
Aberrant Wnt Signaling in Leukemia



Abstract
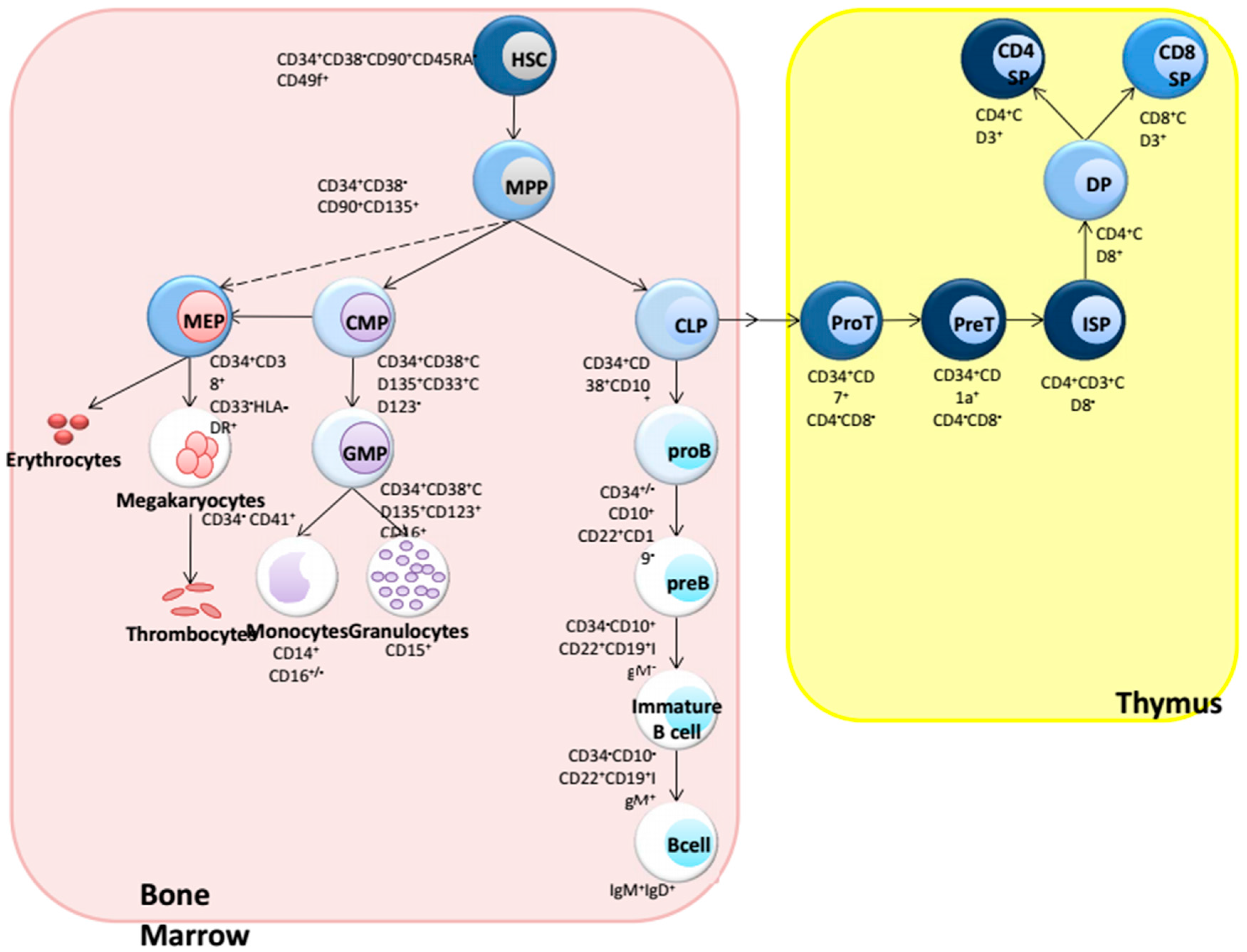
:1. Introduction: Hematopoiesis
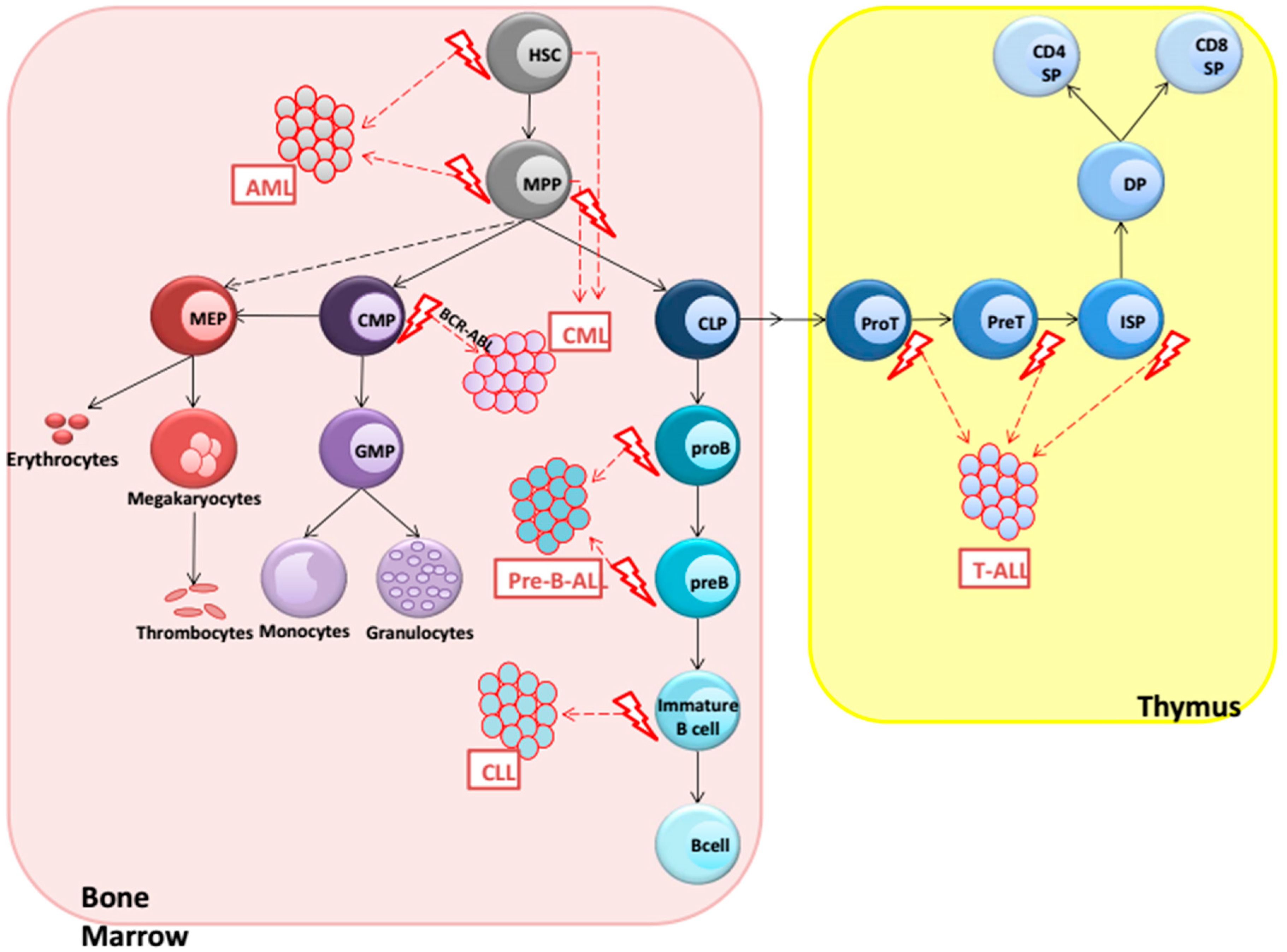
2. Hematological Malignancies
3. AML (Acute Myeloid Leukemia)
4. CML (Chronic Myeloid Leukemia)
5. Precursor B-ALL
6. CLL (Chronic Lymphocytic Leukemia)
7. T-ALL (T-Cell Acute Lymphoblastic Leukemia)
8. Conclusions
Acknowledgments
Conflicts of Interest
References
- Orkin, S.H.; Zon, L.I. Hematopoiesis: An evolving paradigm for stem cell biology. Cell 2008, 132, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Blank, U.; Karlsson, G.; Karlsson, S. Signaling pathways governing stem-cell fate. Blood 2008, 111, 492–503. [Google Scholar] [CrossRef] [PubMed]
- Staal, F.J.; Baum, C.; Cowan, C.; Dzierzak, E.; Hacein-Bey-Abina, S.; Karlsson, S.; Lapidot, T.; Lemischka, I.; Mendez-Ferrer, S.; Mikkers, H.; et al. Stem cell self-renewal: Lessons from bone marrow, gut and IPS toward clinical applications. Leukemia 2011, 25, 1095–1102. [Google Scholar] [CrossRef] [PubMed]
- Luis, T.C.; Killmann, N.M.; Staal, F.J. Signal transduction pathways regulating hematopoietic stem cell biology: Introduction to a series of spotlight reviews. Leukemia 2012, 26, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Eaves, C.J. Hematopoietic stem cells: Concepts, definitions, and the new reality. Blood 2015, 125, 2605–2613. [Google Scholar] [CrossRef] [PubMed]
- Luis, T.C.; Ichii, M.; Brugman, M.H.; Kincade, P.; Staal, F.J. Wnt signaling strength regulates normal hematopoiesis and its deregulation is involved in leukemia development. Leukemia 2012, 26, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Staal, F.J.; Chhatta, A.; Mikkers, H. Caught in a Wnt storm: Complexities of Wnt signalling in hematopoiesis. Exp. Hematol. 2016, 44, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Lento, W.; Congdon, K.; Voermans, C.; Kritzik, M.; Reya, T. Wnt Signaling in Normal and Malignant Hematopoiesis. Cold Spring Harb Perspect Biol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, S.; Kincade, P.W. Wnt-related molecules and signaling pathway equilibrium in hematopoiesis. Cell Stem Cell 2009, 4, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Staal, F.J.; Luis, T.C.; Tiemessen, M.M. Wnt signalling in the immune system: Wnt is spreading its wings. Nat. Rev. Immunol. 2008, 8, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Staal, F.J.; Sen, J.M. The canonical Wnt signaling pathway plays an important role in lymphopoiesis and hematopoiesis. Eur. J. Immunol. 2008, 38, 1788–1794. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. Wnt/beta-catenin signaling in development and disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Fodde, R.; Brabletz, T. Wnt/beta-catenin signaling in cancer stemness and malignant behavior. Curr. Opin. Cell Biol. 2007, 19, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Clevers, H. Wnt signalling in stem cells and cancer. Nature 2005, 434, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Luis, T.C.; Naber, B.A.; Roozen, P.P.; Brugman, M.H.; de Haas, E.F.; Ghazvini, M.; Fibbe, W.E.; van Dongen, J.J.; Fodde, R.; Staal, F.J. Canonical Wnt signaling regulates hematopoiesis in a dosage-dependent fashion. Cell Stem Cell 2011, 9, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Muller-Tidow, C.; Steffen, B.; Cauvet, T.; Tickenbrock, L.; Ji, P.; Diederichs, S.; Sargin, B.; Kohler, G.; Stelljes, M.; Puccetti, E.; et al. Translocation products in acute myeloid leukemia activate the Wnt signaling pathway in hematopoietic cells. Mol. Cell. Biol. 2004, 24, 2890–2904. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Beissert, T.; Kukoc-Zivojnov, N.; Puccetti, E.; Altschmied, J.; Strolz, C.; Boehrer, S.; Gul, H.; Schneider, O.; Ottmann, O.G.; et al. Gamma-catenin contributes to leukemogenesis induced by AML-associated translocation products by increasing the self-renewal of very primitive progenitor cells. Blood 2004, 103, 3535–3543. [Google Scholar] [CrossRef] [PubMed]
- Ysebaert, L.; Chicanne, G.; Demur, C.; de Toni, F.; Prade-Houdellier, N.; Ruidavets, J.B.; Mansat-De Mas, V.; Rigal-Huguet, F.; Laurent, G.; Payrastre, B.; et al. Expression of beta-catenin by acute myeloid leukemia cells predicts enhanced clonogenic capacities and poor prognosis. Leukemia 2006, 20, 1211–1216. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.G.; Pearn, L.; Liddiard, K.; Pumford, S.L.; Burnett, A.K.; Tonks, A.; Darley, R.L. Gamma-catenin is overexpressed in acute myeloid leukemia and promotes the stabilization and nuclear localization of beta-catenin. Leukemia 2013, 27, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, E.A.; Gore, S.D.; Hooker, C.; McDevitt, M.A.; Karp, J.E.; Smith, B.D.; Mohammad, H.P.; Ye, Y.; Herman, J.G.; Carraway, H.E. Acute myeloid leukemia is characterized by Wnt pathway inhibitor promoter hypermethylation. Leuk. Lymphoma 2010, 51, 1711–1719. [Google Scholar] [CrossRef] [PubMed]
- Martin, V.; Agirre, X.; Jimenez-Velasco, A.; Jose-Eneriz, E.S.; Cordeu, L.; Garate, L.; Vilas-Zornoza, A.; Castillejo, J.A.; Heiniger, A.; Prosper, F.; et al. Methylation status of Wnt signaling pathway genes affects the clinical outcome of philadelphia-positive acute lymphoblastic leukemia. Cancer Sci. 2008, 99, 1865–1868. [Google Scholar] [CrossRef] [PubMed]
- Martin, V.; Valencia, A.; Agirre, X.; Cervera, J.; San Jose-Eneriz, E.; Vilas-Zornoza, A.; Rodriguez-Otero, P.; Sanz, M.A.; Herrera, C.; Torres, A.; et al. Epigenetic regulation of the non-canonical Wnt pathway in acute myeloid leukemia. Cancer Sci. 2010, 101, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, R.; Onizuka, M.; Kojima, M.; Shimada, M.; Fukagawa, S.; Tsuboi, K.; Kobayashi, H.; Shintani, A.; Ogawa, Y.; Kawada, H.; et al. Preferential hypermethylation of the Dickkopf-1 promoter in core-binding factor leukaemia. Br. J. Haematol. 2007, 138, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Valencia, A.; Roman-Gomez, J.; Cervera, J.; Such, E.; Barragan, E.; Bolufer, P.; Moscardo, F.; Sanz, G.F.; Sanz, M.A. Wnt signaling pathway is epigenetically regulated by methylation of Wnt antagonists in acute myeloid leukemia. Leukemia 2009, 23, 1658–1666. [Google Scholar] [CrossRef] [PubMed]
- Ying, J.; Li, H.; Chen, Y.W.; Srivastava, G.; Gao, Z.; Tao, Q. Wnt5a is epigenetically silenced in hematologic malignancies and inhibits leukemia cell growth as a tumor suppressor. Blood 2007, 110, 4130–4132. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Chen, Q.; Coles, A.H.; Anderson, S.J.; Pihan, G.; Bradley, A.; Gerstein, R.; Jurecic, R.; Jones, S.N. Wnt5a inhibits B cell proliferation and functions as a tumor suppressor in hematopoietic tissue. Cancer Cell 2003, 4, 349–360. [Google Scholar] [CrossRef]
- Yeung, J.; Esposito, M.T.; Gandillet, A.; Zeisig, B.B.; Griessinger, E.; Bonnet, D.; So, C.W. Beta-catenin mediates the establishment and drug resistance of MLL leukemic stem cells. Cancer Cell 2010, 18, 606–618. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Krivtsov, A.V.; Sinha, A.U.; North, T.E.; Goessling, W.; Feng, Z.; Zon, L.I.; Armstrong, S.A. The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in aml. Science 2010, 327, 1650–1653. [Google Scholar] [CrossRef] [PubMed]
- Lane, S.W.; Wang, Y.J.; Lo Celso, C.; Ragu, C.; Bullinger, L.; Sykes, S.M.; Ferraro, F.; Shterental, S.; Lin, C.P.; Gilliland, D.G.; et al. Differential niche and Wnt requirements during acute myeloid leukemia progression. Blood 2011, 118, 2849–2856. [Google Scholar] [CrossRef] [PubMed]
- Tickenbrock, L.; Schwable, J.; Wiedehage, M.; Steffen, B.; Sargin, B.; Choudhary, C.; Brandts, C.; Berdel, W.E.; Muller-Tidow, C.; Serve, H. Flt3 tandem duplication mutations cooperate with Wnt signaling in leukemic signal transduction. Blood 2005, 105, 3699–3706. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Suzuki, M.; Niwa, Y.; Hiraga, J.; Nagasaka, T.; Ito, M.; Nakamura, S.; Tomita, A.; Abe, A.; Kiyoi, H.; et al. Clinical significance of nuclear non-phosphorylated beta-catenin in acute myeloid leukaemia and myelodysplastic syndrome. Br. J. Haematol. 2008, 140, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Taskesen, E.; Staal, F.J.; Reinders, M.J. An integrated approach of gene expression and DNA-methylation profiles of Wnt signaling genes uncovers novel prognostic markers in acute myeloid leukemia. BMC Bioinform. 2015. [Google Scholar] [CrossRef] [PubMed]
- Corces-Zimmerman, M.R.; Majeti, R. Pre-leukemic evolution of hematopoietic stem cells: The importance of early mutations in leukemogenesis. Leukemia 2014, 28, 2276–2282. [Google Scholar] [CrossRef] [PubMed]
- Guezguez, B.; Almakadi, M.; Benoit, Y.D.; Shapovalova, Z.; Rahmig, S.; Fiebig-Comyn, A.; Casado, F.L.; Tanasijevic, B.; Bresolin, S.; Masetti, R.; et al. GSK3 deficiencies in hematopoietic stem cells initiate pre-neoplastic state that is predictive of clinical outcomes of human acute leukemia. Cancer Cell 2016, 29, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Majeti, R.; Becker, M.W.; Tian, Q.; Lee, T.L.; Yan, X.; Liu, R.; Chiang, J.H.; Hood, L.; Clarke, M.F.; Weissman, I.L. Dysregulated gene expression networks in human acute myelogenous leukemia stem cells. Proc. Natl. Acad. Sci. USA 2009, 106, 3396–3401. [Google Scholar] [CrossRef] [PubMed]
- Beghini, A.; Corlazzoli, F.; del Giacco, L.; Re, M.; Lazzaroni, F.; Brioschi, M.; Valentini, G.; Ferrazzi, F.; Ghilardi, A.; Righi, M.; et al. Regeneration-associated Wnt signaling is activated in long-term reconstituting AC133 bright acute myeloid leukemia cells. Neoplasia 2012, 14, 1236–1248. [Google Scholar] [CrossRef] [PubMed]
- Minke, K.S.; Staib, P.; Puetter, A.; Gehrke, I.; Gandhirajan, R.K.; Schlosser, A.; Schmitt, E.K.; Hallek, M.; Kreuzer, K.A. Small molecule inhibitors of Wnt signaling effectively induce apoptosis in acute myeloid leukemia cells. Eur. J. Haematol. 2009, 82, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Fiskus, W.; Sharma, S.; Saha, S.; Shah, B.; Devaraj, S.G.; Sun, B.; Horrigan, S.; Leveque, C.; Zu, Y.; Iyer, S.; et al. Pre-clinical efficacy of combined therapy with novel beta-catenin antagonist BC2059 and histone deacetylase inhibitor against aml cells. Leukemia 2015, 29, 1267–1278. [Google Scholar] [CrossRef] [PubMed]
- Heidel, F.H.; Arreba-Tutusaus, P.; Armstrong, S.A.; Fischer, T. Evolutionarily conserved signaling pathways: Acting in the shadows of acute myelogenous leukemia’s genetic diversity. Clin. Cancer Res. 2015, 21, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Yang, L.L.; Niu, T.; Cheng, C.; Zhong, L.; Zheng, M.W.; Xiong, Y.; Li, L.L.; Xiang, R.; Chen, L.J.; et al. SKLB-677, an FLT3 and Wnt/beta-catenin signaling inhibitor, displays potent activity in models of FLT3-driven AML. Sci. Rep. 2015. [Google Scholar] [CrossRef]
- Metzeler, K.H.; Heilmeier, B.; Edmaier, K.E.; Rawat, V.P.; Dufour, A.; Dohner, K.; Feuring-Buske, M.; Braess, J.; Spiekermann, K.; Buchner, T.; et al. High expression of lymphoid enhancer-binding factor-1 (Lef1) is a novel favorable prognostic factor in cytogenetically normal acute myeloid leukemia. Blood 2012, 120, 2118–2126. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Zhu, H.; Wu, W.; Xu, J.; Chen, T.; Xu, B.; Qian, S.; Li, J.; Liu, P. Clinical significance of lymphoid enhancer-binding factor 1 expression in acute myeloid leukemia. Leuk. Lymphoma 2014, 55, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Skokowa, J.; Cario, G.; Uenalan, M.; Schambach, A.; Germeshausen, M.; Battmer, K.; Zeidler, C.; Lehmann, U.; Eder, M.; Baum, C.; et al. Lef-1 is crucial for neutrophil granulocytopoiesis and its expression is severely reduced in congenital neutropenia. Nat. Med. 2006, 12, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Tiemessen, M.M.; Baert, M.R.; Schonewille, T.; Brugman, M.H.; Famili, F.; Salvatori, D.C.; Meijerink, J.P.; Ozbek, U.; Clevers, H.; van Dongen, J.J.; et al. The nuclear effector of Wnt-signaling, TCF1, functions as a T-cell specific tumor suppressor for development of lymphomas. PLoS Biol. 2012, 10, e1001430. [Google Scholar] [CrossRef] [PubMed]
- Nowicki, M.O.; Pawlowski, P.; Fischer, T.; Hess, G.; Pawlowski, T.; Skorski, T. Chronic myelogenous leukemia molecular signature. Oncogene 2003, 22, 3952–3963. [Google Scholar] [CrossRef] [PubMed]
- Weerkamp, F.; Dekking, E.; Ng, Y.Y.; van der Velden, V.H.; Wai, H.; Bottcher, S.; Bruggemann, M.; van der Sluijs, A.J.; Koning, A.; Boeckx, N.; et al. Flow cytometric immunobead assay for the detection of BCR-ABL fusion proteins in leukemia patients. Leukemia 2009, 23, 1106–1117. [Google Scholar] [CrossRef] [PubMed]
- Deininger, M.; Buchdunger, E.; Druker, B.J. The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood 2005, 105, 2640–2653. [Google Scholar] [CrossRef] [PubMed]
- Deininger, M.W.; Vieira, S.; Mendiola, R.; Schultheis, B.; Goldman, J.M.; Melo, J.V. BCR-ABL tyrosine kinase activity regulates the expression of multiple genes implicated in the pathogenesis of chronic myeloid leukemia. Cancer Res. 2000, 60, 2049–2055. [Google Scholar] [PubMed]
- Deininger, M.W.; Vieira, S.A.; Parada, Y.; Banerji, L.; Lam, E.W.; Peters, G.; Mahon, F.X.; Kohler, T.; Goldman, J.M.; Melo, J.V. Direct relation between BCR-ABL tyrosine kinase activity and cyclin D2 expression in lymphoblasts. Cancer Res. 2001, 61, 8005–8013. [Google Scholar] [PubMed]
- Druker, B.J.; Guilhot, F.; O’Brien, S.G.; Gathmann, I.; Kantarjian, H.; Gattermann, N.; Deininger, M.W.; Silver, R.T.; Goldman, J.M.; Stone, R.M.; et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N. Engl. J. Med. 2006, 355, 2408–2417. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, C.H.; Ailles, L.E.; Dylla, S.J.; Muijtjens, M.; Jones, C.; Zehnder, J.L.; Gotlib, J.; Li, K.; Manz, M.G.; Keating, A.; et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in Blast-Crisis CML. N. Engl. J. Med. 2004, 351, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Blum, J.; Chen, A.; Kwon, H.Y.; Jung, S.H.; Cook, J.M.; Lagoo, A.; Reya, T. Loss of beta-catenin impairs the renewal of normal and CML stem cells in vivo. Cancer Cell 2007, 12, 528–541. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Li, F.; Xing, S.; Zhao, T.; Peng, W.; Xue, H.H. Hematopoietic and leukemic stem cells have distinct dependence on TCF1 and LEF1 transcription factors. J. Biol. Chem. 2016, 291, 11148–11160. [Google Scholar] [CrossRef] [PubMed]
- Gregory, M.A.; Phang, T.L.; Neviani, P.; Alvarez-Calderon, F.; Eide, C.A.; O’Hare, T.; Zaberezhnyy, V.; Williams, R.T.; Druker, B.J.; Perrotti, D.; et al. Wnt/Ca2+/NFAT signaling maintains survival of Ph+ leukemia cells upon inhibition of Bcr-Abl. Cancer Cell 2010, 18, 74–87. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Li, S. Survival regulation of leukemia stem cells. Cell. Mol. Life Sci. 2016, 73, 1039–1050. [Google Scholar] [CrossRef] [PubMed]
- Heidel, F.H.; Bullinger, L.; Feng, Z.; Wang, Z.; Neff, T.A.; Stein, L.; Kalaitzidis, D.; Lane, S.W.; Armstrong, S.A. Genetic and pharmacologic inhibition of beta-catenin targets imatinib-resistant leukemia stem cells in CML. Cell Stem Cell 2012, 10, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; O’Riordan, M.; Okamura, R.; Devaney, E.; Willert, K.; Nusse, R.; Grosschedl, R. Wnt signaling regulates B lymphocyte proliferation through a LEF-1 dependent mechanism. Immunity 2000, 13, 15–24. [Google Scholar] [CrossRef]
- Ranheim, E.A.; Kwan, H.C.; Reya, T.; Wang, Y.K.; Weissman, I.L.; Francke, U. Frizzled 9 knock-out mice have abnormal B-cell development. Blood 2005, 105, 2487–2494. [Google Scholar] [CrossRef] [PubMed]
- McWhirter, J.R.; Neuteboom, S.T.; Wancewicz, E.V.; Monia, B.P.; Downing, J.R.; Murre, C. Oncogenic homeodomain transcription factor E2A-PBX1 activates a novel Wnt gene in pre-B acute lymphoblastoid leukemia. Proc. Natl. Acad. Sci. USA 1999, 96, 11464–11469. [Google Scholar] [CrossRef] [PubMed]
- Nygren, M.K.; Dosen, G.; Hystad, M.E.; Stubberud, H.; Funderud, S.; Rian, E. Wnt3a activates canonical Wnt signalling in acute lymphoblastic leukaemia (ALL) cells and inhibits the proliferation of B-ALL cell lines. Br. J. Haematol. 2007, 136, 400–413. [Google Scholar] [CrossRef] [PubMed]
- Nygren, M.K.; Dosen-Dahl, G.; Stubberud, H.; Walchli, S.; Munthe, E.; Rian, E. Beta-catenin is involved in N-cadherin-dependent adhesion, but not in canonical Wnt signaling in E2A-PBX1-positive B acute lymphoblastic leukemia cells. Exp. Hematol. 2009, 37, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Saba, N.S.; Angelova, M.; Lobelle-Rich, P.A.; Levy, L.S. Disruption of pre-B-cell receptor signaling jams the Wnt/beta-catenin pathway and induces cell death in B-cell acute lymphoblastic leukemia cell lines. Leuk. Res. 2015, 39, 1220–1228. [Google Scholar] [CrossRef] [PubMed]
- Gandhirajan, R.K.; Staib, P.A.; Minke, K.; Gehrke, I.; Plickert, G.; Schlosser, A.; Schmitt, E.K.; Hallek, M.; Kreuzer, K.A. Small molecule inhibitors of Wnt/beta-catenin/LEF-1 signaling induces apoptosis in chronic lymphocytic leukemia cells in vitro and in vivo. Neoplasia 2010, 12, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Zhao, Y.; Tawatao, R.; Cottam, H.B.; Sen, M.; Leoni, L.M.; Kipps, T.J.; Corr, M.; Carson, D.A. Activation of the Wnt signaling pathway in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2004, 101, 3118–3123. [Google Scholar] [CrossRef] [PubMed]
- Chim, C.S.; Fung, T.K.; Wong, K.F.; Lau, J.S.; Liang, R. Infrequent Wnt inhibitory factor-1 (WIF-1) methylation in chronic lymphocytic leukemia. Leuk. Res. 2006, 30, 1135–1139. [Google Scholar] [CrossRef] [PubMed]
- Chim, C.S.; Pang, R.; Fung, T.K.; Choi, C.L.; Liang, R. Epigenetic dysregulation of Wnt signaling pathway in multiple myeloma. Leukemia 2007, 21, 2527–2536. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Wang, L.; Chen, C.; Liu, Y.; Zhou, P.; Wang, Y.; Wang, X.; Turnbull, J.; Minassian, B.A.; Zheng, P. Laforin negatively regulates cell cycle progression through glycogen synthase kinase 3beta-dependent mechanisms. Mol. Cell. Biol. 2008, 28, 7236–7244. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, N.C.; Ocio, E.M.; de Las Rivas, J.; Maiso, P.; Delgado, M.; Ferminan, E.; Arcos, M.J.; Sanchez, M.L.; Hernandez, J.M.; San Miguel, J.F. Gene expression profiling of B lymphocytes and plasma cells from waldenstrom’s macroglobulinemia: Comparison with expression patterns of the same cell counterparts from chronic lymphocytic leukemia, multiple myeloma and normal individuals. Leukemia 2007, 21, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Janovska, P.; Poppova, L.; Plevova, K.; Plesingerova, H.; Behal, M.; Kaucka, M.; Ovesna, P.; Hlozkova, M.; Borsky, M.; Stehlikova, O.; et al. Autocrine signaling by Wnt-5a deregulates chemotaxis of leukemic cells and predicts clinical outcome in chronic lymphocytic leukemia. Clin. Cancer Res. 2016, 22, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Izon, D.J.; Punt, J.A.; Pear, W.S. Deciphering the role of notch signaling in lymphopoiesis. Curr. Opin. Immunol. 2002, 14, 192–199. [Google Scholar] [CrossRef]
- Jenkinson, E.J.; Jenkinson, W.E.; Rossi, S.W.; Anderson, G. The thymus and T-cell commitment: The right niche for notch? Nat. Rev. Immunol. 2006, 6, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Kumano, K.; Masuda, S.; Hangaishi, A.; Takita, J.; Nakazaki, K.; Kurokawa, M.; Hayashi, Y.; Ogawa, S.; Chiba, S. Mutations of the Notch1 gene in T-cell acute lymphoblastic leukemia: Analysis in adults and children. Leukemia 2005, 19, 1841–1843. [Google Scholar] [CrossRef] [PubMed]
- Weerkamp, F.; van Dongen, J.J.; Staal, F.J. Notch and Wnt signaling in T-lymphocyte development and acute lymphoblastic leukemia. Leukemia 2006, 20, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Weng, A.P.; Ferrando, A.A.; Lee, W.; Morris, J.P.T.; Silverman, L.B.; Sanchez-Irizarry, C.; Blacklow, S.C.; Look, A.T.; Aster, J.C. Activating mutations of Notch1 in human T cell acute lymphoblastic leukemia. Science 2004, 306, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Weng, A.P.; Millholland, J.M.; Yashiro-Ohtani, Y.; Arcangeli, M.L.; Lau, A.; Wai, C.; Del Bianco, C.; Rodriguez, C.G.; Sai, H.; Tobias, J.; et al. C-MYC is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev. 2006, 20, 2096–2109. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Kanakura, Y. Genetic abnormalities associated with acute lymphoblastic leukemia. Cancer Sci. 2016. [Google Scholar] [CrossRef] [PubMed]
- Weber, B.N.; Chi, A.W.; Chavez, A.; Yashiro-Ohtani, Y.; Yang, Q.; Shestova, O.; Bhandoola, A. A critical role for TCF-1 in T-lineage specification and differentiation. Nature 2011, 476, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Dose, M.; Kovalovsky, D.; Chang, R.; O’Neil, J.; Look, A.T.; von Boehmer, H.; Khazaie, K.; Gounari, F. Beta-catenin stabilization stalls the transition from double-positive to single-positive stage and predisposes thymocytes to malignant transformation. Blood 2007, 109, 5463–5472. [Google Scholar] [CrossRef] [PubMed]
- Palomero, T.; Lim, W.K.; Odom, D.T.; Sulis, M.L.; Real, P.J.; Margolin, A.; Barnes, K.C.; O’Neil, J.; Neuberg, D.; Weng, A.P.; et al. Notch1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc. Natl. Acad. Sci. USA 2006, 103, 18261–18266. [Google Scholar] [CrossRef] [PubMed]
- Allman, D.; Karnell, F.G.; Punt, J.A.; Bakkour, S.; Xu, L.; Myung, P.; Koretzky, G.A.; Pui, J.C.; Aster, J.C.; Pear, W.S. Separation of Notch1 promoted lineage commitment and expansion/transformation in developing T cells. J. Exp. Med. 2001, 194, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Ng, O.H.; Erbilgin, Y.; Firtina, S.; Celkan, T.; Karakas, Z.; Aydogan, G.; Turkkan, E.; Yildirmak, Y.; Timur, C.; Zengin, E.; et al. Deregulated Wnt signaling in childhood T-cell acute lymphoblastic leukemia. Blood Cancer J. 2014, 4, e192. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Zhou, X.; Steinke, F.C.; Liu, C.; Chen, S.C.; Zagorodna, O.; Jing, X.; Yokota, Y.; Meyerholz, D.K.; Mullighan, C.G.; et al. The TCF-1 and LEF-1 transcription factors have cooperative and opposing roles in T cell development and malignancy. Immunity 2012, 37, 813–826. [Google Scholar] [CrossRef] [PubMed]
- Staal, F.J.; Clevers, H. Tales of the unexpected: TCF1 functions as a tumor suppressor for leukemias. Immunity 2012, 37, 761–763. [Google Scholar] [CrossRef] [PubMed]
- Spaulding, C.; Reschly, E.J.; Zagort, D.E.; Yashiro-Ohtani, Y.; Beverly, L.J.; Capobianco, A.; Pear, W.S.; Kee, B.L. Notch1 co-opts lymphoid enhancer factor 1 for survival of murine T-cell lymphomas. Blood 2007, 110, 2650–2658. [Google Scholar] [CrossRef] [PubMed]
- Homminga, I.; Pieters, R.; Langerak, A.W.; de Rooi, J.J.; Stubbs, A.; Verstegen, M.; Vuerhard, M.; Buijs-Gladdines, J.; Kooi, C.; Klous, P.; et al. Integrated transcript and genome analyses reveal NKX2-1 and MEF2C as potential oncogenes in T cell acute lymphoblastic leukemia. Cancer Cell 2011, 19, 484–497. [Google Scholar] [CrossRef] [PubMed]
- Giambra, V.; Jenkins, C.E.; Lam, S.H.; Hoofd, C.; Belmonte, M.; Wang, X.; Gusscott, S.; Gracias, D.; Weng, A.P. Leukemia stem cells in T-all require active Hif1alpha and Wnt signaling. Blood 2015, 125, 3917–3927. [Google Scholar] [CrossRef] [PubMed]
- Fleming, H.E.; Janzen, V.; Lo Celso, C.; Guo, J.; Leahy, K.M.; Kronenberg, H.M.; Scadden, D.T. Wnt signaling in the niche enforces hematopoietic stem cell quiescence and is necessary to preserve self-renewal in vivo. Cell Stem Cell 2008, 2, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Scheller, M.; Schonheit, J.; Zimmermann, K.; Leser, U.; Rosenbauer, F.; Leutz, A. Cross talk between Wnt/beta-catenin and IRF8 in leukemia progression and drug resistance. J. Exp. Med. 2013, 210, 2239–2256. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, B.; Meloty-Kapella, L.; Weinmaster, G. Canonical and non-canonical notch ligands. Curr. Top. Dev. Biol. 2010, 92, 73–129. [Google Scholar] [PubMed]
- Wodarz, D.; Garg, N.; Komarova, N.L.; Benjamini, O.; Keating, M.J.; Wierda, W.G.; Kantarjian, H.; James, D.; O’Brien, S.; Burger, J.A. Kinetics of CLL cells in tissues and blood during therapy with the BTK inhibitor ibrutinib. Blood 2014, 123, 4132–4135. [Google Scholar] [CrossRef] [PubMed]
- Kaneta, Y.; Kagami, Y.; Tsunoda, T.; Ohno, R.; Nakamura, Y.; Katagiri, T. Genome-wide analysis of gene-expression profiles in chronic myeloid leukemia cells using a cDNA microarray. Int. J. Oncol. 2003, 23, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Schurch, C.; Riether, C.; Matter, M.S.; Tzankov, A.; Ochsenbein, A.F. CD27 signaling on chronic myelogenous leukemia stem cells activates Wnt target genes and promotes disease progression. J. Clin. Investig. 2012, 122, 624–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazieres, J.; You, L.; He, B.; Xu, Z.; Lee, A.Y.; Mikami, I.; McCormick, F.; Jablons, D.M. Inhibition of Wnt16 in human acute lymphoblastoid leukemia cells containing the t(1;19) translocation induces apoptosis. Oncogene 2005, 24, 5396–5400. [Google Scholar] [CrossRef] [PubMed]
- Rosenwald, A.; Alizadeh, A.A.; Widhopf, G.; Simon, R.; Davis, R.E.; Yu, X.; Yang, L.; Pickeral, O.K.; Rassenti, L.Z.; Powell, J.; et al. Relation of gene expression phenotype to immunoglobulin mutation genotype in B cell chronic lymphocytic leukemia. J. Exp. Med. 2001, 194, 1639–1647. [Google Scholar] [CrossRef] [PubMed]
- Melo, N.; Hobday, C.; Dowsett, M.; Catovsky, D.; Matutes, E.; Morilla, R.; Polliack, A. Oestrogen receptor (ER) analysis in B-cell chronic lymphocytic leukemia: Correlation of biochemical and immunocytochemical methods. Leuk. Res. 1990, 14, 949–952. [Google Scholar] [CrossRef]
- Roman-Gomez, J.; Cordeu, L.; Agirre, X.; Jimenez-Velasco, A.; San Jose-Eneriz, E.; Garate, L.; Calasanz, M.J.; Heiniger, A.; Torres, A.; Prosper, F. Epigenetic regulation of Wnt-signaling pathway in acute lymphoblastic leukemia. Blood 2007, 109, 3462–3469. [Google Scholar] [CrossRef] [PubMed]
- Roman-Gomez, J.; Jimenez-Velasco, A.; Cordeu, L.; Vilas-Zornoza, A.; San Jose-Eneriz, E.; Garate, L.; Castillejo, J.A.; Martin, V.; Prosper, F.; Heiniger, A.; et al. Wnt5a, a putative tumour suppressor of lymphoid malignancies, is inactivated by aberrant methylation in acute lymphoblastic leukaemia. Eur. J. Cancer 2007, 43, 2736–2746. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.; Grandage, V.L.; Linch, D.C.; Khwaja, A. Constitutive activation of the Wnt/beta-catenin signalling pathway in acute myeloid leukaemia. Oncogene 2005, 24, 2410–2420. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; You, L.S.; Ni, W.M.; Ma, Q.L.; Tong, Y.; Mao, L.P.; Qian, J.J.; Jin, J. Beta-catenin and AKT are promising targets for combination therapy in acute myeloid leukemia. Leuk. Res. 2013, 37, 1329–1340. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Li, M.; McDonald, T.; Holyoake, T.L.; Moon, R.T.; Campana, D.; Shultz, L.; Bhatia, R. Microenvironmental protection of CML stem and progenitor cells from tyrosine kinase inhibitors through N-cadherin and Wnt-beta-catenin signaling. Blood 2013, 121, 1824–1838. [Google Scholar] [CrossRef] [PubMed]
- Dorfman, D.M.; Greisman, H.A.; Shahsafaei, A. Loss of expression of the Wnt/beta-catenin-signaling pathway transcription factors lymphoid enhancer factor-1 (LEF-1) and t cell factor-1 (TCF-1) in a subset of peripheral T cell lymphomas. Am. J. Pathol. 2003, 162, 1539–1544. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Possible Mechanism | Hematological Disorder | References |
|---|---|---|
| Wnt protein secretion by tumor cells or microenvironment | AML: tumor cells produce Wnt2B, Wnt6, Wnt 10A, Wnt10B | [35,36] |
| CML: human BM MSC cells secrete Wn1, Wnt2B, Wnt3, Wtn5a, Wtn5B, Wnt6, Wnt8b, Wnt16 | [91,92] | |
| B-ALL: tumor cells produce Wnt16b | [61,93] | |
| CLL: tumor cells express Wnt3a and Wnt5B, Wnt6, Wnt10A, Wnt14 and Wtn16 | [69,94] | |
| Responsiveness of tumor cells to Wnt-signaling | CML: TKI-resistant cells have a high Fzd8 expression | [52] |
| CLL: tumor cells express Fzd3 and LRP5/LRP6 and Ror1 | [63,64,95] | |
| Epigenetic changes (aberrant methylation of Wnt antagonists or Wnt5a) | AML: methylation of sFRP-1, 3, 4, and DKK1, or Wnt5a | [20,22,23,24,32] |
| B-ALL: methylation of DKK3 | [96] | |
| T-ALL: inappropriate methylation of Wnt5a | [97] | |
| CLL: methylation of Wif1, DKK3, sFRP-1, 2, 4, and 5 | [65] | |
| Activating mutations in β-catenin or inactivating mutations in APC or Axin | AML and ALL: inactivating mutations in Axin1 and APC | [16,17,19,98,99] |
| T-ALL: activating mutations in β-catenin, loss of TCF7 tumor suppressor activity | [81,82] | |
| Balance of Tcf/Lef factors in tumor cells | AML: high Lef levels | [41,42] |
| CML: Tcf/Lef factors positiviely regulate ABCB1 | [100] | |
| B-ALL: disbalance of Tcf and Lef levels in tumor cells | [60] | |
| T-ALL: high Lef levels; Tcf1 is tumor suppressor gene | [44,101] | |
| CLL: high Lef levels | [64] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Staal, F.J.T.; Famili, F.; Garcia Perez, L.; Pike-Overzet, K. Aberrant Wnt Signaling in Leukemia. Cancers 2016, 8, 78. https://doi.org/10.3390/cancers8090078
Staal FJT, Famili F, Garcia Perez L, Pike-Overzet K. Aberrant Wnt Signaling in Leukemia. Cancers. 2016; 8(9):78. https://doi.org/10.3390/cancers8090078
Chicago/Turabian StyleStaal, Frank J. T., Farbod Famili, Laura Garcia Perez, and Karin Pike-Overzet. 2016. "Aberrant Wnt Signaling in Leukemia" Cancers 8, no. 9: 78. https://doi.org/10.3390/cancers8090078
APA StyleStaal, F. J. T., Famili, F., Garcia Perez, L., & Pike-Overzet, K. (2016). Aberrant Wnt Signaling in Leukemia. Cancers, 8(9), 78. https://doi.org/10.3390/cancers8090078