
Current Technologies and Recent Developments for Screening of HPV-Associated Cervical and Oropharyngeal Cancers

Abstract
: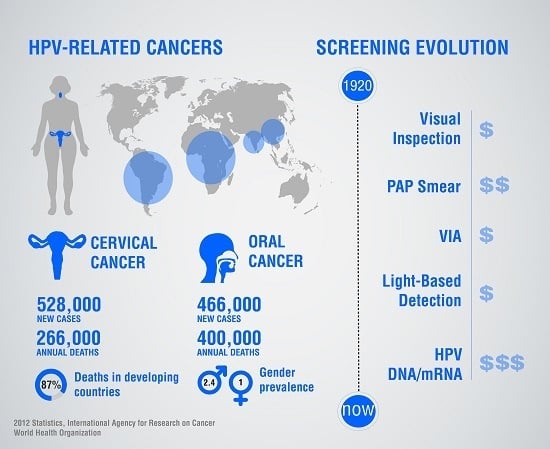
1. Global Burden of Cervical and Oropharyngeal Cancers
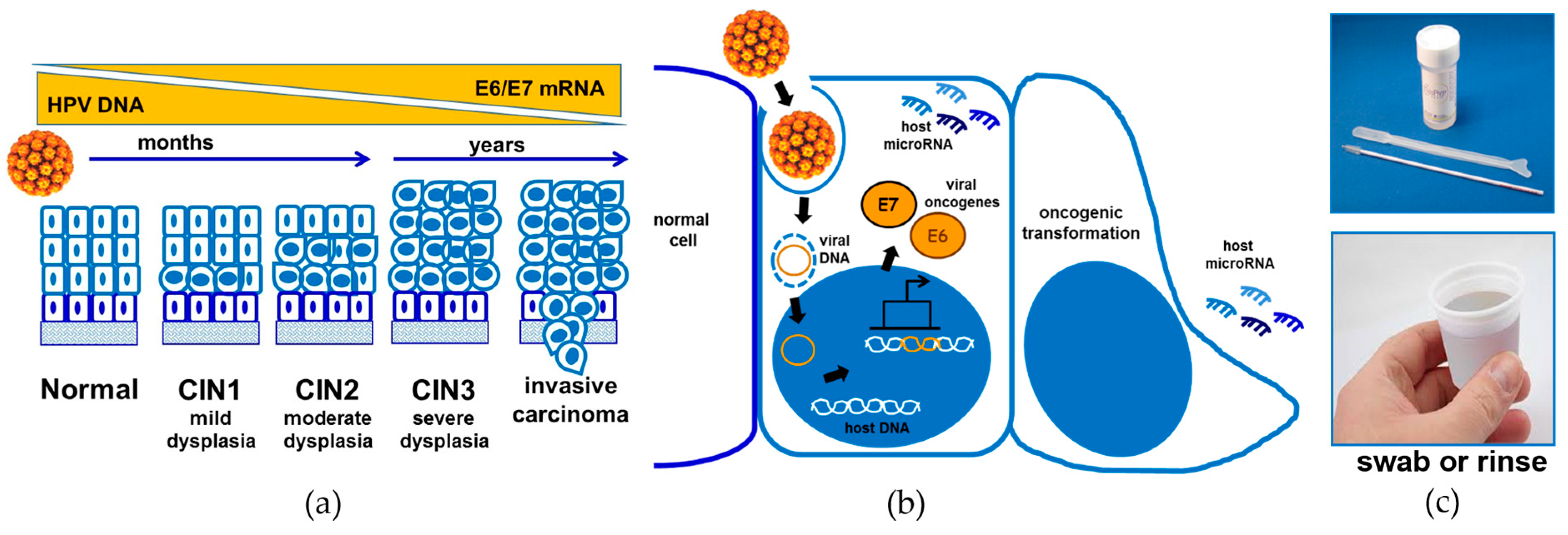
2. Human Papillomaviruses
2.1. Genes Encoded by HPVs
2.2. Role of MicroRNAs in HPV-Associated Cancer
2.3. The Need for Cancer Screening
- Low cost: A sensor that is developed using simple chip fabrication steps can be mass-produced, thus making it amenable for low-resource settings.
- Portability: The platform should be portable and easy to operate with the possibility for battery operation and minimal need of infrastructure.
- Simplicity of assay protocol: An easy-to-use device that is portable will eliminate the need for a sophisticated clinical laboratory facility or highly skilled, trained personnel. The final integrated platform needs to be an automated sample-to-answer device.
- Signal stability: A sensing signal that is stable, reproducible and insensitive to surrounding noise.
- Sensitivity and selectivity: A sensor should be sensitive enough for the target of interest and have the requisite specificity comparable to the gold standard.
- Rapid and robust: The device should generate results rapidly, allowing medical personnel to diagnose disease and start immediate treatment (screen-and-treat). Additionally, the platform should be robust and not require any refrigeration or other external equipment to perform the test.
- Multiplexing: The platform should be flexible enough to detect more than one target biomarker.
3. Established Tools for Screening Cervical and Oropharyngeal Cancers
3.1. Conventional Cytology (Pap Test)
3.2. Liquid-Based Monolayer Cytology
3.3. Visual Inspection with Acetic Acid (VIA)
3.4. Serological Testing of HPV
3.5. Oral Cancer Diagnostic Technologies
4. Nucleic Acid-Based Screening Tools
4.1. Microarray-Based Sensing
4.2. Polymerase Chain Reaction-Based Sensing of HPV DNA
4.3. Next-Generation Sequencing
4.4. Challenges with Current Nucleic Acid Screening Tools
4.5. Advancements in RNA and Electrochemical Sensing
5. Recent Developments in Integrated Lab-on-a-Chip Technologies for Nucleic Acid Screening
6. Future Directions
Acknowledgments
Conflicts of Interest
References
- Bernard, E.; Pons-Salort, M.; Favre, M.; Heard, I.; Delarocque-Astagneau, E.; Guillemot, D.; Thiébaut, A.C. Comparing human papillomavirus prevalences in women with normal cytology or invasive cervical cancer to rank genotypes according to their oncogenic potential: A meta-analysis of observational studies. BMC Infect. Dis. 2013, 13, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- American Cancer Society Cervical Cancer Statistics. Available online: http://www.cancer.org/cancer/cervicalcancer/index (accessed on 27 May 2016).
- Forman, D.; de Martel, C.; Lacey, C.J.; Soerjomataram, I.; Lortet-Tieulent, J.; Bruni, L.; Vignat, J.; Ferlay, J.; Bray, F.; Plummer, M. Global burden of human papillomavirus and related diseases. Vaccine 2012, 30, F12–F23. [Google Scholar] [CrossRef] [PubMed]
- National Institute of Health Cervical Cancer Factsheet. Available online: https://report.nih.gov/nihfactsheets/ViewFactSheet.aspx?csid=76&key=C (accessed on 27 May 2016).
- Ciapponi, A.; Bardach, A.; Glujovsky, D.; Gibbons, L.; Picconi, M.A. Type-specific HPV prevalence in cervical cancer and high-grade lesions in Latin America and the Caribbean: Systematic review and meta-analysis. PLoS ONE 2011, 6, e25493. [Google Scholar] [CrossRef] [PubMed]
- Zahedi, L.; Sizemore, E.; Malcolm, S.; Grossniklaus, E.; Nwosu, O. Knowledge, attitudes and practices regarding cervical cancer and screening among Haitian health care workers. Int. J. Environ. Res. Public Health 2014, 11, 11541–11552. [Google Scholar] [CrossRef] [PubMed]
- Sankaranarayanan, R.; Rajkumar, R.; Esmy, P.; Fayette, J.; Shanthakumary, S.; Frappart, L.; Thara, S.; Cherian, J. Effectiveness, safety and acceptability of ‘see and treat’ with cryotherapy by nurses in a cervical screening study in India. Br. J. Cancer 2007, 96, 738–743. [Google Scholar] [CrossRef] [PubMed]
- Luciani, S.; Gonzales, M.; Munoz, S.; Jeronimo, J.; Robles, S. Effectiveness of cryotherapy treatment for cervical intraepithelial neoplasia. Int. J. Gynecol. Obstet. 2008, 101, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, N.; Bosch, F.X.; de Sanjosé, S.; Herrero, R.; Castellsagué, X.; Shah, K.V.; Snijders, P.J.; Meijer, C.J. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N. Engl. J. Med. 2003, 348, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Kobetz, E.; Kish, J.K.; Campos, N.G.; Koru-Sengul, T.; Bishop, I.; Lipshultz, H.; Barton, B.; Barbee, L. Burden of human papillomavirus among Haitian immigrants in Miami, Florida: Community-based participatory research in action. J. Oncol. 2012, 2012, 728397. [Google Scholar] [CrossRef] [PubMed]
- Cogliano, V.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; WHO International Agency for Research on Cancer. Carcinogenicity of human papillomaviruses. Lancet Oncol. 2005, 6, 204. [Google Scholar] [CrossRef]
- Clifford, G.; Gallus, S.; Herrero, R.; Munoz, N.; Snijders, P.; Vaccarella, S.; Anh, P.; Ferreccio, C.; Hieu, N.; Matos, E. Worldwide distribution of human papillomavirus types in cytologically normal women in the International Agency for Research on Cancer HPV prevalence surveys: A pooled analysis. Lancet 2005, 366, 991–998. [Google Scholar] [CrossRef]
- Wang, S.S.; Wheeler, C.M.; Hildesheim, A.; Schiffman, M.; Herrero, R.; Bratti, M.C.; Sherman, M.E.; Alfaro, M.; Hutchinson, M.L.; Morales, J.; et al. Human leukocyte antigen class I and II alleles and risk of cervical neoplasia: Results from a population-based study in Costa Rica. J. Infect. Dis. 2001, 184, 1310–1314. [Google Scholar] [CrossRef] [PubMed]
- Strickler, H.D.; Palefsky, J.M.; Shah, K.V.; Anastos, K.; Klein, R.S.; Minkoff, H.; Duerr, A.; Massad, L.S.; Celentano, D.D.; Hall, C.; et al. Human papillomavirus type 16 and immune status in human immunodeficiency virus-seropositive women. J. Natl. Cancer Inst. 2003, 95, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Kahn, J.A.; Brown, D.R.; Ding, L.; Widdice, L.E.; Shew, M.L.; Glynn, S.; Bernstein, D.I. Vaccine-type human papillomavirus and evidence of herd protection after vaccine introduction. Pediatrics 2012, 130, e249–e256. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, A.K.; Engels, E.A.; Pfeiffer, R.M.; Hernandez, B.Y.; Xiao, W.; Kim, E.; Jiang, B.; Goodman, M.T.; Sibug-Saber, M.; Cozen, W.; et al. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J. Clin. Oncol. 2011, 29, 4294–4301. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.L.; Puricelli, M.D.; Stack, M.S. Virology and molecular pathogenesis of HPV (human papillomavirus)-associated oropharyngeal squamous cell carcinoma. Biochem. J. 2012, 443, 339–353. [Google Scholar] [CrossRef] [PubMed]
- Ang, K.K.; Harris, J.; Wheeler, R.; Weber, R.; Rosenthal, D.I.; Nguyen-Tan, P.F.; Westra, W.H.; Chung, C.H.; Jordan, R.C.; Lu, C.; et al. Human Papillomavirus and Survival of Patients with Oropharyngeal Cancer. N. Engl. J. Med. 2010, 363, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Gillison, M.L.; Broutian, T.; Pickard, R.K.; Tong, Z.; Xiao, W.; Kahle, L.; Graubard, B.I.; Chaturvedi, A.K. Prevalence of oral HPV infection in the United States, 2009–2010. JAMA 2012, 307, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Duvvuri, U.; Myers, J.N. In Brief. Curr. Probl. Surg. 2009, 46, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Pannone, G.; Santoro, A.; Papagerakis, S.; Lo Muzio, L.; De Rosa, G.; Bufo, P. The role of human papillomavirus in the pathogenesis of head & neck squamous cell carcinoma: An overview. Infect. Agent Cancer 2011, 6, 1–11. [Google Scholar]
- Chaturvedi, A.K.; Engels, E.A.; Anderson, W.F.; Gillison, M.L. Incidence trends for human papillomavirus-related and -unrelated oral squamous cell carcinomas in the United States. J. Clin. Oncol. 2008, 26, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Strati, K.; Lambert, P.F. Human papillomavirus association with head and neck cancers: Understanding virus biology and using it in the development of cancer diagnostics. Expert Opin. Med. Diagn. 2008, 2, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Petersen, P.E. Oral cancer prevention and control–The approach of the World Health Organization. Oral Oncol. 2009, 45, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Syrjanen, S. The role of human papillomavirus infection in head and neck cancers. Ann. Oncol. 2010, 21, 243–245. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.L.; Davis, J.W.; Taylor, K.H.; Johnson, J.; Shi, Z.; Williams, R.; Atasoy, U.; Lewis, J.S.; Stack, M.S. Identification of a human papillomavirus—Associated oncogenic miRNA panel in human oropharyngeal squamous cell carcinoma validated by bioinformatics analysis of the cancer genome atlas. Am. J. Pathol. 2015, 185, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.L.; Stack, M.S. MicroRNA Profiles of HPV-Associated Oropharyngeal Squamous Cell Carcinoma (OPSCC). In Human Papillomavirus (HPV)-Associated Oropharyngeal Cancer; Springer: Cham, Switzerland, 2015; pp. 133–152. [Google Scholar]
- Culleton, S.P.; Androphy, E.J.; Kanginakudru, S. Papillomavirus Replication. In Human Papillomavirus (HPV)-Associated Oropharyngeal Cancer; Springer: Cham, Switzerland, 2015; pp. 103–132. [Google Scholar]
- Smith, E.A.; Matrka, M.C.; Wells, S.I. HPV Virology: Cellular Targets of HPV Oncogenes and Transformation. In Human Papillomavirus (HPV)-Associated Oropharyngeal Cancer; Springer: Cham, Switzerland, 2015; pp. 69–101. [Google Scholar]
- Wise-Draper, T.M.; Wells, S.I. Papillomavirus E6 and E7 proteins and their cellular targets. Front. Biosci. 2008, 13, 1003–1017. [Google Scholar] [CrossRef] [PubMed]
- Kriek, J.; Jaumdally, S.Z.; Masson, L.; Little, F.; Mbulawa, Z.; Gumbi, P.P.; Barnabas, S.L.; Moodley, J.; Denny, L.; Coetzee, D. Female genital tract inflammation, HIV co-infection and persistent mucosal Human Papillomavirus (HPV) infections. Virology 2016, 493, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Pan, X.; Cobb, G.P.; Anderson, T.A. microRNAs as oncogenes and tumor suppressors. Dev. Biol. 2007, 302, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.M.; Raghav, D.; Singh, H.; Raghava, G.P. CCDB: A curated database of genes involved in cervix cancer. Nucleic Acids Res. 2011, 39, D975–D979. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Dua, P.; Agarwal, S.M. A Comprehensive Review of Dysregulated miRNAs Involved in Cervical Cancer. Curr. Genom. 2014, 15, 310–323. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef] [PubMed]
- Botezatu, A.; Goia-Rusanu, C.D.; Iancu, I.V.; Huica, I.; Plesa, A.; Socolov, D.; Ungureanu, C.; Anton, G. Quantitative analysis of the relationship between microRNA‑124a,-34b and-203 gene methylation and cervical oncogenesis. Mol. Med. Rep. 2011, 4, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lu, M.; Qiu, C.; Cui, Q. TransmiR: A transcription factor-microRNA regulation database. Nucleic Acids Res. 2010, 38, D119–D122. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Wang, F.; Wei, Q.; Zhao, Y.; Liu, M.; Li, X.; Tang, H. miR-20a promotes migration and invasion by regulating TNKS2 in human cervical cancer cells. FEBS Lett. 2012, 586, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Köberle, V.; Pleli, T.; Schmithals, C.; Alonso, E.A.; Haupenthal, J.; Bönig, H.; Peveling-Oberhag, J.; Biondi, R.M.; Zeuzem, S.; Kronenberger, B. Differential stability of cell-free circulating microRNAs: Implications for their utilization as biomarkers. PLoS ONE 2013, 8, e75184. [Google Scholar] [CrossRef] [PubMed]
- Jarry, J.; Schadendorf, D.; Greenwood, C.; Spatz, A.; van Kempen, L. The validity of circulating microRNAs in oncology: Five years of challenges and contradictions. Mol. Oncol. 2014, 8, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Turchinovich, A.; Weiz, L.; Langheinz, A.; Burwinkel, B. Characterization of extracellular circulating microRNA. Nucleic Acids Res. 2011, 39, 7223–7233. [Google Scholar] [CrossRef] [PubMed]
- Satterwhite, C.L.; Torrone, E.; Meites, E.; Dunne, E.F.; Mahajan, R.; Ocfemia, M.C.; Su, J.; Xu, F.; Weinstock, H. Sexually transmitted infections among US women and men: Prevalence and incidence estimates, 2008. Sex. Transm. Dis. 2013, 40, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Chesson, H.W.; Dunne, E.F.; Hariri, S.; Markowitz, L.E. The estimated lifetime probability of acquiring human papillomavirus in the United States. Sex. Transm. Dis. 2014, 41, 660–664. [Google Scholar] [CrossRef] [PubMed]
- Hariri, S.; Unger, E.R.; Sternberg, M.; Dunne, E.F.; Swan, D.; Patel, S.; Markowitz, L.E. Prevalence of genital human papillomavirus among females in the United States, the National Health And Nutrition Examination Survey, 2003–2006. J. Infect. Dis. 2011, 204, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Karimi-Zarchi, M.; Peighmbari, F.; Karimi, N.; Rohi, M.; Chiti, Z. A comparison of 3 ways of conventional pap smear, liquid-based cytology and colposcopy vs cervical biopsy for early diagnosis of premalignant lesions or cervical cancer in women with abnormal conventional pap test. Int. J. Biomed. Sci. 2013, 9, 205–210. [Google Scholar] [PubMed]
- Chung, M.H.; McKenzie, K.P.; De Vuyst, H.; Richardson, B.A.; Rana, F.; Pamnani, R.; Njoroge, J.W.; Nyongesa-Malava, E.; Sakr, S.R.; John-Stewart, G.C.; et al. Comparing Papanicolau smear, visual inspection with acetic acid and human papillomavirus cervical cancer screening methods among HIV-positive women by immune status and antiretroviral therapy. AIDS 2013, 27, 2909–2919. [Google Scholar] [CrossRef] [PubMed]
- Rocha-Zavaleta, L.; Ambrosio, J.P.; de Lourdes Mora-Garcia, M.; Cruz-Talonia, F.; Hernandez-Montes, J.; Weiss-Steider, B.; Ortiz-Navarrete, V.; Monroy-Garcia, A. Detection of antibodies against a human papillomavirus (HPV) type 16 peptide that differentiate high-risk from low-risk HPV-associated low-grade squamous intraepithelial lesions. J. Gen. Virol. 2004, 85, 2643–2650. [Google Scholar] [CrossRef] [PubMed]
- Mashberg, A. Reevaluation of toluidine blue application as a diagnostic adjunct in the detection of asymptomatic oral squamous carcinoma: A continuing prospective study of oral cancer III. Cancer 1980, 46, 758–763. [Google Scholar] [CrossRef]
- Torres, A.C.; Márquez, Á.M.; Farhall, J.F.; Fuente, M.J.C.; Castillo, I.G. Efficacy of Chemiluminescence (ViziLite™) as a Screening Method in the Detection of Clinically Suspicious Oral Cancerous and Precancerous Lesions. J. Anal. Oncol. 2013, 2, 176–181. [Google Scholar] [CrossRef]
- Sasieni, P.; Adams, J. Effect of screening on cervical cancer mortality in England and Wales: Analysis of trends with an age period cohort model. BMJ 1999, 318, 1244–1245. [Google Scholar] [CrossRef] [PubMed]
- Sankaranarayanan, R.; Nene, B.M.; Shastri, S.S.; Jayant, K.; Muwonge, R.; Budukh, A.M.; Hingmire, S.; Malvi, S.G.; Thorat, R.; Kothari, A. HPV screening for cervical cancer in rural India. N. Engl. J. Med. 2009, 360, 1385–1394. [Google Scholar] [CrossRef] [PubMed]
- Nanda, K.; McCrory, D.C.; Myers, E.R.; Bastian, L.A.; Hasselblad, V.; Hickey, J.D.; Matchar, D.B. Accuracy of the Papanicolaou test in screening for and follow-up of cervical cytologic abnormalities: A systematic review. Ann. Intern. Med. 2000, 132, 810–819. [Google Scholar] [CrossRef] [PubMed]
- Cuzick, J.; Clavel, C.; Petry, K.; Meijer, C.J.; Hoyer, H.; Ratnam, S.; Szarewski, A.; Birembaut, P.; Kulasingam, S.; Sasieni, P. Overview of the European and North American studies on HPV testing in primary cervical cancer screening. Int. J. Cancer 2006, 119, 1095–1101. [Google Scholar] [CrossRef] [PubMed]
- Raffle, A.E.; Alden, B.; Quinn, M.; Babb, P.J.; Brett, M.T. Outcomes of screening to prevent cancer: Analysis of cumulative incidence of cervical abnormality and modelling of cases and deaths prevented. BMJ 2003, 326, 901. [Google Scholar] [CrossRef] [PubMed]
- Bettigole, C. The thousand-dollar Pap smear. N. Engl. J. Med. 2013, 369, 1486–1487. [Google Scholar] [CrossRef] [PubMed]
- Lozano, R. Comparison of computer-assisted and manual screening of cervical cytology. Gynecol. Oncol. 2007, 104, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Abulafia, O.; Pezzullo, J.C.; Sherer, D.M. Performance of ThinPrep liquid-based cervical cytology in comparison with conventionally prepared Papanicolaou smears: A quantitative survey. Gynecol. Oncol. 2003, 90, 137–144. [Google Scholar] [CrossRef]
- Coste, J.; Cochand-Priollet, B.; de Cremoux, P.; Le Gales, C.; Cartier, I.; Molinie, V.; Labbe, S.; Vacher-Lavenu, M.C.; Vielh, P.; French Society of Clinical Cytology Study Group. Cross sectional study of conventional cervical smear, monolayer cytology, and human papillomavirus DNA testing for cervical cancer screening. BMJ 2003, 326, 733. [Google Scholar] [CrossRef] [PubMed]
- Ronco, G.; Cuzick, J.; Pierotti, P.; Cariaggi, M.P.; Dalla Palma, P.; Naldoni, C.; Ghiringhello, B.; Giorgi-Rossi, P.; Minucci, D.; Parisio, F.; et al. Accuracy of liquid based versus conventional cytology: Overall results of new technologies for cervical cancer screening: Randomised controlled trial. BMJ 2007, 335, 28. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Norman, I.; Elfgren, K.; Gaberi, V.; Hagmar, B.; Hjerpe, A.; Andersson, S. A comparison of liquid-based cytology and Pap smear as a screening method for cervical cancer. Oncol. Rep. 2007, 18, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Davey, E.; d'Assuncao, J.; Irwig, L.; Macaskill, P.; Chan, S.F.; Richards, A.; Farnsworth, A. Accuracy of Reading liquid based cytology slides using the ThinPrep Imager compared with conventional cytology: prospective study. BMJ 2007, 335, 31. [Google Scholar] [CrossRef] [PubMed]
- Sherris, J.; Wittet, S.; Kleine, A.; Sellors, J.; Luciani, S.; Sankaranarayanan, R.; Barone, M.A. Evidence-based, alternative cervical cancer screening approaches in low-resource settings. Int. Perspect. Sex. Reprod. Health 2009, 35, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Sankaranarayanan, R.; Gaffikin, L.; Jacob, M.; Sellors, J.; Robles, S. A critical assessment of screening methods for cervical neoplasia. Int. J. Gynecol. Obstet. 2005, 89, S4–S12. [Google Scholar] [CrossRef] [PubMed]
- Gravitt, P.E.; Paul, P.; Katki, H.A.; Vendantham, H.; Ramakrishna, G.; Sudula, M.; Kalpana, B.; Ronnett, B.M.; Vijayaraghavan, K.; Shah, K.V. Effectiveness of VIA, Pap, and HPV DNA testing in a cervical cancer screening program in a peri-urban community in Andhra Pradesh, India. PLoS ONE 2010, 5, e13711. [Google Scholar] [CrossRef] [PubMed]
- Sankaranarayanan, R.; Wesley, R.S. Testing and reporting the results of visual inspection with 5% acetic acid (VIA). In A Practical Manual on Visual Screening for Cervical Neoplasia; IARC Press: Lyon, France, 2003; pp. 15–26. [Google Scholar]
- Mustafa, M.; Jindal, A.; Singh, P. Visual inspection using acetic acid for cervical cancer in low resource settings. Med. J. Armed Forces India 2010, 66, 382–384. [Google Scholar] [CrossRef]
- Rafati, S. Antibody detection against HPV16 E7 & GP96 fragments as biomarkers in cervical cancer patients. Indian J. Med. Res. 2009, 130, 533–541. [Google Scholar]
- Silins, I.; Avall-Lundqvist, E.; Tadesse, A.; Jansen, K.U.; Stendahl, U.; Lenner, P.; Zumbach, K.; Pawlita, M.; Dillner, J.; Frankendal, B. Evaluation of antibodies to human papillomavirus as prognostic markers in cervical cancer patients. Gynecol. Oncol. 2002, 85, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Sankaranarayanan, R.; Ramadas, K.; Thara, S.; Muwonge, R.; Thomas, G.; Anju, G.; Mathew, B. Long term effect of visual screening on oral cancer incidence and mortality in a randomized trial in Kerala, India. Oral Oncol. 2013, 49, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.; Vigneswaran, N.; Gillenwater, A.; Richards-Kortum, R. Advances in fluorescence imaging techniques to detect oral cancer and its precursors. Future Oncol. 2010, 6, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- Kerr, A.R.; Sirois, D.A.; Epstein, J.B. Clinical evaluation of chemiluminescent lighting: An adjunct for oral mucosal examinations. J. Clin. Dent. 2006, 17, 59–63. [Google Scholar] [PubMed]
- Patton, L.L.; Epstein, J.B.; Kerr, A.R. Adjunctive techniques for oral cancer examination and lesion diagnosis: A systematic review of the literature. J. Am. Dent. Assoc. 2008, 139, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Padda, S.S.; Kaur, Y.; Narang, R.; Kaur, B.; Ramakrishna, P. Chemiluminescent Light: Role in Early Detection of Cancer. J. Indian Acad. Oral Med. Radiol. 2013, 25, 89–92. [Google Scholar]
- Awan, K.; Morgan, P.; Warnakulasuriya, S. Utility of chemiluminescence (ViziLite™) in the detection of oral potentially malignant disorders and benign keratoses. J. Oral Pathol. Med. 2011, 40, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Awan, K.; Morgan, P.; Warnakulasuriya, S. Evaluation of an autofluorescence based imaging system (VELscope™) in the detection of oral potentially malignant disorders and benign keratoses. Oral Oncol. 2011, 47, 274–277. [Google Scholar] [CrossRef] [PubMed]
- Carlson, A.L.; Coghlan, L.G.; Gillenwater, A.M.; Richards-Kortum, R.R. Dual-mode reflectance and fluorescence near-video-rate confocal microscope for architectural, morphological and molecular imaging of tissue. J. Microsc. 2007, 228, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Fedele, S. Diagnostic aids in the screening of oral cancer. Head Neck Oncol. 2009, 1, 1. [Google Scholar] [CrossRef] [PubMed]
- Hasan, S.; Elongovan, S. Conventional and advances diagnostic aids in oral cancer screening—The journey so far. Int. J. Pharm. Pharm. Sci. 2014, 7, 29–33. [Google Scholar]
- Seijas-Naya, F.; García-Carnicero, T.; Gándara-Vila, P.; Couso-Folgueiras, E.; Pérez-Sayáns, M.; Gándara-Vila, R.; García-García, A.; Gándara-Rey, J. Applications of OralCDx® methodology in the diagnosis of oral leukoplakia. Med. Oral Patol. Oral Cir. Bucal 2012, 17, e5–e9. [Google Scholar] [CrossRef] [PubMed]
- Siegel, M.A.; Kahn, M.A.; Palazzolo, M.J. Oral cancer: A prosthodontic diagnosis. J. Prosthodont. 2009, 18, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Petti, S. Oral cancer screening usefulness: Between true and perceived effectiveness. Oral Dis. 2016, 22, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Sankaranarayanan, R.; Swaminathan, R.; Brenner, H.; Chen, K.; Chia, K.S.; Chen, J.G.; Law, S.C.; Ahn, Y.; Xiang, Y.B.; Yeole, B.B. Cancer survival in Africa, Asia, and Central America: A population-based study. Lancet Oncol. 2010, 11, 165–173. [Google Scholar] [CrossRef]
- Koshiol, J.; Wang, E.; Zhao, Y.; Marincola, F.; Landi, M.T. Strengths and limitations of laboratory procedures for microRNA detection. Cancer Epidemiol. Biomark. Prev. 2010, 19, 907–911. [Google Scholar] [CrossRef] [PubMed]
- Hammond, S.M. MicroRNAs as oncogenes. Curr. Opin. Genet. Dev. 2006, 16, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Love, C.; Dave, S. MicroRNA expression profiling using microarrays. Hematol. Malig. 2013, 999, 285–296. [Google Scholar]
- Kim, V.N.; Nam, J. Genomics of microRNA. TRENDS Genet. 2006, 22, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.M.; Parker, J.; Perou, C.M.; Hammond, S.M. A custom microarray platform for analysis of microRNA gene expression. Nat. Methods 2004, 1, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Schena, M.; Shalon, D.; Davis, R.W.; Brown, P.O. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 1995, 270, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.O.; Botstein, D. Exploring the new world of the genome with DNA microarrays. Nat. Genet. 1999, 21, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Han, T.; Melvin, C.D.; Shi, L.; Branham, W.S.; Moland, C.L.; Pine, P.; Thompson, K.L.; Fuscoe, J.C. Improvement in the reproducibility and accuracy of DNA microarray quantification by optimizing hybridization conditions. BMC Bioinform. 2006, 7, 1. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, L.; Soe, M.J.; Snakenborg, D.; Moller, L.B.; Dufva, M. Multi-stringency wash of partially hybridized 60-mer probes reveals that the stringency along the probe decreases with distance from the microarray surface. Nucleic Acids Res. 2008, 36, e132. [Google Scholar] [CrossRef] [PubMed]
- Fesenko, E.E.; Heydarov, R.N.; Stepanova, E.V.; Abramov, M.E.; Chudinov, A.V.; Zasedatelev, A.S.; Mikhailovich, V.M. Microarray with LNA-probes for genotyping of polymorphic variants of Gilbert’s syndrome gene UGT1A1(TA)n. Clin. Chem. Lab. Med. 2013, 51, 1177–1184. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Yuan, Y.; Mu, R.; Shang, H.; Guan, Y. LNA-modified isothermal oligonucleotide microarray for differentiating bacilli of similar origin. J. Biosci. 2014, 39, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Tolstrup, N.; Nielsen, P.S.; Kolberg, J.G.; Frankel, A.M.; Vissing, H.; Kauppinen, S. OligoDesign: Optimal design of LNA (locked nucleic acid) oligonucleotide capture probes for gene expression profiling. Nucleic Acids Res. 2003, 31, 3758–3762. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.W.; Cheng, J.Y.; Huang, C.T.; Yen, M.H.; Young, T.H. Using a microfluidic device for 1 µl DNA microarray hybridization in 500 s. Nucleic Acids Res. 2005, 33, e78. [Google Scholar] [CrossRef] [PubMed]
- Draghici, S.; Khatri, P.; Eklund, A.C.; Szallasi, Z. Reliability and reproducibility issues in DNA microarray measurements. TRENDS Genet. 2006, 22, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Dalma-Weiszhausz, D.D.; Warrington, J.; Tanimoto, E.Y.; Miyada, C.G. The Affymetrix GeneChip® Platform: An Overview. Methods Enzymol. 2006, 410, 3–28. [Google Scholar] [PubMed]
- Wolber, P.K.; Collins, P.J.; Lucas, A.B.; de Witte, A.; Shannon, K.W. The Agilent In Situ-Synthesized Microarray Platform. Methods Enzymol. 2006, 410, 28–57. [Google Scholar] [PubMed]
- Tanić, M.; Yanowski, K.; Andrés, E.; Gómez-López, G.; Socorro, M.R.; Pisano, D.G.; Martinez-Delgado, B.; Benítez, J. miRNA expression profiling of formalin-fixed paraffin-embedded (FFPE) hereditary breast tumors. Genom. Data 2015, 3, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Ach, R.A.; Wang, H.; Curry, B. Measuring microRNAs: Comparisons of microarray and quantitative PCR measurements, and of different total RNA prep methods. BMC Biotechnol. 2008, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Chiang, V.L. Facile means for quantifying microRNA expression by real-time PCR. BioTechniques 2005, 39, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Lundegard, M.; Nylander, K.; Danielsson, K. Difficulties detecting miRNA-203 in human whole saliva by the use of PCR. Med. Oral Patol. Oral Cir. Bucal 2015, 20, e130–134. [Google Scholar] [CrossRef] [PubMed]
- Git, A.; Dvinge, H.; Salmon-Divon, M.; Osborne, M.; Kutter, C.; Hadfield, J.; Bertone, P.; Caldas, C. Systematic comparison of microarray profiling, real-time PCR, and next-generation sequencing technologies for measuring differential microRNA expression. RNA 2010, 16, 991–1006. [Google Scholar] [CrossRef] [PubMed]
- McGlennen, R. OraRisk® HPV 16/18/HR. Available online: http://www.oraldna.com/Resources/HpvNegSampleReport.pdf (accessed on 25 May 2016).
- Naucler, P.; Ryd, W.; Törnberg, S.; Strand, A.; Wadell, G.; Elfgren, K.; Rådberg, T.; Strander, B.; Johansson, B.; Forslund, O. Human papillomavirus and Papanicolaou tests to screen for cervical cancer. N. Engl. J. Med. 2007, 357, 1589–1597. [Google Scholar] [CrossRef] [PubMed]
- Castle, P.E.; Dockter, J.; Giachetti, C.; Garcia, F.A.; McCormick, M.K.; Mitchell, A.L.; Holladay, E.B.; Kolk, D.P. A cross-sectional study of a prototype carcinogenic human papillomavirus E6/E7 messenger RNA assay for detection of cervical precancer and cancer. Clin. Cancer Res. 2007, 13, 2599–2605. [Google Scholar] [CrossRef] [PubMed]
- Lie, A.; Risberg, B.; Borge, B.; Sandstad, B.; Delabie, J.; Rimala, R.; Onsrud, M.; Thoresen, S. DNA-versus RNA-based methods for human papillomavirus detection in cervical neoplasia. Gynecol. Oncol. 2005, 97, 908–915. [Google Scholar] [CrossRef] [PubMed]
- Eder, P.S.; Lou, J.; Huff, J.; Macioszek, J. The next-generation Hybrid Capture® High-Risk HPV DNA assay on a fully automated platform. J. Clin. Virol. 2009, 45, S85–S92. [Google Scholar] [CrossRef]
- Bartholomew, D.A.; Luff, R.D.; Quigley, N.B.; Curtis, M.; Olson, M.C. Analytical performance of Cervista® HPV 16/18 genotyping test for cervical cytology samples. J. Clin. Virol. 2011, 51, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Stoler, M.H.; Wright, T.C., Jr.; Sharma, A.; Apple, R.; Gutekunst, K.; Wright, T.L.; ATHENA (Addressing THE Need for Advanced HPV Diagnostics) HPV Study Group. High-risk human papillomavirus testing in women with ASC-US cytology: Results from the ATHENA HPV study. Am. J. Clin. Pathol. 2011, 135, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Dockter, J.; Schroder, A.; Eaton, B.; Wang, A.; Sikhamsay, N.; Morales, L.; Giachetti, C. Analytical characterization of the APTIMA® HPV Assay. J. Clin. Virol. 2009, 45, S39–S47. [Google Scholar] [CrossRef]
- Pista, A.; Verdasca, N.; Oliveira, A. Clinical performance of the CLART human papillomavirus 2 assay compared with the hybrid capture 2 test. J. Med. Virol. 2011, 83, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Schopp, B.; Holz, B.; Zago, M.; Stubenrauch, F.; Petry, K.; Kjaer, S.K.; Iftner, T. Evaluation of the performance of the novel PapilloCheck® HPV genotyping test by comparison with two other genotyping systems and the HC2 test. J. Med. Virol. 2010, 82, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Otero-Motta, A.P.; Ordóñez, J.L.; Gonzalez-Celador, R.; Rivas, B.; Garcia Macias, M.d.C.; Bullon, A.; Abad, M.D.M. Prevalence of human papillomavirus genotypes in cytologic abnormalities from unvaccinated women living in north-western Spain. APMIS 2011, 119, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Abreu, A.L.; Souza, R.P.; Gimenes, F.; Consolaro, M.E. A review of methods for detect human Papillomavirus infection. Virol. J. 2012, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- Hologic Panther System. Available online: http://www.hologic.com/products/clinical-diagnostics-and-blood-screening/instrument-systems/panther-system (accessed on 25 May 2016).
- Roche cobas 4800 System. Available online: http://molecular.roche.com/instruments/Pages/cobas4800System.aspx (accessed on 25 May 2016).
- Denny, L.; Kuhn, L.; de Souza, M.; Pollack, A.E.; Dupree, W.; Wright, T.C. Screen-and-treat approaches for cervical cancer prevention in low-resource settings: A randomized controlled trial. JAMA 2005, 294, 2173–2181. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Yang, H. Approved assays for detecting HPV DNA—Design, indications, and validation. CAP Today 2012, 26, 40–44. [Google Scholar]
- Levi, A.W.; Bernstein, J.I.; Hui, P.; Duch, K.; Schofield, K.; Chhieng, D.C. A Comparison of the Roche Cobas HPV Test With the Hybrid Capture 2 Test for the Detection of High-Risk Human Papillomavirus Genotypes. Arch. Pathol. Lab. Med. 2016, 140, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Einstein, M.H.; Garcia, F.A.; Mitchell, A.L.; Day, S.P. Age-stratified performance of the Cervista HPV 16/18 genotyping test in women with ASC-US cytology. Cancer Epidemiol. Biomark. Prev. 2011, 20, 1185–1189. [Google Scholar] [CrossRef] [PubMed]
- Boers, A.; Wang, R.; Slagter-Menkema, L.; van Hemel, B.M.; Ghyssaert, H.; van der Zee Ate, G.J.; Wisman, G.B.A.; Schuuring, E. Clinical validation of the Cervista HPV HR test according to the international guidelines for human papillomavirus test requirements for cervical cancer screening. J. Clin. Microbiol. 2014, 52, 4391–4393. [Google Scholar] [CrossRef] [PubMed]
- Hologic Cervista HPV 16/18 Assay. Available online: http://www.hologic.com/products/clinical-diagnostics-blood-screening/assays-and-tests/cervista-hpv-1618-assay (accessed on 25 May 2016).
- Digene HC2 HPV Test. Available online: https://www.qiagen.com/us/shop/detection-solutions/hpv-testing/digene-hc2-hpv-dna-test (accessed on 25 May 2016).
- ThinPrep Cervista HPV HR. Available online: http://www.thinprep.com/hcp/cervista_hpv.html (accessed on 25 May 2016).
- Roche cobas HPV Test. Available online: http://molecular.roche.com/assays/Pages/cobasHPVTest.aspx (accessed on 25 May 2016).
- Hologic Aptima HPV assay. Available online: http://www.hologic.com/products/clinical-diagnostics-blood-screening/assays-and-tests/aptima-hpv-assay (accessed on 25 May 2016).
- Mardis, E.R. The impact of next-generation sequencing technology on genetics. Trends Genet. 2008, 24, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Tam, S.; de Borja, R.; Tsao, M.; McPherson, J.D. Robust global microRNA expression profiling using next-generation sequencing technologies. Lab. Investig. 2014, 94, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Wang, W.; Wilfred, B.R.; Tang, G. Technical variables in high-throughput miRNA expression profiling: Much work remains to be done. Biochim. Biophys. Acta 2008, 1779, 758–765. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Gelfond, J.A.; McManus, L.M.; Shireman, P.K. Reproducibility of quantitative RT-PCR array in miRNA expression profiling and comparison with microarray analysis. BMC Genom. 2009, 10, 407. [Google Scholar] [CrossRef] [PubMed]
- Mestdagh, P.; Feys, T.; Bernard, N.; Guenther, S.; Chen, C.; Speleman, F.; Vandesompele, J. High-throughput stem-loop RT-qPCR miRNA expression profiling using minute amounts of input RNA. Nucleic Acids Res. 2008, 36, e143. [Google Scholar] [CrossRef] [PubMed]
- Avissar, M.; Christensen, B.C.; Kelsey, K.T.; Marsit, C.J. MicroRNA expression ratio is predictive of head and neck squamous cell carcinoma. Clin. Cancer Res. 2009, 15, 2850–2855. [Google Scholar] [CrossRef] [PubMed]
- Baffa, R.; Fassan, M.; Volinia, S.; O'Hara, B.; Liu, C.; Palazzo, J.P.; Gardiman, M.; Rugge, M.; Gomella, L.G.; Croce, C.M. MicroRNA expression profiling of human metastatic cancers identifies cancer gene targets. J. Pathol. 2009, 219, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Mees, S.T.; Mardin, W.A.; Wendel, C.; Baeumer, N.; Willscher, E.; Senninger, N.; Schleicher, C.; Colombo-Benkmann, M.; Haier, J. EP300—A miRNA-regulated metastasis suppressor gene in ductal adenocarcinomas of the pancreas. Int. J. Cancer 2010, 126, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Funes-Huacca, M.E.; Opel, K.; Thompson, R.; McCord, B.R. A comparison of the effects of PCR inhibition in quantitative PCR and forensic STR analysis. Electrophoresis 2011, 32, 1084–1089. [Google Scholar] [CrossRef] [PubMed]
- Marti, A.A.; Jockusch, S.; Stevens, N.; Ju, J.; Turro, N.J. Fluorescent hybridization probes for sensitive and selective DNA and RNA detection. Acc. Chem. Res. 2007, 40, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Sando, S.; Kool, E.T. Imaging of RNA in bacteria with self-ligating quenched probes. J. Am. Chem. Soc. 2002, 124, 9686–9687. [Google Scholar] [CrossRef] [PubMed]
- Socher, E.; Bethge, L.; Knoll, A.; Jungnick, N.; Herrmann, A.; Seitz, O. Low-Noise Stemless PNA Beacons for Sensitive DNA and RNA Detection. Angew. Chem. Int. Ed. 2008, 47, 9555–9559. [Google Scholar] [CrossRef] [PubMed]
- Baeumner, A.J.; Jones, C.; Wong, C.Y.; Price, A. A generic sandwich-type biosensor with nanomolar detection limits. Anal. Bioanal. Chem. 2004, 378, 1587–1593. [Google Scholar] [CrossRef] [PubMed]
- Arata, H.; Komatsu, H.; Hosokawa, K.; Maeda, M. Rapid and sensitive microRNA detection with laminar flow-assisted dendritic amplification on power-free microfluidic chip. PLoS ONE 2012, 7, e48329. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Kong, T.; Zhang, D.; Zhang, J.; Cheng, G. Label-Free MicroRNA Detection Based on Fluorescence Quenching of Gold Nanoparticles with a Competitive Hybridization. Anal. Chem. 2015, 87, 10822–10829. [Google Scholar] [CrossRef] [PubMed]
- Aw, S.S.; Tang, M.X.; Teo, Y.N.; Cohen, S.M. A conformation-induced fluorescence method for microRNA detection. Nucleic Acids Res. 2016, 44, e92. [Google Scholar] [CrossRef] [PubMed]
- Egatz-Gomez, A.; Wang, C.; Klacsmann, F.; Pan, Z.; Marczak, S.; Wang, Y.; Sun, G.; Senapati, S.; Chang, H. Future microfluidic and nanofluidic modular platforms for nucleic acid liquid biopsy in precision medicine. Biomicrofluidics 2016, 10, 032902. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chang, T.; Stoddart, P.R.; Chang, H. Diffraction-limited ultrasensitive molecular nano-arrays with singular nano-cone scattering. Biomicrofluidics 2014, 8, 021101. [Google Scholar] [CrossRef] [PubMed]
- Stern, E.; Wagner, R.; Sigworth, F.J.; Breaker, R.; Fahmy, T.M.; Reed, M.A. Importance of the Debye screening length on nanowire field effect transistor sensors. Nano Lett. 2007, 7, 3405–3409. [Google Scholar] [CrossRef] [PubMed]
- Suni, I.I. Impedance methods for electrochemical sensors using nanomaterials. TrAC Trends Anal. Chem. 2008, 27, 604–611. [Google Scholar] [CrossRef]
- Tinland, B.; Ren, H.; Desruisseaux, C.; McCormick, L.C.; Drouin, G.; Slater, G.W. Diffusion coefficient of DNA molecules during free solution electrophoresis. Electrophoresis 2001, 22, 2424–2432. [Google Scholar]
- Sosnowski, R.G.; Tu, E.; Butler, W.F.; O’Connell, J.P.; Heller, M.J. Rapid determination of single base mismatch mutations in DNA hybrids by direct electric field control. Proc. Natl. Acad. Sci. USA 1997, 94, 1119–1123. [Google Scholar] [CrossRef] [PubMed]
- Leïchlé, T.; Chou, C. Biofunctionalized nanoslits for wash-free and spatially resolved real-time sensing with full target capture. Biomicrofluidics 2015, 9, 034103. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Wang, S.; Wang, L.; Li, F.; Pingguan-Murphy, B.; Lu, T.J.; Xu, F. Advances in paper-based point-of-care diagnostics. Biosens. Bioelectron. 2014, 54, 585–597. [Google Scholar] [CrossRef] [PubMed]
- Cortez, M.A.; Calin, G.A. MicroRNA identification in plasma and serum: A new tool to diagnose and monitor diseases. Expert Opin. Biol. Ther. 2009, 9, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ba, Y.; Ma, L.; Cai, X.; Yin, Y.; Wang, K.; Guo, J.; Zhang, Y.; Chen, J.; Guo, X. Characterization of microRNAs in serum: A novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. 2008, 18, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Hunter, M.P.; Ismail, N.; Zhang, X.; Aguda, B.D.; Lee, E.J.; Yu, L.; Xiao, T.; Schafer, J.; Lee, M.T.; Schmittgen, T.D. Detection of microRNA expression in human peripheral blood microvesicles. PLoS ONE 2008, 3, e3694. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.A.; Baxter, D.H.; Zhang, S.; Huang, D.Y.; Huang, K.H.; Lee, M.J.; Galas, D.J.; Wang, K. The microRNA spectrum in 12 body fluids. Clin. Chem. 2010, 56, 1733–1741. [Google Scholar] [CrossRef] [PubMed]
- Ai, J.; Zhang, R.; Li, Y.; Pu, J.; Lu, Y.; Jiao, J.; Li, K.; Yu, B.; Li, Z.; Wang, R. Circulating microRNA-1 as a potential novel biomarker for acute myocardial infarction. Biochem. Biophys. Res. Commun. 2010, 391, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Moldovan, L.; Batte, K.E.; Trgovcich, J.; Wisler, J.; Marsh, C.B.; Piper, M. Methodological challenges in utilizing miRNAs as circulating biomarkers. J. Cell. Mol. Med. 2014, 18, 371–390. [Google Scholar] [CrossRef] [PubMed]
- Vickers, K.C.; Palmisano, B.T.; Shoucri, B.M.; Shamburek, R.D.; Remaley, A.T. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat. Cell Biol. 2011, 13, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.K.; Zhu, J.Q.; Zhang, J.T.; Li, Q.; Li, Y.; He, J.; Qin, Y.W.; Jing, Q. Circulating microRNA: A novel potential biomarker for early diagnosis of acute myocardial infarction in humans. Eur. Heart J. 2010, 31, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Kroh, E.M.; Parkin, R.K.; Mitchell, P.S.; Tewari, M. Analysis of circulating microRNA biomarkers in plasma and serum using quantitative reverse transcription-PCR (qRT-PCR). Methods 2010, 50, 298–301. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Yuan, Y.; Cho, J.; McClarty, S.; Baxter, D.; Galas, D.J. Comparing the MicroRNA spectrum between serum and plasma. PLoS ONE 2012, 7, e41561. [Google Scholar] [CrossRef] [PubMed]
- McDonald, J.S.; Milosevic, D.; Reddi, H.V.; Grebe, S.K.; Algeciras-Schimnich, A. Analysis of circulating microRNA: Preanalytical and analytical challenges. Clin. Chem. 2011, 57, 833–840. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Qin, Y.; Brewer, G.; Jing, Q. MicroRNA degradation and turnover: Regulating the regulators. Wiley Interdiscip. Rev. 2012, 3, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Chang-Lin, S.; Hung, A.; Chang, D.C.; Lin, Y.W.; Ying, S.Y.; Lin, S.L. Novel glycylated sugar alcohols protect ESC-specific microRNAs from degradation in iPS cells. Nucleic Acids Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Bissonnette, L.; Bergeron, M. Diagnosing infections—Current and anticipated technologies for point-of-care diagnostics and home-based testing. Clin. Microbiol. Infect. 2010, 16, 1044–1053. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.S.; Lee, Y.U.; Kim, H.; Kim, T.; Park, J.; Lee, J.; Kim, J.; Kim, H.; Lee, W.G.; Cho, Y. Fully integrated lab-on-a-disc for simultaneous analysis of biochemistry and immunoassay from whole blood. Lab Chip 2011, 11, 70–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.; Weng, X.; Li, D. Microfluidic whole-blood immunoassays. Microfluid. Nanofluid. 2011, 10, 941–964. [Google Scholar] [CrossRef]
- Lee, Y.; Lien, K.; Lei, H.; Lee, G. An integrated microfluidic system for rapid diagnosis of dengue virus infection. Biosens. Bioelectron. 2009, 25, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Kaigala, G.; Huskins, R.; Preiksaitis, J.; Pang, X.; Pilarski, L.; Backhouse, C. Automated surveillance using microfluidic chip-based PCR thermocycling and product detection to assess risk of polyoma BK virus associated neuropathy in renal transplant recipients. Electrophoresis 2006, 27, 3753–3763. [Google Scholar] [CrossRef] [PubMed]
- Easley, C.J.; Karlinsey, J.M.; Bienvenue, J.M.; Legendre, L.A.; Roper, M.G.; Feldman, S.H.; Hughes, M.A.; Hewlett, E.L.; Merkel, T.J.; Ferrance, J.P.; et al. A fully integrated microfluidic genetic analysis system with sample-in-answer-out capability. Proc. Natl. Acad. Sci. USA 2006, 103, 19272–19277. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Mauk, M.; Qiu, X.; Liu, C.; Kim, J.; Ramprasad, S.; Ongagna, S.; Abrams, W.R.; Malamud, D.; Corstjens, P.L. An integrated, self-contained microfluidic cassette for isolation, amplification, and detection of nucleic acids. Biomed. Microdevices 2010, 12, 705–719. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, B.S.; Buchsbaum, S.F.; Wu, T.; Hsieh, K.; Xiao, Y.; Sun, R.; Soh, H.T. Genetic analysis of H1N1 influenza virus from throat swab samples in a microfluidic system for point-of-care diagnostics. J. Am. Chem. Soc. 2011, 133, 9129–9135. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Qiu, X.; Ongagna, S.; Chen, D.; Chen, Z.; Abrams, W.R.; Malamud, D.; Corstjens, P.L.; Bau, H.H. A timer-actuated immunoassay cassette for detecting molecular markers in oral fluids. Lab Chip 2009, 9, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Mark, D.; Haeberle, S.; Roth, G.; von Stetten, F.; Zengerle, R. Microfluidic lab-on-a-chip platforms: Requirements, characteristics and applications. Chem. Soc. Rev. 2010, 39, 1153–1182. [Google Scholar] [CrossRef] [PubMed]
- Shadfan, B.H.; Simmons, A.R.; Simmons, G.W.; Ho, A.; Wong, J.; Lu, K.H.; Bast, R.C., Jr.; McDevitt, J.T. A multiplexable, microfluidic platform for the rapid quantitation of a biomarker panel for early ovarian cancer detection at the point-of-care. Cancer. Prev. Res. 2015, 8, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.T.; Rotem, A.; Heyman, J.A.; Weitz, D.A. Droplet microfluidics for high-throughput biological assays. Lab Chip 2012, 12, 2146–2155. [Google Scholar] [CrossRef] [PubMed]
- Harshman, D.K.; Reyes, R.; San Park, T.; You, D.J.; Song, J.; Yoon, J. Enhanced nucleic acid amplification with blood in situ by wire-guided droplet manipulation (WDM). Biosens. Bioelectron. 2014, 53, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Lee, J.; Park, J.; Lee, B.; Lee, Y.; Ko, C. One-step pathogen specific DNA extraction from whole blood on a centrifugal microfluidic device. Lab Chip 2007, 7, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Lutz, S.; Weber, P.; Focke, M.; Faltin, B.; Hoffmann, J.; Müller, C.; Mark, D.; Roth, G.; Munday, P.; Armes, N. Microfluidic lab-on-a-foil for nucleic acid analysis based on isothermal recombinase polymerase amplification (RPA). Lab Chip 2010, 10, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Gervais, L.; Delamarche, E. Toward one-step point-of-care immunodiagnostics using capillary-driven microfluidics and PDMS substrates. Lab Chip 2009, 9, 3330–3337. [Google Scholar] [CrossRef] [PubMed]
- Yager, P.; Domingo, G.J.; Gerdes, J. Point-of-care diagnostics for global health. Annu. Rev. Biomed. Eng. 2008, 10, 107–144. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A.W.; Phillips, S.T.; Whitesides, G.M.; Carrilho, E. Diagnostics for the developing world: Microfluidic paper-based analytical devices. Anal. Chem. 2009, 82, 3–10. [Google Scholar] [CrossRef] [PubMed]
- San Park, T.; Li, W.; McCracken, K.E.; Yoon, J. Smartphone quantifies Salmonella from paper microfluidics. Lab Chip 2013, 13, 4832–4840. [Google Scholar] [CrossRef] [PubMed]
- Sauer-Budge, A.F.; Mirer, P.; Chatterjee, A.; Klapperich, C.M.; Chargin, D.; Sharon, A. Low cost and manufacturable complete microTAS for detecting bacteria. Lab Chip 2009, 9, 2803–2810. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Yu, M.; Sun, X.; Woolley, A.T. Microdevices integrating affinity columns and capillary electrophoresis for multibiomarker analysis in human serum. Lab Chip 2010, 10, 2527–2533. [Google Scholar] [CrossRef] [PubMed]
- Pike, J.; Godbert, S.; Johnson, S. Comparison of volunteers’ experience of using, and accuracy of reading, different types of home pregnancy test formats. Expert Opin. Med. Diagn. 2013, 7, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Anek-vorapong, R.; Sinthuwattanawibool, C.; Podewils, L.J.; McCarthy, K.; Ngamlert, K.; Promsarin, B.; Varma, J.K. Validation of the GenoType® MTBDR plus assay for detection of MDR-TB in a public health laboratory in Thailand. BMC Infect. Dis. 2010, 10, 1. [Google Scholar] [CrossRef] [PubMed]
- Chinchai, T.; Chansaenroj, J.; Swangvaree, S.; Junyangdikul, P.; Poovorawan, Y. Prevalence of human papillomavirus genotypes in cervical cancer. Int. J. Gynecol. Cancer 2012, 22, 1063–1068. [Google Scholar] [CrossRef] [PubMed]
- Kingma, D.W.; Allen, R.A.; Moore, W.; Caughron, S.K.; Melby, M.; Gillies, E.M.; Marlar, R.A.; Dunn, S.T. HPV genotype distribution in oral and oropharyngeal squamous cell carcinoma using seven in vitro amplification assays. Anticancer Res. 2010, 30, 5099–5104. [Google Scholar] [PubMed]
- Su, W.; Gao, X.; Jiang, L.; Qin, J. Microfluidic platform towards point-of-care diagnostics in infectious diseases. J. Chromatogr. A 2015, 1377, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.D.; Linder, V.; Sia, S.K. Commercialization of microfluidic point-of-care diagnostic devices. Lab Chip 2012, 12, 2118–2134. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.D.; Chin, S.Y.; Laksanasopin, T.; Sia, S.K. Low-Cost Microdevices for Point-of-Care Testing; Point-of-Care Diagnostics on a Chip; Springer: Berlin, Germany, 2013; pp. 3–21. [Google Scholar]
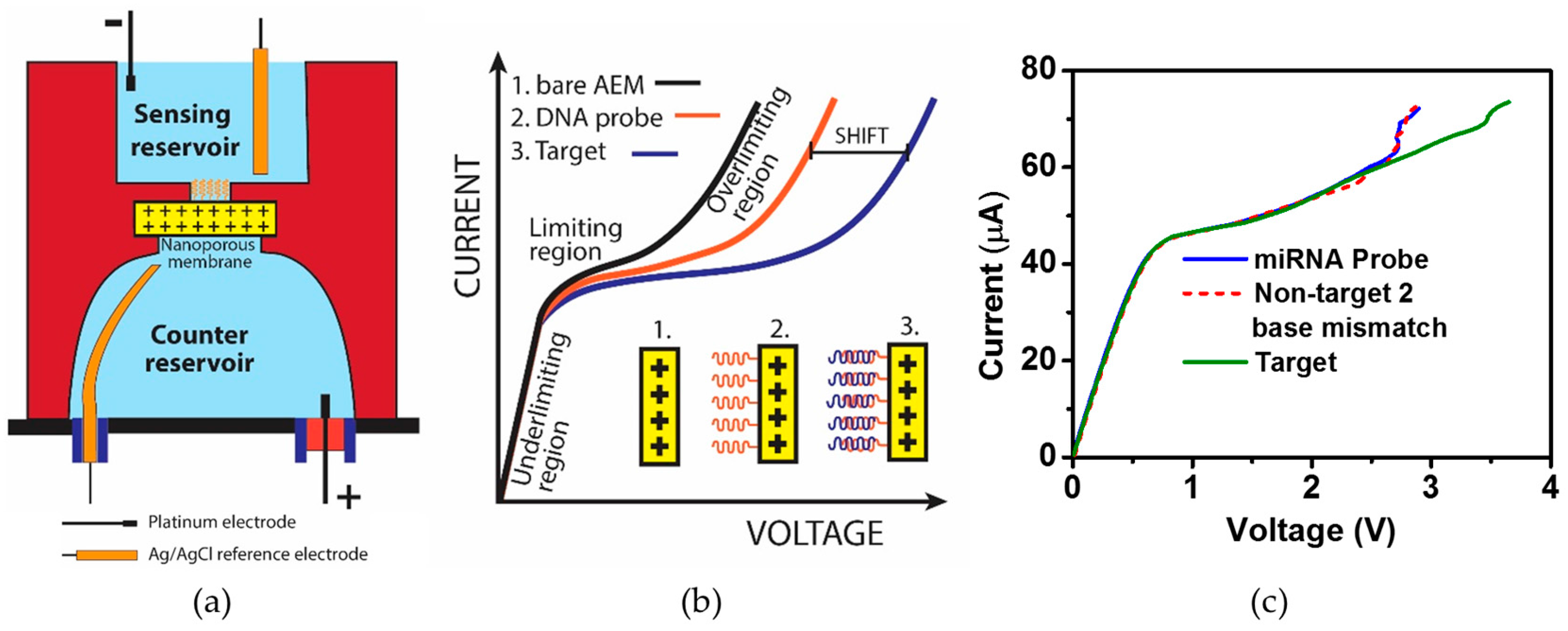
- Senapati, S.; Slouka, Z.; Shah, S.S.; Behura, S.K.; Shi, Z.; Stack, M.S.; Severson, D.W.; Chang, H. An ion-exchange nanomembrane sensor for detection of nucleic acids using a surface charge inversion Phenomenon. Biosens. Bioelectron. 2014, 60, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Slouka, Z.; Senapati, S.; Shah, S.; Lawler, R.; Shi, Z.; Stack, M.S.; Chang, H. Integrated, DC voltage-driven nucleic acid diagnostic platform for real sample analysis: Detection of oral cancer. Talanta 2015, 145, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Yossifon, G.; Demekhin, E.A. Nanoscale electrokinetics and microvortices: How microhydrodynamics affects nanofluidic ion flux. Annu. Rev. Fluid Mech. 2012, 44, 401–426. [Google Scholar] [CrossRef]
- Slouka, Z.; Senapati, S.; Chang, H. Microfluidic systems with ion-selective membranes. Annu. Rev. Anal. Chem. 2014, 7, 317–335. [Google Scholar] [CrossRef] [PubMed]
- Slouka, Z.; Senapati, S.; Yan, Y.; Chang, H. Charge Inversion, Water Splitting, and Vortex Suppression Due to DNA Sorption on Ion-Selective Membranes and Their Ion-Current Signatures. Langmuir 2013, 29, 8275–8283. [Google Scholar] [CrossRef] [PubMed]
- Senapati, S.; Basuray, S.; Slouka, Z.; Cheng, L.; Chang, H. A Nanomembrane-Based Nucleic Acid Sensing Platform for Portable Diagnostics. Microfluid. Technol. Appl. 2011, 304, 153–169. [Google Scholar]
- Taller, D.; Richards, K.; Slouka, Z.; Senapati, S.; Hill, R.; Go, D.B.; Chang, H. On-chip surface acoustic wave lysis and ion-exchange nanomembrane detection of exosomal RNA for pancreatic cancer study and diagnosis. Lab Chip 2015, 15, 1656–1666. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
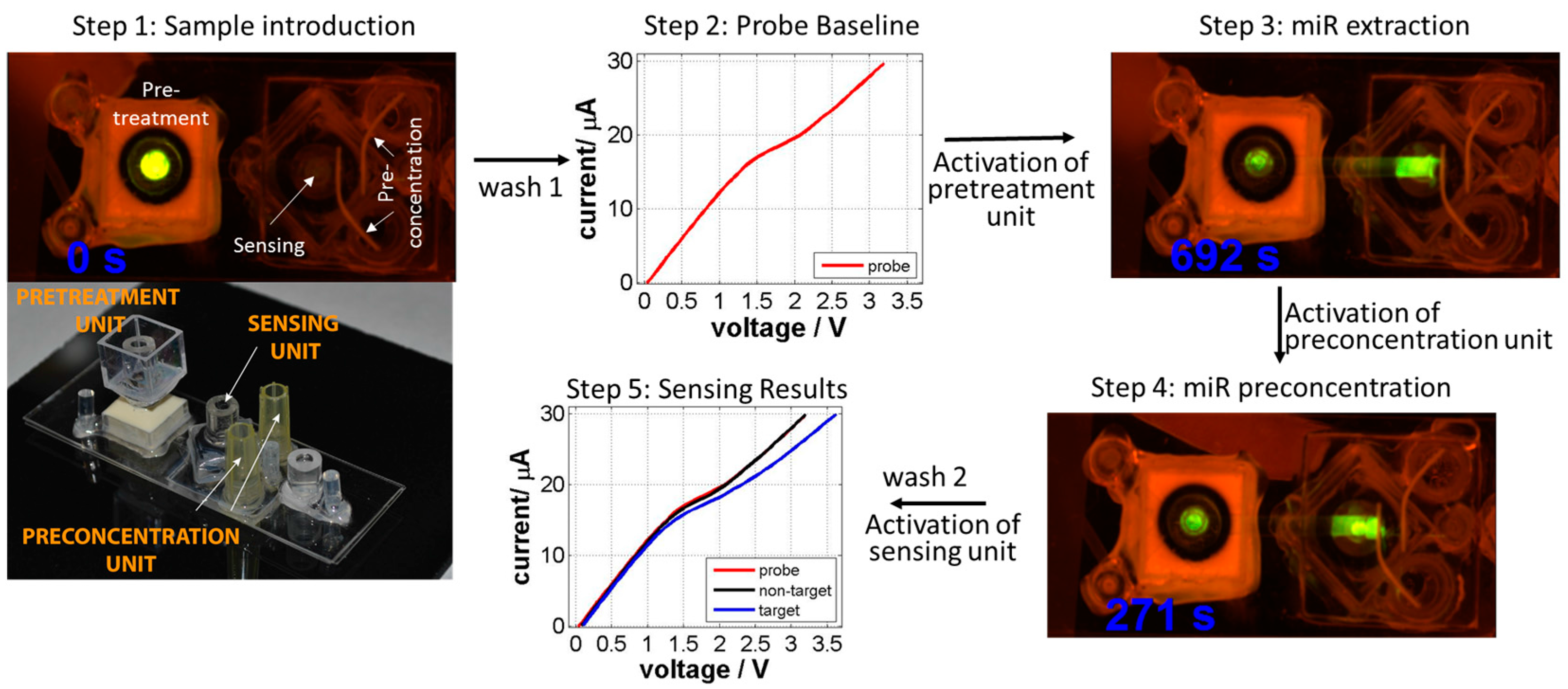{kind=link}
| Screening Tools | Cancer | Advantages | Disadvantages | Performance |
|---|---|---|---|---|
| Pap smear | Cervical |
|
|
|
| Liquid-based cytology | Cervical |
|
|
|
| VIA | Cervical |
|
|
|
| HPV antibody detection | Cervical/Oral |
|
|
|
| Visual Inspection | Oral |
|
|
|
| Tissue biopsy | Cervical/Oral |
|
|
|
| Light-based detection systems | Oral |
|
|
|
| Technique | Throughput | Specificity | Sensitivity | Cost/miR [84] | Assay Time |
|---|---|---|---|---|---|
| Microarray | High | Moderate | Low | Low | 1–2 days |
| qRT-PCR | Low | High | High | High | Few hours |
| Next generation sequencing | High | High | High | Low (High/sample) | 2–5 days |
| Performance Specification | Qiagen® HC2 Assay | Cervista® HPV HR | Cervista® HPV 16/18 | Cobas® HPV | Aptima® |
|---|---|---|---|---|---|
| Detection Target | 13 high-risk HPV DNA | 14 high-risk HPV DNA | HPV 16 and 18 | 14 high-risk HPV DNA | E6/E7 mRNA of high risk HPV |
| Detection Mechanism | Antibody hybridizes to HPV DNA–RNA | Invader Chemistry | Invader Chemistry | PCR + Fluorescence | Transcription-mediated amplification HPV 16/18 |
| Sample Processing | Additional pretreatment needed | Additional pretreatment needed | Additional pretreatment needed | Automated sample extraction | Automated sample extraction |
| Clinical Sensitivity [112,121,122,123] | ≈94% | ≈89% | 68%–70% | ≈93% | >92% |
| Specificity [112,121,122,123] | >89% | ≈91% | 62%–80% | ≈99% | ≈99% |
| Assay vs. System [117,118,124,125,126,127,128] |  Assay |  Assay |  Assay |  Cobas® HPV System |  Panther System |
| Cost [124,125,126,127,128] | $71 per test° | $30 per test° | $30 per test° | $35 per test° + capital equipment | $30 per test° + capital equipment |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shah, S.S.; Senapati, S.; Klacsmann, F.; Miller, D.L.; Johnson, J.J.; Chang, H.-C.; Stack, M.S. Current Technologies and Recent Developments for Screening of HPV-Associated Cervical and Oropharyngeal Cancers. Cancers 2016, 8, 85. https://doi.org/10.3390/cancers8090085
Shah SS, Senapati S, Klacsmann F, Miller DL, Johnson JJ, Chang H-C, Stack MS. Current Technologies and Recent Developments for Screening of HPV-Associated Cervical and Oropharyngeal Cancers. Cancers. 2016; 8(9):85. https://doi.org/10.3390/cancers8090085
Chicago/Turabian StyleShah, Sunny S., Satyajyoti Senapati, Flora Klacsmann, Daniel L. Miller, Jeff J. Johnson, Hsueh-Chia Chang, and M. Sharon Stack. 2016. "Current Technologies and Recent Developments for Screening of HPV-Associated Cervical and Oropharyngeal Cancers" Cancers 8, no. 9: 85. https://doi.org/10.3390/cancers8090085
APA StyleShah, S. S., Senapati, S., Klacsmann, F., Miller, D. L., Johnson, J. J., Chang, H. -C., & Stack, M. S. (2016). Current Technologies and Recent Developments for Screening of HPV-Associated Cervical and Oropharyngeal Cancers. Cancers, 8(9), 85. https://doi.org/10.3390/cancers8090085