
RASSF1A Site-Specific Methylation Hotspots in Cancer and Correlation with RASSF1C and MOAP-1


 ,
,  ,
,
Abstract
:1. Introduction
2. Results
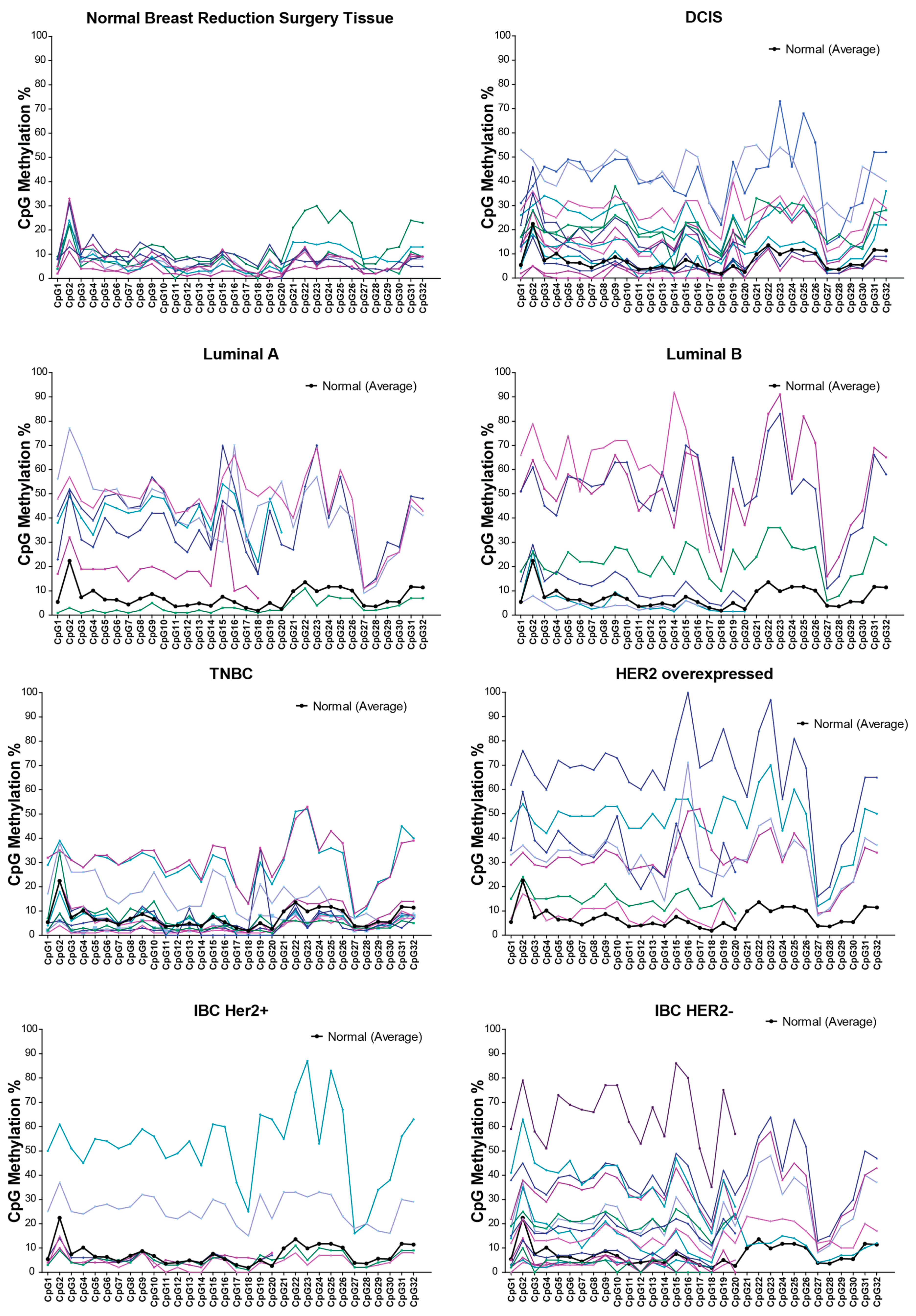
2.1. RASSF1A Promoter Methylation in Cancer Tumor Tissue
2.2. RASSF1A Promoter Methylation in Normal and Cancer Cell Lines
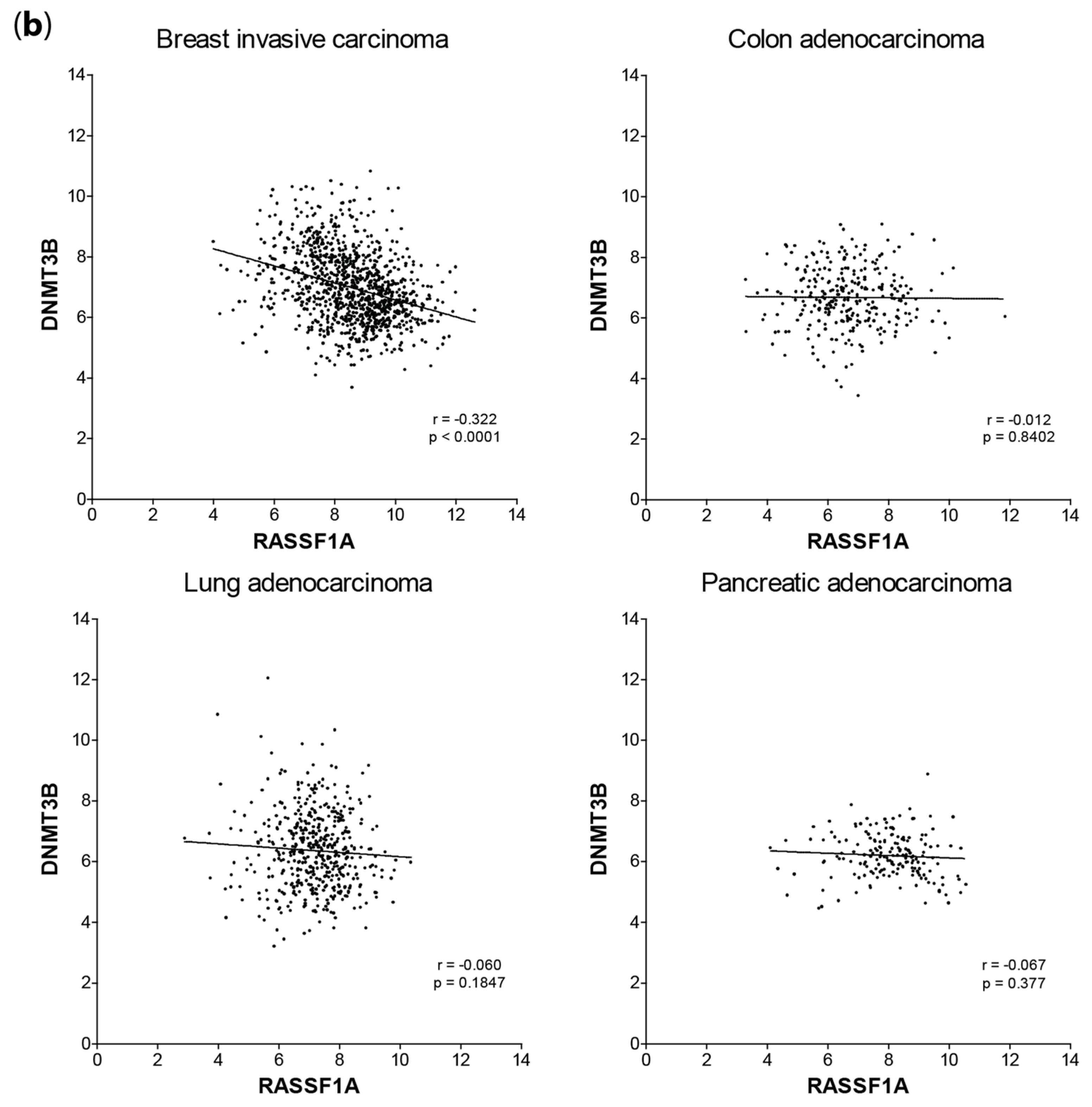
2.3. mRNA Expression Correlation of DNMT1and DNMT3B with RASSF1A
2.4. mRNA Expression Correlation of MOAP-1 with RASSF1A
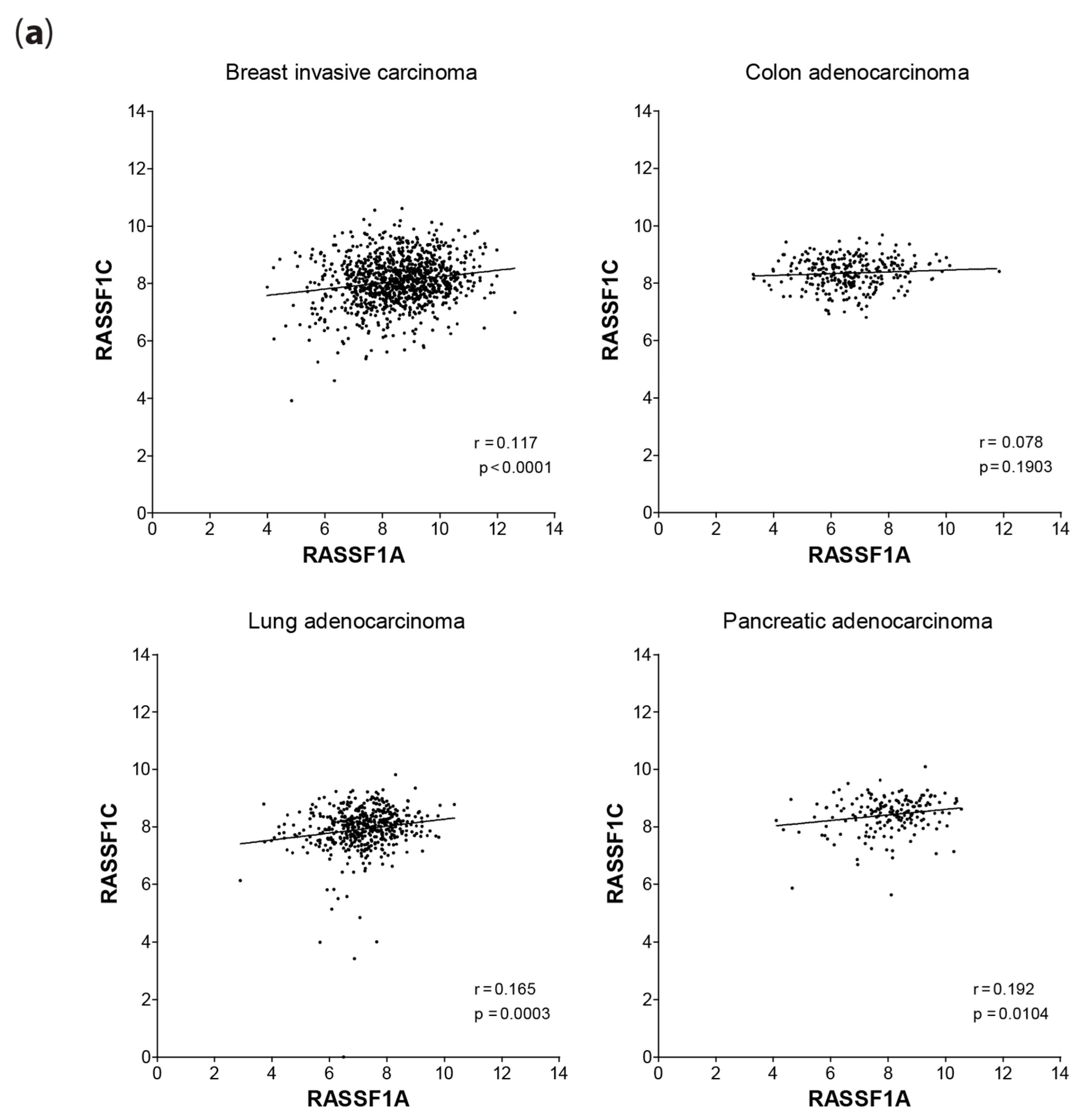
2.5. mRNA Expression Correlation of RASSF1C with RASSF1A
3. Discussion
4. Materials and Methods
4.1. Cancer Samples
4.2. Cell Lines
4.3. DNA Isolation and Bisulfite Treatment
4.4. Methylation Analysis by Pyrosequencing
4.5. Quantitative Real-Time PCR
4.6. Statistical Analysis
4.7. TCGA Database Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Donninger, H.; Vos, M.D.; Clark, G.J. The rassf1a tumor suppressor. J. Cell Sci. 2007, 120, 3163–3172. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; He, J.; Li, J.; Tian, D.; Gu, L.; Zhou, M. Methylation of rassf1a gene promoter is regulated by p53 and daxx. FASEB J. 2013, 27, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Volodko, N.; Gordon, M.; Salla, M.; Ghazaleh, H.A.; Baksh, S. Rassf tumor suppressor gene family: Biological functions and regulation. FEBS Lett. 2014, 588, 2671–2684. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.H.; Yazici, H.; Wu, H.C.; Terry, M.B.; Gonzalez, K.; Qu, M.; Dalay, N.; Santella, R.M. Aberrant promoter hypermethylation and genomic hypomethylation in tumor, adjacent normal tissues and blood from breast cancer patients. Anticancer Res. 2010, 30, 2489–2496. [Google Scholar] [PubMed]
- Zhong, X.; Isharwal, S.; Naples, J.M.; Shiff, C.; Veltri, R.W.; Shao, C.; Bosompem, K.M.; Sidransky, D.; Hoque, M.O. Hypermethylation of genes detected in urine from ghanaian adults with bladder pathology associated with schistosoma haematobium infection. PLoS ONE 2013, 8, e59089. [Google Scholar] [CrossRef] [PubMed]
- Dammann, R.; Schagdarsurengin, U.; Seidel, C.; Strunnikova, M.; Rastetter, M.; Baier, K.; Pfeifer, G.P. The tumor suppressor rassf1a in human carcinogenesis: An update. Histol. Histopathol. 2005, 20, 645–663. [Google Scholar] [PubMed]
- Zhang, C.Y.; Zhao, Y.X.; Xia, R.H.; Han, J.; Wang, B.S.; Tian, Z.; Wang, L.Z.; Hu, Y.H.; Li, J. Rassf1a promoter hypermethylation is a strong biomarker of poor survival in patients with salivary adenoid cystic carcinoma in a chinese population. PLoS ONE 2014, 9, e110159. [Google Scholar] [CrossRef] [PubMed]
- Kuzmina, N.S.; Lapteva, N.; Rubanovich, A.V. Hypermethylation of gene promoters in peripheral blood leukocytes in humans long term after radiation exposure. Environ. Res. 2016, 146, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Foley, C.J.; Freedman, H.; Choo, S.L.; Onyskiw, C.; Fu, N.Y.; Yu, V.C.; Tuszynski, J.; Pratt, J.C.; Baksh, S. Dynamics of rassf1a/moap-1 association with death receptors. Mol. Cell. Biol. 2008, 28, 4520–4535. [Google Scholar] [CrossRef] [PubMed]
- Richter, A.M.; Pfeifer, G.P.; Dammann, R.H. The rassf proteins in cancer; from epigenetic silencing to functional characterization. Biochim. Biophys. Acta 2009, 1796, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Moshnikova, A.; Kuznetsov, S.; Khokhlatchev, A.V. Interaction of the growth and tumour suppressor nore1a with microtubules is not required for its growth-suppressive function. BMC Res. Notes 2008, 1. [Google Scholar] [CrossRef] [PubMed]
- Song, M.S.; Chang, J.S.; Song, S.J.; Yang, T.H.; Lee, H.; Lim, D.S. The centrosomal protein ras association domain family protein 1a (rassf1a)-binding protein 1 regulates mitotic progression by recruiting rassf1a to spindle poles. J. Biol. Chem. 2005, 280, 3920–3927. [Google Scholar] [CrossRef] [PubMed]
- Gordon, M.; El-Kalla, M.; Zhao, Y.; Fiteih, Y.; Law, J.; Volodko, N.; Mohamed, A.; El-Kadi, A.O.; Liu, L.; Odenbach, J.; et al. The tumor suppressor gene, rassf1a, is essential for protection against inflammation—Induced injury. PLoS ONE 2013, 8, e75483. [Google Scholar] [CrossRef] [PubMed]
- Tommasi, S.; Dammann, R.; Zhang, Z.; Wang, Y.; Liu, L.; Tsark, W.M.; Wilczynski, S.P.; Li, J.; You, M.; Pfeifer, G.P. Tumor susceptibility of rassf1a knockout mice. Cancer Res. 2005, 65, 92–98. [Google Scholar] [PubMed]
- Van der Weyden, L.; Tachibana, K.K.; Gonzalez, M.A.; Adams, D.J.; Ng, B.L.; Petty, R.; Venkitaraman, A.R.; Arends, M.J.; Bradley, A. The rassf1a isoform of rassf1 promotes microtubule stability and suppresses tumorigenesis. Mol. Cell. Biol. 2005, 25, 8356–8367. [Google Scholar] [CrossRef] [PubMed]
- Said, A.; Salla, M.; Gordon, M.; El-Kalla, M.; Baksh, S. Spontaneous and Age-related Inflammation in the Rassf1a−/− Mice; Departments of Pediatrics and Biochemistry, University of Alberta: Edmonton, AB, Canada, Canada Unpublished Observations; 2016. [Google Scholar]
- Van der Weyden, L.; Arends, M.J.; Dovey, O.M.; Harrison, H.L.; Lefebvre, G.; Conte, N.; Gergely, F.V.; Bradley, A.; Adams, D.J. Loss of rassf1a cooperates with apc(min) to accelerate intestinal tumourigenesis. Oncogene 2008, 27, 4503–4508. [Google Scholar] [CrossRef] [PubMed]
- Tommasi, S.; Besaratinia, A.; Wilczynski, S.P.; Pfeifer, G.P. Loss of rassf1a enhances p53-mediated tumor predisposition and accelerates progression to aneuploidy. Oncogene 2010, 30, 690–700. [Google Scholar] [CrossRef] [PubMed]
- Law, J.; Yu, V.C.; Baksh, S. Modulator of apoptosis 1: A highly regulated rassf1a-interacting bh3-like protein. Mol. Biol. Int. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Law, J.; Salla, M.; Zare, A.; Wong, Y.; Luong, L.; Volodko, N.; Svystun, O.; Flood, K.; Lim, J.; Sung, M.; et al. Modulator of apoptosis 1 (moap-1) is a tumor suppressor protein linked to the rassf1a protein. J. Biol. Chem. 2015, 290, 24100–24118. [Google Scholar] [CrossRef] [PubMed]
- Li, L.C.; Dahiya, R. Methprimer: Designing primers for methylation pcrs. Bioinformatics 2002, 18, 1427–1431. [Google Scholar] [CrossRef] [PubMed]
- Volodko, N.; Baksh, S. Analysis of Moap-1 Promoter-Specific Methylation in Cancer; Department of Pediatrics, University of Alberta: Edmonton, AB, Canada, Canada Unpublished Observations; 2016. [Google Scholar]
- Koch, A.; De Meyer, T.; Jeschke, J.; Van Criekinge, W. Mexpress: Visualizing expression, DNA methylation and clinical tcga data. BMC Genomics 2015, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donninger, H.; Clark, J.A.; Monaghan, M.K.; Schmidt, M.L.; Vos, M.; Clark, G.J. Cell cycle restriction is more important than apoptosis induction for rassf1a protein tumor suppression. J. Biol. Chem. 2014, 289, 31287–31295. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Rong, R.; Sheikh, M.S.; Huang, Y. Cullin-4a.DNA damage-binding protein 1 e3 ligase complex targets tumor suppressor rassf1a for degradation during mitosis. J. Biol. Chem. 2010, 286, 6971–6978. [Google Scholar] [CrossRef] [PubMed]
- Dhiab, M.B.; Ziadi, S.; Mestiri, S.; Gacem, R.B.; Ksiaa, F.; Trimeche, M. DNA methylation patterns in ebv-positive and ebv-negative hodgkin lymphomas. Cell. Oncol. 2015, 38, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.L.; Tian, G.L.; Chen, S.J.; Xu, L.I.; Wang, H.Q. Promoter methylation of p16 and rassf1a genes may contribute to the risk of papillary thyroid cancer: A meta-analysis. Exp. Ther. Med. 2015, 10, 1549–1555. [Google Scholar] [CrossRef] [PubMed]
- Gonzalo, V.; Lozano, J.J.; Munoz, J.; Balaguer, F.; Pellise, M.; Rodriguez de Miguel, C.; Andreu, M.; Jover, R.; Llor, X.; Giraldez, M.D.; et al. Aberrant gene promoter methylation associated with sporadic multiple colorectal cancer. PloS ONE 2010, 5, e8777. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, N.; Terai, T.; Ajioka, Y.; Abe, S.; Kobayasi, O.; Hirai, S.; Hino, O.; Watanabe, H.; Sato, N.; Shimoda, T.; et al. Frequent hypermethylation of rassf1a in early flat-type colorectal tumors. Oncogene 2004, 23, 8900–8907. [Google Scholar] [CrossRef] [PubMed]
- Michalowski, M.B.; de Fraipont, F.; Plantaz, D.; Michelland, S.; Combaret, V.; Favrot, M.C. Methylation of tumor-suppressor genes in neuroblastoma: The rassf1a gene is almost always methylated in primary tumors. Pediatr. Blood Cancer 2008, 50, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Palakurthy, R.K.; Wajapeyee, N.; Santra, M.K.; Gazin, C.; Lin, L.; Gobeil, S.; Green, M.R. Epigenetic silencing of the rassf1a tumor suppressor gene through hoxb3-mediated induction of dnmt3b expression. Mol. Cell 2009, 36, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Shukla, N.; Ameur, N.; Yilmaz, I.; Nafa, K.; Lau, C.Y.; Marchetti, A.; Borsu, L.; Barr, F.G.; Ladanyi, M. Oncogene mutation profiling of pediatric solid tumors reveals significant subsets of embryonal rhabdomyosarcoma and neuroblastoma with mutated genes in growth signaling pathways. Clin. Cancer Res. 2012, 18, 748–757. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.S.; Fu, N.Y.; Sukumaran, S.K.; Wan, K.F.; Wan, Q.; Yu, V.C. Trim39 is a moap-1-binding protein that stabilizes moap-1 through inhibition of its poly-ubiquitination process. Exp. Cell Res. 2009, 315, 1313–1325. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.J.; Zhang, L.; Tang, W.; Chen, C.; Yang, C.S.; Kornbluth, S. The trim39 ubiquitin ligase inhibits apc/ccdh1-mediated degradation of the bax activator moap-1. J. Cell. Biol. 2012, 197, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Rebhan, M.; Chalifa-Caspi, V.; Prilusky, J.; Lancet, D. Genecards: Integrating information about genes, proteins and diseases. Trends Genet. 1997, 13. [Google Scholar] [CrossRef]
- Yan, B.; Zhao, J.L. Mir-1228 prevents cellular apoptosis through targeting of moap1 protein. Apoptosis 2012, 17, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Chen, W.; Kong, D.; Li, X.; Lu, H.; Liu, S.; Wang, J.; Du, L.; Kong, Q.; Huang, X.; et al. Mir-25 targets the modulator of apoptosis 1 gene in lung cancer. Carcinogenesis 2015, 36, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Dai, J.; Deng, H.; Wan, H.; Liu, M.; Wang, J.; Li, S.; Li, X.; Tang, H. Mir-1228 promotes the proliferation and metastasis of hepatoma cells through a p53 forward feedback loop. Br. J. Cancer 2015, 112, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Razumilava, N.; Bronk, S.F.; Smoot, R.L.; Fingas, C.D.; Werneburg, N.W.; Roberts, L.R.; Mott, J.L. Mir-25 targets tnf-related apoptosis inducing ligand (trail) death receptor-4 and promotes apoptosis resistance in cholangiocarcinoma. Hepatology 2012, 55, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.; Lee, E.M.; Cha, H.J.; Kim, K.; Yoon, Y.; Lee, H.; Kim, J.; Kim, Y.J.; Lee, H.G.; Jeung, H.K.; et al. Resveratrol alters microrna expression profiles in a549 human non-small cell lung cancer cells. Mol. Cells 2011, 32, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Reeves, M.E.; Firek, M.; Chen, S.T.; Amaar, Y. The rassf1 gene and the opposing effects of the rassf1a and rassf1c isoforms on cell proliferation and apoptosis. Mol. Biol. Int. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Li, T.T.; Feng, X.; Hsiang, E.; Xiong, Y.; Guan, K.L.; Lei, Q.Y. Targeted polyubiquitylation of rassf1c by the mule and scfbeta-trcp ligases in response to DNA damage. Biochem. J. 2010, 441, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Vlahov, N.; Scrace, S.; Soto, M.S.; Grawenda, A.M.; Bradley, L.; Pankova, D.; Papaspyropoulos, A.; Yee, K.S.; Buffa, F.; Goding, C.R.; et al. Alternate rassf1 transcripts control src activity, e-cadherin contacts, and yap-mediated invasion. Curr. Biol. 2015, 25, 3019–3034. [Google Scholar] [CrossRef] [PubMed]
- The Protein Atlas. Available online: http://www.Proteinatlas.Org/ensg00000068028-rassf1/tissue (accessed on 1 June 2016).
- The Genecards. Available online: http://www.Genecards.Org/cgi-bin/carddisp.Pl?Gene=rassf1 (accessed on 1 June 2016).
- Chang, J.W.; Hsu, H.S.; Ni, H.J.; Chuang, C.T.; Hsiung, C.H.; Huang, T.H.; Wang, Y.C. Distinct epigenetic domains separated by a ctcf bound insulator between the tandem genes, blu and rassf1a. PLoS ONE 2010, 5, e12847. [Google Scholar] [CrossRef] [PubMed]
- Zare-Abdollahi, D.; Safari, S.; Movafagh, A.; Ghadiani, M.; Riazi-Isfahani, S.; Omrani, M.D. Intact expression status of rassf1a in acute myeloid leukemia. Med. Oncol. 2014, 31. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Zhang, X.H.; Zhang, K.; Li, W.; Wang, W.J.; Luo, D.X.; Gao, L. 5-aza-2″-deoxycytidine inhibits retinoblastoma cell by reactivating epigenetically silenced rassf1a gene. Int. J. Ophthalmol. 2014, 7, 51–56. [Google Scholar] [PubMed]
- Montenegro, M.F.; Saez-Ayala, M.; Pinero-Madrona, A.; Cabezas-Herrera, J.; Rodriguez-Lopez, J.N. Reactivation of the tumour suppressor rassf1a in breast cancer by simultaneous targeting of DNA and e2f1 methylation. PLoS ONE 2012, 7, e52231. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhu, F.; Chen, P. Mir-7 and mir-218 epigenetically control tumor suppressor genes rassf1a and claudin-6 by targeting hoxb3 in breast cancer. Biochem. Biophys. Res. Commun. 2012, 424, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Beckedorff, F.C.; Ayupe, A.C.; Crocci-Souza, R.; Amaral, M.S.; Nakaya, H.I.; Soltys, D.T.; Menck, C.F.; Reis, E.M.; Verjovski-Almeida, S. The intronic long noncoding rna anrassf1 recruits prc2 to the rassf1a promoter, reducing the expression of rassf1a and increasing cell proliferation. PLoS Genet. 2013, 9, e1003705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.D.; Zheng, W.W.; Wang, G.X.; Kang, X.C.; Qin, L.; Ji, J.J.; Hao, S. Epigenetic silencing of mir-181b contributes to tumorigenicity in colorectal cancer by targeting rassf1a. Int. J. Oncol. 2016, 48, 1977–1984. [Google Scholar] [CrossRef] [PubMed]
- Steinmann, K.; Sandner, A.; Schagdarsurengin, U.; Dammann, R.H. Frequent promoter hypermethylation of tumor-related genes in head and neck squamous cell carcinoma. Oncol. Rep. 2009, 22, 1519–1526. [Google Scholar] [PubMed]
- Google Patents. Available online: http://www.Google.Com/patents/us20110152339 (accessed on 1 June 2016).
- Google Patents. Available online: http://www.Google.Fm/patents/cn101512014b?Cl=fr (accessed on 1 June 2016).
- Biometrx. Available online: http://www.Biometrxllc.Com/our-products (accessed on 1 June 2016).
- Delaney, C.; Garg, S.K.; Yung, R. Analysis of DNA methylation by pyrosequencing. Methods Mol. Biol. 2015, 1343, 249–264. [Google Scholar] [PubMed]
- Radonic, A.; Thulke, S.; Mackay, I.M.; Landt, O.; Siegert, W.; Nitsche, A. Guideline to reference gene selection for quantitative real-time pcr. Biochem. Biophys. Res. Commun. 2004, 313, 856–862. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas. Available online: http://cancergenome.Nih.Gov/ (accessed on 20 August 2014).













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assay | Primer Sequence 5’-3’ | |
|---|---|---|
| Pyrosequencing | ||
| Assay 1 (20 CpGs) | Forward | GGATGTGGGGATTTTTTTTTTTTAGTATAG |
| Reverse | Biotin-ACTTCAACCCCTCCCCCAAAA | |
| Sequencing | TTTTTTAGTATAGTAAAGTTGGT | |
| Assay 2 (12 CpGs) | Forward | TTTCGGGTTTTATAGTTTTTGTATTTAGGT |
| Reverse | Biotin-ACTCCCGCAACTCAATAAACTCAAACTC | |
| Sequencing | TTTAGTAGTTTAGTTTGGATTTTGG | |
| MOAP1 | Forward | GAGTGTTAGTTAGAGTTTAGGGGAGTTT |
| Reverse | Biotin-CTCACCCCTCCCAACCCT | |
| Sequencing | AGGGGAGTTTGTTTT | |
| qRT-PCR | ||
| GAPDH | Forward | CATGACAACTTTGGTATCGTG |
| Reverse | GTGTCGCTGTTGAAGTCAGA | |
| RASSF1a | Forward | CCTCTGTGGCGACTTCATCTG |
| Reverse | CAACAGTCCAGGCAGACGAG | |
| MOAP-1 | Forward | CTCAATTGCTCCTCTCTGTACC |
| Reverse | CATGACAACTTTGGTATCGTG | |
| RASSF1c | Forward | GCTACTGCAGCCAAGAGGAC |
| Reverse | AGGTGTCTCCCACTCCACAG | |
| PPIA | Forward | GCCGAGGAAAACCGTGTACT |
| Reverse | TGTCTGCAAACAGCTCAAAGG |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Volodko, N.; Salla, M.; Zare, A.; Abulghasem, E.-A.; Vincent, K.; Benesch, M.G.K.; McMullen, T.P.W.; Bathe, O.F.; Postovit, L.; Baksh, S. RASSF1A Site-Specific Methylation Hotspots in Cancer and Correlation with RASSF1C and MOAP-1. Cancers 2016, 8, 55. https://doi.org/10.3390/cancers8060055
Volodko N, Salla M, Zare A, Abulghasem E-A, Vincent K, Benesch MGK, McMullen TPW, Bathe OF, Postovit L, Baksh S. RASSF1A Site-Specific Methylation Hotspots in Cancer and Correlation with RASSF1C and MOAP-1. Cancers. 2016; 8(6):55. https://doi.org/10.3390/cancers8060055
Chicago/Turabian StyleVolodko, Natalia, Mohamed Salla, Alaa Zare, El-Arbi Abulghasem, Krista Vincent, Matthew G.K. Benesch, Todd P.W. McMullen, Oliver F. Bathe, Lynne Postovit, and Shairaz Baksh. 2016. "RASSF1A Site-Specific Methylation Hotspots in Cancer and Correlation with RASSF1C and MOAP-1" Cancers 8, no. 6: 55. https://doi.org/10.3390/cancers8060055
APA StyleVolodko, N., Salla, M., Zare, A., Abulghasem, E. -A., Vincent, K., Benesch, M. G. K., McMullen, T. P. W., Bathe, O. F., Postovit, L., & Baksh, S. (2016). RASSF1A Site-Specific Methylation Hotspots in Cancer and Correlation with RASSF1C and MOAP-1. Cancers, 8(6), 55. https://doi.org/10.3390/cancers8060055