1. Introduction
The Wnt/β-catenin signaling pathway controls cellular proliferation and differentiation in the intestinal crypt microenvironment [
1]. Mutations in components of this pathway are highly prevalent in spontaneously arising colorectal cancers (CRCs) and lead to deregulated expression of genes controlled by the β-catenin transcriptional co-activator [
1,
2]. β-Catenin is recruited to Wnt responsive DNA regulatory elements (WREs) through interaction with members of the T-cell factor/Lymphoid enhancer factor (TCF/Lef; hereafter TCF) family of sequence specific transcription factors [
3]. The TCF7L2 family member is highly expressed in intestinal cells [
4,
5]. An understanding of colorectal carcinogenesis requires the identification of Wnt/β-catenin target genes and the WREs that control their expression.
The
c-MYC proto-oncogene (
MYC) encodes a transcription factor that stimulates cellular proliferation and growth by controlling expression of genes whose products regulate metabolism, ribosome biogenesis, and cell cycle progression [
6,
7]. He
et al. identified
MYC as a direct Wnt/β-catenin target gene in a human CRC cell line and mapped a 5′ WRE within the
MYC proximal promoter region [
8]. Two consensus TCF-binding elements (TBEs) were shown to contribute to 5′ WRE activity [
8]. Following this seminal report, a model was proposed, whereby deregulated
MYC gene expression by oncogenic Wnt/β-catenin signaling was an underlying cause of CRC. However, it was unknown at that time whether the 5′ WRE was the only WRE that controlled
MYC in CRC, or whether there were additional, yet unidentified
MYC WREs.
By sequencing DNA isolated in β-catenin chromatin immunoprecipitation (ChIP) assays conducted in the HCT116 human CRC cell line, we previously identified a robust β-catenin binding site that mapped 1.4-kb downstream from the
MYC transcription stop site [
9,
10]. This binding site demarcated a
MYC 3′ WRE that, like the 5′ WRE, also contained two consensus TBEs [
11]. Mutation of either TBE reduced
MYC 3′ WRE enhancer activity when assayed using standard luciferase reporter plasmids [
11]. Furthermore, we demonstrated that TCF7L2/β-catenin complexes bound to the
MYC 3′ WRE coordinated a chromatin loop with the
MYC 5′ proximal promoter [
12]. This chromatin conformation required TCF7L2/β-catenin transcription complexes and correlated with
MYC expression in these cells.
In this report, we tested whether the MYC 3′ WRE controls oncogenic MYC expression in the HCT116 human CRC cell line. Using CRISPR/Cas9 gene editing, we isolated a clonal population of cells containing a mutation that deleted the first of two TBEs within the MYC 3′ WRE. We found that mutating TBE1 reduced TCF7L2 and β-catenin binding to the 3′ WRE and compromised MYC expression. RNA-Seq analysis of control and 3′ WRE-Mut cells, found genes controlling metabolic processes were differentially expressed in knockout cells, most of which are known MYC targets. Finally, when compared to control cells, 3′ WRE-Mut cells displayed reduced cellular proliferation, clonogenic growth, and growth as tumors in a mouse xenograft model. Thus, our findings indicate that the MYC 3′ WRE is required for oncogenic MYC expression in a human CRC cell line.
3. Discussion
Data from The Cancer Genome Atlas (TCGA) project has confirmed that mutations in components of the Wnt/β-catenin signaling pathway occur frequently in CRCs [
20]. In most cases, these mutations target the
APC tumor suppressor leading to inappropriate expression of genes controlled by TCF/β-catenin transcription complexes [
2]. Since the discovery of
MYC as a direct Wnt/β-catenin target gene [
8], significant effort has been put forth to identify the WREs that control its expression in human CRC cells. Numerous studies have correlated TCF/β-catenin occupancy at WREs with
MYC expression in these cells; however, what has been lacking in the field, due to difficulties in genetically engineering somatic cell lines, is whether any of these WREs are required for oncogenic
MYC expression. In this report, we used CRISPR/Cas9 to delete TBE1 within the
MYC 3′ WRE in the HCT116 human CRC cell line. We demonstrate that a single TCF binding element embedded within the
MYC 3′ WRE is a critical regulator of
MYC expression and that it is required to sustain the full oncogenic potential of these cells.
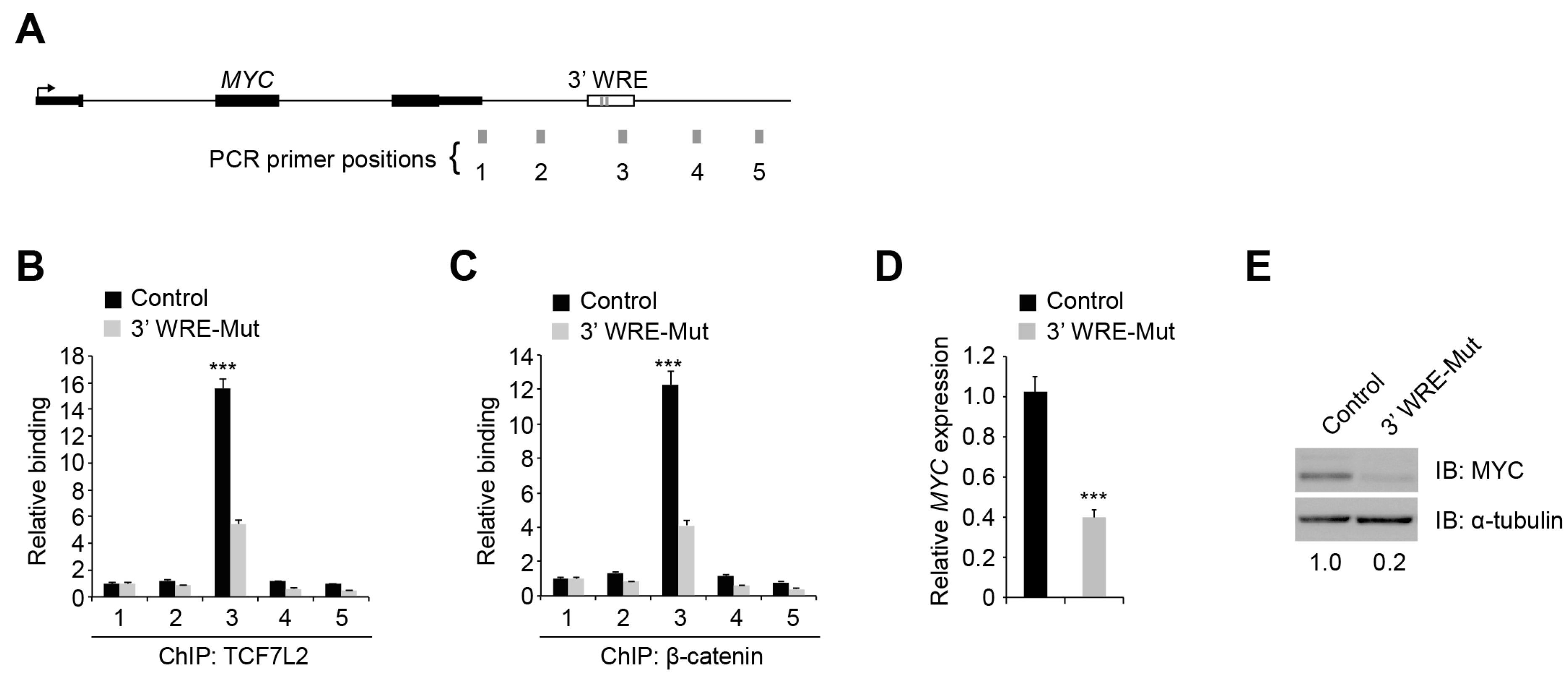
In our previous β-catenin ChIP-seq screen, nearly half of the high-confidence β-catenin binding regions contained a canonical TCF motif, with most regions containing more than one [
10]. Whether each motif contributed to β-catenin recruitment to that region was unknown. Here, we demonstrate that TBEs within a single WRE are not redundant as deletion of TBE1 diminished TCF7L2 and β-catenin occupancy at the
MYC 3′ WRE by 50% (
Figure 2B,C). This indicates that TBE2 is unable to compensate for loss of TBE1. Moreover, sequence analysis of the regions flanking the
MYC 3′ WRE identified multiple consensus TCF motifs downstream. Our ChIP-qPCR analysis in 3′ WRE-Mut cells failed to detect TCF7L2 or β-catenin binding to these regions (
Figure 2B,C). These results clearly demonstrate that multiple TBEs within a single WRE are not functionally redundant and that TCF7L2/β-catenin complexes do not simply shift to occupy other TBEs to compensate for loss of function.
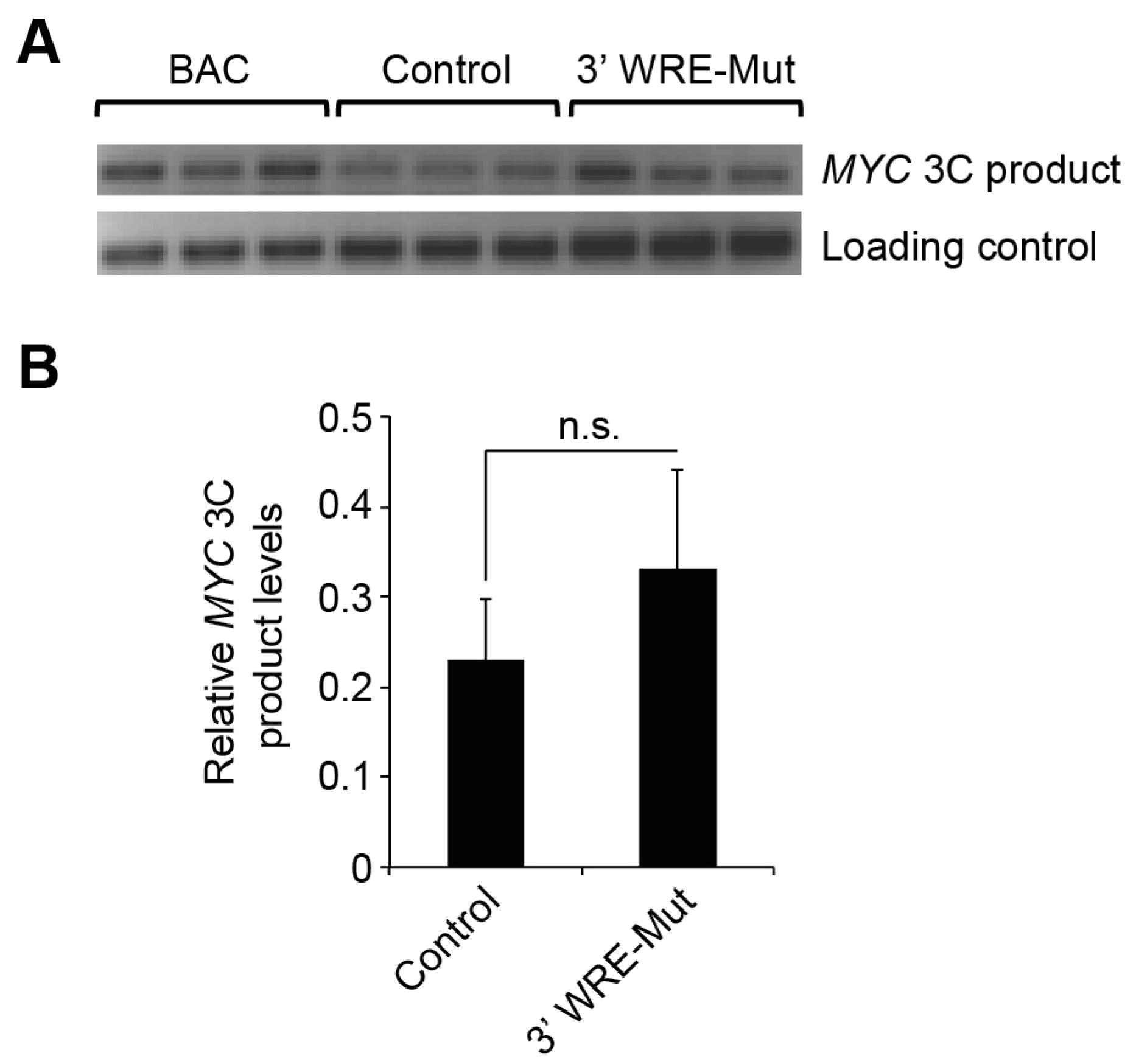
In our 3C analysis, we found that deletion of TBE1 did not affect formation of the
MYC 5′3′ chromatin loop even though
MYC expression is significantly reduced in these cells (
Figure 3). Thus, this chromatin conformation is not a critical determinant of oncogenic
MYC expression. However, it is also possible that our 3C analysis lacks the sensitivity to detect small changes in the frequency of the 5′3′ chromatin loop or that the residual levels of TCF7L2/β-catenin binding to the
MYC 3′ WRE through TBE2 are sufficient to maintain the chromatin architecture. It is also intriguing to speculate that the
MYC 3′ WRE may affect other aspects of
MYC expression such as 3′ termination and message processing. Future work will examine this possibility, as well as others, and the 3′ WRE-Mut cells will provide an important resource to tease out the mechanistic details for how this element is operating
in vivo.
In a previous study, Yao
et al. reported that deletion of enhancer 7, which is also known as the
MYC -335 WRE, reduced
MYC expression 1.5-fold in HCT116 cells [
21]. Through RNA-Seq analysis, they found that expression of 105 genes was down regulated in cells lacking this enhancer. We found that deleting TBE1 within the
MYC 3′ WRE reduced
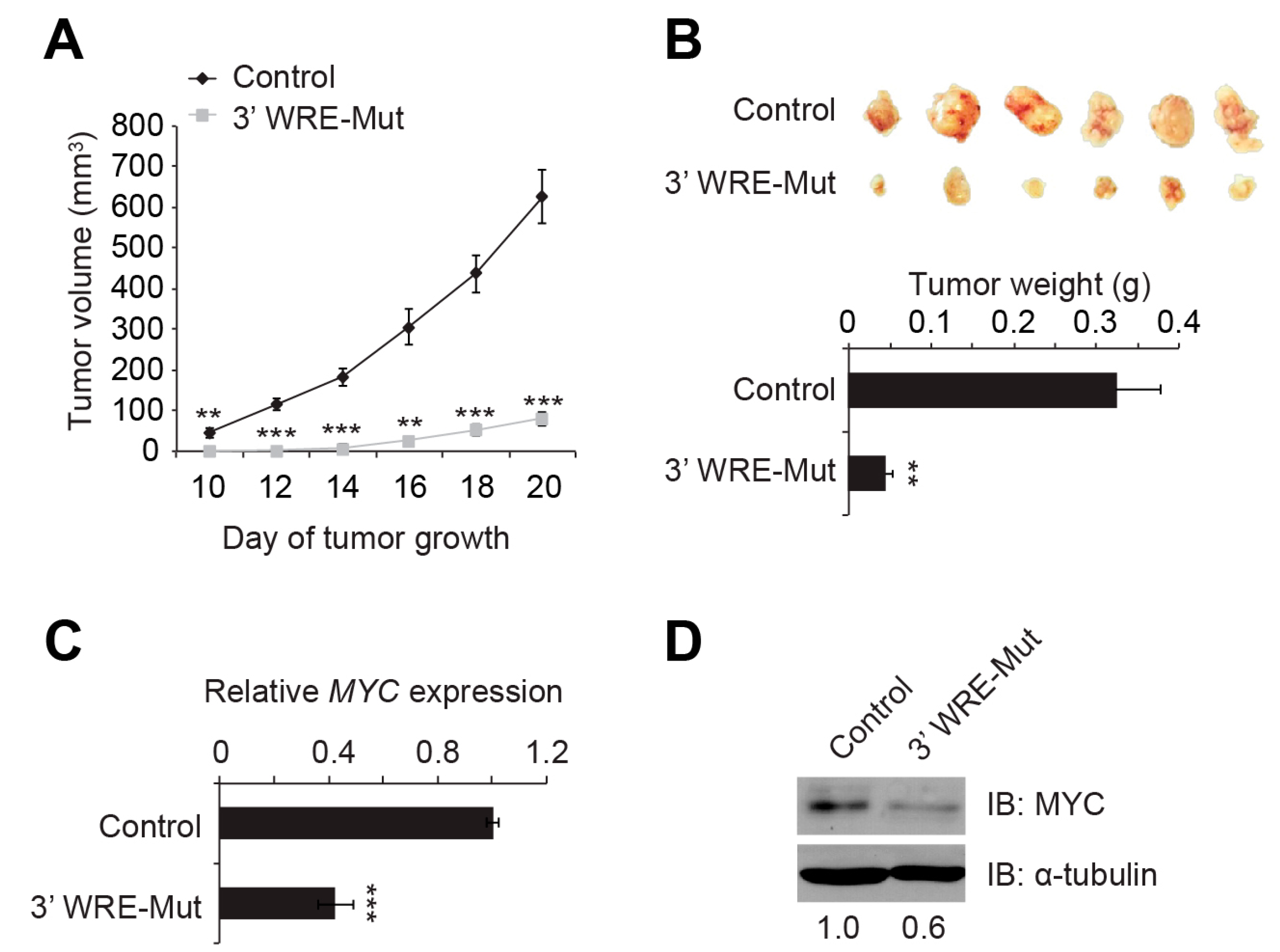
MYC expression 2.5-fold and MYC protein levels 5-fold (
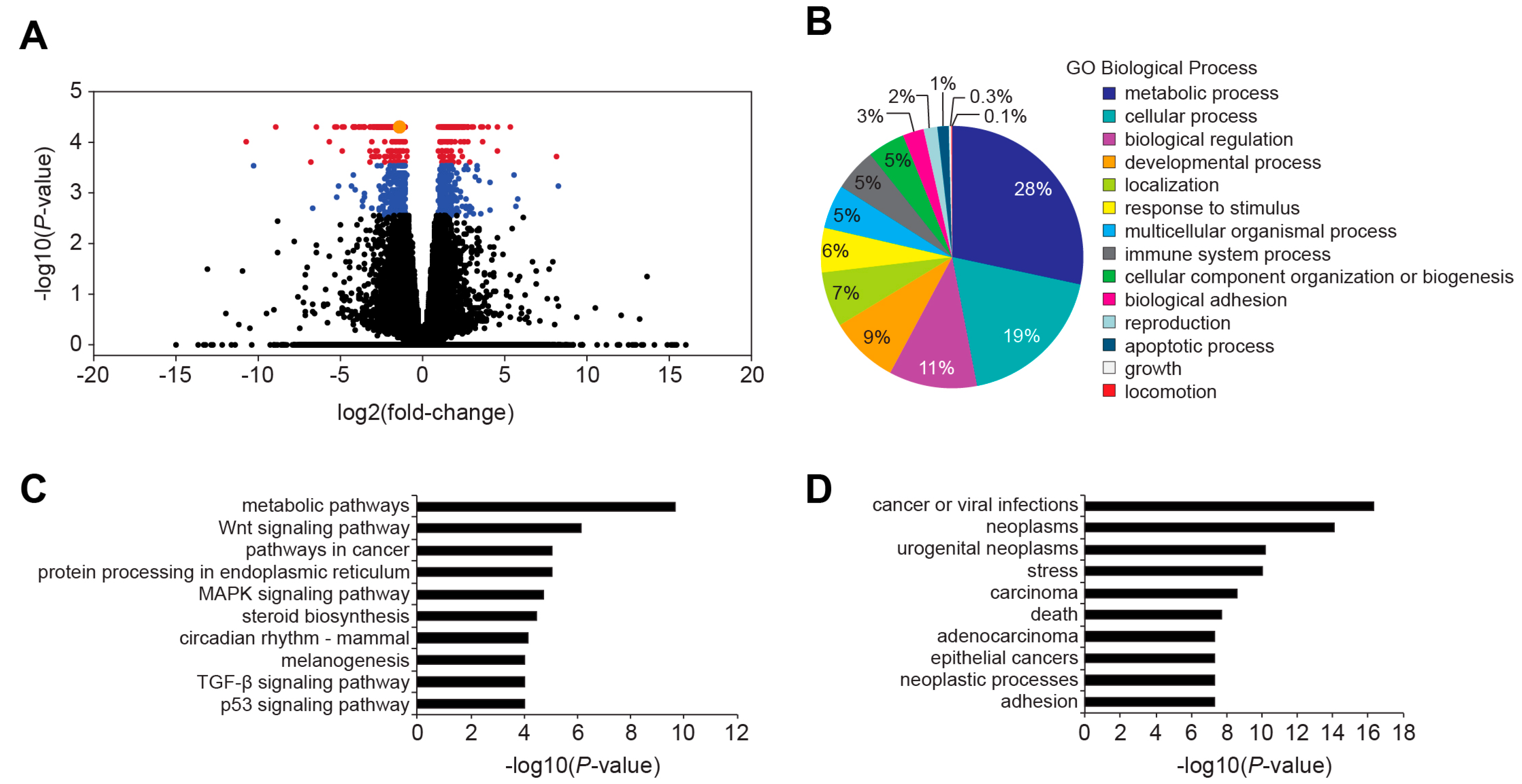
Figure 2D,E). In our RNA-Seq analysis of control and 3′ WRE-Mut cells, expression of 476 genes was down regulated (
Figure 4 and
Table S2). Because mutations in TBE1 had a more dramatic effect on
MYC expression than deletion of enhancer 7, this likely accounts for the discrepancy in the number of targets whose expression levels changed. In addition, it is likely that down-regulated genes are either direct or indirect targets of the MYC transcription factor. Our finding that expression of 215 known or predicted MYC targets was down regulated in 3′ WRE-Mut cells support this conclusion. Together, these results indicate that in comparison to enhancer 7, the
MYC 3′ WRE is a stronger regulatory element that controls
MYC expression and the transcriptome in HCT116 cells.
It is also possible that enhancer 7 and the
MYC 3′ WRE are directly juxtaposed to target genes outside the
MYC locus through long-range chromatin loops to influence their expression. Indeed, Cai
et al. found that this is likely the case for enhancer 7 in a circular chromosome conformation capture analysis (4C) analysis conducted in prostate cancer cells [
22]. A more thorough investigation of the chromatin architecture of control and 3′ WRE-Mut or enhancer 7-deleted cells using 4C is required to address the potential for these WREs to directly regulate expression of alternative genes in CRC.
Recently, Tak
et al. found that deleting enhancer 7 compromised HCT116 cell growth [
23]. We found that deleting TBE1 reduced HCT116 cell proliferation, clonogenic growth, anchorage-independent growth in soft agar, and tumor growth when these cells were injected into the flanks of immune-compromised animals (
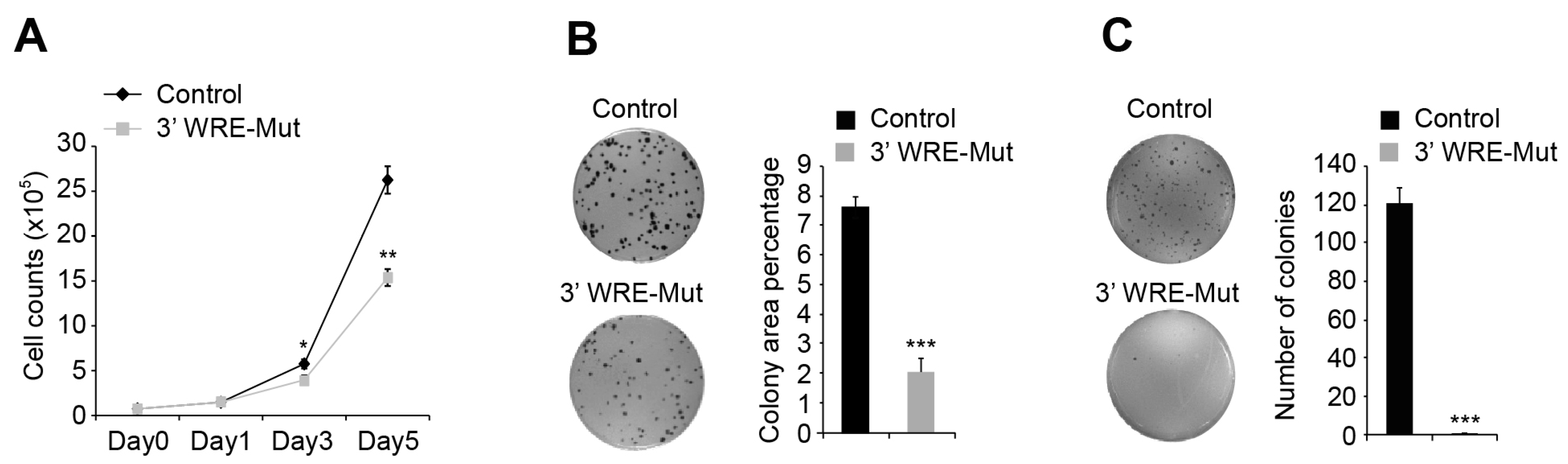
Figure 5 and
Figure 6). In our RNA-seq analysis of control and 3′ WRE-Mut cells, 215 of the 949 differentially expressed genes are known or predicted MYC target genes (
Supplementary Materials Figure S2). These results suggest that
MYC 3′ TBE1 deletion, and subsequent reduction in MYC protein levels, alters expression of the MYC target gene network. GO and KEGG analyses of differentially expressed targets in control and TBE1-deleted cells found that genes comprising metabolic pathways or processes were the most significantly altered (
Figure 4B,C). As MYC has been shown to promote metabolic reprogramming of cancer cells [
24], these findings suggest that the reduced oncogenic potential of 3′ WRE-Mut cells is due to an inability to sustain deregulated cellular metabolism.
Studies using mouse models of CRC have shown that MYC is required for intestinal tumorigenesis caused by deregulated Wnt/β-catenin signaling [
17]. In the first report that targeted a WRE in the mouse genome, Sur
et al. found that deleting the
Myc −335 WRE did not alter intestinal homeostasis [
25]. However,
Myc −335 WRE
−/− ApcMin/+ mice contained a reduced number of tumors in both the small intestines and colons when compared to
ApcMin/+ mice [
25]. In contrast, deleting the
Myc 3′ WRE in mice altered the crypt microenvironment and increased
Myc transcript and MYC protein levels in their colons [
26].
Myc 3′ WRE
−/− ApcMin/+ mice contained an elevated number of tumors in the colons in comparison to
ApcMin/+ mice [
27]. Moreover, deletion of the
Myc 3′ WRE promoted colitis-associated colorectal cancer in a mouse model of this disease [
27]. These findings are seemingly at odds with those reported here, which indicate that the
MYC 3′ WRE is critical for maintaining the oncogenic potential of a human CRC cell line. There are several possible explanations that may account for these different results. First, while the
MYC 3′ WRE is highly conserved in mouse overall, TBE1 is not conserved, perhaps indicating that the function of this regulatory element differs between mouse and human [
26]. Second, tumors that develop in
ApcMin/+ mice and in the colitis-associated cancer model are primarily adenomas, whereas human CRC cell lines are predominantly adenocarcinomas, thereby complicating direct comparisons between the two model systems [
28,
29]. Third,
Myc 3′ WRE
−/− mice harbor germ-line mutations and deregulated
MYC expression in cells other than intestinal epithelial cells and these cells may contribute to tumorigenesis. Perhaps the most important difference between these two systems is that in our mouse studies, MYC expression was deregulated prior to the development of tumors whereas in our human cell line studies, these cells were already addicted to high levels of MYC. Regardless, these studies indicate that when considering the function of a regulatory element, interpretation of results obtained from the model system used must be carefully taken into consideration.
With the advancement of gene-editing technologies, the possibility of targeting enhancer elements for therapeutic purposes is within reach [
30]. While additional work is required to ensure the specificity of CRISPR/Cas9 complexes in order to reduce potential off target effects [
31], our findings indicate that targeting the
MYC 3′ WRE in human CRCs may be of therapeutic value. While introducing an active DNA nuclease (Cas9) into cells may raise concern, the CRISPR system may be harnessed in other ways to silence an oncogenic element [
32,
33]. For instance, by introducing multiple guide RNAs that tile the
MYC 3′ WRE, and a catalytically inactive Cas9 protein fused to the Sin3 interaction domain (SID), one could target the mSin3A/HDAC corepressor complex to this site to repress oncogenic
MYC expression [
34,
35]. The key will be to develop specific delivery strategies that target cancer and not the uninvolved colonic epithelium. As colorectal cancer remains a serious worldwide health problem, more creative measures such as these are required to treat this disease.
4. Materials and Methods
4.1. Cell Culture
HEK293T (CRL-3216) and HCT116 (CCL-247) were obtained from the American Type Culture Collection (Rockville, MD, USA). Cell lines were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% Fetal Bovine Serum (FBS, Atlantic Biologicals, S11150, Frederick, MD, USA), 50 units/mL penicillin (Corning Mediatech, 30-002-CI, Tewksbury, MA, USA) 2 mM Glutamax (ThermoFisher Scientific, 35050-061, Waltham, MA, USA), and 0.1 mg/mL streptomycin (Corning Mediatech, 30-002-CI) at 37 °C in 5% CO2.
4.2. Plasmids
The CRISPR Design tool [
15] was first used to identify the CRISPR guide RNA (crRNA) sequence within the
MYC 3′ WRE. DNA oligonucleotides corresponding to this sequence were annealed, phosphorylated using T4 polynucleotide kinase (New England Biolabs, M0201S, Ipswich, MA, USA), and ligated into the BbsI site within the pX260 CRISPR/Cas9 plasmid (Addgene 42229) using instructions on the Genome Engineering Resources website [
36]. This plasmid is referred to as pX260-
MYC 3′ TBE1. The oligonucleotide sequences corresponding to the guide RNA are listed in
Table S1.
To design the
MYC 3′ WRE-specific CRISPR reporter plasmid, we employed a stepwise approach using standard PCR and sub-cloning procedures. The pEGFP-N1 plasmid (Clontech, 6085-1, Mountain View, CA, USA) served as the vector backbone.
BFP was PCR-amplified from pRSET-BFP (ThermoFisher, V354-20) and the amplified product was inserted as an NheI-EcoRI fragment into pEGFP-N1. Next, the
EGFP sequence was removed by digesting the vector with BamHI and NotI and replaced with an out-of-frame
EGFP sequence that was PCR-amplified using an upstream primer that disrupted the reading frame. Finally, a 43-bp oligonucleotide containing the
MYC 3′ TBE1 CRISPR/Cas9 target sequence was annealed and inserted in between
BFP and the out-of-frame
EGFP as an EcoRI-BamHI fragment. This plasmid is referred to as pCRISPR-reporter and the oligonucleotide sequences used for its generation are listed in
Table S1.
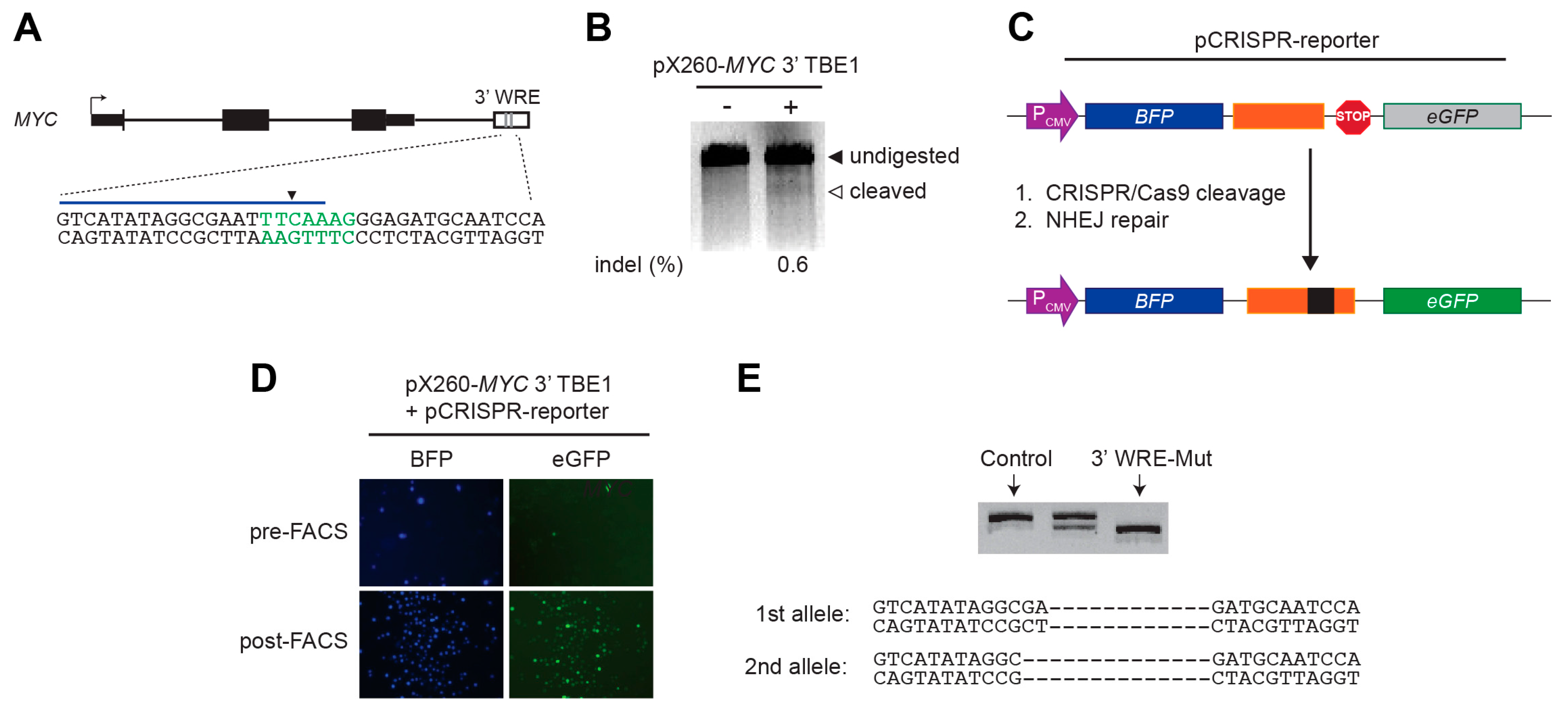
4.3. Mismatch Cleavage Assays
The SURVEYOR mutation detection kit (Transgenomics, 706025, Omaha, NE, USA) was used to evaluate the cleavage frequency of
MYC 3′ TBE1 CRISPR/Cas9 complexes. Approximately 3 × 10
5 HEK293T cells were seeded in each well of a six-well dish and transfected with either 500 ng of pX260-
MYC 3′ TBE1, or 500 ng of pmaxGFP (Lonza, VDF-1012, Köln, Germany) as a control, using Lipofectamine 2000 (ThermoFisher, 11668). The following day, the growth media was supplemented with 0.5 μg/mL puromycin. After two days in selection media, genomic DNA was isolated using the Genomic DNA Purification Kit (ThermoFisher, K0512). A 200 ng quantity of genomic DNA was used as the template in PCR reactions, with primer sequences provided in
Table S1, to amplify a 1049-bp region flanking the CRISPR/Cas9 target site within the
MYC 3′ WRE. The amplicons were purified using the QIAquick PCR purification kit (Qiagen, 28104, Gaithersburg, MD, USA). A total of 400 ng purified PCR product was subjected to rapid denaturation and re-annealing using guidelines provided in the SURVEYOR kit (Transgenomics, 706025). The heteroduplexes were digested with SURVEYOR nuclease and the products were resolved on a 3% synergel/agarose gel (Diversified Biotech, SYN-100, Dedham, MA, USA). ImageJ software was used to measure the band intensities of both undigested PCR products and cleaved fragments. The percentage of insertions/deletions (indels) was estimated using the formula 100 × (1 − [(1 − (
b +
c)/(
a +
b +
c)]
1/2) as described by Ran
et al. [
37].
4.4. Generation and Evaluation of HCT116 MYC 3′ WRE-Mut Cells
Approximately 2.5 × 105 HCT116 cells were seeded into a single well of a 12-well dish and the cells were transfected the following day with 375 ng pX260-MYC 3′ TBE1, 187.5 ng of pCRISPR-reporter, and 562.5 ng pBlueScript (as carrier DNA) using ViaFect reagent (Promega, E4981, Madison, WI, USA) at a 3:1 ratio of reagent to DNA. Three days later, a BD fluorescent-activated cell sorting (FACS) Aria instrument (BD Biosciences, San Jose, CA, USA) was used to sort single GFP+ cells into each well of three 96-well plates. After expansion of single-cell clones, genomic DNA was isolated from each clone using Lyse-and-Go reagent (ThermoFisher Scientific, 78882) and the region flanking the CRISPR/Cas9 target sequence within the MYC 3′ WRE was PCR-amplified. The PCR products were analyzed on a non-denaturing polyacrylamide gel containing 8% acrylamide-bisacrylamide (29:1, w/w) and 0.5× Tris-borate EDTA (TBE) to detect differences in band sizes relative to products generated from non-transfected HCT116 parental cells. A single clone was identified that contained a smaller product and sequencing the product identified a 12-bp deletion that removed the entire TBE1 sequence motif on one allele. This cell line was expanded, and the transfection/sorting procedure was repeated to isolate the 3′ WRE-Mut cell line. These cells contained a 14-bp deletion that removed TBE1 on the second allele.
4.5. Chromatin Immunoprecipitation (ChIP)
ChIP assays were performed as previously described [
10] using approximately 5 × 10
6 cells per sample. The cross-linked and sheared chromatin was precipitated with 3 μg of anti-TCF4 (TCF7L2) (Millipore, 05-511, Darmstadt, Germany) or anti-β-catenin (BD Transduction, 610154, San Jose, CA, USA) antibodies. The precipitated chromatin was analyzed by quantitative real-time PCR (qPCR) using the primer sets listed in
Table S1 and a MyIQ real-time PCR machine (Bio-rad, Hercules, CA, USA). A standard curve was generated using serial dilutions of sonicated and purified input DNA to ensure that ChIP signals were within the linear range of detection. The data is presented as relative levels that were obtained using the standard curves.
4.6. Real-Time Reverse Transcription PCR (qRT-PCR)
For experiments measuring
MYC transcript levels in control and 3′ WRE-Mut cells, 7.5 × 10
5 cells were plated in a 10 cm dish in a 7 mL volume of DMEM. Total RNA was isolated 5 days later using TRIzol reagent (ThermoFisher, 15596), and cDNAs were synthesized from 500 ng of RNA using the iScript cDNA synthesis kit (Bio-Rad, 1708891) according to the manufacturer’s instructions. To evaluate
MYC expression in tumors explanted from xenografted mice, a slice of the tumor was homogenized in TRIzol reagent and RNAs were isolated from these samples using instructions provided by the manufacturer. To assess
MYC transcript levels, cDNA samples were diluted 1:100, of which 3 μL were analyzed in 15 μL qPCR reactions containing 2× Sensimix (Bioline, BIO-96020,Taunton, MA, USA), and 10 pmol each of forward and reverse primers (
Table S1). The reactions were cycled using parameters previously described [
10].
GAPDH levels were measured as an internal reference and the data is presented as relative expression (2
−ΔΔCt).
4.7. Western Blot
Five days after plating 7.5 × 105 control and 3′ WRE-Mut cells in a 10 cm dish with 7 mL DMEM, the cells were collected and the pellet was re-suspended in 400 μL of RIPA buffer (50 mM Tris HCl, pH 8.0; 150 mM NaCl; 1% NP-40; 0.5% sodium deoxycholate; 0.1% SDS) containing freshly added protease inhibitors (1 mM PMSF, 10 μL/mL aprotinin, 10 μg/mL leupeptin). The cells were swelled on ice and briefly sonicated using a Misonix XL-2000 liquid processor (2 × 5 s, output wattage 4, 30 s rest time between pulses). After clarifying the lysates by centrifugation at 16,000× g for 10 min at 4 °C, the protein content was quantified using Bradford analysis (Bio-Rad, 500-0006), and 20 μg was subjected to PAGE using an 8% denaturing polyacrylamide gel. Standard Western blotting procedures were followed to detect proteins using anti-MYC Y69 (Abcam, ab32072, 1:1000 dilution, Cambridge, UK) and anti-α-tubulin (Sigma, T9026, 1:1000 dilution, St. Louis, MO, USA) antibodies. To detect MYC levels in explanted tumors, a section of the tumor was cut and homogenized in RIPA buffer containing protease inhibitors. Preparation of protein lysates and analysis was conducted as above except blots were probed with anti-MYC N-262 antibodies (Santa Cruz, sc-764, 1:250 dilution, Santa Cruz, CA, USA).
4.8. Chromosome Conformation Capture
Chromosome conformation capture (3C) analysis was performed on control and 3′ WRE-Mut cell populations as previously described with minor modifications [
38]. A bacterial artificial chromosome harboring the
MYC genomic locus served as a control for the 3C-qPCR analysis. Formaldehyde cross-linked chromatin was digested with 40 μL (400 units) of BstYI (New England Biolabs) overnight at 37 °C. Primer sequences used to detect the
MYC 5′3′ chromatin loop, and those to amplify the control region, are available in
Table S1. 3C products were amplified for 32 cycles in triplicate reactions using DreamTaq polymerase (ThermoFisher, K1081). Products were resolved on a 1.5% agarose gel, imaged, and band intensities were quantified using ImageJ software. Each
MYC 3C product was first normalized to the average value obtained from the three BAC 3C products, then to the average value of the three internal control bands.
4.9. Analysis of Potential Off-Targets
To screen for potential off target effects of CRISPR cleavage in 3′ WRE-Mut cells, genomic DNA was isolated from 2.5 × 10
6 parental control HCT116 cells and 3′ WRE-Mut cells using TNES buffer. Following phenol:chloroform:isoamyl alcohol extraction and chloroform back-extraction, genomic DNA was precipitated in ethanol. Potential off-targets were identified using the CRISPR Design tool [
15]. These sites were PCR-amplified using primers that flanked the target site and the products were analyzed on an 8% non-denaturing polyacrylamide gel. Primer sequences used in these reactions are listed in
Table S1.
4.10. RNA-Seq
RNA was extracted from two biological replicates containing 7.5 × 10
5 control or 3′ WRE-Mut cells using the mirVana kit (ThermoFisher, AM1560). The RNAs were quantified using a NanoDrop apparatus (ThermoFisher) which also ensured that the A
260:A
280 ratio was 1.9 or above. The RNA integration number (RIN) for each sample was measured using an RNA 6000 Nano Kit (Agilent, 5067-1511, Santa Clara, CA, USA), and a RIN value of above 7 was required to proceed. The cDNA libraries were prepared using the NEXTflex™ Rapid RNA Sequencing Kit (BioO Scientific, 5138-02, Austin, TX, USA) and NEXTflex™ RNA-Seq Barcodes–24 Kit (BioO Scientific, 512980) following the manufacturer’s instructions. The processed libraries were assessed for fragment length size distribution and were subsequently concentrated using a BioAnalyzer High Sensitivity DNA kit (Agilent, 5067-4626). The libraries were pooled, diluted to 2 nM in elution buffer (Qiagen, 19086), and then denatured following Illumina sequencing guidelines. The denatured libraries were diluted to 10 pM in pre-chilled hybridization buffer, and loaded into a TruSeq SR v3 flow cell (TruSeq SR Cluster Kit v3-cBot™ –HS, GD-401-3001, Illumina, San Diego, CA, USA). The samples were sequenced using a HiSeq 2500 that was set to run for 50 cycles using a single-read recipe (TruSeq SBS Kit v3, FC-401-3002) according to the manufacturer’s instructions. The sequence reads that passed the default purifying setting of the Illumina CASAVA pipeline (released version 1.8, Illumina) were further trimmed/filtered by requiring a quality score cutoff of 20 using the FASTX-Toolkit [
39]. The filtered reads were aligned to the human reference genome (build hg19) using Tophat v2.0.9 [
40], allowing up to two mismatches. Fragments Per Kilobase Of Exon Per Million Fragments Mapped (FPKM) values were calculated using Cufflinks v2.0.2 [
41] provided with the Gencode v19 gene annotation. The Gene Ontology (GO) [
42] and WebGestalt [
43] databases were used to classify genes that were differentially expressed in 3′ WRE-Mut
versus control cells. The RNA-Seq data was deposited on the Gene Expression Omnibus database [
44] and can be accessed using reference number, GSE70833.
4.11. Cell Proliferation
Approximately 7.5 × 104 parental or 3′ WRE-Mut cells were seeded in duplicate wells of a 6-well plate. On days one, three, and five after plating, the cells were pelleted, stained with 0.2% trypan blue, and 20 μL of the cell suspension was loaded into a Cellometer cell-counting chamber (Nexcelom Bioscience, CHT4-SD100, Lawrence, MA, USA). Viable cells were quantified using a Cellometer Auto T4 Cell Counter (Nexcelom Bioscience).
4.12. Clonogenic Growth Assays
Two hundred control or 3′ WRE-Mut cells were seeded in triplicate wells of a 6-well dish and ten days later, the cells were washed twice with 1× PBS and fixed for 5 min at room temperature in 500 μL of a methanol:acetic acid solution (3:1 ratio). The cells were then stained with a 0.5% solution of crystal violet dissolved in methanol for 15 min at room temperature and washed with distilled water to remove excess stain. Following guidelines provided in Guzman
et al. [
45], stained colonies were quantified using the ColonyArea ImageJ plug-in application.
4.13. Anchorage-Independent Growth Assays
Anchorage-independent growth assays were performed as described previously [
46] with minor modifications. Briefly, 5 × 10
3 control or 3′ WRE-Mut cells were seeded within a 0.4% layer of agar on top of a 0.5% layer in triplicate wells of a 6-well dish. After 21 days, colonies were stained with 0.01% crystal violet and quantified using the particle analyzing function of ImageJ software.
4.14. Mouse Xenograft Assays
Approximately 5 × 10
6 control or 3′ WRE-Mut cells were resuspended in 150 μL of 1X PBS and injected subcutaneously into the flanks of NU/NU nude mice (strain #088, Charles River Labs, Wilmington, MA, USA). Starting on day 10 after the injection, tumors were measured every two days using standard calipers. Volumes were determined using the formula width
2 × length/2 [
47]. At the conclusion of the experiment (day 20), the tumors were removed, weighed on a standard balance, photographed, and minced into small portions for RNA and protein analysis. The Pennsylvania State University College of Medicine Institutional Animal Care and Use Committee (IACUC) approved the animal protocols used in this study.
4.15. Statistical Analysis
Each experiment was repeated at least three times. For ChIP-qPCR and qRT-PCR, each sample was amplified in quadruplicate wells in each experiment. A Student’s t-test was used to calculate statistical significance in these experiments as well as those involving cell counts, colony growth assays, anchorage-independent growth assays, and xenograft tumor assays.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





