
Targeting PDK1 for Chemosensitization of Cancer Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. The Journey to PDK1 Discovery
1.2. Physiological Roles of PDK1 and Its Correlation with Malignancies
2. PDK1 in Chemoresistance
2.1. Ovarian Cancer
2.2. Breast Cancer
2.3. Acute Myeloid Leukaemia
2.4. PDK1 and Chemoresistance in Multiple Types of Cancer
3. PDK1 Oncogenic Signaling in Chemoresistance: Beyond AKT
3.1. PDK1-PLK1-MYC Axis
3.2. PDK1-YAP/Hippo Pathway Axis
3.3. PDK1-SGK Axis
3.4. Inhibitors of PDK1
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 3D | Three dimensional |
| 5-FU | 5-fluorouracil |
| AML | acute myeloid leukaemia |
| ATP | adenosine triphosphate |
| AurA | Aurora Kinase A |
| BAD | Bcl-2-associated death promoter |
| c/EBPβ | CCAAT/Enhancer Binding Protein Beta |
| CAMKK2 | Ca2+/calmodulin-dependent protein kinase kinase 2 |
| cDDP | cis-diamminedichloroplatinum(II)/cisplatin/cisplatinum |
| COL11A1 | collagen type XI aplha1 |
| COX2 | cyclooxygenase-2 |
| CTGF | connective tissue growth factor |
| EGF | epidermal growth factor |
| EMT | epithelial-to-mesenchymal transition |
| EOC | epithelial ovarian carcinoma |
| ERα | oestrogen receptor α |
| FABP5 | fatty acid-binding protein 5 |
| Foxo | forkhead box O |
| GAP | GTPase activating protein |
| GBM | glioblastoma multiforme |
| GPCR | G-protein-coupled receptor |
| HDAC | histone deacetylase |
| Hpo | Hippo |
| Hsp90 | heat shock protein 90 |
| IC50 | half maximal inhibitory concentration |
| IGF1 | insulin growth factor 1 |
| K-RAS | Kirsten Rat Sarcoma Viral Oncogene Homolog |
| Lats | Large Tumor Suppressor Kinase 1 |
| LPA | lysophosphatidic acid |
| LSC | leukaemia stem cells |
| Mats | Mob-as-tumour-suppressor |
| MLL-AF9 | mixed lineage leukemia-ALL1- fused gene from chromosome 9 protein |
| MM | Multiple myeloma |
| MOBKL1A/1B | Mps one binder kinase activator-like 1A |
| mRNA | messenger ribonucleic acid |
| mSIN1 | mammalian stress-activated protein kinase interacting protein 1 |
| MTDH | metadherin |
| mTOR | mammalian/mechanistic target of rapamycin |
| MYC | v-myc myelocytomatosis viral oncogene homolog (avian) |
| NDRG1 | N-Myc Downstream Regulated 1 |
| nM | nanomolar |
| p21Cip1 | cyclin-dependent kinase inhibitor 1 |
| PDAC | pancreatic ductal adenocarcinoma |
| PDK1 | 3-phosphoinositide-dependent protein kinase 1 |
| PH | pleckstrin homology |
| PI3K | phosphoinositide 3-kinase |
| PIF | PDK1-interacting fragment |
| PKB/Akt | protein kinase B |
| PKC | protein kinase C |
| PLCε | phospholipase Cε |
| PLK1 | Polo-like kinase 1 |
| PPAR β | peroxisome proliferator-activated receptor β |
| PRAS40 | inhibitory subunit 40-kDa proline-rich |
| PtdIns(3,4,5)P3 | phosphatidylinositol (3,4,5)-trisphosphate |
| PtdIns(4,5)P2 | phosphatidylinositol (4,5)-bisphosphate |
| PTEN | phosphate and tensin homolog |
| RA | retinoic acid |
| RTK | receptor tyrosine kinase |
| Rheb | Ras homolog enriched in brain |
| S6K1 | Ribosomal protein S6 kinase beta-1 |
| Sav | Salvador |
| Ser | serine |
| sfRon | short-form Ron |
| SGK | Serum and glucocorticoid-induced protein kinase |
| Src | V-Src Avian Sarcoma (Schmidt-RuppinA-2) Viral Oncogene |
| SWH | Salvador/Warts/Hippo |
| TCRP1 | tongue cancer resistance-related protein-1 |
| TEADs | TEA domain transcription factors |
| Thr | threonine |
| TIMP1 | tissue inhibitor of metalloproteinase-1 |
| TNBC | triple negative breast cancer |
| TRAIL | tumour necrosis factor-α-related apoptosis-inducing ligand |
| TSC | tuberous sclerosis complex |
| TUSC4 | tumour suppressor candidate 4 |
| UPP | ubiquitin (Ub)-proteasome pathway |
| VEGF-A | vascular endothelial growth factor-A |
| VP-16 | Etoposide Phosphate |
| Wts | Warts |
| YAP | Yes-associated protein 1 |
| μΜ | micromolar |
References
- Alessi, D. Discovery of PDKI, one of the missing links in insulin signal transduction. Biochem. Soc. Trans. 2001, 29, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Alessi, D.R.; James, S.R.; Downes, C.P.; Holmes, A.B.; Gaffney, P.R.; Reese, C.B.; Cohen, P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Bα. Curr. Biol. 1997, 7, 261–269. [Google Scholar] [CrossRef]
- Alessi, D.R.; Deak, M.; Casamayor, A.; Caudwell, F.B.; Morrice, N.; Norman, D.G.; Gaffney, P.; Reese, C.B.; MacDougall, C.N.; Harbison, D. 3-Phosphoinositide-dependent protein kinase-1 (PDK1): Structural and functional homology with the Drosophila DSTPK61 kinase. Curr. Biol. 1997, 7, 776–789. [Google Scholar] [CrossRef]
- Casamayor, A.; Morrice, N.A.; Alessi, D.R. Phosphorylation of Ser-241 is essential for the activity of 3-phosphoinositide-dependent protein kinase-1: Identification of five sites of phosphorylation in vivo. Biochem. J. 1999, 342, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, M.A.; Mora, A.; Ashby, P.R.; Williams, M.R.; Murray-Tait, V.; Malone, L.; Prescott, A.R.; Lucocq, J.M.; Alessi, D.R. Essential role of PDK1 in regulating cell size and development in mice. EMBO J. 2002, 21, 3728–3738. [Google Scholar] [CrossRef] [PubMed]
- Collins, B.J.; Deak, M.; Arthur, J.S.; Armit, L.J.; Alessi, D.R. In vivo role of the PIF-binding docking site of PDK1 defined by knock-in mutation. EMBO J. 2003, 22, 4202–4211. [Google Scholar] [CrossRef] [PubMed]
- McManus, E.J.; Collins, B.J.; Ashby, P.R.; Prescott, A.R.; Murray-Tait, V.; Armit, L.J.; Arthur, J.S.; Alessi, D.R. The in vivo role of PtdIns(3,4,5)P3 binding to PDK1 PH domain defined by knockin mutation. EMBO J. 2004, 23, 2071–2082. [Google Scholar] [CrossRef] [PubMed]
- Mora, A.; Davies, A.M.; Bertrand, L.; Sharif, I.; Budas, G.R.; Jovanović, S.; Mouton, V.; Kahn, C.R.; Lucocq, J.M.; Gray, G.A. Deficiency of PDK1 in cardiac muscle results in heart failure and increased sensitivity to hypoxia. EMBO J. 2003, 22, 4666–4676. [Google Scholar] [CrossRef] [PubMed]
- Alfonso, M.; Lipina, C.; Tronche, F.; Sutherland, C.; Alessi, D.R. Deficiency of PDK1 in liver results in glucose intolerance, impairment of insulin-regulated gene expression and liver failure. Biochem. J. 2005, 385, 639–648. [Google Scholar]
- Okamoto, Y.; Ogawa, W.; Nishizawa, A.; Inoue, H.; Teshigawara, K.; Kinoshita, S.; Matsuki, Y.; Watanabe, E.; Hiramatsu, R.; Sakaue, H. Restoration of glucokinase expression in the liver normalizes postprandial glucose disposal in mice with hepatic deficiency of PDK1. Diabetes 2007, 56, 1000–1009. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, N.; Kido, Y.; Uchida, T.; Asahara, S.-i.; Shigeyama, Y.; Matsuda, T.; Takeda, A.; Tsuchihashi, D.; Nishizawa, A.; Ogawa, W. Ablation of PDK1 in pancreatic [beta] cells induces diabetes as a result of loss of [beta] cell mass. Nat. Genet. 2006, 38, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Bayascas, J.R.; Sakamoto, K.; Armit, L.; Arthur, J.S.C.; Alessi, D.R. Evaluation of approaches to generation of tissue-specific knock-in mice. J. Biol. Chem. 2006, 281, 28772–28781. [Google Scholar] [CrossRef] [PubMed]
- Bayascas, J.R.; Wullschleger, S.; Sakamoto, K.; García-Martínez, J.M.; Clacher, C.; Komander, D.; Van Aalten, D.M.; Boini, K.M.; Lang, F.; Lipina, C. Mutation of the PDK1 PH domain inhibits protein kinase B/Akt, leading to small size and insulin resistance. Mol. Cell. Biol. 2008, 28, 3258–3272. [Google Scholar] [CrossRef] [PubMed]
- Rotte, A.; Bhandaru, M.; Ackermann, T.F.; Boini, K.M.; Lang, F. Role of PDK1 in regulation of gastric acid secretion. Cell. Physiol. Biochem. 2008, 22, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Sandu, C.; Artunc, F.; Palmada, M.; Rexhepaj, R.; Grahammer, F.; Hussain, A.; Yun, C.; Alessi, D.R.; Lang, F. Impaired intestinal NHE3 activity in the PDK1 hypomorphic mouse. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 291, G868–G876. [Google Scholar] [CrossRef] [PubMed]
- Muranen, T.A.; Greco, D.; Fagerholm, R.; Kilpivaara, O.; Kämpjärvi, K.; Aittomäki, K.; Blomqvist, C.; Heikkilä, P.; Borg, Å.; Nevanlinna, H. Breast tumors from CHEK2 1100delC-mutation carriers: Genomic landscape and clinical implications. Breast Cancer Res. 2011, 13, R90. [Google Scholar] [CrossRef] [PubMed]
- Choucair, K.A.; Guérard, K.-P.; Ejdelman, J.; Chevalier, S.; Yoshimoto, M.; Scarlata, E.; Fazli, L.; Sircar, K.; Squire, J.A.; Brimo, F. The 16p13. 3 (PDPK1) genomic gain in prostate cancer: A potential role in disease progression. Transl. Oncol. 2012, 5, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Maurer, M.; Su, T.; Saal, L.H.; Koujak, S.; Hopkins, B.D.; Barkley, C.R.; Wu, J.; Nandula, S.; Dutta, B.; Xie, Y. 3-Phosphoinositide-dependent kinase 1 potentiates upstream lesions on the phosphatidylinositol 3-kinase pathway in breast carcinoma. Cancer Res. 2009, 69, 6299–6306. [Google Scholar] [CrossRef] [PubMed]
- Di Blasio, L.; Gagliardi, P.A.; Puliafito, A.; Primo, L. Serine/Threonine Kinase 3-Phosphoinositide-Dependent Protein Kinase-1 (PDK1) as a key regulator of cell migration and cancer dissemination. Cancers 2017, 9, E25. [Google Scholar] [CrossRef] [PubMed]
- Pinner, S.; Sahai, E. PDK1 regulates cancer cell motility by antagonising inhibition of ROCK1 by RhoE. Nat. Cell Biol. 2008, 10, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, C.; Falasca, M. Targeting PDK1 in cancer. Curr. Med. Chem. 2011, 18, 2763–2769. [Google Scholar] [CrossRef] [PubMed]
- Gagliardi, P.A.; di Blasio, L.; Primo, L. PDK1: A signaling hub for cell migration and tumor invasion. Biochim. Biophys. Acta 2015, 1856, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Bayascas, J.R. PDK1: The major transducer of PI 3-kinase actions. In Phosphoinositide 3-Kinase in Health and Disease; Springer: Berlin, Germany, 2010; pp. 9–29. [Google Scholar]
- Toricelli, M.; Melo, F.H.; Hunger, A.; Zanatta, D.; Strauss, B.E.; Jasiulionis, M.G. Timp1 promotes cell survival by activating the PDK1 signaling pathway in melanoma. Cancers 2017, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Fu, L.; Cui, M.; Wang, Y.; Xu, Y.; Li, M.; Mi, J. Amino acid transporter SLC38A3 promotes metastasis of non-small cell lung cancer cells by activating PDK1. Cancer Lett. 2017, 393, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, F.; Ao, P.; Li, X.; Zheng, H.; Wu, D.; Zhang, N.; She, J.; Yuan, J.; Wu, X. Correlation of PDK1 expression with clinicopathologic features and prognosis of hepatocellular carcinoma. Onco Targets Ther. 2016, 9, 5597–5602. [Google Scholar] [PubMed]
- Vasudevan, K.M.; Barbie, D.A.; Davies, M.A.; Rabinovsky, R.; McNear, C.J.; Kim, J.J.; Hennessy, B.T.; Tseng, H.; Pochanard, P.; Kim, S.Y. AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell 2009, 16, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Samuels, Y.; Wang, Z.; Bardelli, A.; Silliman, N.; Ptak, J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, S.M.; Riggins, G.J. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004, 304, 554. [Google Scholar] [CrossRef] [PubMed]
- Samuels, Y.; Diaz, L.A.; Schmidt-Kittler, O.; Cummins, J.M.; DeLong, L.; Cheong, I.; Rago, C.; Huso, D.L.; Lengauer, C.; Kinzler, K.W. Mutant PIK3CA promotes cell growth and invasion of human cancer cells. Cancer Cell 2005, 7, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Gagliardi, P.A.; di Blasio, L.; Orso, F.; Seano, G.; Sessa, R.; Taverna, D.; Bussolino, F.; Primo, L. 3-phosphoinositide-dependent kinase 1 controls breast tumor growth in a kinase-dependent but Akt-independent manner. Neoplasia 2012, 14, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Dobbin, Z.C.; Landen, C.N. The importance of the PI3K/AKT/MTOR pathway in the progression of ovarian cancer. Int. J. Mol. Sci. 2013, 14, 8213–8227. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Riley, C.; Quinn, M.A. An immunohistochemical perspective of PPAR beta and one of its putative targets PDK1 in normal ovaries, benign and malignant ovarian tumours. Br. J. Cancer 2008, 98, 1415–1424. [Google Scholar] [CrossRef] [PubMed]
- Moxley, K.M.; Wang, L.; Welm, A.L.; Bieniasz, M. Short-form Ron is a novel determinant of ovarian cancer initiation and progression. Genes Cancer 2016, 7, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhao, L.; Derose, Y.S.; Lin, Y.C.; Bieniasz, M.; Eyob, H.; Buys, S.S.; Neumayer, L.; Welm, A.L. Short-Form Ron Promotes Spontaneous Breast Cancer Metastasis through Interaction with Phosphoinositide 3-Kinase. Genes Cancer 2011, 2, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Lian, S.; Shao, Y.; Liu, H.; He, J.; Lu, W.; Zhang, Y.; Jiang, Y.; Zhu, J. PDK1 induces JunB, EMT, cell migration and invasion in human gallbladder cancer. Oncotarget 2015, 6, 29076–29086. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-H.; Chang, T.-H.; Huang, Y.-F.; Chen, C.-C.; Chou, C.-Y. COL11A1 confers chemoresistance on ovarian cancer cells through the activation of Akt/c/EBPβ pathway and PDK1 stabilization. Oncotarget 2015, 6, 23748–23763. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lacerda, D.; Warman, M.; Beier, D.; Yoshioka, H.; Ninomiya, Y.; Oxford, J.; Morris, N.; Andrikopoulos, K.; Ramirez, F. A fibrillar collagen gene, Col11a1, is essential for skeletal morphogenesis. Cell 1995, 80, 423–430. [Google Scholar] [CrossRef]
- Nakamura, A.; Naito, M.; Tsuruo, T.; Fujita, N. Freud-1/Aki1, a novel PDK1-interacting protein, functions as a scaffold to activate the PDK1/Akt pathway in epidermal growth factor signaling. Mol. Cell. Biol. 2008, 28, 5996–6009. [Google Scholar] [CrossRef] [PubMed]
- Falasca, M.; Chiozzotto, D.; Godage, H.; Mazzoletti, M.; Riley, A.; Previdi, S.; Potter, B.; Broggini, M.; Maffucci, T. A novel inhibitor of the PI3K/Akt pathway based on the structure of inositol 1, 3, 4, 5, 6-pentakisphosphate. Br. J. Cancer 2010, 102, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Gocher, A.M.; Azabdaftari, G.; Euscher, L.M.; Dai, S.; Karacosta, L.G.; Franke, T.F.; Edelman, A.M. Akt activation by Ca2+/calmodulin-dependent protein kinase kinase 2 (CaMKK2) in ovarian cancer cells. J. Biol. Chem. 2017, 292, 14188–14204. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; George, J.; Deb, S.; Degoutin, J.L.; Takano, E.A.; Fox, S.B.; Bowtell, D.D.; Harvey, K.F. The Hippo pathway transcriptional co-activator, YAP, is an ovarian cancer oncogene. Oncogene 2011, 30, 2810–2822. [Google Scholar] [CrossRef] [PubMed]
- Fyffe, C.; Falasca, M. 3-Phosphoinositide-dependent protein kinase-1 as an emerging target in the management of breast cancer. Cancer Manag. Res. 2013, 5, 271–280. [Google Scholar] [PubMed]
- Iorns, E.; Lord, C.J.; Ashworth, A. Parallel RNAi and compound screens identify the PDK1 pathway as a target for tamoxifen sensitization. Biochem. J. 2009, 417, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Peifer, C.; Alessi, D.R. New anti-cancer role for PDK1 inhibitors: Preventing resistance to tamoxifen. Biochem. J. 2009, 417, e5–e7. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Huang, J.-W.; Tseng, P.-H.; Yang, Y.-T.; Fowble, J.; Shiau, C.-W.; Shaw, Y.-J.; Kulp, S.K.; Chen, C.-S. From the cyclooxygenase-2 inhibitor celecoxib to a novel class of 3-phosphoinositide-dependent protein kinase-1 inhibitors. Cancer Res. 2004, 64, 4309–4318. [Google Scholar] [CrossRef] [PubMed]
- Weng, S.-C.; Kashida, Y.; Kulp, S.K.; Wang, D.; Brueggemeier, R.W.; Shapiro, C.L.; Chen, C.-S. Sensitizing estrogen receptor-negative breast cancer cells to tamoxifen with OSU-03012, a novel celecoxib-derived phosphoinositide-dependent protein kinase-1/Akt signaling inhibitor. Mol. Cancer Ther. 2008, 7, 800–808. [Google Scholar] [CrossRef] [PubMed]
- Fry, M.J. Phosphoinositide 3-kinase signalling in breast cancer: How big a role might it play? Breast Cancer Res. 2001, 3, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Hsieh, F.; Song, H.; Lin, J. Elevated phosphorylation and activation of PDK-1/AKT pathway in human breast cancer. Br. J. Cancer 2005, 93, 1372–1381. [Google Scholar] [CrossRef] [PubMed]
- Liang, K.; Lu, Y.; Li, X.; Zeng, X.; Glazer, R.I.; Mills, G.B.; Fan, Z. Differential roles of phosphoinositide-dependent protein kinase-1 and akt1 expression and phosphorylation in breast cancer cell resistance to Paclitaxel, Doxorubicin, and gemcitabine. Mol. Pharmacol. 2006, 70, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Aranha, O.; Li, Y.; Pettit, G.R.; Sarkar, F.H.; Philip, P.A. Sensitization of human breast cancer cells to gemcitabine by the protein kinase C modulator bryostatin 1. Cancer Chemother. Pharmacol. 2003, 52, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Xu, H.; Glazer, R.I. Transformation of mammary epithelial cells by 3-phosphoinositide-dependent protein kinase-1 (PDK1) is associated with the induction of protein kinase Cα. Cancer Res. 2002, 62, 3538–3543. [Google Scholar] [PubMed]
- Thulasiraman, P.; McAndrews, D.J.; Mohiudddin, I.Q. Curcumin restores sensitivity to retinoic acid in triple negative breast cancer cells. BMC Cancer 2014, 14, 724. [Google Scholar] [CrossRef] [PubMed]
- Meads, M.B.; Gatenby, R.A.; Dalton, W.S. Environment-mediated drug resistance: A major contributor to minimal residual disease. Nat. Rev. Cancer 2009, 9, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Li, C.; Zhang, Y.; Wang, L.; Peng, L.; Cheng, H.; Wang, W.; Chu, Y.; Xu, M.; Cheng, T. Phosphoinositide-dependent kinase 1 regulates leukemia stem cell maintenance in MLL-AF9-induced murine acute myeloid leukemia. Biochem. Biophys. Res. Commun. 2015, 459, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Zabkiewicz, J.; Pearn, L.; Hills, R.K.; Morgan, R.G.; Tonks, A.; Burnett, A.K.; Darley, R.L. The PDK1 master kinase is over-expressed in acute myeloid leukemia and promotes PKC-mediated survival of leukemic blasts. Haematologica 2014, 99, 858–864. [Google Scholar] [CrossRef] [PubMed]
- Cheong, J.W.; Eom, J.I.; Maeng, H.Y.; Lee, S.T.; Hahn, J.S.; Ko, Y.W.; Min, Y.H. Phosphatase and tensin homologue phosphorylation in the C-terminal regulatory domain is frequently observed in acute myeloid leukaemia and associated with poor clinical outcome. Br. J. Haematol. 2003, 122, 454–456. [Google Scholar] [CrossRef] [PubMed]
- Kornblau, S.M.; Womble, M.; Qiu, Y.H.; Jackson, C.E.; Chen, W.; Konopleva, M.; Estey, E.H.; Andreeff, M. Simultaneous activation of multiple signal transduction pathways confers poor prognosis in acute myelogenous leukemia. Blood 2006, 108, 2358–2365. [Google Scholar] [CrossRef] [PubMed]
- Grandage, V.; Gale, R.; Linch, D.; Khwaja, A. PI3-kinase/Akt is constitutively active in primary acute myeloid leukaemia cells and regulates survival and chemoresistance via NF-kB, MAPkinase and p53 pathways. Leukemia 2005, 19, 586–594. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef]
- Miyashita, T.; Krajewski, S.; Krajewska, M.; Wang, H.G.; Lin, H.; Liebermann, D.A.; Hoffman, B.; Reed, J.C. Tumor suppressor p53 is a regulator of bcl-2 and bax gene expression in vitro and in vivo. Oncogene 1994, 9, 1799–1805. [Google Scholar] [PubMed]
- Lowe, S.W.; Bodis, S.; McClatchey, A.; Remington, L.; Ruley, H.E.; Fisher, D.E.; Housman, D.E.; Jacks, T. p53 status and the efficacy of cancer therapy in vivo. Science 1994, 266, 807–810. [Google Scholar] [CrossRef] [PubMed]
- Prokop, A.; Wieder, T.; Sturm, I.; Ebmann, F.; Seeger, K.; Wuchter, C.; Ludwig, W.; Henze, G.; Dörken, B.; Daniel, P. Relapse in childhood acute lymphoblastic leukemia is associated with a decrease of the Bax/Bcl-2 ratio and loss of spontaneous caspase-3 processing in vivo. Leukemia 2000, 14, 1606–1613. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.; Steelman, L.; Abrams, S.; Bertrand, F.; Ludwig, D.; Bäsecke, J.; Libra, M.; Stivala, F.; Milella, M.; Tafuri, A. Targeting survival cascades induced by activation of Ras/Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways for effective leukemia therapy. Leukemia 2008, 22, 708–722. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Brachova, P.; Yang, S.; Xiong, Z.; Zhang, Y.; Thiel, K.W.; Leslie, K.K. Knockdown of MTDH sensitizes endometrial cancer cells to cell death induction by death receptor ligand TRAIL and HDAC inhibitor LBH589 co-treatment. PLoS ONE 2011, 6, e20920. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Fujita, N.; Tsuruo, T. Regulation of kinase activity of 3-phosphoinositide-dependent protein kinase-1 by binding to 14–3-3. J. Biol. Chem. 2002, 277, 39360–39367. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Sato, S.; Ishida, A.; Tsuruo, T. Involvement of Hsp90 in signaling and stability of 3-phosphoinositide-dependent kinase-1. J. Biol. Chem. 2002, 277, 10346–10353. [Google Scholar] [CrossRef] [PubMed]
- Kurata, A.; Katayama, R.; Watanabe, T.; Tsuruo, T.; Fujita, N. TUSC4/NPRL2, a novel PDK1-interacting protein, inhibits PDK1 tyrosine phosphorylation and its downstream signaling. Cancer Sci. 2008, 99, 1827–1834. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Liu, H.; Qiu, Q.; Zhang, Z.; Gu, Y.; He, Z. TCRP1 promotes NIH/3T3 cell transformation by over-activating PDK1 and AKT1. Oncogenesis 2017, 6, e323. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, C.; Gu, Y.; Zhang, Z.; Zheng, G.; He, Z. TCRP1 contributes to cisplatin resistance by preventing Pol β degradation in lung cancer cells. Mol. Cell. Biochem. 2015, 398, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Fan, S.; Liu, B.; Zheng, G.; Yu, Y.; Ouyang, Y.; He, Z. TCRP1 promotes radioresistance of oral squamous cell carcinoma cells via Akt signal pathway. Mol. Cell. Biochem. 2011, 357, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Ericson, K.; Gan, C.; Cheong, I.; Rago, C.; Samuels, Y.; Velculescu, V.E.; Kinzler, K.W.; Huso, D.L.; Vogelstein, B.; Papadopoulos, N. Genetic inactivation of AKT1, AKT2, and PDPK1 in human colorectal cancer cells clarifies their roles in tumor growth regulation. Proc. Natl. Acad. Sci. USA 2010, 107, 2598–2603. [Google Scholar] [CrossRef] [PubMed]
- Ellwood-Yen, K.; Keilhack, H.; Kunii, K.; Dolinski, B.; Connor, Y.; Hu, K.; Nagashima, K.; O’Hare, E.; Erkul, Y.; Di Bacco, A. PDK1 attenuation fails to prevent tumor formation in PTEN-deficient transgenic mouse models. Cancer Res. 2011, 71, 3052–3065. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Li, Z.; Lee, P.L.; Guan, P.; Aau, M.Y.; Lee, S.T.; Feng, M.; Lim, C.Z.; Lee, E.Y.J.; Wee, Z.N. PDK1 signaling toward PLK1-MYC activation confers oncogenic transformation, tumor-initiating cell activation, and resistance to mTOR-targeted therapy. Cancer Discov. 2013, 3, 1156–1171. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.J.; Liu, H.; Ridky, T.W.; Cassarino, D.; Segal, E.; Chang, H.Y. Module map of stem cell genes guides creation of epithelial cancer stem cells. Cell Stem Cell 2008, 2, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Pan, D. The hippo signaling pathway in development and cancer. Dev. Cell 2010, 19, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Ye, X.; Yu, J.; Li, L.; Li, W.; Li, S.; Yu, J.; Lin, J.D.; Wang, C.Y.; Chinnaiyan, A.M.; et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008, 22, 1962–1971. [Google Scholar] [CrossRef] [PubMed]
- Fan, R.; Kim, N.G.; Gumbiner, B.M. Regulation of Hippo pathway by mitogenic growth factors via phosphoinositide 3-kinase and phosphoinositide-dependent kinase-1. Proc. Natl. Acad. Sci. USA 2013, 110, 2569–2574. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A. Targeting PI3K signalling in cancer: Opportunities, challenges and limitations. Nat. Rev. Cancer 2009, 9, 550–562. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef] [PubMed]
- Juric, D.; Rodon, J.; Gonzalez-Angulo, A.; Burris, H.; Bendell, J.; Berlin, J.; Middleton, M.; Bootle, D.; Boehm, M.; Schmitt, A. BYL719, a next generation PI3K alpha specific inhibitor: Preliminary safety. In Proceedings of the AACR 103rd Annual Meeting, Chicago, IL, USA, 6–10 April 2013. [Google Scholar]
- Juric, D.; Krop, I.; Ramanathan, R.; Xiao, J.; Sanabria, S.; Wilson, T.; Choi, Y.; Parmar, H.; Hsu, J.; Baselga, J. GDC-0032, a beta isoform-sparing PI3K inhibitor: Results of a first-in-human dose escalation study. In Proceedings of the AACR 104th Annual Meeting, Washington, DC, USA, 31 March–4 April 2012. [Google Scholar]
- Elkabets, M.; Vora, S.; Juric, D.; Morse, N.; Mino-Kenudson, M.; Muranen, T.; Tao, J.; Campos, A.B.; Rodon, J.; Ibrahim, Y.H.; et al. mTORC1 inhibition is required for sensitivity to PI3K p110alpha inhibitors in PIK3CA-mutant breast cancer. Sci. Transl. Med. 2013, 5, 196ra99. [Google Scholar] [CrossRef] [PubMed]
- Dibble, C.C.; Cantley, L.C. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 2015, 25, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Castel, P.; Ellis, H.; Bago, R.; Toska, E.; Razavi, P.; Carmona, F.J.; Kannan, S.; Verma, C.S.; Dickler, M.; Chandarlapaty, S. PDK1-SGK1 signaling sustains AKT-independent mTORC1 activation and confers resistance to PI3Kα inhibition. Cancer Cell 2016, 30, 229–242. [Google Scholar] [CrossRef] [PubMed]
- Webb, A.E.; Brunet, A. FOXO transcription factors: Key regulators of cellular quality control. Trends Biochem. Sci. 2014, 39, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.T.; Campbell, D.G.; Morrice, N.; Auld, G.C.; Shpiro, N.; Marquez, R.; Peggie, M.; Bain, J.; Bloomberg, G.B.; Grahammer, F.; et al. Exploitation of KESTREL to identify NDRG family members as physiological substrates for SGK1 and GSK3. Biochem. J. 2004, 384, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Sommer, E.M.; Dry, H.; Cross, D.; Guichard, S.; Davies, B.R.; Alessi, D.R. Elevated SGK1 predicts resistance of breast cancer cells to Akt inhibitors. Biochem. J. 2013, 452, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Zurashvili, T.; Cordón-Barris, L.; Ruiz-Babot, G.; Zhou, X.; Lizcano, J.M.; Gómez, N.; Giménez-Llort, L.; Bayascas, J.R. Interaction of PDK1 with phosphoinositides is essential for neuronal differentiation but dispensable for neuronal survival. Mol. Cell. Biol. 2013, 33, 1027–1040. [Google Scholar] [CrossRef] [PubMed]
- Scortegagna, M.; Lau, E.; Zhang, T.; Feng, Y.; Sereduk, C.; Yin, H.; De, S.K.; Meeth, K.; Platt, J.T.; Langdon, C.G.; et al. PDK1 and SGK3 Contribute to the Growth of BRAF-Mutant Melanomas and Are Potential Therapeutic Targets. Cancer Res. 2015, 75, 1399–1412. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.M.; Bulman, C.; McMahon, M. BRAFV600E cooperates with PI3K signaling, independent of AKT, to regulate melanoma cell proliferation. Mol. Cancer Res. 2014, 12, 447–463. [Google Scholar] [CrossRef] [PubMed]
- Bago, R.; Sommer, E.; Castel, P.; Crafter, C.; Bailey, F.P.; Shpiro, N.; Baselga, J.; Cross, D.; Eyers, P.A.; Alessi, D.R. The hVps34-SGK3 pathway alleviates sustained PI3K/Akt inhibition by stimulating mTORC1 and tumour growth. EMBO J. 2016, 35, 1902–1922. [Google Scholar] [CrossRef] [PubMed]
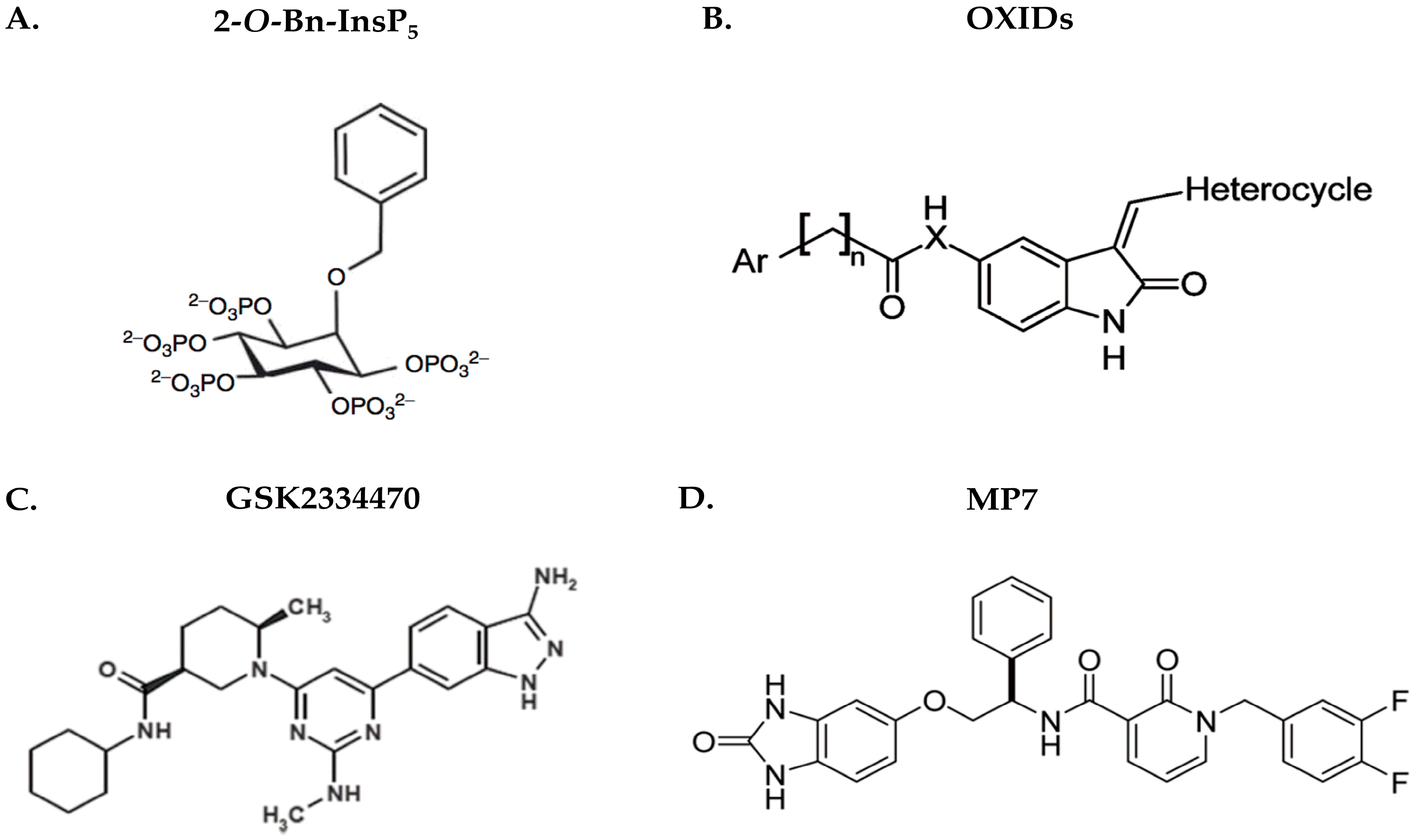
- Barile, E.; De, S.K.; Pellecchia, M. PDK1 inhibitors. Pharm. Pat. Anal. 2012, 1, 145–163. [Google Scholar] [CrossRef] [PubMed]
- Peifer, C.; Alessi, D.R. Small-Molecule Inhibitors of PDK1. ChemMedChem 2008, 3, 1810–1838. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Fairservice, A.; Deak, M.; Kular, G.S.; Prescott, A.R.; Downes, C.P.; Safrany, S.T.; Alessi, D.R.; van Aalten, D.M. Structural insights into the regulation of PDK1 by phosphoinositides and inositol phosphates. EMBO J. 2004, 23, 3918–3928. [Google Scholar] [CrossRef] [PubMed]
- Maffucci, T.; Piccolo, E.; Cumashi, A.; Iezzi, M.; Riley, A.M.; Saiardi, A.; Godage, H.Y.; Rossi, C.; Broggini, M.; Iacobelli, S. Inhibition of the phosphatidylinositol 3-kinase/Akt pathway by inositol pentakisphosphate results in antiangiogenic and antitumor effects. Cancer Res. 2005, 65, 8339–8349. [Google Scholar] [CrossRef] [PubMed]
- Nesi, G.; Sestito, S.; Mey, V.; Ricciardi, S.; Falasca, M.; Danesi, R.; Lapucci, A.; Breschi, M.C.; Fogli, S.; Rapposelli, S. Synthesis of novel 3, 5-disubstituted-2-oxindole derivatives as antitumor agents against human nonsmall cell lung cancer. ACS Med. Chem. Lett. 2013, 4, 1137–1141. [Google Scholar] [CrossRef] [PubMed]
- Sestito, S.; Nesi, G.; Daniele, S.; Martelli, A.; Digiacomo, M.; Borghini, A.; Pietra, D.; Calderone, V.; Lapucci, A.; Falasca, M. Design and synthesis of 2-oxindole based multi-targeted inhibitors of PDK1/Akt signaling pathway for the treatment of glioblastoma multiforme. Eur. J. Med. Chem. 2015, 105, 274–288. [Google Scholar] [CrossRef] [PubMed]
- Najafov, A.; Sommer, E.M.; Axten, J.M.; Deyoung, M.P.; Alessi, D.R. Characterization of GSK2334470, a novel and highly specific inhibitor of PDK1. Biochem. J. 2011, 433, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Leroux, A.E.; Schulze, J.; Biondi, R.M. AGC kinases, mechanisms of regulation and innovative drug development. Semin. Cancer Biol. 2017, in press. [Google Scholar] [CrossRef] [PubMed]
- Biondi, R.M. Phosphoinositide-dependent protein kinase 1, a sensor of protein conformation. Trends Biochem. Sci. 2004, 29, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Arencibia, J.M.; Pastor-Flores, D.; Bauer, A.F.; Schulze, J.O.; Biondi, R.M. AGC protein kinases: From structural mechanism of regulation to allosteric drug development for the treatment of human diseases. Biochim. Biophys. Acta 2013, 1834, 1302–1321. [Google Scholar] [CrossRef] [PubMed]
- Najafov, A.; Shpiro, N.; Alessi, D.R. Akt is efficiently activated by PIF-pocket-and PtdIns (3, 4, 5) P3-dependent mechanisms leading to resistance to PDK1 inhibitors. Biochem. J. 2012, 448, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Jansen, V.M.; Bhola, N.E.; Bauer, J.A.; Formisano, L.; Lee, K.-M.; Hutchinson, K.E.; Witkiewicz, A.K.; Moore, P.D.; Estrada, M.V.; Sánchez, V. Kinome-wide RNA interference screen reveals a role for PDK1 in acquired resistance to CDK4/6 inhibition in ER-positive breast cancer. Cancer Res. 2017, 77, 2488–2499. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Huang, X.; Liu, H.; Xiao, F.; Wei, J.; You, L.; Qian, W. PDK1 inhibitor GSK2334470 exerts antitumor activity in multiple myeloma and forms a novel multitargeted combination with dual mTORC1/C2 inhibitor PP242. Oncotarget 2017, 8, 39185–39197. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.; Enquist, J.; Iwig, J.; Binnerts, M.E.; Jamieson, G.; Fox, J.A.; Craig, A.R. Abstract C198: PDK1 inhibitors SNS-229 and SNS-510 cause pathway modulation, apoptosis and tumor regression in hematologic cancer models in addition to solid tumors. Mol. Cancer. Ther. 2015, 14, C198. [Google Scholar] [CrossRef]
- Carceller, V. AACR-NCI-EORTC—27th International Symposium—Molecular Targets and Cancer Therapeutics (November 5–9, 2015—Boston, Massachusetts, USA). Drugs Today 2015, 51, 669–675. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, K.; Shumway, S.D.; Sathyanarayanan, S.; Chen, A.H.; Dolinski, B.; Xu, Y.; Keilhack, H.; Nguyen, T.; Wiznerowicz, M.; Li, L. Genetic and pharmacological inhibition of PDK1 in cancer cells characterization of a selective allosteric kinase inhibitor. J. Biol. Chem. 2011, 286, 6433–6448. [Google Scholar] [CrossRef] [PubMed]
- Daniele, S.; Sestito, S.; Pietrobono, D.; Giacomelli, C.; Chiellini, G.; Di Maio, D.; Marinelli, L.; Novellino, E.; Martini, C.; Rapposelli, S. Dual inhibition of PDK1 and Aurora kinase A: An effective strategy to induce differentiation and apoptosis of human glioblastoma multiforme stem cells. ACS Chem. Neurosci. 2016, 8, 100–114. [Google Scholar] [CrossRef] [PubMed]
- Biddinger, S.B.; Kahn, C.R. From mice to men: Insights into the insulin resistance syndromes. Annu. Rev. Physiol. 2006, 68, 123–158. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Leevers, S.J.; Ahmadi, K.; Timms, J.; Katso, R.; Driscoll, P.C.; Woscholski, R.; Parker, P.J.; Waterfield, M.D. Synthesis and function of 3-phosphorylated inositol lipids. Annu. Rev. Biochem. 2001, 70, 535–602. [Google Scholar] [CrossRef] [PubMed]
- Bayascas, J.R.; Leslie, N.R.; Parsons, R.; Fleming, S.; Alessi, D.R. Hypomorphic mutation of PDK1 suppresses tumorigenesis in PTEN(+/−) mice. Curr. Biol. 2005, 15, 1839–1846. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, C.; Calleja, V.; Ferro, R.; Fantin, A.; Riley, A.M.; Potter, B.V.L.; Brennan, C.H.; Maffucci, T.; Larijani, B.; Falasca, M. A Small Molecule Inhibitor of PDK1/PLCγ1 Interaction Blocks Breast and Melanoma Cancer Cell Invasion. Sci. Rep. 2016, 6, 26142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, L.; Voskas, D.; Woodgett, J. Activation of PDK-1 maintains mouse embryonic stem cell self-renewal in a PKB-dependent manner. Oncogene 2013, 32, 5397–5408. [Google Scholar] [CrossRef] [PubMed]
- Signore, M.; Pelacchi, F.; Di Martino, S.; Runci, D.; Biffoni, M.; Giannetti, S.; Morgante, L.; De Majo, M.; Petricoin, E.; Stancato, L. Combined PDK1 and CHK1 inhibition is required to kill glioblastoma stem-like cells in vitro and in vivo. Cell Death Dis. 2014, 5, e1223. [Google Scholar] [CrossRef] [PubMed]
- Ferro, R.; Falasca, M. Emerging role of the KRAS-PDK1 axis in pancreatic cancer. World J. Gastroenterol. 2014, 20, 10752–10757. [Google Scholar] [CrossRef] [PubMed]
- Eser, S.; Reiff, N.; Messer, M.; Seidler, B.; Gottschalk, K.; Dobler, M.; Hieber, M.; Arbeiter, A.; Klein, S.; Kong, B. Selective requirement of PI3K/PDK1 signaling for Kras oncogene-driven pancreatic cell plasticity and cancer. Cancer Cell 2013, 23, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Gagliardi, P.A.; Puliafito, A.; Primo, L. PDK1: At the crossroad of cancer signaling pathways. Semin. Cancer Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Sargeant, A.M.; Klein, R.D.; Rengel, R.C.; Clinton, S.K.; Kulp, S.K.; Kashida, Y.; Yamaguchi, M.; Wang, X.; Chen, C.-S. Chemopreventive and bioenergetic signaling effects of PDK1/Akt pathway inhibition in a transgenic mouse model of prostate cancer. Toxicol. Pathol. 2007, 35, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.X.; Packer, M.D.; Huang, J.; Akhmametyeva, E.M.; Kulp, S.K.; Chen, C.-S.; Giovannini, M.; Jacob, A.; Welling, D.B.; Chang, L.-S. Growth inhibitory and anti-tumour activities of OSU-03012, a novel PDK-1 inhibitor, on vestibular schwannoma and malignant schwannoma cells. Eur. J. Cancer 2009, 45, 1709–1720. [Google Scholar] [CrossRef] [PubMed]
- Pietri, M.; Dakowski, C.; Hannaoui, S.; Alleaume-Butaux, A.; Hernandez-Rapp, J.; Ragagnin, A.; Mouillet-Richard, S.; Haik, S.; Bailly, Y.; Peyrin, J.-M. PDK1 decreases TACE-mediated [alpha]-secretase activity and promotes disease progression in prion and Alzheimer's diseases. Nat. Med. 2013, 19, 1124–1131. [Google Scholar] [CrossRef] [PubMed]
- Feldman, R.I.; Wu, J.M.; Polokoff, M.A.; Kochanny, M.J.; Dinter, H.; Zhu, D.; Biroc, S.L.; Alicke, B.; Bryant, J.; Yuan, S. Novel small molecule inhibitors of 3-phosphoinositide-dependent kinase-1. J. Biol. Chem. 2005, 280, 19867–19874. [Google Scholar] [CrossRef] [PubMed]
- Paweletz, C.P.; Andersen, J.N.; Pollock, R.; Nagashima, K.; Hayashi, M.L.; Shangshuan, U.Y.; Guo, H.; Bobkova, E.V.; Xu, Z.; Northrup, A. Identification of direct target engagement biomarkers for kinase-targeted therapeutics. PLoS ONE 2011, 6, e26459. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Harris, T.K. Role of the PH domain in regulating in vitro autophosphorylation events required for reconstitution of PDK1 catalytic activity. Bioorg. Chem. 2006, 34, 200–223. [Google Scholar] [CrossRef] [PubMed]




© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Emmanouilidi, A.; Falasca, M. Targeting PDK1 for Chemosensitization of Cancer Cells. Cancers 2017, 9, 140. https://doi.org/10.3390/cancers9100140
Emmanouilidi A, Falasca M. Targeting PDK1 for Chemosensitization of Cancer Cells. Cancers. 2017; 9(10):140. https://doi.org/10.3390/cancers9100140
Chicago/Turabian StyleEmmanouilidi, Aikaterini, and Marco Falasca. 2017. "Targeting PDK1 for Chemosensitization of Cancer Cells" Cancers 9, no. 10: 140. https://doi.org/10.3390/cancers9100140
APA StyleEmmanouilidi, A., & Falasca, M. (2017). Targeting PDK1 for Chemosensitization of Cancer Cells. Cancers, 9(10), 140. https://doi.org/10.3390/cancers9100140