
The Receptor Tyrosine Kinase AXL in Cancer Progression

Abstract
:1. Introduction
2. The Physiologic Role of GAS6/AXL Signaling
3. GAS6 and AXL Expression in Cancer
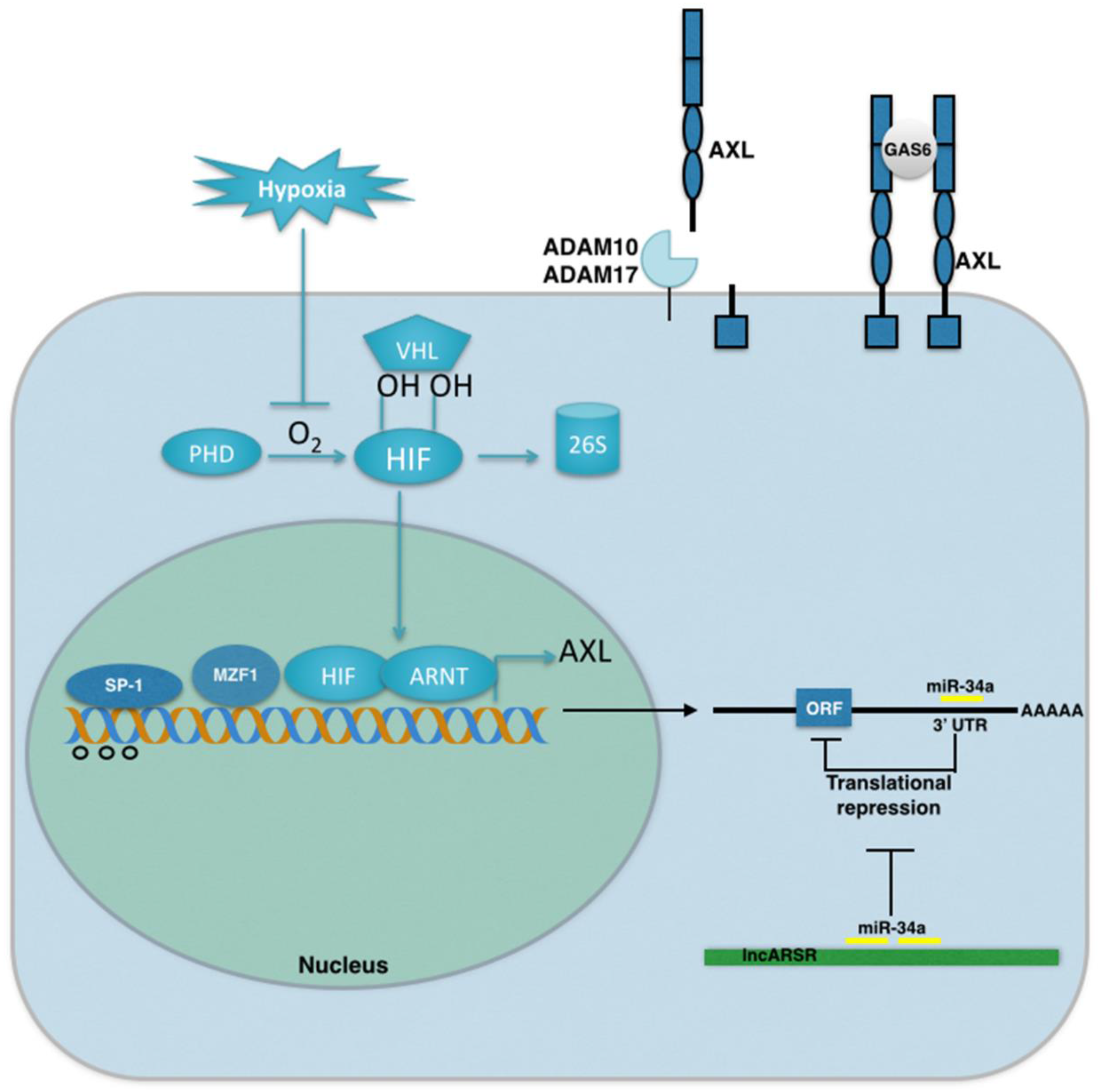
4. Mechanisms of AXL Regulation in Cancer
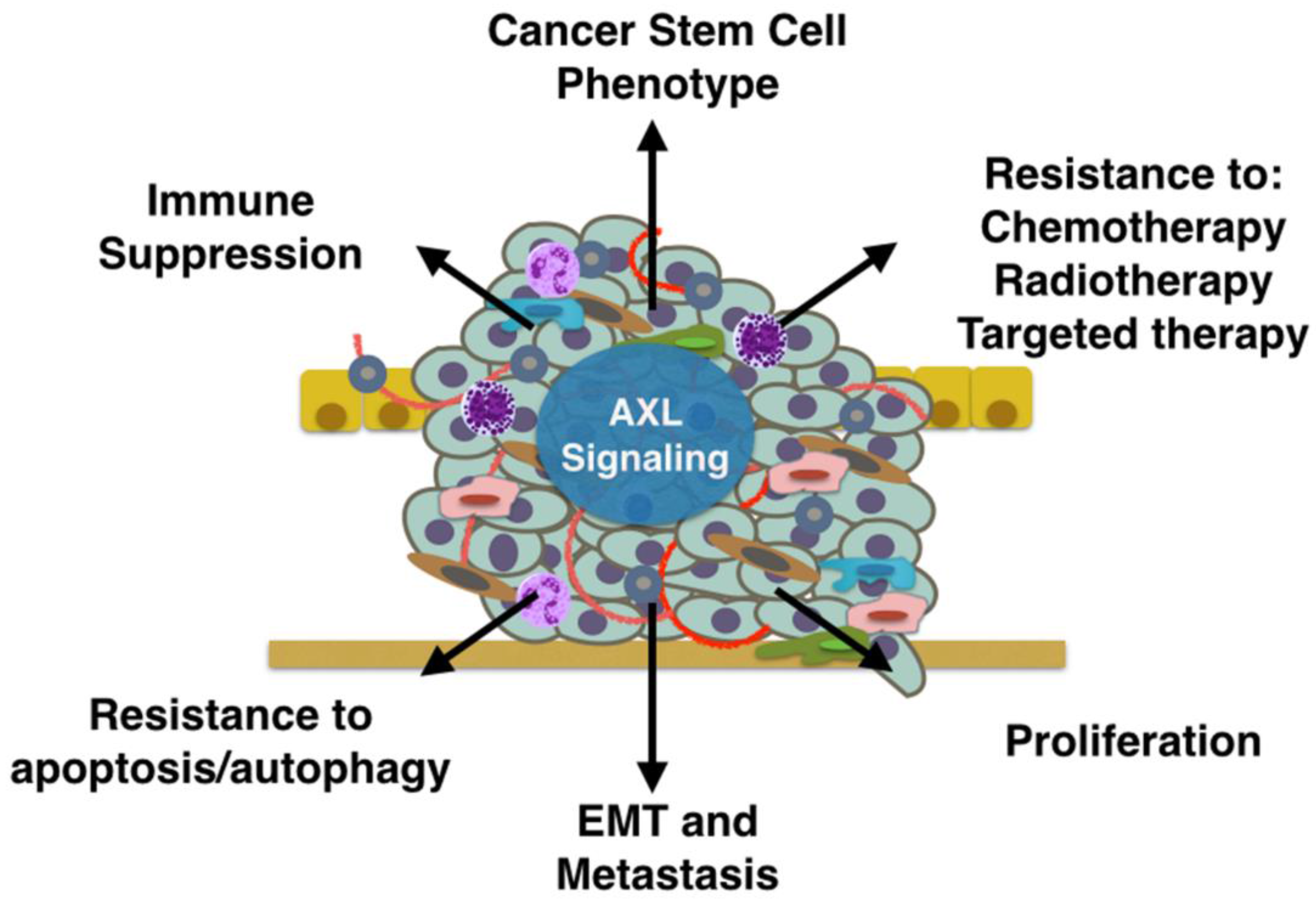
5. Mechanisms of AXL-Mediated Tumor Progression and Metastasis
5.1. Proliferation, Survival, Anti-Apoptosis
5.2. Drug Resistance
5.3. Stem Cell Phenotype
5.4. Metastasis
5.5. Immune Suppression
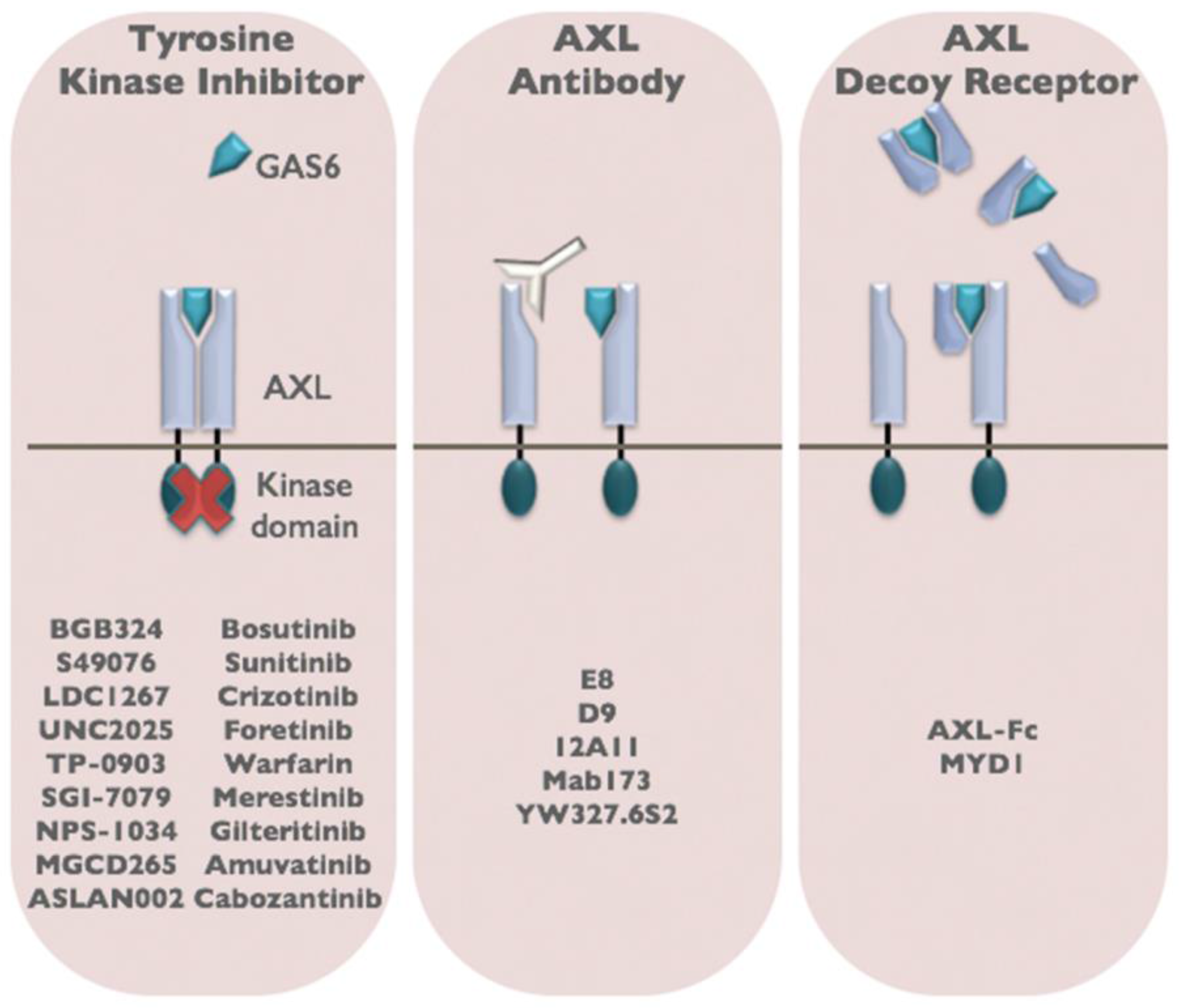
6. Therapeutic Targeting of AXL
7. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- O’Bryan, J.P.; Frye, R.A.; Cogswell, P.C.; Neubauer, A.; Kitch, B.; Prokop, C.; Espinosa, R., 3rd; Le Beau, M.M.; Earp, H.S.; Liu, E.T. Axl, a transforming gene isolated from primary human myeloid leukemia cells, encodes a novel receptor tyrosine kinase. Mol. Cell. Biol. 1991, 11, 5016–5031. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.K.; Dawson, T.L.; Mullaney, D.L.; Snodgrass, H.R.; Earp, H.S. Cloning and mRNA expression analysis of a novel human protooncogene, c-mer. Cell Growth Differ. 1994, 5, 647–657. [Google Scholar] [PubMed]
- Lai, C.; Gore, M.; Lemke, G. Structure, expression, and activity of tyro 3, a neural adhesion-related receptor tyrosine kinase. Oncogene 1994, 9, 2567–2578. [Google Scholar] [PubMed]
- Mark, M.R.; Chen, J.; Hammonds, R.G.; Sadick, M.; Godowsk, P.J. Characterization of Gas6, a member of the superfamily of G domain-containing proteins, as a ligand for Rse and Axl. J. Biol. Chem. 1996, 271, 9785–9789. [Google Scholar] [PubMed]
- Nagata, K.; Ohashi, K.; Nakano, T.; Arita, H.; Zong, C.; Hanafusa, H.; Mizuno, K. Identification of the product of growth arrest-specific gene 6 as a common ligand for Axl, Sky, and Mer receptor tyrosine kinases. J. Biol. Chem. 1996, 271, 30022–30027. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, K.; Nagata, K.; Toshima, J.; Nakano, T.; Arita, H.; Tsuda, H.; Suzuki, K.; Mizuno, K. Stimulation of sky receptor tyrosine kinase by the product of growth arrest-specific gene 6. J. Biol. Chem. 1995, 270, 22681–22684. [Google Scholar] [CrossRef] [PubMed]
- Stitt, T.N.; Conn, G.; Gore, M.; Lai, C.; Bruno, J.; Radziejewski, C.; Mattsson, K.; Fisher, J.; Gies, D.R.; Jones, P.F.; et al. The anticoagulation factor protein S and its relative, Gas6, are ligands for the Tyro 3/Axl family of receptor tyrosine kinases. Cell 1995, 80, 661–670. [Google Scholar] [CrossRef]
- Lemke, G. Biology of the TAM receptors. Cold Spring Harb. Perspect. Biol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Gore, M.; Zhang, Q.; Camenisch, T.; Boast, S.; Casagranda, F.; Lai, C.; Skinner, M.K.; Klein, R.; Matsushima, G.K.; et al. Tyro-3 family receptors are essential regulators of mammalian spermatogenesis. Nature 1999, 398, 723–728. [Google Scholar] [PubMed]
- Angelillo-Scherrer, A.; de Frutos, P.; Aparicio, C.; Melis, E.; Savi, P.; Lupu, F.; Arnout, J.; Dewerchin, M.; Hoylaerts, M.; Herbert, J.; et al. Deficiency or inhibition of Gas6 causes platelet dysfunction and protects mice against thrombosis. Nat. Med. 2001, 7, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Angelillo-Scherrer, A.; Burnier, L.; Flores, N.; Savi, P.; DeMol, M.; Schaeffer, P.; Herbert, J.M.; Lemke, G.; Goff, S.P.; Matsushima, G.K.; et al. Role of Gas6 receptors in platelet signaling during thrombus stabilization and implications for antithrombotic therapy. J. Clin. Investig. 2005, 115, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Burstyn-Cohen, T.; Heeb, M.J.; Lemke, G. Lack of protein s in mice causes embryonic lethal coagulopathy and vascular dysgenesis. J. Clin. Investig. 2009, 119, 2942–2953. [Google Scholar] [CrossRef] [PubMed]
- Shieh, Y.S.; Lai, C.Y.; Kao, Y.R.; Shiah, S.G.; Chu, Y.W.; Lee, H.S.; Wu, C.W. Expression of Axl in lung adenocarcinoma and correlation with tumor progression. Neoplasia 2005, 7, 1058–1064. [Google Scholar] [PubMed]
- Hutterer, M.; Knyazev, P.; Abate, A.; Reschke, M.; Maier, H.; Stefanova, N.; Knyazeva, T.; Barbieri, V.; Reindl, M.; Muigg, A.; et al. Axl and growth arrest-specific gene 6 are frequently overexpressed in human gliomas and predict poor prognosis in patients with glioblastoma multiforme. Clin. Cancer Res. 2008, 14, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Koorstra, J.B.; Karikari, C.A.; Feldmann, G.; Bisht, S.; Rojas, P.L.; Offerhaus, G.J.; Alvarez, H.; Maitra, A. The Axl receptor tyrosine kinase confers an adverse prognostic influence in pancreatic cancer and represents a new therapeutic target. Cancer Biol. Ther. 2009, 8, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, A.; Martuszewska, D.; Johansson, M.; Ekman, C.; Hafizi, S.; Ljungberg, B.; Dahlback, B. Differential expression of Axl and Gas6 in renal cell carcinoma reflecting tumor advancement and survival. Clin. Cancer Res. 2009, 15, 4742–4749. [Google Scholar] [CrossRef] [PubMed]
- Hector, A.; Montgomery, E.A.; Karikari, C.; Canto, M.; Dunbar, K.B.; Wang, J.S.; Feldmann, G.; Hong, S.M.; Haffner, M.C.; Meeker, A.K.; et al. The Axl receptor tyrosine kinase is an adverse prognostic factor and a therapeutic target in esophageal adenocarcinoma. Cancer Biol. Ther. 2010, 10, 1009–1018. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Yen, C.Y.; Liu, S.Y.; Chen, C.K.; Chiang, C.F.; Shiah, S.G.; Chen, P.H.; Shieh, Y.S. Axl is a prognostic marker in oral squamous cell carcinoma. Ann. Surg. Oncol. 2012, 19, S500–S508. [Google Scholar] [CrossRef] [PubMed]
- Pinato, D.J.; Mauri, F.A.; Lloyd, T.; Vaira, V.; Casadio, C.; Boldorini, R.L.; Sharma, R. The expression of Axl receptor tyrosine kinase influences the tumour phenotype and clinical outcome of patients with malignant pleural mesothelioma. Br. J. Cancer 2013, 108, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.X.; Li, Q.Y.; Yang, Z. Axl and prostasin are biomarkers for prognosis of ovarian adenocarcinoma. Ann. Diagn. Pathol. 2013, 17, 425–429. [Google Scholar] [CrossRef] [PubMed]
- Dunne, P.D.; McArt, D.G.; Blayney, J.K.; Kalimutho, M.; Greer, S.; Wang, T.; Srivastava, S.; Ong, C.W.; Arthur, K.; Loughrey, M.; et al. Axl is a key regulator of inherent and chemotherapy-induced invasion and predicts a poor clinical outcome in early-stage colon cancer. Clin. Cancer Res. 2014, 20, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Brand, T.M.; Iida, M.; Stein, A.P.; Corrigan, K.L.; Braverman, C.M.; Coan, J.P.; Pearson, H.E.; Bahrar, H.; Fowler, T.L.; Bednarz, B.P.; et al. Axl is a logical molecular target in head and neck squamous cell carcinoma. Clin. Cancer Res. 2015, 21, 2601–2612. [Google Scholar] [CrossRef] [PubMed]
- Hattori, S.; Kikuchi, E.; Kosaka, T.; Miyazaki, Y.; Tanaka, N.; Miyajima, A.; Mikami, S.; Oya, M. Relationship between increased expression of the Axl/Gas6 signal cascade and prognosis of patients with upper tract urothelial carcinoma. Ann. Surg. Oncol. 2016, 23, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Rea, K.; Pinciroli, P.; Sensi, M.; Alciato, F.; Bisaro, B.; Lozneanu, L.; Raspagliesi, F.; Centritto, F.; Cabodi, S.; Defilippi, P.; et al. Novel Axl-driven signaling pathway and molecular signature characterize high-grade ovarian cancer patients with poor clinical outcome. Oncotarget 2015, 6, 30859–30875. [Google Scholar] [PubMed]
- Hsieh, M.S.; Yang, P.W.; Wong, L.F.; Lee, J.M. The Axl receptor tyrosine kinase is associated with adverse prognosis and distant metastasis in esophageal squamous cell carcinoma. Oncotarget 2016, 7, 36956–36970. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, K.; Yan, Z.; Xia, Y.; Li, J.; Shi, L.; Zou, Q.; Wan, X.; Jiao, B.; Wang, H.; et al. Axl expression stratifies patients with poor prognosis after hepatectomy for hepatocellular carcinoma. PLoS ONE 2016, 11, e0154767. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Tian, R.; Yong, B.; Luo, C.; Tan, P.; Shen, J.; Peng, T. Gas6/Axl mediates tumor cell apoptosis, migration and invasion and predicts the clinical outcome of osteosarcoma patients. Biochem. Biophys. Res. Commun. 2013, 435, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Gjerdrum, C.; Tiron, C.; Hoiby, T.; Stefansson, I.; Haugen, H.; Sandal, T.; Collett, K.; Li, S.; McCormack, E.; Gjertsen, B.T.; et al. Axl is an essential epithelial-to-mesenchymal transition-induced regulator of breast cancer metastasis and patient survival. Proc. Natl. Acad. Sci. USA 2010, 107, 1124–1129. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H., 2nd; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.C.; Lay, J.D.; Huang, J.S.; Cheng, A.L.; Tang, J.L.; Lin, M.T.; Lai, G.M.; Chuang, S.E. Receptor tyrosine kinase Axl is induced by chemotherapy drugs and overexpression of Axl confers drug resistance in acute myeloid leukemia. Cancer Lett. 2008, 268, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Lee, J.C.; Lin, L.; Olivas, V.; Au, V.; LaFramboise, T.; Abdel-Rahman, M.; Wang, X.; Levine, A.D.; Rho, J.K.; et al. Activation of the Axl kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat. Genet. 2012, 44, 852–860. [Google Scholar] [CrossRef] [PubMed]
- Konieczkowski, D.J.; Johannessen, C.M.; Abudayyeh, O.; Kim, J.W.; Cooper, Z.A.; Piris, A.; Frederick, D.T.; Barzily-Rokni, M.; Straussman, R.; Haq, R.; et al. A melanoma cell state distinction influences sensitivity to mapk pathway inhibitors. Cancer Discov. 2014, 4, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Muller, J.; Krijgsman, O.; Tsoi, J.; Robert, L.; Hugo, W.; Song, C.; Kong, X.; Possik, P.A.; Cornelissen-Steijger, P.D.; Geukes Foppen, M.H.; et al. Low mitf/axl ratio predicts early resistance to multiple targeted drugs in melanoma. Nat. Commun. 2014. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Liu, X.D.; Sun, M.; Zhang, X.; German, P.; Bai, S.; Ding, Z.; Tannir, N.; Wood, C.G.; Matin, S.F.; et al. Targeting met and Axl overcomes resistance to sunitinib therapy in renal cell carcinoma. Oncogene 2016, 35, 2687–2697. [Google Scholar] [CrossRef] [PubMed]
- Whitman, S.P.; Kohlschmidt, J.; Maharry, K.; Volinia, S.; Mrozek, K.; Nicolet, D.; Schwind, S.; Becker, H.; Metzeler, K.H.; Mendler, J.H.; et al. Gas6 expression identifies high-risk adult aml patients: Potential implications for therapy. Leukemia 2014, 28, 1252–1258. [Google Scholar] [CrossRef] [PubMed]
- Buehler, M.; Tse, B.; Leboucq, A.; Jacob, F.; Caduff, R.; Fink, D.; Goldstein, D.R.; Heinzelmann-Schwarz, V. Meta-analysis of microarray data identifies Gas6 expression as an independent predictor of poor survival in ovarian cancer. BioMed Res. Int. 2013. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, M.; Sonobe, M.; Nakayama, E.; Kobayashi, M.; Kikuchi, R.; Kitamura, J.; Imamura, N.; Date, H. Higher expression of receptor tyrosine kinase Axl, and differential expression of its ligand, Gas6, predict poor survival in lung adenocarcinoma patients. Ann. Surg. Oncol. 2013, 20, S467–S476. [Google Scholar] [CrossRef] [PubMed]
- Sawabu, T.; Seno, H.; Kawashima, T.; Fukuda, A.; Uenoyama, Y.; Kawada, M.; Kanda, N.; Sekikawa, A.; Fukui, H.; Yanagita, M.; et al. Growth arrest-specific gene 6 and Axl signaling enhances gastric cancer cell survival via Akt pathway. Mol. Carcinog. 2007, 46, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Liu, G.; Wang, L.; Liu, H. Elevated serum gas6 is a novel prognostic biomarker in patients with oral squamous cell carcinoma. PLoS ONE 2015, 10, e0133940. [Google Scholar] [CrossRef] [PubMed]
- Rankin, E.B.; Giaccia, A.J. Hypoxic control of metastasis. Science 2016, 352, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Rankin, E.B.; Fuh, K.C.; Castellini, L.; Viswanathan, K.; Finger, E.C.; Diep, A.N.; LaGory, E.L.; Kariolis, M.S.; Chan, A.; Lindgren, D.; et al. Direct regulation of Gas6/Axl signaling by hif promotes renal metastasis through src and met. Proc. Natl. Acad. Sci. USA 2014, 111, 13373–13378. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Wang, J.; Shiozawa, Y.; McGee, S.; Kim, J.; Jung, Y.; Joseph, J.; Berry, J.E.; Havens, A.; Pienta, K.J.; et al. Hypoxia stabilizes Gas6/Axl signaling in metastatic prostate cancer. Mol. Cancer Res. 2012, 10, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Nalwoga, H.; Ahmed, L.; Arnes, J.B.; Wabinga, H.; Akslen, L.A. Strong expression of hypoxia-inducible factor-1α (HIF-1α) is associated with Axl expression and features of aggressive tumors in african breast cancer. PLoS ONE 2016, 11, e0146823. [Google Scholar] [CrossRef] [PubMed]
- Manfioletti, G.; Brancolini, C.; Avanzi, G.; Schneider, C. The protein encoded by a growth arrest-specific gene (Gas6) is a new member of the vitamin k-dependent proteins related to protein s, a negative coregulator in the blood coagulation cascade. Mol. Cell. Biol. 1993, 13, 4976–4985. [Google Scholar] [CrossRef] [PubMed]
- Goruppi, S.; Ruaro, E.; Schneider, C. Gas6, the ligand of axl tyrosine kinase receptor, has mitogenic and survival activities for serum starved nih3t3 fibroblasts. Oncogene 1996, 12, 471–480. [Google Scholar] [PubMed]
- Aghourian, M.N.; Lemarie, C.A.; Bertin, F.R.; Blostein, M.D. Prostaglandin E synthase is upregulated by Gas6 during cancer-induced venous thrombosis. Blood 2016, 127, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Waizenegger, J.S.; Ben-Batalla, I.; Weinhold, N.; Meissner, T.; Wroblewski, M.; Janning, M.; Riecken, K.; Binder, M.; Atanackovic, D.; Taipaleenmaeki, H.; et al. Role of growth arrest-specific gene 6-mer axis in multiple myeloma. Leukemia 2015, 29, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Ben-Batalla, I.; Schultze, A.; Wroblewski, M.; Erdmann, R.; Heuser, M.; Waizenegger, J.S.; Riecken, K.; Binder, M.; Schewe, D.; Sawall, S.; et al. Axl, a prognostic and therapeutic target in acute myeloid leukemia mediates paracrine crosstalk of leukemia cells with bone marrow stroma. Blood 2013, 122, 2443–2452. [Google Scholar] [CrossRef] [PubMed]
- Loges, S.; Schmidt, T.; Tjwa, M.; van Geyte, K.; Lievens, D.; Lutgens, E.; Vanhoutte, D.; Borgel, D.; Plaisance, S.; Hoylaerts, M.; et al. Malignant cells fuel tumor growth by educating infiltrating leukocytes to produce the mitogen gas6. Blood 2010, 115, 2264–2273. [Google Scholar] [CrossRef] [PubMed]
- Mudduluru, G.; Allgayer, H. The human receptor tyrosine kinase axl gene—Promoter characterization and regulation of constitutive expression by sp1, sp3 and cpg methylation. Biosci. Rep. 2008, 28, 161–176. [Google Scholar] [CrossRef] [PubMed]
- Mudduluru, G.; Vajkoczy, P.; Allgayer, H. Myeloid zinc finger 1 induces migration, invasion, and in vivo metastasis through axl gene expression in solid cancer. Mol. Cancer Res. 2010, 8, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Li, X.J.; Ren, Z.J.; Tang, J.H. Microrna-34a: A potential therapeutic target in human cancer. Cell Death Dis. 2014, 5, e1327. [Google Scholar] [CrossRef] [PubMed]
- Mudduluru, G.; Ceppi, P.; Kumarswamy, R.; Scagliotti, G.V.; Papotti, M.; Allgayer, H. Regulation of axl receptor tyrosine kinase expression by mir-34a and mir-199a/b in solid cancer. Oncogene 2011, 30, 2888–2899. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Shi, X.; Ling, F.; Wang, C.; Liu, J.; Wang, W.; Li, M. Mir-34a suppresses ovarian cancer proliferation and motility by targeting axl. Tumour Biol. 2015, 36, 7277–7283. [Google Scholar] [CrossRef] [PubMed]
- Mackiewicz, M.; Huppi, K.; Pitt, J.J.; Dorsey, T.H.; Ambs, S.; Caplen, N.J. Identification of the receptor tyrosine kinase axl in breast cancer as a target for the human mir-34a microrna. Breast Cancer Res. Treat. 2011, 130, 663–679. [Google Scholar] [CrossRef] [PubMed]
- Qu, L.; Ding, J.; Chen, C.; Wu, Z.J.; Liu, B.; Gao, Y.; Chen, W.; Liu, F.; Sun, W.; Li, X.F.; et al. Exosome-transmitted lncarsr promotes sunitinib resistance in renal cancer by acting as a competing endogenous rna. Cancer Cell 2016, 29, 653–668. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Oudin, M.J.; Sullivan, R.J.; Wang, S.J.; Meyer, A.S.; Im, H.; Frederick, D.T.; Tadros, J.; Griffith, L.G.; Lee, H.; et al. Reduced proteolytic shedding of receptor tyrosine kinases is a post-translational mechanism of kinase inhibitor resistance. Cancer Discov. 2016, 6, 382–399. [Google Scholar] [CrossRef] [PubMed]
- Johansson, G.; Peng, P.C.; Huang, P.Y.; Chien, H.F.; Hua, K.T.; Kuo, M.L.; Chen, C.T.; Lee, M.J. Soluble axl: A possible circulating biomarker for neurofibromatosis type 1 related tumor burden. PLoS ONE 2014, 9, e115916. [Google Scholar] [CrossRef] [PubMed]
- Reichl, P.; Fang, M.; Starlinger, P.; Staufer, K.; Nenutil, R.; Muller, P.; Greplova, K.; Valik, D.; Dooley, S.; Brostjan, C.; et al. Multicenter analysis of soluble axl reveals diagnostic value for very early stage hepatocellular carcinoma. Int. J. Cancer 2015, 137, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.P.; Wen, Y.; Varnum, B.; Hung, M.C. Akt is required for axl-gas6 signaling to protect cells from e1a-mediated apoptosis. Oncogene 2002, 21, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.P.; Liao, Y.; Robinson, D.; Kung, H.J.; Liu, E.T.; Hung, M.C. Axl-gas6 interaction counteracts e1a-mediated cell growth suppression and proapoptotic activity. Mol. Cell. Biol. 1999, 19, 8075–8082. [Google Scholar] [CrossRef] [PubMed]
- Paccez, J.D.; Duncan, K.; Vava, A.; Correa, R.G.; Libermann, T.A.; Parker, M.I.; Zerbini, L.F. Inactivation of gsk3beta and activation of NF-κB pathway via axl represents an important mediator of tumorigenesis in esophageal squamous cell carcinoma. Mol. Biol. Cell 2015, 26, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Gong, M.; Li, X.; Zhou, Y.; Gao, W.; Tulpule, A.; Chaudhary, P.M.; Jung, J.; Gill, P.S. Induction, regulation, and biologic function of Axl receptor tyrosine kinase in kaposi sarcoma. Blood 2010, 116, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Ou, W.B.; Corson, J.M.; Flynn, D.L.; Lu, W.P.; Wise, S.C.; Bueno, R.; Sugarbaker, D.J.; Fletcher, J.A. Axl regulates mesothelioma proliferation and invasiveness. Oncogene 2011, 30, 1643–1652. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Secreto, C.; Boysen, J.; Sassoon, T.; Shanafelt, T.D.; Mukhopadhyay, D.; Kay, N.E. The novel receptor tyrosine kinase Axl is constitutively active in B-cell chronic lymphocytic leukemia and acts as a docking site of nonreceptor kinases: Implications for therapy. Blood 2011, 117, 1928–1937. [Google Scholar] [CrossRef] [PubMed]
- Van Ginkel, P.R.; Gee, R.L.; Shearer, R.L.; Subramanian, L.; Walker, T.M.; Albert, D.M.; Meisner, L.F.; Varnum, B.C.; Polans, A.S. Expression of the receptor tyrosine kinase Axl promotes ocular melanoma cell survival. Cancer Res. 2004, 64, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Vajkoczy, P.; Knyazev, P.; Kunkel, A.; Capelle, H.H.; Behrndt, S.; von Tengg-Kobligk, H.; Kiessling, F.; Eichelsbacher, U.; Essig, M.; Read, T.A.; et al. Dominant-negative inhibition of the axl receptor tyrosine kinase suppresses brain tumor cell growth and invasion and prolongs survival. Proc. Natl. Acad. Sci. USA 2006, 103, 5799–5804. [Google Scholar] [CrossRef] [PubMed]
- Rankin, E.B.; Fuh, K.C.; Taylor, T.E.; Krieg, A.J.; Musser, M.; Yuan, J.; Wei, K.; Kuo, C.J.; Longacre, T.A.; Giaccia, A.J. Axl is an essential factor and therapeutic target for metastatic ovarian cancer. Cancer Res. 2010, 70, 7570–7579. [Google Scholar] [CrossRef] [PubMed]
- Ammoun, S.; Provenzano, L.; Zhou, L.; Barczyk, M.; Evans, K.; Hilton, D.A.; Hafizi, S.; Hanemann, C.O. Axl/Gas6/NFκB signalling in schwannoma pathological proliferation, adhesion and survival. Oncogene 2014, 33, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Ye, X.; Pham, T.; Lin, E.; Chan, S.; McNamara, E.; Neve, R.M.; Belmont, L.; Koeppen, H.; Yauch, R.L.; et al. Axl inhibition sensitizes mesenchymal cancer cells to antimitotic drugs. Cancer Res. 2014, 74, 5878–5890. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Peng, D.; Chen, Z.; Sehdev, V.; Belkhiri, A. Abl regulation by axl promotes cisplatin resistance in esophageal cancer. Cancer Res. 2013, 73, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Giles, K.M.; Kalinowski, F.C.; Candy, P.A.; Epis, M.R.; Zhang, P.M.; Redfern, A.D.; Stuart, L.M.; Goodall, G.J.; Leedman, P.J. Axl mediates acquired resistance of head and neck cancer cells to the epidermal growth factor receptor inhibitor erlotinib. Mol. Cancer Ther. 2013, 12, 2541–2558. [Google Scholar] [CrossRef] [PubMed]
- Brand, T.M.; Iida, M.; Stein, A.P.; Corrigan, K.L.; Braverman, C.M.; Luthar, N.; Toulany, M.; Gill, P.S.; Salgia, R.; Kimple, R.J.; et al. Axl mediates resistance to cetuximab therapy. Cancer Res. 2014, 74, 5152–5164. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.S.; Miller, M.A.; Gertler, F.B.; Lauffenburger, D.A. The receptor Axl diversifies egfr signaling and limits the response to egfr-targeted inhibitors in triple-negative breast cancer cells. Sci. Signal. 2013, 6, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Elkabets, M.; Pazarentzos, E.; Juric, D.; Sheng, Q.; Pelossof, R.A.; Brook, S.; Benzaken, A.O.; Rodon, J.; Morse, N.; Yan, J.J.; et al. Axl mediates resistance to PI3Kα inhibition by activating the EGFR/PKC/Mtor axis in head and neck and esophageal squamous cell carcinomas. Cancer Cell 2015, 27, 533–546. [Google Scholar] [CrossRef] [PubMed]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Cichon, M.A.; Szentpetery, Z.; Caley, M.P.; Papadakis, E.S.; Mackenzie, I.C.; Brennan, C.H.; O’Toole, E.A. The receptor tyrosine kinase Axl regulates cell-cell adhesion and stemness in cutaneous squamous cell carcinoma. Oncogene 2014, 33, 4185–4192. [Google Scholar] [CrossRef] [PubMed]
- Asiedu, M.K.; Beauchamp-Perez, F.D.; Ingle, J.N.; Behrens, M.D.; Radisky, D.C.; Knutson, K.L. Axl induces epithelial-to-mesenchymal transition and regulates the function of breast cancer stem cells. Oncogene 2014, 33, 1316–1324. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.; Phillips, E.; Kim, S.H.; Taylor, D.; Hielscher, T.; Puccio, L.; Hjelmeland, A.B.; Lichter, P.; Nakano, I.; Goidts, V. Kinome-wide shRNA screen identifies the receptor tyrosine kinase Axl as a key regulator for mesenchymal glioblastoma stem-like cells. Stem Cell Rep. 2015, 4, 899–913. [Google Scholar] [CrossRef] [PubMed]
- Tai, K.Y.; Shieh, Y.S.; Lee, C.S.; Shiah, S.G.; Wu, C.W. Axl promotes cell invasion by inducing mmp-9 activity through activation of NF-κB and brg-1. Oncogene 2008, 27, 4044–4055. [Google Scholar] [CrossRef] [PubMed]
- Abu-Thuraia, A.; Gauthier, R.; Chidiac, R.; Fukui, Y.; Screaton, R.A.; Gratton, J.P.; Cote, J.F. Axl phosphorylates elmo scaffold proteins to promote rac activation and cell invasion. Mol. Cell. Biol. 2015, 35, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Fingleton, B.; Matrisian, L.M. Matrix metalloproteinase inhibitors and cancer: Trials and tribulations. Science 2002, 295, 2387–2392. [Google Scholar] [CrossRef] [PubMed]
- Ruffell, B.; Coussens, L.M. Macrophages and therapeutic resistance in cancer. Cancer Cell 2015, 27, 462–472. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.K.; DeRyckere, D.; Davies, K.D.; Earp, H.S. The TAM family: Phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat. Rev. Cancer 2014, 14, 769–785. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Lemke, G. Homeostatic regulation of the immune system by receptor tyrosine kinases of the Tyro 3 family. Science 2001, 293, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Lemke, G.; Rothlin, C.V. Immunobiology of the tam receptors. Nat. Rev. Immunol. 2008, 8, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Li, Y.; Stawicki, S.; Couto, S.; Eastham-Anderson, J.; Kallop, D.; Weimer, R.; Wu, Y.; Pei, L. An anti-axl monoclonal antibody attenuates xenograft tumor growth and enhances the effect of multiple anticancer therapies. Oncogene 2010, 29, 5254–5264. [Google Scholar] [CrossRef] [PubMed]
- Chiu, K.C.; Lee, C.H.; Liu, S.Y.; Chou, Y.T.; Huang, R.Y.; Huang, S.M.; Shieh, Y.S. Polarization of tumor-associated macrophages and Gas6/Axl signaling in oral squamous cell carcinoma. Oral Oncol. 2015, 51, 683–689. [Google Scholar] [CrossRef] [PubMed]
- Geiger, T.L.; Sun, J.C. Development and maturation of natural killer cells. Curr. Opin. Immunol. 2016, 39, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Paolino, M.; Choidas, A.; Wallner, S.; Pranjic, B.; Uribesalgo, I.; Loeser, S.; Jamieson, A.M.; Langdon, W.Y.; Ikeda, F.; Fededa, J.P.; et al. The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature 2014, 507, 508–512. [Google Scholar] [CrossRef] [PubMed]
- Park, I.K.; Giovenzana, C.; Hughes, T.L.; Yu, J.; Trotta, R.; Caligiuri, M.A. The Axl/Gas6 pathway is required for optimal cytokine signaling during human natural killer cell development. Blood 2009, 113, 2470–2477. [Google Scholar] [CrossRef] [PubMed]
- Caraux, A.; Lu, Q.; Fernandez, N.; Riou, S.; Di Santo, J.P.; Raulet, D.H.; Lemke, G.; Roth, C. Natural killer cell differentiation driven by tyro3 receptor tyrosine kinases. Nat. Immunol. 2006, 7, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Linger, R.M.; Keating, A.K.; Earp, H.S.; Graham, D.K. Taking aim at mer and Axl receptor tyrosine kinases as novel therapeutic targets in solid tumors. Expert Opin. Ther. Targets 2010, 14, 1073–1090. [Google Scholar] [CrossRef] [PubMed]
- Holland, S.J.; Pan, A.; Franci, C.; Hu, Y.; Chang, B.; Li, W.; Duan, M.; Torneros, A.; Yu, J.; Heckrodt, T.J.; et al. R428, a selective small molecule inhibitor of axl kinase, blocks tumor spread and prolongs survival in models of metastatic breast cancer. Cancer Res. 2010, 70, 1544–1554. [Google Scholar] [CrossRef] [PubMed]
- Vaishampayan, U. Cabozantinib as a novel therapy for renal cell carcinoma. Curr. Oncol. Rep. 2013, 15, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Kariolis, M.S.; Miao, Y.R.; Jones, D.S., 2nd; Kapur, S.; Mathews, I.I.; Giaccia, A.J.; Cochran, J.R. An engineered Axl ‘decoy receptor’ effectively silences the Gas6-Axl signaling axis. Nat. Chem. Biol. 2014, 10, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Holland, S.J.; Powell, M.J.; Franci, C.; Chan, E.W.; Friera, A.M.; Atchison, R.E.; McLaughlin, J.; Swift, S.E.; Pali, E.S.; Yam, G.; et al. Multiple roles for the receptor tyrosine kinase Axl in tumor formation. Cancer Res. 2005, 65, 9294–9303. [Google Scholar] [CrossRef] [PubMed]
- Tape, C.J.; Ling, S.; Dimitriadi, M.; McMahon, K.M.; Worboys, J.D.; Leong, H.S.; Norrie, I.C.; Miller, C.J.; Poulogiannis, G.; Lauffenburger, D.A.; et al. Oncogenic KRAS regulates tumor cell signaling via stromal reciprocation. Cell 2016, 165, 910–920. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Tumor Type | References |
|---|---|
| Acute myeloid leukemia | [30] |
| Breast | [28] |
| Colorectal | [21] |
| Esophageal adenocarcinoma | [17,25] |
| Glioblastoma multiforme | [14] |
| Head and neck squamous cell carcinoma | [22] |
| Hepatocellular carcinoma | [26] |
| Lung adenocarcinoma | [13,31,37] |
| Melanoma | [29,32,33] |
| Oral squamous carcinoma | [18] |
| Osteosarcoma | [27] |
| Ovarian adenocarcinoma | [20,24] |
| Pancreatic ductal adenocarcinoma | [15] |
| Pleural mesothelioma | [19] |
| Renal cell carcinoma | [16,34] |
| Urothelial carcinoma | [23] |
| Tumor Type | References |
|---|---|
| Acute myeloid leukemia | [35] |
| Gastric | [38] |
| Glioblastoma multiforme | [14] |
| Lung adenocarcinoma | [37] |
| Oral squamous carcinoma | [39] |
| Ovarian adenocarcinoma | [36] |
| Renal cell carcinoma | [16] |
| Urothelial carcinoma | [23] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rankin, E.B.; Giaccia, A.J. The Receptor Tyrosine Kinase AXL in Cancer Progression. Cancers 2016, 8, 103. https://doi.org/10.3390/cancers8110103
Rankin EB, Giaccia AJ. The Receptor Tyrosine Kinase AXL in Cancer Progression. Cancers. 2016; 8(11):103. https://doi.org/10.3390/cancers8110103
Chicago/Turabian StyleRankin, Erinn B., and Amato J. Giaccia. 2016. "The Receptor Tyrosine Kinase AXL in Cancer Progression" Cancers 8, no. 11: 103. https://doi.org/10.3390/cancers8110103
APA StyleRankin, E. B., & Giaccia, A. J. (2016). The Receptor Tyrosine Kinase AXL in Cancer Progression. Cancers, 8(11), 103. https://doi.org/10.3390/cancers8110103