]
- Rybak, A.P.; He, L.; Kapoor, A.; Cutz, J.C.; Tang, D. Characterization of sphere-propagating cells with stem-like properties from DU145 prostate cancer cells. Biochim. Biophys. Acta 2011, 1813, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Hamburger, A.W.; Wang, L.; Khan, M.A.; Hussain, A. Androgen deprivation and stem cell markers in prostate cancers. Int. J. Clin. Exp. Pathol. 2009, 3, 128–138. [Google Scholar] [PubMed]
- Shang, Z.; Cai, Q.; Zhang, M.; Zhu, S.; Ma, Y.; Sun, L.; Jiang, N.; Tian, J.; Niu, X.; Chen, J.; et al. A switch from CD44(+) cell to emt cell drives the metastasis of prostate cancer. Oncotarget 2015, 6, 1202–1216. [Google Scholar] [CrossRef] [PubMed]
- Roudier, M.P.; True, L.D.; Higano, C.S.; Vesselle, H.; Ellis, W.; Lange, P.; Vessella, R.L. Phenotypic heterogeneity of end-stage prostate carcinoma metastatic to bone. Hum. Pathol. 2003, 34, 646–653. [Google Scholar] [CrossRef]
- Shah, R.B.; Mehra, R.; Chinnaiyan, A.M.; Shen, R.; Ghosh, D.; Zhou, M.; Macvicar, G.R.; Varambally, S.; Harwood, J.; Bismar, T.A.; et al. Androgen-independent prostate cancer is a heterogeneous group of diseases: Lessons from a rapid autopsy program. Cancer Res. 2004, 64, 9209–9216. [Google Scholar] [CrossRef] [PubMed]
- Klarmann, G.J.; Hurt, E.M.; Mathews, L.A.; Zhang, X.; Duhagon, M.A.; Mistree, T.; Thomas, S.B.; Farrar, W.L. Invasive prostate cancer cells are tumor initiating cells that have a stem cell-like genomic signature. Clin. Exp. Metastasis 2009, 26, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Wang, B.E.; Leong, K.G.; Yue, P.; Li, L.; Jhunjhunwala, S.; Chen, D.; Seo, K.; Modrusan, Z.; Gao, W.Q.; et al. Androgen deprivation causes epithelial-mesenchymal transition in the prostate: Implications for androgen-deprivation therapy. Cancer Res. 2012, 72, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Kahn, B.; Collazo, J.; Kyprianou, N. Androgen receptor as a driver of therapeutic resistance in advanced prostate cancer. Int. J. Biol. Sci. 2014, 10, 588–595. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Kono, E.; Tran, C.P.; Miyazaki, H.; Yamashiro, J.; Shimomura, T.; Fazli, L.; Wada, R.; Huang, J.; Vessella, R.L.; et al. Monoclonal antibody targeting of N-cadherin inhibits prostate cancer growth, metastasis and castration resistance. Nat. Med. 2010, 16, 1414–1420. [Google Scholar] [CrossRef] [PubMed]
- Nouri, M.; Ratther, E.; Stylianou, N.; Nelson, C.C.; Hollier, B.G.; Williams, E.D. Androgen-targeted therapy-induced epithelial mesenchymal plasticity and neuroendocrine transdifferentiation in prostate cancer: An opportunity for intervention. Front. Oncol. 2014, 4, 370. [Google Scholar] [CrossRef] [PubMed]
- Dethlefsen, C.; Hojfeldt, G.; Hojman, P. The role of intratumoral and systemic IL-6 in breast cancer. Breast Cancer Res. Treat. 2013, 138, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, P.C.; Behrmann, I.; Haan, S.; Hermanns, H.M.; Muller-Newen, G.; Schaper, F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 2003, 374, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lathia, J.D.; Wu, Q.; Wang, J.; Li, Z.; Heddleston, J.M.; Eyler, C.E.; Elderbroom, J.; Gallagher, J.; Schuschu, J.; et al. Targeting interleukin 6 signaling suppresses glioma stem cell survival and tumor growth. Stem Cells 2009, 27, 2393–2404. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, C.L.; Dillon, R.; Devakumar, A.; Shi, S.D.; Greig, M.; Rogers, J.C.; Krastins, B.; Rosenblatt, M.; Kilmer, G.; Major, M.; et al. Quantitative phosphoproteomic analysis of the STAT3/IL-6/hif1alpha signaling network: An initial study in GSC11 glioblastoma stem cells. J. Proteome Res. 2010, 9, 430–443. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.; Cho, H.J.; Cho, S.M.; Jo, K.; Park, J.A.; Kim, N.H.; Amidon, G.L.; Kim, J.S.; Shin, H.C. Blockade of interleukin-6 receptor suppresses the proliferation of H460 lung cancer stem cells. Int. J. Oncol. 2012, 41, 310–316. [Google Scholar] [PubMed]
- Lin, L.; Liu, A.; Peng, Z.; Lin, H.J.; Li, P.K.; Li, C.; Lin, J. STAT3 is necessary for proliferation and survival in colon cancer-initiating cells. Cancer Res. 2011, 71, 7226–7237. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D.; Hirsch, H.A.; Struhl, K. An epigenetic switch involving nf-kappab, LIN28, LET-7 microrna, and il6 links inflammation to cell transformation. Cell 2009, 139, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D.; Hirsch, H.A.; Wang, G.; Struhl, K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc. Natl. Acad. Sci. USA 2011, 108, 1397–1402. [Google Scholar] [CrossRef] [PubMed]
- Marotta, L.L.; Almendro, V.; Marusyk, A.; Shipitsin, M.; Schemme, J.; Walker, S.R.; Bloushtain-Qimron, N.; Kim, J.J.; Choudhury, S.A.; Maruyama, R.; et al. The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(−) stem cell-like breast cancer cells in human tumors. J. Clin. Investig. 2011, 121, 2723–2735. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.T.; Wang, J.Y.; Chen, M.K.; Chen, H.C.; Chang, T.H.; Su, B.W.; Chang, P.J. Colon cancer mesenchymal stem cells modulate the tumorigenicity of colon cancer through interleukin 6. Exp. Cell Res. 2013, 319, 2216–2229. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Lamura, L.; Gallo, M.; Maffia, V.; Normanno, N. Mesenchymal stem cell-derived interleukin-6 and vascular endothelial growth factor promote breast cancer cell migration. J. Cell. Biochem. 2012, 113, 3363–3370. [Google Scholar] [CrossRef] [PubMed]
- Korkaya, H.; Kim, G.I.; Davis, A.; Malik, F.; Henry, N.L.; Ithimakin, S.; Quraishi, A.A.; Tawakkol, N.; D’Angelo, R.; Paulson, A.K.; et al. Activation of an IL6 inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. Mol. Cell 2012, 47, 570–584. [Google Scholar] [CrossRef] [PubMed]
- Tam, L.; McGlynn, L.M.; Traynor, P.; Mukherjee, R.; Bartlett, J.M.; Edwards, J. Expression levels of the JAK/STAT pathway in the transition from hormone-sensitive to hormone-refractory prostate cancer. Br. J. Cancer 2007, 97, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Kwon, E.M.; Holt, S.K.; Fu, R.; Kolb, S.; Williams, G.; Stanford, J.L.; Ostrander, E.A. Androgen metabolism and JAK/STAT pathway genes and prostate cancer risk. Cancer Epidemiol. 2012, 36, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Akimoto, S.; Okumura, A.; Fuse, H. Relationship between serum levels of interleukin-6, tumor necrosis factor-alpha and bone turnover markers in prostate cancer patients. Endocr. J. 1998, 45, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Adler, H.L.; McCurdy, M.A.; Kattan, M.W.; Timme, T.L.; Scardino, P.T.; Thompson, T.C. Elevated levels of circulating interleukin-6 and transforming growth factor-beta1 in patients with metastatic prostatic carcinoma. J. Urol. 1999, 161, 182–187. [Google Scholar] [CrossRef]
- Drachenberg, D.E.; Elgamal, A.A.; Rowbotham, R.; Peterson, M.; Murphy, G.P. Circulating levels of interleukin-6 in patients with hormone refractory prostate cancer. Prostate 1999, 41, 127–133. [Google Scholar] [CrossRef]
- Nakashima, J.; Tachibana, M.; Horiguchi, Y.; Oya, M.; Ohigashi, T.; Asakura, H.; Murai, M. Serum interleukin 6 as a prognostic factor in patients with prostate cancer. Clin. Cancer Res. 2000, 6, 2702–2706. [Google Scholar] [PubMed]
- Qu, Y.; Oyan, A.M.; Liu, R.; Hua, Y.; Zhang, J.; Hovland, R.; Popa, M.; Liu, X.; Brokstad, K.A.; Simon, R.; et al. Generation of prostate tumor-initiating cells is associated with elevation of reactive oxygen species and IL-6/STAT3 signaling. Cancer Res. 2013, 73, 7090–7100. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhu, Y.; Lou, W.; Cui, Y.; Evans, C.P.; Gao, A.C. Inhibition of constitutively active STAT3 reverses enzalutamide resistance in lncap derivative prostate cancer cells. Prostate 2014, 74, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Kroon, P.; Berry, P.A.; Stower, M.J.; Rodrigues, G.; Mann, V.M.; Simms, M.; Bhasin, D.; Chettiar, S.; Li, C.; Li, P.K.; et al. JAK-STAT blockade inhibits tumor initiation and clonogenic recovery of prostate cancer stem-like cells. Cancer Res. 2013, 73, 5288–5298. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, A.; Herrmann, A.; Cherryholmes, G.; Kowolik, C.; Buettner, R.; Pal, S.; Yu, H.; Muller-Newen, G.; Jove, R. Loss of androgen receptor expression promotes a stem-like cell phenotype in prostate cancer through STAT3 signaling. Cancer Res. 2014, 74, 1227–1237. [Google Scholar] [CrossRef] [PubMed]
- Kregel, S.; Kiriluk, K.J.; Rosen, A.M.; Cai, Y.; Reyes, E.E.; Otto, K.B.; Tom, W.; Paner, G.P.; Szmulewitz, R.Z.; Vander Griend, D.J. SOX2 is an androgen receptor-repressed gene that promotes castration-resistant prostate cancer. PLoS ONE 2013, 8, e53701. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.H.; Izumi, K.; Lee, S.O.; Lin, W.J.; Yeh, S.; Chang, C. Anti-androgen receptor ASC-J9 vs. anti-androgens MDV3100 (Enzalutamide) or Casodex (Bicalutamide) leads to opposite effects on prostate cancer metastasis via differential modulation of macrophage infiltration and STAT3-CCL2 signaling. Cell Death Dis. 2013, 4, e764. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.H.; Lee, S.O.; Niu, Y.; Xu, D.; Liang, L.; Li, L.; Yeh, S.D.; Fujimoto, N.; Yeh, S.; Chang, C. Differential androgen deprivation therapies with anti-androgens casodex/bicalutamide or MDV3100/enzalutamide versus anti-androgen receptor ASC-J9(r) lead to promotion versus suppression of prostate cancer metastasis. J. Biol. Chem. 2013, 288, 19359–19369. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.Y.; Izumi, K.; Lai, K.P.; Liang, L.; Li, L.; Miyamoto, H.; Lin, W.J.; Chang, C. Infiltrating macrophages promote prostate tumorigenesis via modulating androgen receptor-mediated CCL4-STAT3 signaling. Cancer Res. 2013, 73, 5633–5646. [Google Scholar] [CrossRef] [PubMed]
- Jia, S.; Gao, X.; Lee, S.H.; Maira, S.M.; Wu, X.; Stack, E.C.; Signoretti, S.; Loda, M.; Zhao, J.J.; Roberts, T.M. Opposing effects of androgen deprivation and targeted therapy on prostate cancer prevention. Cancer Discov. 2013, 3, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Kahn, M. Can we safely target the wnt pathway? Nat. Rev. Drug Discov. 2014, 13, 513–532. [Google Scholar] [CrossRef] [PubMed]
- Krausova, M.; Korinek, V. Wnt signaling in adult intestinal stem cells and cancer. Cell. Signal. 2014, 26, 570–579. [Google Scholar] [CrossRef] [PubMed]
- Boman, B.M.; Fields, J.Z. An APC:Wnt counter-current-like mechanism regulates cell division along the human colonic crypt axis: A mechanism that explains how APC mutations induce proliferative abnormalities that drive colon cancer development. Front. Oncol. 2013, 3, 244. [Google Scholar] [CrossRef] [PubMed]
- Atlasi, Y.; Looijenga, L.; Fodde, R. Cancer stem cells, pluripotency, and cellular heterogeneity: A wnter perspective. Curr. Top. Dev. Biol. 2014, 107, 373–404. [Google Scholar] [PubMed]
- Rajan, P.; Sudbery, I.M.; Villasevil, M.E.; Mui, E.; Fleming, J.; Davis, M.; Ahmad, I.; Edwards, J.; Sansom, O.J.; Sims, D.; et al. Next-generation sequencing of advanced prostate cancer treated with androgen-deprivation therapy. Eur. Urol. 2014, 66, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; White, T.A.; MacKenzie, A.P.; Clegg, N.; Lee, C.; Dumpit, R.F.; Coleman, I.; Ng, S.B.; Salipante, S.J.; Rieder, M.J.; et al. Exome sequencing identifies a spectrum of mutation frequencies in advanced and lethal prostate cancers. Proc. Natl. Acad. Sci. USA 2011, 108, 17087–17092. [Google Scholar] [CrossRef] [PubMed]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Dai, J.; Zhang, H.; Sottnik, J.L.; Keller, J.M.; Escott, K.J.; Sanganee, H.J.; Yao, Z.; McCauley, L.K.; Keller, E.T. Activation of the wnt pathway through AR79, a GSK3beta inhibitor, promotes prostate cancer growth in soft tissue and bone. Mol. Cancer Res. 2013, 11, 1597–1610. [Google Scholar] [CrossRef] [PubMed]
- Bisson, I.; Prowse, D.M. Wnt signaling regulates self-renewal and differentiation of prostate cancer cells with stem cell characteristics. Cell Res. 2009, 19, 683–697. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, I.S.; Chang, K.C.; Tsai, Y.T.; Ke, J.Y.; Lu, P.J.; Lee, K.H.; Yeh, S.D.; Hong, T.M.; Chen, Y.L. Microrna-320 suppresses the stem cell-like characteristics of prostate cancer cells by downregulating the wnt/beta-catenin signaling pathway. Carcinogenesis 2013, 34, 530–538. [Google Scholar] [CrossRef] [PubMed]
- Arnold, K.; Sarkar, A.; Yram, M.A.; Polo, J.M.; Bronson, R.; Sengupta, S.; Seandel, M.; Geijsen, N.; Hochedlinger, K. SOX2(+) adult stem and progenitor cells are important for tissue regeneration and survival of mice. Cell Stem Cell 2011, 9, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawamura, T.; Suzuki, J.; Wang, Y.V.; Menendez, S.; Morera, L.B.; Raya, A.; Wahl, G.M.; Izpisua Belmonte, J.C. Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature 2009, 460, 1140–1144. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Collado, M.; Villasante, A.; Strati, K.; Ortega, S.; Canamero, M.; Blasco, M.A.; Serrano, M. The INK4/Arf locus is a barrier for ips cell reprogramming. Nature 2009, 460, 1136–1139. [Google Scholar] [CrossRef] [PubMed]
- Marion, R.M.; Strati, K.; Li, H.; Murga, M.; Blanco, R.; Ortega, S.; Fernandez-Capetillo, O.; Serrano, M.; Blasco, M.A. A p53-mediated DNA damage response limits reprogramming to ensure ips cell genomic integrity. Nature 2009, 460, 1149–1153. [Google Scholar] [CrossRef] [PubMed]
- Utikal, J.; Polo, J.M.; Stadtfeld, M.; Maherali, N.; Kulalert, W.; Walsh, R.M.; Khalil, A.; Rheinwald, J.G.; Hochedlinger, K. Immortalization eliminates a roadblock during cellular reprogramming into iPS cells. Nature 2009, 460, 1145–1148. [Google Scholar] [CrossRef] [PubMed]
- Weina, K.; Utikal, J. SOX2 and cancer: Current research and its implications in the clinic. Clin. Transl. Med. 2014, 3, 19. [Google Scholar] [CrossRef] [PubMed]
- Leis, O.; Eguiara, A.; Lopez-Arribillaga, E.; Alberdi, M.J.; Hernandez-Garcia, S.; Elorriaga, K.; Pandiella, A.; Rezola, R.; Martin, A.G. SOX2 expression in breast tumours and activation in breast cancer stem cells. Oncogene 2012, 31, 1354–1365. [Google Scholar] [CrossRef] [PubMed]
- Rybak, A.P.; Tang, D. SOX2 plays a critical role in egfr-mediated self-renewal of human prostate cancer stem-like cells. Cell. Signal. 2013, 25, 2734–2742. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhu, H.H.; Chu, M.; Liu, Y.; Zhang, C.; Liu, G.; Yang, X.; Yang, R.; Gao, W.Q. Symmetrical and asymmetrical division analysis provides evidence for a hierarchy of prostate epithelial cell lineages. Nat. Commun. 2014, 5, 4758. [Google Scholar] [CrossRef] [PubMed]
- Jeter, C.R.; Liu, B.; Liu, X.; Chen, X.; Liu, C.; Calhoun-Davis, T.; Repass, J.; Zaehres, H.; Shen, J.J.; Tang, D.G. Nanog promotes cancer stem cell characteristics and prostate cancer resistance to androgen deprivation. Oncogene 2011, 30, 3833–3845. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Murayama, Y.; Hashimoto, K.; Nakamura, Y.; Lin, C.S.; Yokoyama, K.K.; Saito, S. Role of tumor suppressor genes in the cancer-associated reprogramming of human induced pluripotent stem cells. Stem Cell Res. Ther. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, K.; Tanaka, T.; Nakai, D.; Morita, N.; Suzuki, K. Immunohistochemical expression of four different stem cell markers in prostate cancer: High expression of nanog in conjunction with hypoxia-inducible factor-1alpha expression is involved in prostate epithelial malignancy. Oncol. Lett. 2014, 8, 985–992. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, J.; Zhang, Z.; Zhou, W.; Wang, A.J.; Heddleston, J.M.; Pinna, C.M.; Hubaud, A.; Stadler, B.; Choi, M.; Bar, M.; et al. Hif induces human embryonic stem cell markers in cancer cells. Cancer Res. 2011, 71, 4640–4652. [Google Scholar] [CrossRef] [PubMed]
- Mimeault, M.; Batra, S.K. Hypoxia-inducing factors as master regulators of stemness properties and altered metabolism of cancer- and metastasis-initiating cells. J. Cell. Mol. Med. 2013, 17, 30–54. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, N.; Nimura, K.; Nagano, H.; Yamaguchi, S.; Nonomura, N.; Kaneda, Y. CRISPR/Cas9-mediated gene knockout of NANOG and NANOGP8 decreases the malignant potential of prostate cancer cells. Oncotarget 2015, 6, 22361–22374. [Google Scholar] [CrossRef] [PubMed]
- Nyquist, M.D.; Dehm, S.M. Interplay between genomic alterations and androgen receptor signaling during prostate cancer development and progression. Horm. Cancer 2013, 4, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Haffner, M.C.; Aryee, M.J.; Toubaji, A.; Esopi, D.M.; Albadine, R.; Gurel, B.; Isaacs, W.B.; Bova, G.S.; Liu, W.; Xu, J.; et al. Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements. Nat. Genet. 2010, 42, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Mani, R.S.; Tomlins, S.A.; Callahan, K.; Ghosh, A.; Nyati, M.K.; Varambally, S.; Palanisamy, N.; Chinnaiyan, A.M. Induced chromosomal proximity and gene fusions in prostate cancer. Science 2009, 326, 1230. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.F.; Lawrence, M.S.; Demichelis, F.; Drier, Y.; Cibulskis, K.; Sivachenko, A.Y.; Sboner, A.; Esgueva, R.; Pflueger, D.; Sougnez, C.; et al. The genomic complexity of primary human prostate cancer. Nature 2011, 470, 214–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreso, A.; Dick, J.E. Evolution of the cancer stem cell model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Ruizeveld de Winter, J.A.; Janssen, P.J.; Sleddens, H.M.; Verleun-Mooijman, M.C.; Trapman, J.; Brinkmann, A.O.; Santerse, A.B.; Schroder, F.H.; van der Kwast, T.H. Androgen receptor status in localized and locally progressive hormone refractory human prostate cancer. Am. J. Pathol. 1994, 144, 735–746. [Google Scholar] [PubMed]
- Masai, M.; Sumiya, H.; Akimoto, S.; Yatani, R.; Chang, C.S.; Liao, S.S.; Shimazaki, J. Immunohistochemical study of androgen receptor in benign hyperplastic and cancerous human prostates. Prostate 1990, 17, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Van der Kwast, T.H.; Schalken, J.; Ruizeveld de Winter, J.A.; van Vroonhoven, C.C.; Mulder, E.; Boersma, W.; Trapman, J. Androgen receptors in endocrine-therapy-resistant human prostate cancer. Int. J. Cancer 1991, 48, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, T.; Watanabe, M.; Suzuki, H.; Toyota, M.; Sekita, N.; Hirokawa, Y.; Mizokami, A.; Ito, H.; Yatani, R.; Shiraishi, T. Epigenetic regulation of androgen receptor gene expression in human prostate cancers. Lab. Investig. 2000, 80, 1789–1796. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chen, X.; Rycaj, K.; Chao, H.P.; Deng, Q.; Jeter, C.; Liu, C.; Honorio, S.; Li, H.; Davis, T.; et al. Systematic dissection of phenotypic, functional, and tumorigenic heterogeneity of human prostate cancer cells. Oncotarget 2015, 6, 23959–23986. [Google Scholar] [CrossRef] [PubMed]
- Castagnetta, L.A.; Miceli, M.D.; Sorci, C.M.; Pfeffer, U.; Farruggio, R.; Oliveri, G.; Calabro, M.; Carruba, G. Growth of LNCaP human prostate cancer cells is stimulated by estradiol via its own receptor. Endocrinology 1995, 136, 2309–2319. [Google Scholar] [PubMed]
- Marcelli, M.; Haidacher, S.J.; Plymate, S.R.; Birnbaum, R.S. Altered growth and insulin-like growth factor-binding protein-3 production in PC3 prostate carcinoma cells stably transfected with a constitutively active androgen receptor complementary deoxyribonucleic acid. Endocrinology 1995, 136, 1040–1048. [Google Scholar] [CrossRef] [PubMed]
- Sadi, M.V.; Barrack, E.R. Image analysis of androgen receptor immunostaining in metastatic prostate cancer. Heterogeneity as a predictor of response to hormonal therapy. Cancer 1993, 71, 2574–2580. [Google Scholar] [CrossRef]
- Wang, G.; Wang, J.; Sadar, M.D. Crosstalk between the androgen receptor and beta-catenin in castrate-resistant prostate cancer. Cancer Res. 2008, 68, 9918–9927. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, N.N.; Shao, S.; Hoang, B.H.; Mercola, D.; Zi, X. Wnt signaling in castration-resistant prostate cancer: Implications for therapy. Am. J. Clin. Exp. Urol. 2014, 2, 27–44. [Google Scholar] [PubMed]
- Kregel, S.; Szmulewitz, R.Z.; Vander Griend, D.J. The pluripotency factor nanog is directly upregulated by the androgen receptor in prostate cancer cells. Prostate 2014, 74, 1530–1543. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Sethi, S.; Li, Y.; Chen, W.; Sakr, W.A.; Heath, E.; Sarkar, F.H. Androgen receptor splice variants contribute to prostate cancer aggressiveness through induction of emt and expression of stem cell marker genes. Prostate 2015, 75, 161–174. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ojo, D.; Lin, X.; Wong, N.; Gu, Y.; Tang, D. Prostate Cancer Stem-like Cells Contribute to the Development of Castration-Resistant Prostate Cancer. Cancers 2015, 7, 2290-2308. https://doi.org/10.3390/cancers7040890
Ojo D, Lin X, Wong N, Gu Y, Tang D. Prostate Cancer Stem-like Cells Contribute to the Development of Castration-Resistant Prostate Cancer. Cancers. 2015; 7(4):2290-2308. https://doi.org/10.3390/cancers7040890
Chicago/Turabian StyleOjo, Diane, Xiaozeng Lin, Nicholas Wong, Yan Gu, and Damu Tang. 2015. "Prostate Cancer Stem-like Cells Contribute to the Development of Castration-Resistant Prostate Cancer" Cancers 7, no. 4: 2290-2308. https://doi.org/10.3390/cancers7040890
APA StyleOjo, D., Lin, X., Wong, N., Gu, Y., & Tang, D. (2015). Prostate Cancer Stem-like Cells Contribute to the Development of Castration-Resistant Prostate Cancer. Cancers, 7(4), 2290-2308. https://doi.org/10.3390/cancers7040890