
Molecular Genetics of Intracranial Meningiomas with Emphasis on Canonical Wnt Signalling



Abstract
:1. Introduction
2. Epidemiology and Histopathological Classification
3. Genetics and Signalling Pathways
3.1. Chromosome Aberrations
3.2. Gene Mutations and Gene Expression Analysis
3.3. Microsatellite Instability
3.4. Epigenetic Studies in Meningioma
3.5. Signalling Pathways
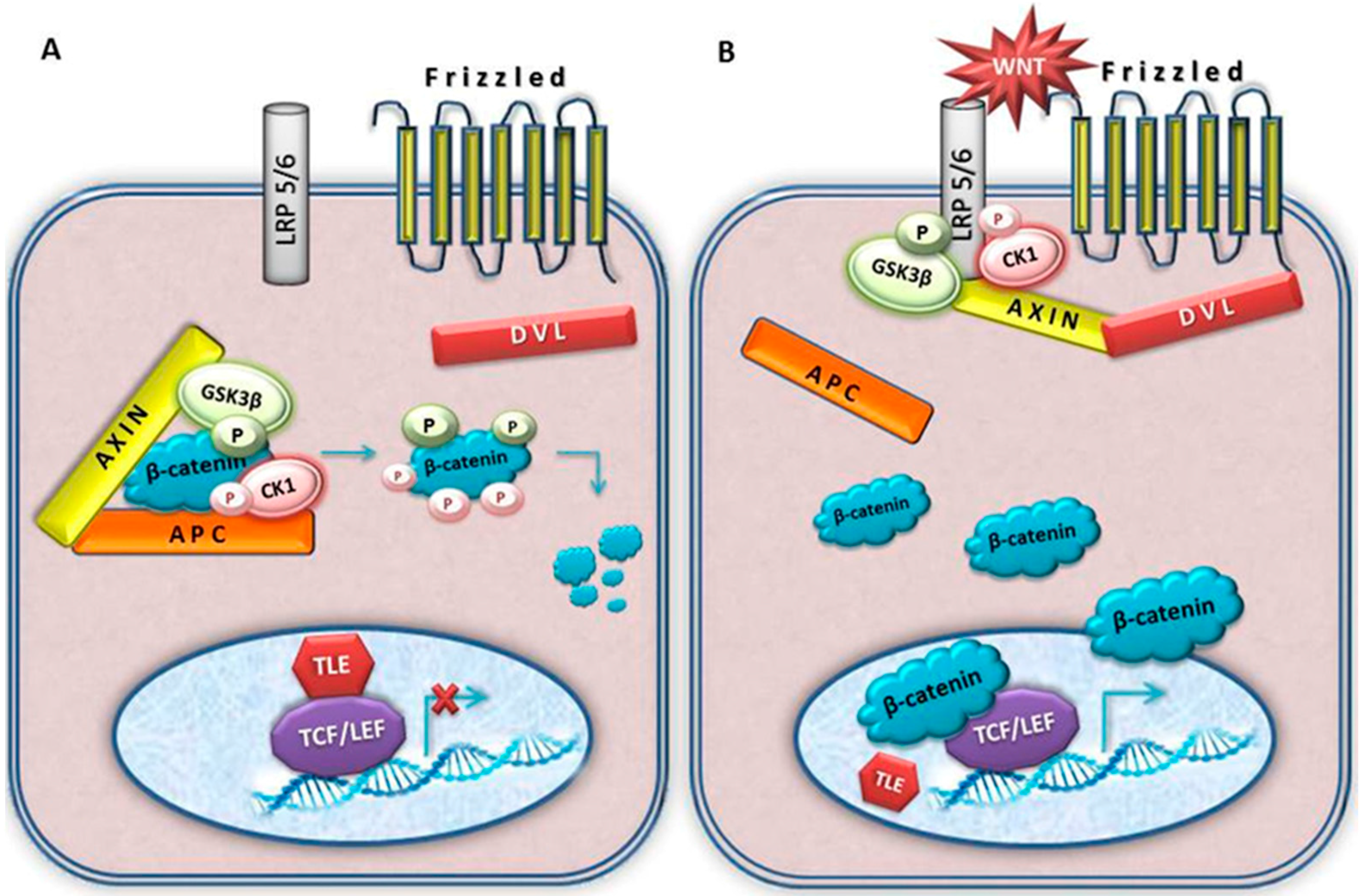
4. Wnt Signalling
5. Key Wnt Signalling Molecules Involved in Meningioma
6. Epithelial-to-Mesenchymal Transduction
7. Conclusions and Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Domingues, P.H.; Teodósio, C.; Otero, Á.; Sousa, P.; Gonçalves, J.M.; Nieto, A.B.; Lopes, M.C.; de Oliveira, C.; Orfao, A.; Tabernero, M.D. The protein expression profile of meningioma cells is associated with distinct cytogenetic tumour subgroups. Neuropathol. Appl. Neurobiol. 2015, 41, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H. Meningiomas, Diagnosis Treatment and Outcome, 1st ed.; Springer: London, UK, 2009. [Google Scholar]
- Mehta, B.C.; Holman, D.W.; Grzybowski, D.M.; Chalmers, J.J. Characterization of Arachnoidal Cells Cultured on Three-Dimensional Nonwoven PET Matrix. Tissue Eng. 2007, 13, 1269–1279. [Google Scholar] [CrossRef] [PubMed]
- Riemenschneider, M.J.; Perry, A.; Reifenberger, G. Histological classification and molecular genetics of meningiomas. Lancet Neurol. 2006, 5, 1045–1054. [Google Scholar] [CrossRef]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheihauer, B.W.; Kleihues, P. The 2007 WHO Classification of Tumors of the Central Nervous System. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Mawrin, C.; Perry, A. Pathological classification and molecular genetics of meningiomas. J. Neurooncol. 2010, 99, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Claus, E.B.; Bondy, M.L.; Wiemels, J.L.; Wrensch, M.; Black, P.M. Epidemiology of intracranial meningioma. Neurosurgery 2005, 57, 1088–1094. [Google Scholar] [CrossRef] [PubMed]
- Mawrin, C.; Chung, C.; Preusser, M. Biology and clinical management challenges in meningioma. Am. Soc. Clin. Oncol. Educ. Book 2015, e106–e115. [Google Scholar] [CrossRef] [PubMed]
- Fathi, A.R.; Roelcke, U. Meningioma. Curr. Neurol. Neurosci. Rep. 2013. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Gittleman, H.; Farah, P.; Ondracek, A.; Chen, Y.; Wolinsky, Y.; Stroup, N.E.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2006–2010. Neuro-Oncology 2013, 15, ii1–ii56. [Google Scholar] [CrossRef] [PubMed]
- Speirs, V.; Boyle-Walsh, E.; Fraser, W.D. Constitutive co-expression of estrogen and progesterone receptor mRNA in human meningiomas by RT-PCR and response of in vitro cell cultures to steroid hormones. Int. J. Cancer 1997, 72, 714–719. [Google Scholar] [CrossRef]
- Chang, Z.N.; Guo, C.-L.; Ahronowitz, I.; Stemmer-Rachamimov, A.O.; MacCollin, M.; Nunes, F.P. A role for the p53 pathway in the pathology of meningiomas with NF2 loss. J. Neurooncol. 2009, 91, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Trott, G.; Pereira-Lima, J.F.S.; Leaes, C.G.S.; Ferreira, N.P.; Barbosa-Coutinho, L.M.; Oliveira, M.C. Abundant immunohistochemical expression of dopamine D2 receptor and p53 protein in meningiomas: Follow-up, relation to gender, age, tumor grade, and recurrence. Braz. J. Med. Biol. Res. 2015, 48, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Benson, V.S.; Kirichek, O.; Beral, V.; Green, J. Menopausal hormone therapy and central nervous system tumor risk: Large UK prospective study and meta-analysis. Int. J. Cancer 2015, 136, 2369–2377. [Google Scholar] [CrossRef] [PubMed]
- Ter Wengel, P.V.; Martin, E.; Gooren, L.; Den Heijer, M.; Peerdeman, S.M. Meningiomas in three male-to-female transgender subjects using oestrogens/progestogens and review of the literature. Andrologia 2016. [Google Scholar] [CrossRef] [PubMed]
- Tabernero, M.D.; Maillo, A.; Gil-Bellosta, C.J.; Castrillo, A.; Sousa, P.; Merino, M.; Orfao, A. Gene expression profiles of meningiomas are associated with tumor cytogenetics and patient outcome. Brain Pathol. 2009, 19, 409–420. [Google Scholar] [CrossRef] [PubMed]
- Lamszus, K. Meningioma pathology, genetics, and biology. J. Neuropathol. Exp. Neurol. 2004, 63, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Marosi, C.; Hassler, M.; Roessler, K.; Reni, M.; Sant, M.; Mazzab, E.; Vecht, C. Meningioma. Crit. Rev. Oncol. Hematol. 2008, 67, 153–171. [Google Scholar] [CrossRef] [PubMed]
- Vranic, A.; Peyre, M.; Kalamarides, M. New insights into meningioma: From genetics to trials. Curr. Opin. Oncol. 2012, 24, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Murnyák, B.; Bognár, L.; Klekner, Á.; Hortobágyi, T. Epigenetics of meningiomas. BioMed Res. Int. 2015, 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, M. Pathology and molecular genetics of meningioma: recent advances. Neurol. Med. Chir. 2015. [Google Scholar] [CrossRef] [PubMed]
- Champeaux, C.; Wilson, E.; Brandner, S.; Shieff, C.; Thorne, L. World Health Organization grade III meningiomas. A retrospective study for outcome and prognostic factors assessment. Br. J. Neurosurg. 2015, 29, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Perry, A.; Scheithauer, B.W.; Stafford, S.L.; Lohse, C.M.; Wollean, P.C. “Malignancy” in meningiomas: A clinicopathologic study of 116 patients, with grading implications. Cancer 1999, 85, 2046–2056. [Google Scholar] [PubMed]
- Skiriute, D.; Tamasauskas, S.; Asmoniene, V.; Saferis, V.; Skauminas, K.; Deltuva, V.; Tamasauskas, A. Tumor grade-related NDRG2 gene expression in primary and recurrent intracranial meningiomas. J. Neurooncol. 2011, 102, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, A.B.; Tabernero, M.D.; Maíllo, A.; Sayagués, J.M.; Ciudad, J.; Merino, M.; Alguero, M.C.; Lubombo, A.M.; Sousa, P.; Santos-Briz, A.; et al. The cytogenetic relationship between primary and recurrent meningiomas points to the need for new treatment strategies in cases at high risk of relapse. Clin. Cancer Res. 2006, 12, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Xiang, C.X.; Zhou, Y.; Ao, X.S.; Zhou, D.Q.; Peng, P.; Zhang, H.Q.; Liu, H.D.; Huang, X. Gene expression profile for predicting survival of patients with meningioma. Int. J. Oncol. 2015, 46, 791–797. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Liu, J.; Patel, S.; Cloughesy, T.; Lai, A.; Farooqi, H.; Seligson, D.; Dong, J.; Liau, L.; Becker, D.; Mischel, P.; Shams, S.; Nelson, S. Genomic landscape of meningiomas. Brain Pathol. 2010, 20, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Finkelstein, S.; Hamilton, R.L.; Rekha, R.; King, J.T., Jr.; Omalu, B. Loss of heterozygosity analysis of benign, atypical, and anaplastic meningiomas. Neurosurgery 2004, 55, 1163–1173. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.; Boström, J.P.; Hartmann, C. Molecular genetics of meningiomas: From basic research to potential clinical applications. Neurosurgery 2007, 60, 787–798. [Google Scholar] [CrossRef] [PubMed]
- Barnholtz-Sloan, J.S.; Kruchko, C. Meningiomas: Causes and risk factors. Neurosurg. Focus 2007. [Google Scholar] [CrossRef] [PubMed]
- Weber, R.G.; Bostrom, J.; Wolter, M.; Baudis, M.; Collins, V.P.; Reifenberger, G.; Lichter, P. Analysis of genomic alterations in benign, atypical, and anaplastic meningiomas: Toward a genetic model of meningioma progression. Proc. Natl. Acad. Sci. USA 1997, 94, 14719–14724. [Google Scholar] [CrossRef] [PubMed]
- Sayagués, J.M.; Tabernero, M.D.; Maíllo, A.; Espinosa, A.; Rasillo, A.; Díaz, P.; Ciudad, J.; López, A.; Merino, M.; Gonçalves, J.M.; et al. Intratumoral patterns of clonal evolution in meningiomas as defined by multicolor interphase fluorescence in situ hybridization (FISH): Is there a relationship between histopathologically benign and atypical/anaplastic lesions? J. Mol. Diagn. 2004, 6, 316–325. [Google Scholar] [CrossRef]
- Rouleau, G.A.; Merel, P.; Lutchman, M.; Sanson, M.; Zucman, J.; Marineau, C.; Hoang-Xuan, K.; Demczuk, S.; Desmaze, C.; Plougastel, B.; et al. Alteration in a new gene encoding a putative membrane-organizing protein causes neurofibromatosis type 2. Nature 1993, 363, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Trofatter, J.A.; MacCollin, M.M.; Rutter, J.L.; Murrell, J.R.; Duyao, M.P.; Parry, D.M.; Eldridge, R.; Kley, N.; Menon, A.G.; Pulaski, K.; et al. A novel moesin-, ezrin-, radixin-like gene is a candidate for the neurofibromatosis 2 tumor suppressor. Cell 1993, 72, 791–800. [Google Scholar] [CrossRef]
- Gusella, J.F.; Ramesh, V.; MacCollin, M.; Jacoby, L.B. Merlin: The neurofibromatosis 2 tumor suppressor. Biochim. Biophys. Acta 1999, 1423, M29–M36. [Google Scholar] [CrossRef]
- Fuller, C.E.; Perry, A. Molecular diagnostics in central nervous system tumors. Adv. Anat. Pathol. 2005, 12, 180–194. [Google Scholar] [CrossRef] [PubMed]
- Ragel, B.T.; Jensen, R.L. Molecular genetics of meningiomas. Neurosurg. Focus 2005, 119, 1–8. [Google Scholar] [CrossRef]
- Pećina-Šlaus, N. Merlin the NF2 gene product. Pathol. Oncol. Res. 2013, 19, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Tabernero, M.; Jara-Acevedo, M.; Nieto, A.B.; Caballero, A.R.; Otero, A.; Sousa, P.; Gonçalves, J.; Domingues, P.H.; Orfao, A. Association between mutation of the NF2 gene and monosomy 22 in menopausal women with sporadic meningiomas. BMC Med. Genet. 2013, 14, 114. [Google Scholar] [CrossRef] [PubMed]
- Miller, R., Jr.; DeCandio, M.L.; Dixon-Mah, Y.; Giglio, P.; Vandergrift, W.A., 3rd; Banik, N.L.; Patel, S.J.; Varma, A.K.; Das, A. Molecular Targets and Treatment of Meningioma. J. Neurol. Neurosurg. 2014, 1, PMC4255716. [Google Scholar]
- Pavelin, S.; Bečić, K.; Forempoher, G.; Tomić, S.; Capkun, V.; Drmić-Hofman, I.; Mrklić, I.; Lušić, I.; Pogorelić, Z. The Significance of Immunohistochemical Expression of Merlin, Ki-67, and p53 in Meningiomas. Appl. Immunohistochem. Mol. Morphol. 2014, 22, 46–49. [Google Scholar] [CrossRef] [PubMed]
- Bell, D.W. Our changing view of the genomic landscape of cancer. J. Pathol. 2010, 220, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.; Shi, L.; Gao, F.; Russin, J.; Zeng, L.; He, S.; Chen, T.C.; Giannotta, S.L.; Weisenberger, D.J.; Zada, G.; et al. Genomic and transcriptome analysis revealing an oncogenic functional module in meningiomas. Neurosurg. Focus 2013. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zhang, K.; Zhao, Y.; Guo, Q.; Guo, D.; Zhang, J. Evidence for involvement of steroid receptors and coactivators in neuroepithelial and meningothelial tumors. Tumour Biol. 2015, 36, 3251–3261. [Google Scholar] [CrossRef] [PubMed]
- Aarhus, M.; Lund-Johansen, M.; Knappskog, P.M. Gene expression profiling of meningiomas: Current status after a decade of microarray-based transcriptomic studies. Acta Neurochir. 2011, 153, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Brastianos, P.K.; Horowitz, P.M.; Santagata, S.; Jones, R.T.; McKenna, A.; Getz, G.; Ligon, K.L.; Palescandolo, E.; van Hummelen, P.; Ducar, M.D.; et al. Genomic sequencing of meningiomas identifies oncogenic SMO and AKT1 mutations. Nat. Genet. 2013, 45, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Clark, V.E.; Erson-Omay, E.Z.; Serin, A.; Yin, J.; Cotney, J.; Ozduman, K.; Avşar, T.; Li, J.; Murray, P.B.; Henegariu, O.; et al. Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science 2013, 339, 1077–1080. [Google Scholar] [CrossRef] [PubMed]
- Bi, W.L.; Abedalthagafi, M.; Horowitz, P.; Agarwalla, P.K.; Mei, Y.; Aizer, A.A.; Brewster, R.; Dunn, G.P.; Al-Mefty, O.; Alexander, B.M.; et al. Genomic landscape of intracranial meningiomas. J. Neurosurg. 2016, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sahm, F.; Bissel, J.; Koelsche, C.; Schweizer, L.; Capper, D.; Reuss, D.; Böhmer, K.; Lass, U.; Göck, T.; Kalis, K.; et al. AKT1E17K mutations cluster with meningothelial and transitional meningiomas and can be detected by SFRP1 immunohistochemistry. Acta Neuropathol. 2013, 126, 757–762. [Google Scholar] [CrossRef] [PubMed]
- Abedalthagafi, M.; Bi, W.L.; Aizer, A.A.; Merrill, P.H.; Brewster, R.; Agarwalla, P.K.; Listewnik, M.L.; Dias-Santagata, D.; Thorner, A.R.; Van Hummelen, P.; et al. Oncogenic PI3K mutations are as common as AKT1 and SMO mutations in meningioma. Neuro-Oncology 2016, 18, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Glez, V.; Bello, M.J.; Franco-Hernandez, C.; de Campos, J.M.; Isla, A.; Vaquero, J.; Rey, J.A. Mutational analysis of the DAL-1/4.1B tumour-suppressor gene locus in meningiomas. Int. J. Mol. Med. 2005, 16, 771–774. [Google Scholar] [CrossRef] [PubMed]
- Peyrard, M.; Fransson, I.; Xie, Y.G.; Han, F.Y.; Ruttledge, M.H.; Swahn, S.; Collins, J.E.; Dunham, I.; Collins, V.P.; Dumanski, J.P. Characterization of a new member of the human beta-adaptin gene family from chromosome 22q12, a candidate meningioma gene. Hum. Mol. Genet. 1994, 3, 1393–1399. [Google Scholar] [CrossRef] [PubMed]
- Lekanne Deprez, R.H.; Riegman, P.H.; Groen, N.A.; Warringa, U.L.; van Biezen, N.A.; Molijn, A.C.; Bootsma, D.; de Jong, P.J.; Menon, A.G.; Kley, N.A.; et al. Cloning and characterization of MN1, a gene from chromosome 22q11, which is disrupted by a balanced translocation in a meningioma. Oncogene 1995, 10, 1521–1528. [Google Scholar] [PubMed]
- Zhang, X.; Jia, H.; Lu, Y.; Dong, C.; Hou, J.; Wang, Z.; Wang, F.; Zhong, H.; Wang, L.; Wang, K. Exome sequencing on malignant meningiomas identified mutations in neurofibromatosis type 2 (NF2) and meningioma 1 (MN1) genes. Discov. Med. 2014, 18, 301–311. [Google Scholar] [PubMed]
- Mashiyama, S.; Murakami, Y.; Yoshimoto, T.; Sekiya, T.; Hayashi, K. Detection of p53 gene mutations in human brain tumors by single-strand conformation polymorphism analysis of polymerase chain reaction products. Oncogene 1991, 6, 1313–1318. [Google Scholar] [PubMed]
- Wang, J.-L.; Zhang, Z.-J.; Hartman, M.; Smits, A.; Westermark, B.; Muhr, C.; Nistér, M. Detection of TP53 gene mutation in human meningiomas: A study using immunohistochemistry, polymerase chain reaction/single-strand conformation polymorphism and DNA sequencing techniques on paraffin-embedded samples. Int. J. Cancer 1995, 64, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Joachim, T.; Ram, Z.; Rappaport, Z.H.; Simon, M.; Schramm, J.; Wiestler, O.D.; von Deimling, A. Comparative analysis of the NF2, TP53, PTEN, KRAS, NRAS and HRAS genes in sporadic and radiation-induced human meningiomas. Int. J. Cancer 2001, 94, 218–221. [Google Scholar] [CrossRef] [PubMed]
- Lusis, E.A.; Watson, M.A.; Chicoine, M.R.; Lyman, M.; Roerig, P.; Reifenberger, G.; Gutmann, D.H.; Perry, A. Integrative genomic analysis identifies NDRG2 as a candidate tumor suppressor gene frequently inactivated in clinically aggressive meningioma. Cancer Res. 2005, 65, 7121–7126. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.T.; Kim, J.W.; Kang, Y.H.; Kim, K.D.; Lee, S.J.; Choi, S.C.; Kim, K.S.; Chae, S.K.; Kim, J.W.; Lim, J.S.; et al. NDRG2 and PRA1 interact and synergistically inhibit T-cell factor/β-catenin signaling. FEBS Lett. 2012, 586, 3962–3968. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Lim, J.; Yang, Y.; Lee, M.S.; Lim, J.S. N-myc downstream-regulated gene 2 (NDRG2) suppresses the epithelial-mesenchymal transition (EMT) in breast cancer cells via STAT3/Snail signaling. Cancer Lett. 2014, 354, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Pećina-Šlaus, N.; Kafka, A.; Vladušić, T.; Tomas, D.; Logara, M.; Skoko, J.; Hrašćan, R. Loss of p53 expression is accompanied with upregulation of beta-catenin in meningiomas: A concomitant reciprocal expression. Int. J. Exp. Pathol. 2016, 97, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Ionov, Y.; Peinado, M.A.; Malkhosyan, S.; Shibata, D.; Perucho, M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature 1993, 363, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Pećina-Šlaus, N.; Nikuševa Martić, T.; Deak, A.J.; Zeljko, M.; Hrašćan, R.; Tomas, D.; Musani, V. Genetic and protein changes of E-cadherin in meningiomas. J. Cancer Res. Clin. Oncol. 2010, 136, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Pykett, M.J.; Murphy, M.; Harnish, P.R.; George, D.L. Identification of a microsatellite instability phenotype in meningiomas. Cancer Res. 1994, 54, 6340–6343. [Google Scholar] [PubMed]
- Sobrido, M.J.; Pereira, C.R.; Barros, F.; Forteza, J.; Carracedo, A.; Lema, M. Low frequency of replication errors in primary nervous system tumours. J. Neurol. Neurosurg. Psychiatry 2000, 69, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Guo, S.Z.; Beggs, A.H.; Maruyama, T.; Santarius, T.; Dashner, K.; Olsen, N.; Wu, J.K.; Black, P. Microsatellite instability analysis of primary human brain tumors. Oncogene 1996, 12, 1417–1423. [Google Scholar] [PubMed]
- Bethke, L.; Murray, A.; Webb, E.; Schoemaker, M.; Muir, K.; McKinney, P.; Hepworth, S.; Dimitropoulou, P.; Lophatananon, A.; Feychting, M.; et al. Comprehensive analysis of DNA repair gene variants and risk of meningioma. J. Natl. Cancer Inst. 2008, 100, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Rajaraman, P.; Hutchinson, A.; Wichner, S.; Black, P.M.; Fine, H.A.; Loeffler, J.S.; Selker, R.G.; Shapiro, W.R.; Rothman, N.; Linet, M.S.; et al. DNA repair gene polymorphisms and risk of adult meningioma, glioma, and acoustic neuroma. Neuro-Oncology 2010, 12, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.N.; Wang, P.; Zhang, J.; Zhou, B.Y.; Mao, Q.; Liu, Y.H. Analysis of the role of hMLH1 hypermethylation and microsatellite instability in meningioma progression. Genet. Mol. Res. 2012, 11, 3933–3941. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.W.; Kim, T.M.; Song, S.S.; Shrinath, N.; Park, R.; Kalamarides, M.; Park, P.J.; Black, P.M.; Carroll, R.S.; Johnson, M.D. Alternative splicing of CHEK2 and codeletion with NF2 promote chromosomal instability in meningioma. Neoplasia 2012, 14, 20–28. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Pham, M.H.; Pease, M.; Zada, G.; Giannotta, S.L.; Wang, K.; Mack, W.J. A review of epigenetic and gene expression alterations associated with intracranial meningiomas. Neurosurg. Focus 2013. [Google Scholar] [CrossRef] [PubMed]
- Hilton, D.A.; Shivane, A.; Kirk, L.; Bassiri, K.; Enki, D.G.; Hanemann, C.O. Activation of multiple growth factor signalling pathways is frequent in meningiomas. Neuropathology 2015, 36, 250–261. [Google Scholar] [CrossRef] [PubMed]
- Cimino, P.J. Malignant progression to anaplastic meningioma: Neuropathology, molecular pathology, and experimental models. Exp. Mol. Pathol. 2015, 99, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Zali, H.; Rezaei Tavirani, M. Meningioma protein-protein interaction network. Arch. Iran. Med. 2014, 17, 262–272. [Google Scholar] [PubMed]
- Sharma, S.; Ray, S.; Mukherjee, S.; Moiyadi, A.; Sridhar, E.; Srivastava, S. Multipronged quantitative proteomic analyses indicate modulation of various signal transduction pathways in human meningiomas. Proteomics 2015, 15, 394–407. [Google Scholar] [CrossRef] [PubMed]
- Watson, M.A.; Gutmann, D.H.; Peterson, K.; Chicoine, M.R.; Kleinschmidt-DeMasters, B.K.; Brown, H.G.; Perry, A. Molecular characterization of human meningiomas by gene expression profiling using high-density oligonucleotide microarrays. Am. J. Pathol. 2002, 161, 665–672. [Google Scholar] [CrossRef]
- Stamenkovic, I.; Yu, Q. Merlin, a “magic” linker between the extracellular cues and intracellular signaling pathways that regulate cell motility, proliferation, and survival. Curr. Protein Pept. Sci. 2010, 11, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Sughrue, M.E.; Yeung, A.H.; Rutkowski, M.J.; Cheung, S.W.; Parsa, A.T. Molecular biology of familial and sporadic vestibular schwannomas: Implications for novel therapeutics. J. Neurosurg. 2011, 114, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Ammoun, S.; Hanemann, C.O. Emerging therapeutic targets in schwannomas and other merlin-deficient tumors. Nat. Rev. Neurol. 2011, 7, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Hanemann, C.O. Merlin, a multi-suppressor from cell membrane to the nucleus. FEBS Lett. 2012, 586, 1403–1408. [Google Scholar] [CrossRef] [PubMed]
- Sherman, L.S.; Gutmann, D.H. Merlin: Hanging tumor suppression on the Rac. Trends Cell Biol. 2001, 11, 442–444. [Google Scholar] [CrossRef]
- Huveneers, S.; Danen, E.H. Adhesion signalling—Crosstalk between integrins, Src and Rho. J. Cell Sci. 2009, 122, 1059–1069. [Google Scholar] [CrossRef] [PubMed]
- Nordqvist, A.C.; Mathiesen, T. Expression of IGF-II, IGFBP-2, -5, and -6 in meningiomas with different brain invasiveness. J. Neurooncol. 2002, 57, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Cuevas, I.C.; Slocum, A.L.; Jun, P.; Costello, J.F.; Bollen, A.W.; Riggins, G.J.; McDermott, M.W.; Lal, A. Meningioma transcript profiles reveal deregulated Notch signaling pathway. Cancer Res. 2005, 65, 5070–5075. [Google Scholar] [CrossRef] [PubMed]
- Logan, C.Y.; Nusse, R. The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 2004, 20, 781–810. [Google Scholar] [CrossRef] [PubMed]
- Lien, W.H.; Fuchs, E. Wnt some lose some: Transcriptional governance of stem cells by Wnt/β-catenin signaling. Genes Dev. 2014, 28, 1517–1532. [Google Scholar] [CrossRef] [PubMed]
- Harrison-Uy, S.J.; Pleasure, S.J. Wnt Signaling and Forebrain Development. Cold Spring Harb. Perspect. Biol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Salinas, P.C. Wnt signaling in the vertebrate central nervous system: From axon guidance to synaptic function. Cold Spring Harb. Perspect. Biol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Malenka, R.C. Beta-catenin is critical for dendritic morphogenesis. Nat. Neurosci. 2003, 6, 1169–1177. [Google Scholar] [CrossRef] [PubMed]
- Chenn, A. Wnt/β-catenin signaling in cerebral cortical development. Organogenesis 2008, 4, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Lang, J.; Maeda, Y.; Bannerman, P.; Xu, J.; Horiuchi, M.; Pleasure, D.; Guo, F. Adenomatous Polyposis Coli regulates oligodendroglial development. J. Neurosci. 2013, 33, 3113–3130. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Wu, S.F.; Goering, L.M.; Dorsky, R.I. Canonical Wnt signaling through Lef1 is required for hypothalamic neurogenesis. Development 2006, 133, 4451–4461. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Deng, Y.; Liu, L.; Yu, K.; Zhang, L.; Wang, H.; He, X.; Wang, J.; Lu, C.; Wu, L.N.; et al. Dual regulatory switch through interactions of Tcf7l2/Tcf4 with stage-specific partners propels oligodendroglial maturation. Nat. Commun. 2016. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhang, Q.; Song, N.N.; Zhang, L.; Sun, Y.L.; Hu, L.; Chen, J.Y.; Zhu, W.; Li, J.; Ding, Y.Q. Lrp5/6 are required for cerebellar development and for suppressing TH expression in Purkinje cells via β-catenin. Mol. Brain 2016. [Google Scholar] [CrossRef] [PubMed]
- Kafka, A.; Bašić-Kinda, S.; Pećina-Šlaus, N. The cellular story of dishevelleds. Croat. Med. J. 2014, 55, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Hart, M.J.; de los Santos, R.; Albert, I.N.; Rubinfeld, B.; Polakis, P. Downregulation of β-catenin by human Axin and its association with the APC tumor suppressor, β-catenin and GSK-3β. Curr. Biol. 1998, 8, 573–581. [Google Scholar] [CrossRef]
- Cliffe, A.; Hamada, F.; Bienz, M. A role of Dishevelled in relocating Axin to the plasma membrane during wingless signaling. Curr. Biol. 2003, 13, 960–966. [Google Scholar] [CrossRef]
- Polakis, P. Wnt signaling in cancer. Cold Spring Harb. Perspect. Biol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Fagotto, F. Looking beyond the Wnt pathway for the deep nature of β-catenin. EMBO Rep. 2013, 14, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Wrobel, G.; Roerig, P.; Kokocinski, F.; Neben, K.; Hahn, M.; Reifenberger, G.; Lichter, P. Microarray-based gene expression profiling of benign, atypical and anaplastic meningiomas identified novel genes associated with meningioma progression. Int. J. Cancer 2005, 114, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Magán, E.; Rodríguez de Lope, A.; Ribalta, T.; Ruano, Y.; Campos-Martín, Y.; Pérez-Bautista, G.; García, J.F.; García-Claver, A.; Fiaño, C.; Hernández-Moneo, J.-L.; et al. Differential expression profiling analyses identifies downregulation of 1p, 6q, and 14q genes and overexpression of 6p histone cluster 1 genes as markers of recurrence in meningiomas. Neuro-Oncology 2010, 12, 1278–1290. [Google Scholar] [CrossRef] [PubMed]
- Pećina-Šlaus, N.; Kafka, A.; Varošanec, A.M.; Marković, L.; Krsnik, Ž.; Njiric, N.; Mrak, G. Expression patterns of Wnt signaling component sFRP3 in astrocytoma and glioblastoma. Mol. Med. Rep. 2016, 13, 4245–4251. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, N.; Kim, Y.J.; Mueller, S.C.; Backes, C.; Werner, T.V.; Galata, V.; Sartorius, E.; Bohle, R.M.; Keller, A.; Meese, E. Posttranscriptional deregulation of signaling pathways in meningioma subtypes by differential expression of miRNAs. Neuro-Oncology 2015, 17, 1250–1260. [Google Scholar] [CrossRef] [PubMed]
- Pećina-Šlaus, N.; Nikuševa Martić, T.; Tomas, D.; Beroš, V.; Zeljko, M.; Čupić, H. Meningiomas exhibit loss of heterozygosity of the APC gene. J. Neurooncol. 2008, 87, 63–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cong, F.; Varmus, H. Nuclear-cytoplasmic shuttling of Axin regulates subcellular localization of beta-catenin. Proc. Natl. Acad. Sci. USA 2004, 101, 2882–2887. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Fu, A.K.Y.; Ip, N.Y. Axin: An emerging key scaffold at the synapse. IUBMB Life 2013, 65, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Pećina-Šlaus, N.; Kafka, A.; Vladušić, T.; Pećina, H.I.; Hrašćan, R. AXIN1’s expression and localization in meningiomas and association to changes of APC and E-cadherin. 2016, in press. [Google Scholar]
- Schwechheimer, K.; Zhou, L.; Birchmeier, W. E-Cadherin in human brain tumours: Loss of immunoreactivity in malignant meningiomas. Virchows Arch. 1998, 432, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Utsuki, S.; Oka, H.; Sato, Y.; Kawano, N.; Tsuchiya, B.; Kobayashi, I.; Fujii, K. Invasive meningioma is associated with a low expression of E-cadherin and beta-catenin. Clin. Neuropathol. 2005, 24, 8–12. [Google Scholar] [PubMed]
- Brunner, E.C.; Romeike, B.F.; Jung, M.; Comtesse, N.; Meese, E. Altered expression of beta-catenin/E-cadherin in meningiomas. Histopathology 2006, 49, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Saydam, O.; Shen, Y.; Würdinger, T.; Senol, O.; Boke, E.; James, M.F.; Tannous, B.A.; Stemmer-Rachamimov, A.O.; Yi, M.; Stephens, R.M.; et al. Downregulated microRNA-200a in meningiomas promotes tumor growth by reducing E-cadherin and activating the Wnt/beta-catenin signaling pathway. Mol. Cell. Biol. 2009, 29, 5923–5940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levina, E.; Oren, M.; Ben-Ze’ev, A. Down-regulation of beta-catenin by p53 involves changes in the rate of beta-catenin phosphorylation and Axin dynamics. Oncogene 2004, 23, 4444–4453. [Google Scholar] [CrossRef] [PubMed]
- Sadot, E.; Geiger, B.; Oren, M.; Ben-Ze’ev, A. Down-regulation of beta-catenin by activated p53. Mol. Cell. Biol. 2001, 21, 6768–6781. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.Y.; Kim, K.M.; Kim, B.K.; Shim, J.K.; Lee, J.H.; Huh, Y.M.; Kim, S.H.; Kim, E.H.; Park, E.K.; Shim, K.W.; et al. Isolation of mesenchymal stem-like cells in meningioma specimens. Int. J. Oncol. 2013, 43, 1260–1268. [Google Scholar] [CrossRef] [PubMed]
- Kida, S.; Yamashima, T.; Kubota, T.; Ito, H.; Yamamoto, S. A light and electron microscopic and immunohistochemical study of human arachnoid villi. J. Neurosurg. 1988, 69, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Lehembre, F.; Yilmaz, M.; Wicki, A.; Schomber, T.; Strittmatter, K.; Ziegler, D.; Kren, A.; Went, P.; Derksen, P.W.; Berns, A.; et al. NCAM-induced focal adhesion assembly: A functional switch upon loss of E-cadherin. EMBO J. 2008, 27, 2603–2615. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhou, B.P. New insights of epithelial-mesenchymal transition in cancer metastasis. Acta Biochim. Biophys. Sin. 2008, 40, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Howard, S.; Deroo, T.; Fujita, Y.; Itasaki, N. A positive role of cadherin in Wnt/β-catenin signalling during epithelial-mesenchymal transition. PLoS ONE 2011, 6, e23899. [Google Scholar] [CrossRef] [PubMed]
- Howng, S.L.; Wu, C.H.; Cheng, T.S.; Sy, W.D.; Lin, P.C.; Wang, C.; Hong, Y.R. Differential expression of Wnt genes, beta-catenin and E-cadherin in human brain tumors. Cancer Lett. 2002, 183, 95–101. [Google Scholar] [CrossRef]
- Li, X.; Xu, Y.; Chen, Y.; Chen, S.; Jia, X.; Sun, T.; Liu, Y.; Li, X.; Xiang, R.; Li, N. SOX2 promotes tumor metastasis by stimulating epithelial-to-mesenchymal transition via regulation of WNT/β-catenin signal network. Cancer Lett. 2013, 336, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tian, X.J.; Xing, J. Signal Transduction Pathways of EMT Induced by TGF-β, SHH, and WNT and Their Crosstalks. J. Clin. Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Pećina-Šlaus, N.; Cicvara-Pecina, T.; Kafka, A. Epithelial-to-mesenchymal transition: Possible role in meningiomas. Front. Biosci. 2012, 4, 889–896. [Google Scholar]



{kind=link}
{kind=link}
| Affected Genes and Their Locations | MA or ES ** | Expressional Changes * | Meningioma Grade | Tumorigenesis | Citations |
|---|---|---|---|---|---|
| PI3K/3q26 | MA | ↑ | Grade I | Early event | [52] |
| SMO/7q32.1 | MA | ↑ | Grade I | Early event | [17,22,28,47,48,49,50] |
| KLF4/9q31 | MA | ↓↑ | Grade I | Early event | [17,22,28,47,48,49,50] |
| AKT1/14q32.33 | MA | ↑ | Grade I | Early event | [17,22,28,47,48,49,50] |
| TRAF7/16p13 | MA | unknown | Grade I | Early event | [17,22,28,47,48,49,50] |
| DAL1/18p11.32 | MA; ES | ↓ | Grade I, II, III | Early event/Progression | [4,6,9,18] |
| SMARCB1/22q11.23 | MA | ↓ | Multiple meningioma | Early event | [20,22] |
| NF2/22q12.2 | MA; ES | ↓ | Grade I, II, III | Early event | [9,22,34,35,36,37] |
| BAM22/22q12.2 | MA | ↓ | Grade I, II, III | Early event | [6,38,54] |
| CDKN2A/9p21 | MA | ↓ | Grade III | Progression | [4,6,9,30,38] |
| ARF/9p21 | MA | ↓ | Grade III | Progression | [4,6,9,30,38] |
| CDKN2B/9p21 | MA | ↓ | Grade III | Progression | [4,6,9,30,38] |
| NDRG2/14q11.2 | MA; ES | ↓ | Grade II, III | Progression | [6,25,60] |
| MEG3/14q32 | MA; ES | ↓ | Grade III | Progression | [6] |
| TP53/17p13.1 | MA; ES | ↓↑ | Grade I, II, III | Progression | [57,58,59,63] |
| MN1/22q12.1 | MA; ES | ↑↓ | Grade I, II, III | Progression | [38,44,55,56] |
| LARGE/22q12.3 | MA | ↓ | Grade I, II, III | Progression | [4,38] |
| TIMP3/22q12 | MA; ES | ↓ | Grade III | Progression | [6] |
| Gene | Locus | Product | Function | Deregulation | Meningioma Effect | Citation |
|---|---|---|---|---|---|---|
| FZD2 | 17q21.1 | Frizzled class receptor 2 | receptor for Wnt signaling proteins | upregulation | tumorigenesis | [78] |
| FZD7 | 2q33 | Frizzled class receptor 7 | receptor for Wnt signaling proteins | upregulation | [44,73] | |
| CSNK1A1 | 5q32 | Casein kinase 1, alpha 1 | transferring phosphorus-containing groups protein tyrosine kinase activity | upregulation | tumorigenesis | [77] |
| APC | 5q22.2 | Adenomatous polyposis coli | negative regulator of Wnt signaling tumor suppressor | loss of heterozygosity | tumorigenesis | [106] |
| AXIN1 | 16q13.3 | Axin1 | negative regulator of Wnt signaling tumor suppressor | gross deletions, downregulation, MSI | cell growth and tumor progression | [107,108] |
| CTNNB1 | 3p22.1 | β-catenin | key downstream component of the canonical Wnt signaling transcription cofactor | upregulation | cell growth and tumor progression associated to complex karyotype meningiomas | [1,63,73,102,114,115] |
| PPP2CA | 4q24 | Serine/threonine protein phosphatase 2B | negative control of cell growth and division | downregulation | tumorigenesis | [77] |
| TCF3 | 19p13.3 | Transcription factor 3 (T cell factor 3) | transcription factor | upregulation | tumorigenesis | [44,73] |
| CCND1 | 11q13.3 | Cyclin D1 | regulator (progression through) of cell cycle | upregulation | cell growth and tumor progression | [28,102] |
| ENC1 | 5q13.3 | Ectodermal-neural cortex 1 | role in the oxidative stress response a role in malignant transformation | upregulation | cell growth and tumor progression | [102] |
| FRZB (SFRP3) | 2q32.1 | Secreted frizzled-related protein 3 | modulator of Wnt signaling | upregulation | tumorigenesis | [44] |
| SFRP1 | 8p11.21 | Secreted frizzled-related protein 1 | tumor suppressor | downregulation, | recurrence | [28,41,44,73,102,103] |
| upregulation | when AKT1E17K mutation is present | |||||
| CDH1 | 16q22.1 | E-cadherin | regulator of cell-cell adhesions | Downregulation loss of function, gross deletion, MSI | cell growth and tumor progression | [65,73,78,110,111,112,113] |
| NDRG2 | 14q11.2 | N-myc downstream regulator 2 | transcription factor tumor suppressor | Downregulation, promoter hyperventilation | cell growth and tumor progression | [6,25,61,62] |
| CDK5R1 | 17q11.2 | Cyclin-dependent kinase 5, regulatory subunit 1 | G1/S transition of mitotic cell cycle, development of the central nervous system | upregulation | cell growth and tumor progression | [73,102] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pećina-Šlaus, N.; Kafka, A.; Lechpammer, M. Molecular Genetics of Intracranial Meningiomas with Emphasis on Canonical Wnt Signalling. Cancers 2016, 8, 67. https://doi.org/10.3390/cancers8070067
Pećina-Šlaus N, Kafka A, Lechpammer M. Molecular Genetics of Intracranial Meningiomas with Emphasis on Canonical Wnt Signalling. Cancers. 2016; 8(7):67. https://doi.org/10.3390/cancers8070067
Chicago/Turabian StylePećina-Šlaus, Nives, Anja Kafka, and Mirna Lechpammer. 2016. "Molecular Genetics of Intracranial Meningiomas with Emphasis on Canonical Wnt Signalling" Cancers 8, no. 7: 67. https://doi.org/10.3390/cancers8070067
APA StylePećina-Šlaus, N., Kafka, A., & Lechpammer, M. (2016). Molecular Genetics of Intracranial Meningiomas with Emphasis on Canonical Wnt Signalling. Cancers, 8(7), 67. https://doi.org/10.3390/cancers8070067