
Aberrant GLI1 Activation in DNA Damage Response, Carcinogenesis and Chemoresistance

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Molecular Mechanism of the HH Signaling Cascade
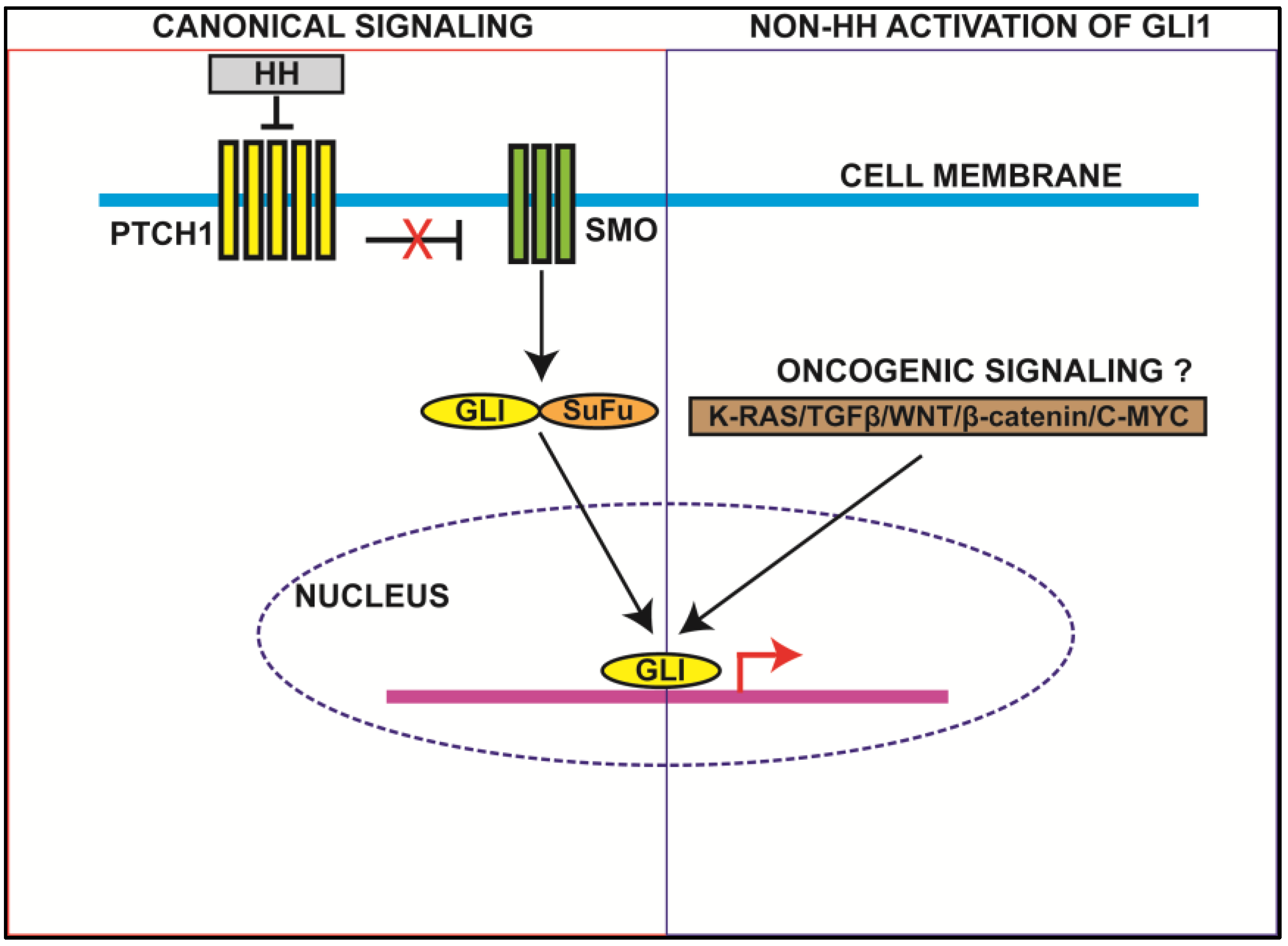
2.1. Canonical HH Signaling
2.2. Non-HH Activation of GLI1

2.3. Molecular Properties of GLI1
3. Role of Aberrant GLI1 in DNA Damage and Repair
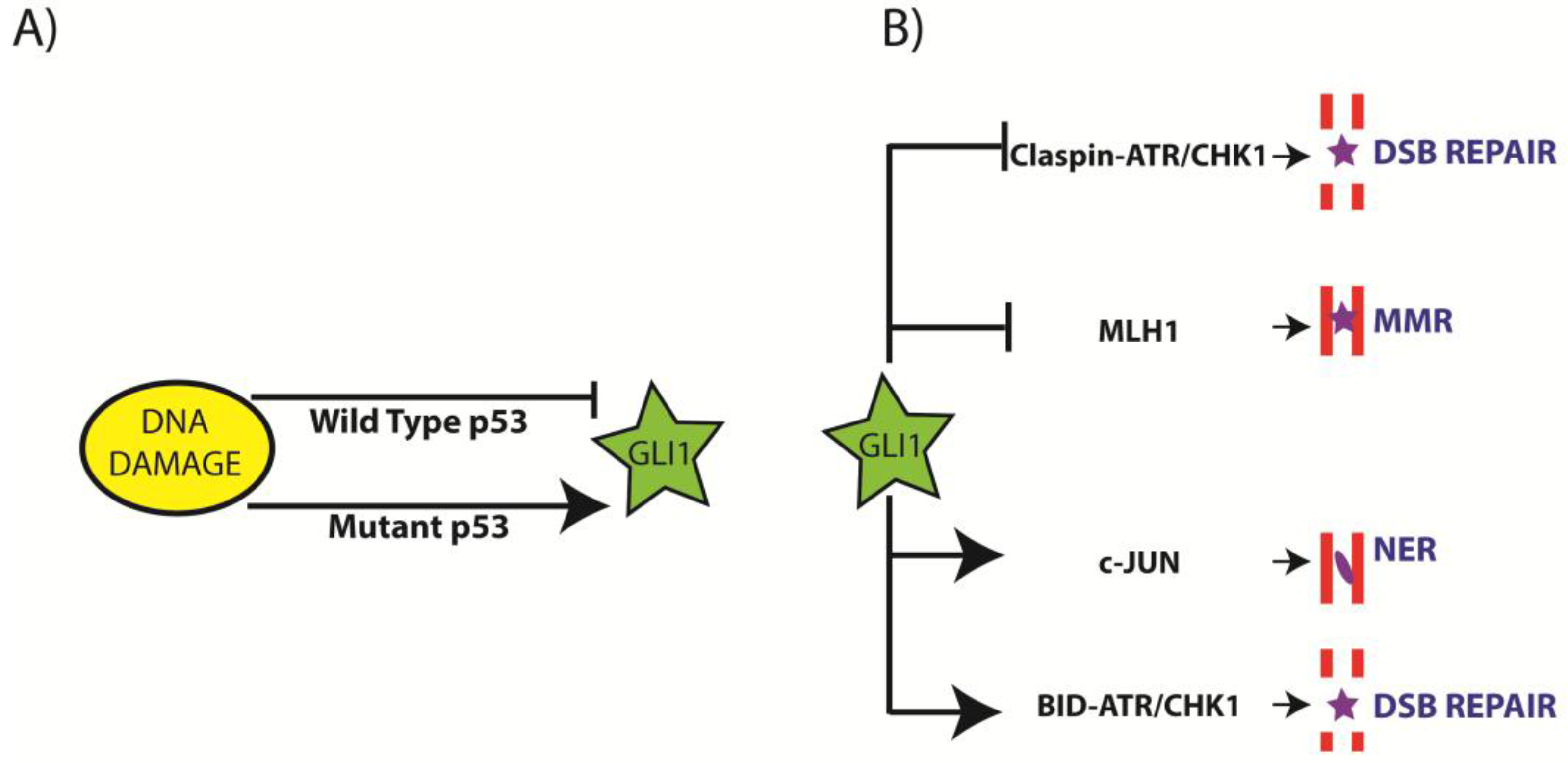
3.1. Regulation of GLI1 by DNA Damage

3.2. Inhibition of GLI1 Induces DNA Damage
3.3. Regulation of NER by Aberrant GLI1
3.4. Regulation of Mis-Match Repair (MMR) by Aberrant GLI1
3.5. Regulation of DSB Repair by Aberrant GLI1
3.6. Section Summary
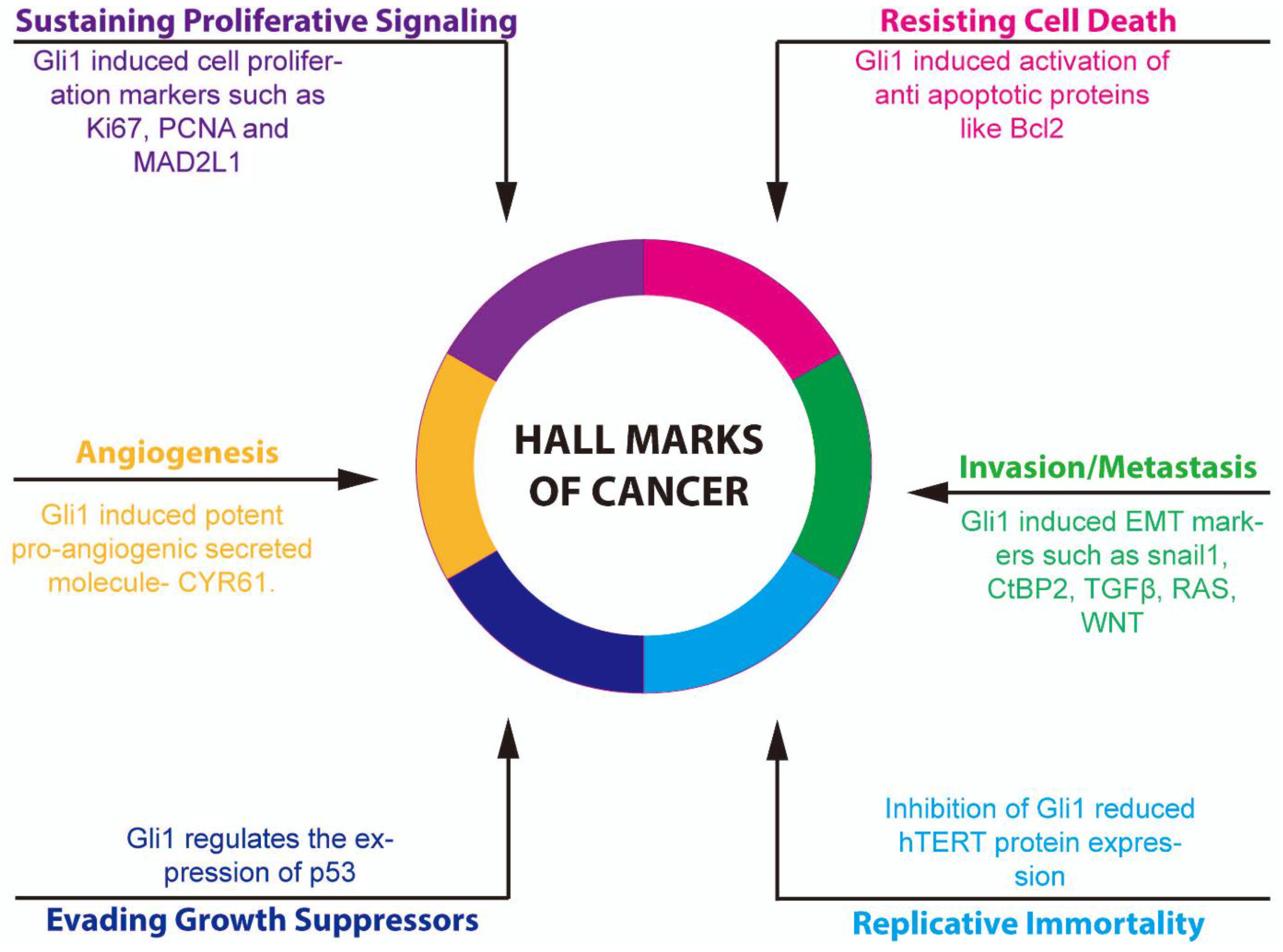
4. Role of Aberrant GLI1 in Carcinogenesis
4.1. Aberrant GLI1 in Multiple Cancers
4.2. Role of Aberrant GLI1 in Pancreatic Cancer

4.3. Role of Aberrant GLI1 in Medulloblastoma
4.4. Role of Aberrant GLI1 in Breast Cancer
4.5. Role of Aberrant GLI1 in Inducing EMT
4.6. Role of Aberrant GLI1 in Regulating hTERT
4.7. Role of Aberrant GLI1 in Epigenetic Regulation
4.8. Role of HH-Independent Aberrant GLI1 in Carcinogenesis
4.9. Role of Aberrant GLI1 in Tumor Recurrence
4.10. Section Summary
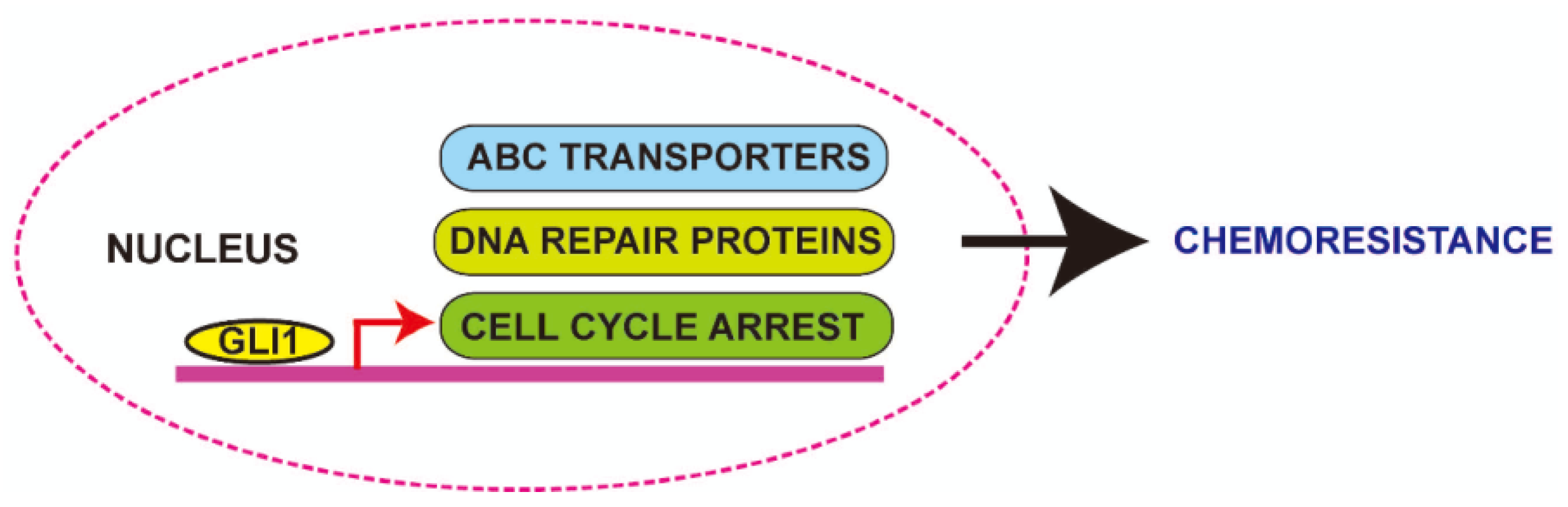
5. Role of Aberrant GLI1 in Chemoresistance
5.1. Role of Aberrant GLI1 in MDR

5.2. Role of Aberrant GLI1 in Modifying the Drug Activity
5.3. Role of Aberrant GLI1 in Radio-Resistance
5.4. Inhibitors of Aberrant GLI1
5.5. Section Summary
6. Conclusions and Future Perspectives
Acknowledgments
Conflicts of Interest
References
- Nüsslein-Volhard, C.; Wieschaus, E. Mutations affecting segment number and polarity in Drosophila. Nature 1980, 287, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Belloni, E.; Muenke, M.; Roessler, E.; Traverso, G.; Siegel-Bartelt, J.; Frumkin, A.; Mitchell, H.F.; Donis-Keller, H.; Helms, C.; Hing, A.V.; et al. Identification of Sonic hedgehog as a candidate gene responsible for holoprosencephaly. Nat. Genet. 1996, 14, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Gritli-Linde, A.; Bei, M.; Maas, R.; Zhang, X.M.; Linde, A.; McMahon, A.P. Shh signaling within the dental epithelium is necessary for cell proliferation, growth and polarization. Dev. Camb. Engl. 2002, 129, 5323–5337. [Google Scholar] [CrossRef]
- Ingham, P.W.; McMahon, A.P. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev. 2001, 15, 3059–3087. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, L.V.; Milenković, L.; Higgins, K.M.; Scott, M.P. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 1997, 277, 1109–1113. [Google Scholar] [CrossRef] [PubMed]
- Dahmane, N.; Lee, J.; Robins, P.; Heller, P.; Ruiz i Altaba, A. Activation of the transcription factor Gli1 and the Sonic hedgehog signalling pathway in skin tumours. Nature 1997, 389, 876–881. [Google Scholar] [PubMed]
- COSMIC: Catalogue of Somatic Mutations in Cancer—Home Page. Available online: http://cancer.sanger.ac.uk/cosmic (accessed on 16 October 2015).
- Zhu, H.; Lo, H.-W. The human glioma-associated oncogene homolog 1 (GLI1) family of transcription factors in gene regulation and diseases. Curr. Genom. 2010, 11, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Aberger, F.; Ruiz I Altaba, A. Context-dependent signal integration by the GLI code: The oncogenic load, pathways, modifiers and implications for cancer therapy. Semin. Cell Dev. Biol. 2014, 33, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Mahindroo, N.; Punchihewa, C.; Fujii, N. Hedgehog-Gli signaling pathway inhibitors as anticancer agents. J. Med. Chem. 2009, 52, 3829–3845. [Google Scholar] [CrossRef] [PubMed]
- Yauch, R.L.; Dijkgraaf, G.J.P.; Alicke, B.; Januario, T.; Ahn, C.P.; Holcomb, T.; Pujara, K.; Stinson, J.; Callahan, C.A.; Tang, T.; et al. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science 2009, 326, 572–574. [Google Scholar] [CrossRef] [PubMed]
- Jimeno, A.; Weiss, G.J.; Miller, W.H.; Gettinger, S.; Eigl, B.J.C.; Chang, A.L.S.; Dunbar, J.; Devens, S.; Faia, K.; Skliris, G.; et al. Phase I study of the Hedgehog pathway inhibitor IPI-926 in adult patients with solid tumors. Clin. Cancer Res. 2013, 19, 2766–2774. [Google Scholar] [CrossRef] [PubMed]
- McMillan, R.; Matsui, W. Molecular pathways: The hedgehog signaling pathway in cancer. Clin. Cancer Res. 2012, 18, 4883–4888. [Google Scholar] [CrossRef] [PubMed]
- Hui, C.-C.; Angers, S. Gli proteins in development and disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 513–537. [Google Scholar] [CrossRef] [PubMed]
- Corbit, K.C.; Aanstad, P.; Singla, V.; Norman, A.R.; Stainier, D.Y.R.; Reiter, J.F. Vertebrate Smoothened functions at the primary cilium. Nature 2005, 437, 1018–1021. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Takebe, N.; Lorusso, P. Targeting the Hedgehog pathway in cancer. Ther. Adv. Med. Oncol. 2010, 2, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Dennler, S.; André, J.; Alexaki, I.; Li, A.; Magnaldo, T.; ten Dijke, P.; Wang, X.-J.; Verrecchia, F.; Mauviel, A. Induction of sonic hedgehog mediators by transforming growth factor-beta: Smad3-dependent activation of Gli2 and Gli1 expression in vitro and in vivo. Cancer Res. 2007, 67, 6981–6986. [Google Scholar] [CrossRef] [PubMed]
- Javelaud, D.; Alexaki, V.I.; Dennler, S.; Mohammad, K.S.; Guise, T.A.; Mauviel, A. TGF-β/SMAD/GLI2 signaling axis in cancer progression and metastasis. Cancer Res. 2011, 71, 5606–5610. [Google Scholar] [CrossRef] [PubMed]
- Kriegshäuser, G.; Auner, V.; Zeillinger, R. New and potential clinical applications of KRAS as a cancer biomarker. Expert Opin. Med. Diagn. 2010, 4, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.; Mei, F.C.; Xie, J.; Cheng, X. Oncogenic KRAS activates hedgehog signaling pathway in pancreatic cancer cells. J. Biol. Chem. 2007, 282, 14048–14055. [Google Scholar] [CrossRef] [PubMed]
- Rajurkar, M.; De Jesus-Monge, W.E.; Driscoll, D.R.; Appleman, V.A.; Huang, H.; Cotton, J.L.; Klimstra, D.S.; Zhu, L.J.; Simin, K.; Xu, L.; et al. The activity of Gli transcription factors is essential for Kras-induced pancreatic tumorigenesis. Proc. Natl. Acad. Sci. USA 2012, 109, E1038–E1047. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.W.; Gallant, M.; Lamm, M.L.G.; Iannaccone, S.; Vieux, K.-F.; Proytcheva, M.; Hyjek, E.; Iannaccone, P.; Walterhouse, D. Noncanonical regulation of the Hedgehog mediator GLI1 by c-MYC in Burkitt lymphoma. Mol. Cancer Res. MCR 2013, 11, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Beauchamp, E.; Bulut, G.; Abaan, O.; Chen, K.; Merchant, A.; Matsui, W.; Endo, Y.; Rubin, J.S.; Toretsky, J.; Uren, A. GLI1 is a direct transcriptional target of EWS-FLI1 oncoprotein. J. Biol. Chem. 2009, 284, 9074–9082. [Google Scholar] [CrossRef] [PubMed]
- Stecca, B.; Ruiz i Altaba, A. A GLI1-p53 inhibitory loop controls neural stem cell and tumour cell numbers. EMBO J. 2009, 28, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Scales, S.J.; de Sauvage, F.J. Mechanisms of Hedgehog pathway activation in cancer and implications for therapy. Trends Pharmacol. Sci. 2009, 30, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Kinzler, K.W.; Vogelstein, B. The GLI gene encodes a nuclear protein which binds specific sequences in the human genome. Mol. Cell. Biol. 1990, 10, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef] [PubMed]
- Krauthammer, M.; Kong, Y.; Ha, B.H.; Evans, P.; Bacchiocchi, A.; McCusker, J.P.; Cheng, E.; Davis, M.J.; Goh, G.; Choi, M.; et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat. Genet. 2012, 44, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.-P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Pickering, C.R.; Zhou, J.H.; Lee, J.J.; Drummond, J.A.; Peng, S.A.; Saade, R.E.; Tsai, K.Y.; Curry, J.L.; Tetzlaff, M.T.; Lai, S.Y.; et al. Mutational landscape of aggressive cutaneous squamous cell carcinoma. Clin. Cancer Res. 2014, 20, 6582–6592. [Google Scholar] [CrossRef] [PubMed]
- Dahlén, A.; Mertens, F.; Mandahl, N.; Panagopoulos, I. Molecular genetic characterization of the genomic ACTB-GLI fusion in pericytoma with t(7;12). Biochem. Biophys. Res. Commun. 2004, 325, 1318–1323. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.A.-J.; Lai, F.P.-L.; Gui, H.-S.; Sham, M.-H.; Tam, P.K.-H.; Garcia-Barcelo, M.-M.; Hui, C.-C.; Ngan, E.S.-W. Identification of GLI Mutations in Patients with Hirschsprung Disease That Disrupt Enteric Nervous System Development in Mice. Gastroenterology 2015. [Google Scholar] [CrossRef] [PubMed]
- Lees, C.W.; Zacharias, W.J.; Tremelling, M.; Noble, C.L.; Nimmo, E.R.; Tenesa, A.; Cornelius, J.; Torkvist, L.; Kao, J.; Farrington, S.; et al. Analysis of germline GLI1 variation implicates hedgehog signalling in the regulation of intestinal inflammatory pathways. PLoS Med. 2008, 5, e239. [Google Scholar] [CrossRef] [PubMed]
- Shimokawa, T.; Tostar, U.; Lauth, M.; Palaniswamy, R.; Kasper, M.; Toftgard, R.; Zaphiropoulos, P.G. Novel human glioma-associated oncogene 1 (GLI1) splice variants reveal distinct mechanisms in the terminal transduction of the hedgehog signal. J. Biol. Chem. 2008, 283, 14345–14354. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.-W.; Zhu, H.; Cao, X.; Aldrich, A.; Ali-Osman, F. A novel splice variant of GLI1 that promotes glioblastoma cell migration and invasion. Cancer Res. 2009, 69, 6790–6798. [Google Scholar] [CrossRef] [PubMed]
- Harrison, W.; Cochrane, B.; Neill, G.; Philpott, M. The oncogenic GLI transcription factors facilitate keratinocyte survival and transformation upon exposure to genotoxic agents. Oncogene 2014, 33, 2432–2440. [Google Scholar] [CrossRef] [PubMed]
- Frappart, P.-O.; Lee, Y.; Russell, H.R.; Chalhoub, N.; Wang, Y.-D.; Orii, K.E.; Zhao, J.; Kondo, N.; Baker, S.J.; McKinnon, P.J. Recurrent genomic alterations characterize medulloblastoma arising from DNA double-strand break repair deficiency. Proc. Natl. Acad. Sci. USA 2009, 106, 1880–1885. [Google Scholar] [CrossRef] [PubMed]
- Mazzà, D.; Infante, P.; Colicchia, V.; Greco, A.; Alfonsi, R.; Siler, M.; Antonucci, L.; Po, A.; De Smaele, E.; Ferretti, E.; et al. PCAF ubiquitin ligase activity inhibits Hedgehog/Gli1 signaling in p53-dependent response to genotoxic stress. Cell Death Differ. 2013, 20, 1688–1697. [Google Scholar] [CrossRef] [PubMed]
- Mazumdar, T.; Devecchio, J.; Agyeman, A.; Shi, T.; Houghton, J.A. Blocking Hedgehog survival signaling at the level of the GLI genes induces DNA damage and extensive cell death in human colon carcinoma cells. Cancer Res. 2011, 71, 5904–5914. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Mazumdar, T.; Devecchio, J.; Duan, Z.-H.; Agyeman, A.; Aziz, M.; Houghton, J.A. cDNA microarray gene expression profiling of hedgehog signaling pathway inhibition in human colon cancer cells. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Agyeman, A.; Mazumdar, T.; Houghton, J.A. Regulation of DNA damage following termination of Hedgehog (HH) survival signaling at the level of the GLI genes in human colon cancer. Oncotarget 2012, 3, 854–868. [Google Scholar] [CrossRef] [PubMed]
- Kudo, K.; Gavin, E.; Das, S.; Amable, L.; Shevde, L.A.; Reed, E. Inhibition of Gli1 results in altered c-Jun activation, inhibition of cisplatin-induced upregulation of ERCC1, XPD and XRCC1, and inhibition of platinum-DNA adduct repair. Oncogene 2012, 31, 4718–4724. [Google Scholar] [CrossRef] [PubMed]
- Amable, L.; Gavin, E.; Kudo, K.; Meng, E.; Rocconi, R.P.; Shevde, L.A.; Reed, E. GLI1 upregulates C-JUN through a specific 130-kDa isoform. Int. J. Oncol. 2014, 44, 655–661. [Google Scholar] [PubMed]
- Inaguma, S.; Riku, M.; Hashimoto, M.; Murakami, H.; Saga, S.; Ikeda, H.; Kasai, K. GLI1 interferes with the DNA mismatch repair system in pancreatic cancer through BHLHE41-mediated suppression of MLH1. Cancer Res. 2013, 73, 7313–7323. [Google Scholar] [CrossRef] [PubMed]
- Leonard, J.M.; Ye, H.; Wetmore, C.; Karnitz, L.M. Sonic Hedgehog signaling impairs ionizing radiation-induced checkpoint activation and induces genomic instability. J. Cell Biol. 2008, 183, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, K.; Mani, C.; Barnett, R.; Nalluri, S.; Bachaboina, L.; Rocconi, R.P.; Athar, M.; Owen, L.B.; Palle, K. Gli1 protein regulates the S-phase checkpoint in tumor cells via Bid protein, and its inhibition sensitizes to DNA topoisomerase 1 inhibitors. J. Biol. Chem. 2014, 289, 31513–31525. [Google Scholar] [CrossRef] [PubMed]
- Schnidar, H.; Eberl, M.; Klingler, S.; Mangelberger, D.; Kasper, M.; Hauser-Kronberger, C.; Regl, G.; Kroismayr, R.; Moriggl, R.; Sibilia, M.; et al. Epidermal growth factor receptor signaling synergizes with Hedgehog/GLI in oncogenic transformation via activation of the MEK/ERK/JUN pathway. Cancer Res. 2009, 69, 1284–1292. [Google Scholar] [CrossRef] [PubMed]
- Santini, R.; Vinci, M.C.; Pandolfi, S.; Penachioni, J.Y.; Montagnani, V.; Olivito, B.; Gattai, R.; Pimpinelli, N.; Gerlini, G.; Borgognoni, L.; et al. Hedgehog-GLI signaling drives self-renewal and tumorigenicity of human melanoma-initiating cells. Stem Cells Dayt. Ohio 2012, 30, 1808–1818. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.-K.; Kim, H.-J.; Lee, Y.-S.; Han, M.-E.; Yoon, S.; Baek, S.-Y.; Kim, B.-S.; Kim, J.-B.; Oh, S.-O. Hedgehog signaling regulates proliferation of prostate cancer cells via stathmin1; Clin. Exp. Med. 2010, 10, 51–57. [Google Scholar]
- Yoon, J.W.; Kita, Y.; Frank, D.J.; Majewski, R.R.; Konicek, B.A.; Nobrega, M.A.; Jacob, H.; Walterhouse, D.; Iannaccone, P. Gene expression profiling leads to identification of GLI1-binding elements in target genes and a role for multiple downstream pathways in GLI1-induced cell transformation. J. Biol. Chem. 2002, 277, 5548–5555. [Google Scholar] [CrossRef] [PubMed]
- Eichberger, T.; Sander, V.; Schnidar, H.; Regl, G.; Kasper, M.; Schmid, C.; Plamberger, S.; Kaser, A.; Aberger, F.; Frischauf, A.-M. Overlapping and distinct transcriptional regulator properties of the GLI1 and GLI2 oncogenes. Genomics 2006, 87, 616–632. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.-L.; Sun, H.-J.; Wang, Y.-S.; Huang, S.-H.; Xie, J.-W.; Zhang, H.-W. Study of Sonic hedgehog signaling pathway related molecules in gastric carcinoma. World J. Gastroenterol. 2006, 12, 3965–3969. [Google Scholar] [PubMed]
- Yoshizaki, A.; Nakayama, T.; Naito, S.; Wen, C.-Y.; Sekine, I. Expressions of sonic hedgehog, patched, smoothened and Gli-1 in human intestinal stromal tumors and their correlation with prognosis. World J. Gastroenterol. 2006, 12, 5687–5691. [Google Scholar] [PubMed]
- Brunner, M.; Thurnher, D.; Pammer, J.; Heiduschka, G.; Petzelbauer, P.; Schmid, C.; Schneider, S.; Erovic, B.M. Expression of hedgehog signaling molecules in Merkel cell carcinoma. Head Neck 2010, 32, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Shen, S.S.; Zhou, S.; Ni, J.; Chen, D.; Wang, G.; Li, Y. STAT3 activation and aberrant ligand-dependent sonic hedgehog signaling in human pulmonary adenocarcinoma. Exp. Mol. Pathol. 2012, 93, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Fei, D.L.; Sanchez-Mejias, A.; Wang, Z.; Flaveny, C.; Long, J.; Singh, S.; Rodriguez-Blanco, J.; Tokhunts, R.; Giambelli, C.; Briegel, K.J.; et al. Hedgehog signaling regulates bladder cancer growth and tumorigenicity. Cancer Res. 2012, 72, 4449–4458. [Google Scholar] [CrossRef] [PubMed]
- Dimitrova, K.; Stoehr, M.; Dehghani, F.; Dietz, A.; Wichmann, G.; Bertolini, J.; Mozet, C. Overexpression of the Hedgehog signalling pathway in head and neck squamous cell carcinoma. Onkologie 2013, 36, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Bakry, O.A.; Samaka, R.M.; Shoeib, M.A.M.; Megahed, D.M. Immunolocalization of glioma-associated oncogene homolog 1 in non melanoma skin cancer. Ultrastruct. Pathol. 2015, 39, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Duan, F.; Lin, M.; Li, C.; Ding, X.; Qian, G.; Zhang, H.; Ge, S.; Fan, X.; Li, J. Effects of inhibition of hedgehog signaling on cell growth and migration of uveal melanoma cells. Cancer Biol. Ther. 2014, 15, 544–559. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Xu, X.; Xu, A.; Liu, C.; Liang, F.; Xue, M.; Bai, L. Aberrant activation of Sonic hedgehog signaling in chronic cholecystitis and gallbladder carcinoma. Hum. Pathol. 2014, 45, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Hong, Z.; Bi, A.; Chen, D.; Gao, L.; Yin, Z.; Luo, L. Activation of hedgehog signaling pathway in human non-small cell lung cancers. Pathol. Oncol. Res. 2014, 20, 917–922. [Google Scholar] [CrossRef] [PubMed]
- Inaguma, S.; Kasai, K.; Ikeda, H. GLI1 facilitates the migration and invasion of pancreatic cancer cells through MUC5AC-mediated attenuation of E-cadherin. Oncogene 2011, 30, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Xu, L.; Guo, C.; Ke, A.; Hu, G.; Xu, X.; Mo, W.; Yang, L.; Huang, Y.; He, S.; et al. Identification of RegIV as a novel GLI1 target gene in human pancreatic cancer. PLoS ONE 2011, 6, e18434. [Google Scholar] [CrossRef] [PubMed]
- Mills, L.D.; Zhang, Y.; Marler, R.J.; Herreros-Villanueva, M.; Zhang, L.; Almada, L.L.; Couch, F.; Wetmore, C.; Pasca di Magliano, M.; Fernandez-Zapico, M.E. Loss of the transcription factor GLI1 identifies a signaling network in the tumor microenvironment mediating KRAS oncogene-induced transformation. J. Biol. Chem. 2013, 288, 11786–11794. [Google Scholar] [CrossRef] [PubMed]
- Mills, L.D.; Zhang, L.; Marler, R.; Svingen, P.; Fernandez-Barrena, M.G.; Dave, M.; Bamlet, W.; McWilliams, R.R.; Petersen, G.M.; Faubion, W.; et al. Inactivation of the transcription factor GLI1 accelerates pancreatic cancer progression. J. Biol. Chem. 2014, 289, 16516–16525. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.Y.; Ji, H.; Ouyang, Z.; Zhou, B.; Ma, W.; Vokes, S.A.; McMahon, A.P.; Wong, W.H.; Scott, M.P. Hedgehog pathway-regulated gene networks in cerebellum development and tumorigenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 9736–9741. [Google Scholar] [CrossRef] [PubMed]
- Ayrault, O.; Zhao, H.; Zindy, F.; Qu, C.; Sherr, C.J.; Roussel, M.F. Atoh1 inhibits neuronal differentiation and collaborates with Gli1 to generate medulloblastoma-initiating cells. Cancer Res. 2010, 70, 5618–5627. [Google Scholar] [CrossRef] [PubMed]
- Colvin Wanshura, L.E.; Galvin, K.E.; Ye, H.; Fernandez-Zapico, M.E.; Wetmore, C. Sequential activation of Snail1 and N-Myc modulates sonic hedgehog-induced transformation of neural cells. Cancer Res. 2011, 71, 5336–5345. [Google Scholar] [CrossRef] [PubMed]
- Uchida, H.; Arita, K.; Yunoue, S.; Yonezawa, H.; Shinsato, Y.; Kawano, H.; Hirano, H.; Hanaya, R.; Tokimura, H. Role of sonic hedgehog signaling in migration of cell lines established from CD133-positive malignant glioma cells. J. Neurooncol. 2011, 104, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Frolova, N.; Sadlonova, A.; Novak, Z.; Steg, A.; Page, G.P.; Welch, D.R.; Lobo-Ruppert, S.M.; Ruppert, J.M.; Johnson, M.R.; et al. Hedgehog signaling and response to cyclopamine differ in epithelial and stromal cells in benign breast and breast cancer. Cancer Biol. Ther. 2006, 5, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Thomas, Z.I.; Gibson, W.; Sexton, J.Z.; Aird, K.M.; Ingram, S.M.; Aldrich, A.; Lyerly, H.K.; Devi, G.R.; Williams, K.P. Targeting GLI1 expression in human inflammatory breast cancer cells enhances apoptosis and attenuates migration. Br. J. Cancer 2011, 104, 1575–1586. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, W.; Yang, Q.; Zhou, S. Nuclear localization of GLI1 and elevated expression of FOXC2 in breast cancer is associated with the basal-like phenotype. Histol. Histopathol. 2012, 27, 475–484. [Google Scholar] [PubMed]
- Fiaschi, M.; Rozell, B.; Bergström, A.; Toftgård, R. Development of mammary tumors by conditional expression of GLI1. Cancer Res. 2009, 69, 4810–4817. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Wang, L.-H.; Wen, Y.-Y.; Song, M.; Li, B.-L.; Chen, X.-L.; Xu, M.; An, S.-X.; Zhao, J.; Lu, Y.-Y.; et al. Expression and regulation mechanisms of Sonic Hedgehog in breast cancer. Cancer Sci. 2010, 101, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Harris, L.G.; Pannell, L.K.; Singh, S.; Samant, R.S.; Shevde, L.A. Increased vascularity and spontaneous metastasis of breast cancer by hedgehog signaling mediated upregulation of cyr61. Oncogene 2012, 31, 3370–3380. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Deng, W.; Nail, C.D.; Bailey, S.K.; Kraus, M.H.; Ruppert, J.M.; Lobo-Ruppert, S.M. Snail induction is an early response to Gli1 that determines the efficiency of epithelial transformation. Oncogene 2006, 25, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Song, T.; Dou, C.; Jia, Y.; Liu, Q. CtBP2 is an independent prognostic marker that promotes GLI1 induced epithelial-mesenchymal transition in hepatocellular carcinoma. Oncotarget 2015, 6, 3752–3769. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Deng, W.; Lobo-Ruppert, S.M.; Ruppert, J.M. Gli1 acts through Snail and E-cadherin to promote nuclear signaling by beta-catenin. Oncogene 2007, 26, 4489–4498. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Siu, M.K.Y.; Au, C.W.H.; Chan, Q.K.Y.; Chan, H.Y.; Wong, E.S.Y.; Ip, P.P.C.; Ngan, H.Y.S.; Cheung, A.N.Y. Aberrant activation of hedgehog signaling pathway contributes to endometrial carcinogenesis through beta-catenin. Mod. Pathol. 2009, 22, 839–847. [Google Scholar] [PubMed]
- Xu, X.; Zhou, Y.; Xie, C.; Wei, S.; Gan, H.; He, S.; Wang, F.; Xu, L.; Lu, J.; Dai, W.; et al. Genome-wide screening reveals an EMT molecular network mediated by Sonic hedgehog-Gli1 signaling in pancreatic cancer cells. PLoS ONE 2012, 7, e43119. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Su, B.; Xie, C.; Wei, S.; Zhou, Y.; Liu, H.; Dai, W.; Cheng, P.; Wang, F.; Xu, X.; et al. Sonic hedgehog-Gli1 signaling pathway regulates the epithelial mesenchymal transition (EMT) by mediating a new target gene, S100A4, in pancreatic cancer cells. PLoS ONE 2014, 9, e96441. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Siu, M.K.Y.; Au, C.W.H.; Wong, E.S.Y.; Chan, H.Y.; Ip, P.P.C.; Ngan, H.Y.S.; Cheung, A.N.Y. Aberrant activation of hedgehog signaling pathway in ovarian cancers: Effect on prognosis, cell invasion and differentiation. Carcinogenesis 2009, 30, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Neill, G.W.; Harrison, W.J.; Ikram, M.S.; Williams, T.D.L.; Bianchi, L.S.; Nadendla, S.K.; Green, J.L.; Ghali, L.; Frischauf, A.-M.; O’Toole, E.A.; et al. GLI1 repression of ERK activity correlates with colony formation and impaired migration in human epidermal keratinocytes. Carcinogenesis 2008, 29, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Mazumdar, T.; Sandhu, R.; Qadan, M.; DeVecchio, J.; Magloire, V.; Agyeman, A.; Li, B.; Houghton, J.A. Hedgehog signaling regulates telomerase reverse transcriptase in human cancer cells. PLoS ONE 2013, 8, e75253. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Wang, F.; Yang, L.; Guo, C.; Wan, R.; Ke, A.; Xu, L.; Hu, G.; Xu, X.; Shen, J.; et al. Expression of DNMT1 and DNMT3a are regulated by GLI1 in human pancreatic cancer. PLoS ONE 2011, 6, e27684. [Google Scholar] [CrossRef] [PubMed]
- Bian, Y.-H.; Huang, S.-H.; Yang, L.; Ma, X.-L.; Xie, J.-W.; Zhang, H.-W. Sonic hedgehog-Gli1 pathway in colorectal adenocarcinomas. World J. Gastroenterol. 2007, 13, 1659–1665. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.; Undèn, A.B.; Krause, D.; Malmqwist, U.; Raza, K.; Zaphiropoulos, P.G.; Toftgård, R. Induction of basal cell carcinomas and trichoepitheliomas in mice overexpressing GLI-1. Proc. Natl. Acad. Sci. USA 2000, 97, 3438–3443. [Google Scholar] [CrossRef]
- Huang, S.; He, J.; Zhang, X.; Bian, Y.; Yang, L.; Xie, G.; Zhang, K.; Tang, W.; Stelter, A.A.; Wang, Q.; et al. Activation of the hedgehog pathway in human hepatocellular carcinomas. Carcinogenesis 2006, 27, 1334–1340. [Google Scholar] [CrossRef] [PubMed]
- Che, L.; Yuan, Y.-H.; Jia, J.; Ren, J. Activation of sonic hedgehog signaling pathway is an independent potential prognosis predictor in human hepatocellular carcinoma patients. Chin. J. Cancer Res. Chung-Kuo Yen Cheng Yen Chiu 2012, 24, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Jeng, K.-S.; Sheen, I.-S.; Jeng, W.-J.; Lin, C.-C.; Lin, C.-K.; Su, J.-C.; Yu, M.-C.; Fang, H.-Y. High expression of patched homolog-1 messenger RNA and glioma-associated oncogene-1 messenger RNA of sonic hedgehog signaling pathway indicates a risk of postresection recurrence of hepatocellular carcinoma. Ann. Surg. Oncol. 2013, 20, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Cui, D.; Xu, Q.; Wang, K.; Che, X. Gli1 is a potential target for alleviating multidrug resistance of gliomas. J. Neurol. Sci. 2010, 288, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Yuan, X.; Zeng, Z.; Tunici, P.; Ng, H.; Abdulkadir, I.R.; Lu, L.; Irvin, D.; Black, K.L.; Yu, J.S. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol. Cancer 2006, 5, 67. [Google Scholar] [CrossRef] [PubMed]
- Ulasov, I.V.; Nandi, S.; Dey, M.; Sonabend, A.M.; Lesniak, M.S. Inhibition of Sonic hedgehog and Notch pathways enhances sensitivity of CD133+ glioma stem cells to temozolomide therapy. Mol. Med. Camb. Mass 2011, 17, 103–112. [Google Scholar] [PubMed]
- Steg, A.D.; Katre, A.A.; Bevis, K.S.; Ziebarth, A.; Dobbin, Z.C.; Shah, M.M.; Alvarez, R.D.; Landen, C.N. Smoothened antagonists reverse taxane resistance in ovarian cancer. Mol. Cancer Ther. 2012, 11, 1587–1597. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jiao, M.; Li, L.; Wu, D.; Wu, K.; Li, X.; Zhu, G.; Dang, Q.; Wang, X.; Hsieh, J.-T.; et al. Tumorspheres derived from prostate cancer cells possess chemoresistant and cancer stem cell properties. J. Cancer Res. Clin. Oncol. 2012, 138, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, L.; Jiao, M.; Wu, D.; Wu, K.; Li, X.; Zhu, G.; Yang, L.; Wang, X.; Hsieh, J.-T.; et al. Genistein inhibits the stemness properties of prostate cancer cells through targeting Hedgehog-Gli1 pathway. Cancer Lett. 2012, 323, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Sims-Mourtada, J.; Izzo, J.G.; Ajani, J.; Chao, K.S.C. Sonic Hedgehog promotes multiple drug resistance by regulation of drug transport. Oncogene 2007, 26, 5674–5679. [Google Scholar] [CrossRef] [PubMed]
- Queiroz, K.C.S.; Ruela-de-Sousa, R.R.; Fuhler, G.M.; Aberson, H.L.; Ferreira, C.V.; Peppelenbosch, M.P.; Spek, C.A. Hedgehog signaling maintains chemoresistance in myeloid leukemic cells. Oncogene 2010, 29, 6314–6322. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.R.; Kunkalla, K.; Qu, C.; Schlette, E.; Neelapu, S.S.; Samaniego, F.; Vega, F. ABCG2 is a direct transcriptional target of hedgehog signaling and involved in stroma-induced drug tolerance in diffuse large B-cell lymphoma. Oncogene 2011, 30, 4874–4886. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.-T.; Zhuan-Sun, Y.-X.; Zhuang, Y.-Y.; Wei, S.-L.; Tang, J.; Chen, W.-B.; Zhang, S.-N. Inhibition of hedgehog signaling depresses self-renewal of pancreatic cancer stem cells and reverses chemoresistance. Int. J. Oncol. 2012, 41, 1707–1714. [Google Scholar] [PubMed]
- Amable, L.; Fain, J.; Gavin, E.; Reed, E. Gli1 contributes to cellular resistance to cisplatin through altered cellular accumulation of the drug. Oncol. Rep. 2014, 32, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.; Demars, C.J.; Comba, A.; Gainullin, V.G.; Rizvi, Z.; Almada, L.L.; Wang, K.; Lomberk, G.; Fernández-Zapico, M.E.; Buttar, N.S. Combinatorial chemoprevention reveals a novel smoothened-independent role of GLI1 in esophageal carcinogenesis. Cancer Res. 2010, 70, 6787–6796. [Google Scholar] [CrossRef] [PubMed]
- Dormoy, V.; Béraud, C.; Lindner, V.; Coquard, C.; Barthelmebs, M.; Brasse, D.; Jacqmin, D.; Lang, H.; Massfelder, T. Vitamin D3 triggers antitumor activity through targeting hedgehog signaling in human renal cell carcinoma. Carcinogenesis 2012, 33, 2084–2093. [Google Scholar] [CrossRef] [PubMed]
- Zahreddine, H.A.; Culjkovic-Kraljacic, B.; Assouline, S.; Gendron, P.; Romeo, A.A.; Morris, S.J.; Cormack, G.; Jaquith, J.B.; Cerchietti, L.; Cocolakis, E.; et al. The sonic hedgehog factor GLI1 imparts drug resistance through inducible glucuronidation. Nature 2014, 511, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wu, K.; Gao, D.; Zhu, G.; Wu, D.; Wang, X.; Chen, Y.; Du, Y.; Song, W.; Ma, Z.; et al. Reciprocal regulation of hypoxia-inducible factor 2α and GLI1 expression associated with the radioresistance of renal cell carcinoma. Int. J. Radiat. Oncol. Biol. Phys. 2014, 90, 942–951. [Google Scholar] [CrossRef] [PubMed]
- Stanton, B.Z.; Peng, L.F.; Maloof, N.; Nakai, K.; Wang, X.; Duffner, J.L.; Taveras, K.M.; Hyman, J.M.; Lee, S.W.; Koehler, A.N.; et al. A small molecule that binds Hedgehog and blocks its signaling in human cells. Nat. Chem. Biol. 2009, 5, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Athar, M.; Li, C.; Tang, X.; Chi, S.; Zhang, X.; Kim, A.L.; Tyring, S.K.; Kopelovich, L.; Hebert, J.; Epstein, E.H.; et al. Inhibition of smoothened signaling prevents ultraviolet B-induced basal cell carcinomas through regulation of Fas expression and apoptosis. Cancer Res. 2004, 64, 7545–7552. [Google Scholar] [CrossRef] [PubMed]
- Taipale, J.; Chen, J.K.; Cooper, M.K.; Wang, B.; Mann, R.K.; Milenkovic, L.; Scott, M.P.; Beachy, P.A. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature 2000, 406, 1005–1009. [Google Scholar] [PubMed]
- Dlugosz, A.; Agrawal, S.; Kirkpatrick, P. Vismodegib. Nat. Rev. Drug Discov. 2012, 11, 437–438. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Samant, R.S.; Shevde, L.A. Nonclassical activation of Hedgehog signaling enhances multidrug resistance and makes cancer cells refractory to Smoothened-targeting Hedgehog inhibition. J. Biol. Chem. 2013, 288, 11824–11833. [Google Scholar] [CrossRef] [PubMed]
- Kaylani, S.Z.; Xu, J.; Srivastava, R.K.; Kopelovich, L.; Pressey, J.G.; Athar, M. Rapamycin targeting mTOR and hedgehog signaling pathways blocks human rhabdomyosarcoma growth in xenograft murine model. Biochem. Biophys. Res. Commun. 2013, 435, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R.K.; Kaylani, S.Z.; Edrees, N.; Li, C.; Talwelkar, S.S.; Xu, J.; Palle, K.; Pressey, J.G.; Athar, M. GLI inhibitor GANT-61 diminishes embryonal and alveolar rhabdomyosarcoma growth by inhibiting Shh/AKT-mTOR axis. Oncotarget 2014, 5, 12151–12165. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Chen, X.; Zhang, P.; Fan, Y.; Ma, A.; Pang, T.; Song, Z.; Jin, Y.; Hao, W.; Liu, F.; et al. Inhibition of hedgehog signaling by GANT58 induces apoptosis and shows synergistic antitumor activity with AKT inhibitor in acute T cell leukemia cells. Biochimie 2014, 101, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Yu, K.; Zhang, L.; Li, Y.; Li, Q.; Yang, Z.; Shen, T.; Duan, L.; Xiong, W.; Wang, W. Synergistic inhibition of colon carcinoma cell growth by Hedgehog-Gli1 inhibitor arsenic trioxide and phosphoinositide 3-kinase inhibitor LY294002. OncoTargets Ther. 2015, 8, 877–883. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- LoRusso, P.M.; Rudin, C.M.; Reddy, J.C.; Tibes, R.; Weiss, G.J.; Borad, M.J.; Hann, C.L.; Brahmer, J.R.; Chang, I.; Darbonne, W.C.; et al. Phase I trial of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with refractory, locally advanced or metastatic solid tumors. Clin. Cancer Res. 2011, 17, 2502–2511. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palle, K.; Mani, C.; Tripathi, K.; Athar, M. Aberrant GLI1 Activation in DNA Damage Response, Carcinogenesis and Chemoresistance. Cancers 2015, 7, 2330-2351. https://doi.org/10.3390/cancers7040894
Palle K, Mani C, Tripathi K, Athar M. Aberrant GLI1 Activation in DNA Damage Response, Carcinogenesis and Chemoresistance. Cancers. 2015; 7(4):2330-2351. https://doi.org/10.3390/cancers7040894
Chicago/Turabian StylePalle, Komaraiah, Chinnadurai Mani, Kaushlendra Tripathi, and Mohammad Athar. 2015. "Aberrant GLI1 Activation in DNA Damage Response, Carcinogenesis and Chemoresistance" Cancers 7, no. 4: 2330-2351. https://doi.org/10.3390/cancers7040894
APA StylePalle, K., Mani, C., Tripathi, K., & Athar, M. (2015). Aberrant GLI1 Activation in DNA Damage Response, Carcinogenesis and Chemoresistance. Cancers, 7(4), 2330-2351. https://doi.org/10.3390/cancers7040894