
Evolving Significance and Future Relevance of Anti-Angiogenic Activity of mTOR Inhibitors in Cancer Therapy


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
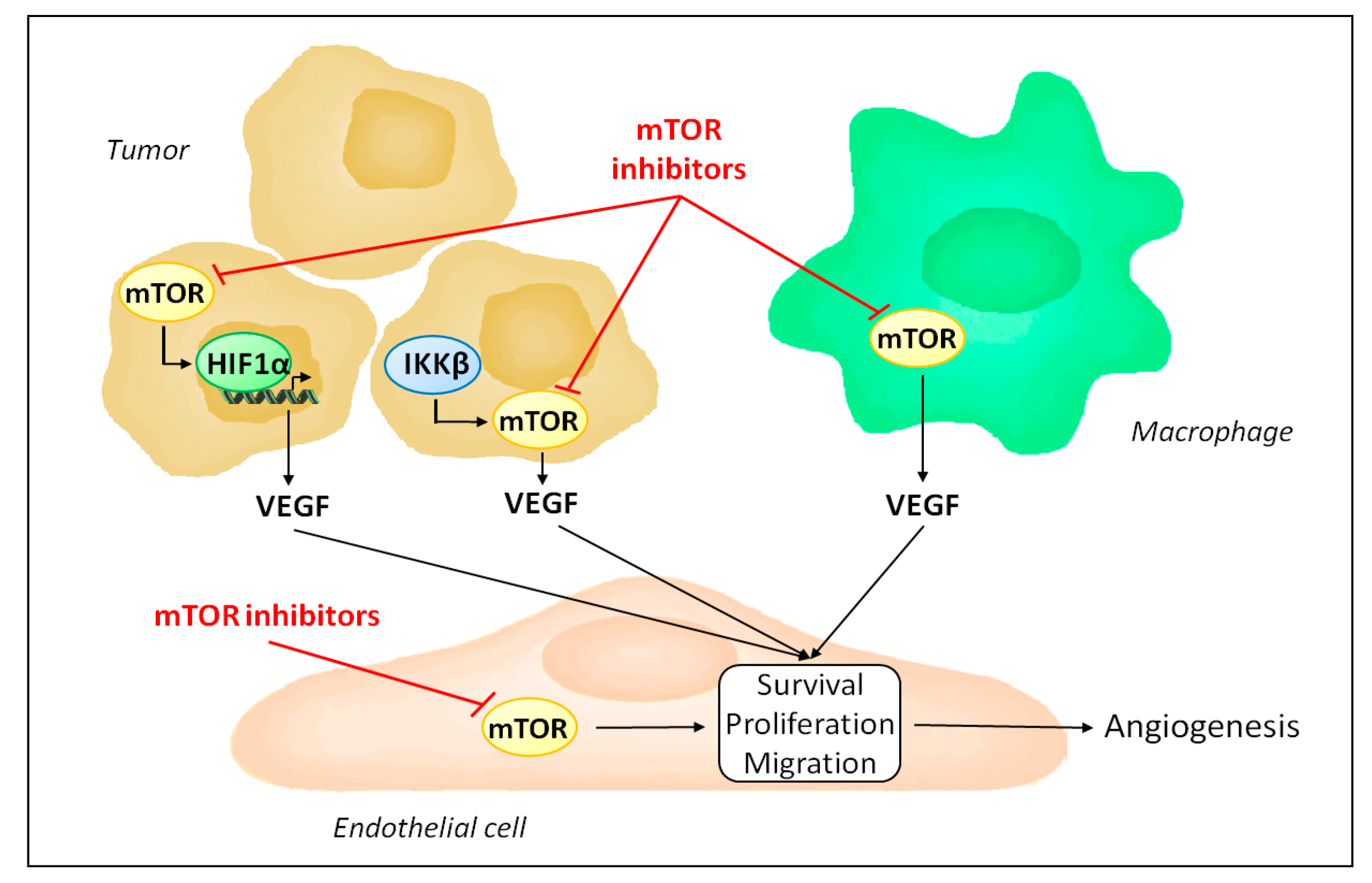
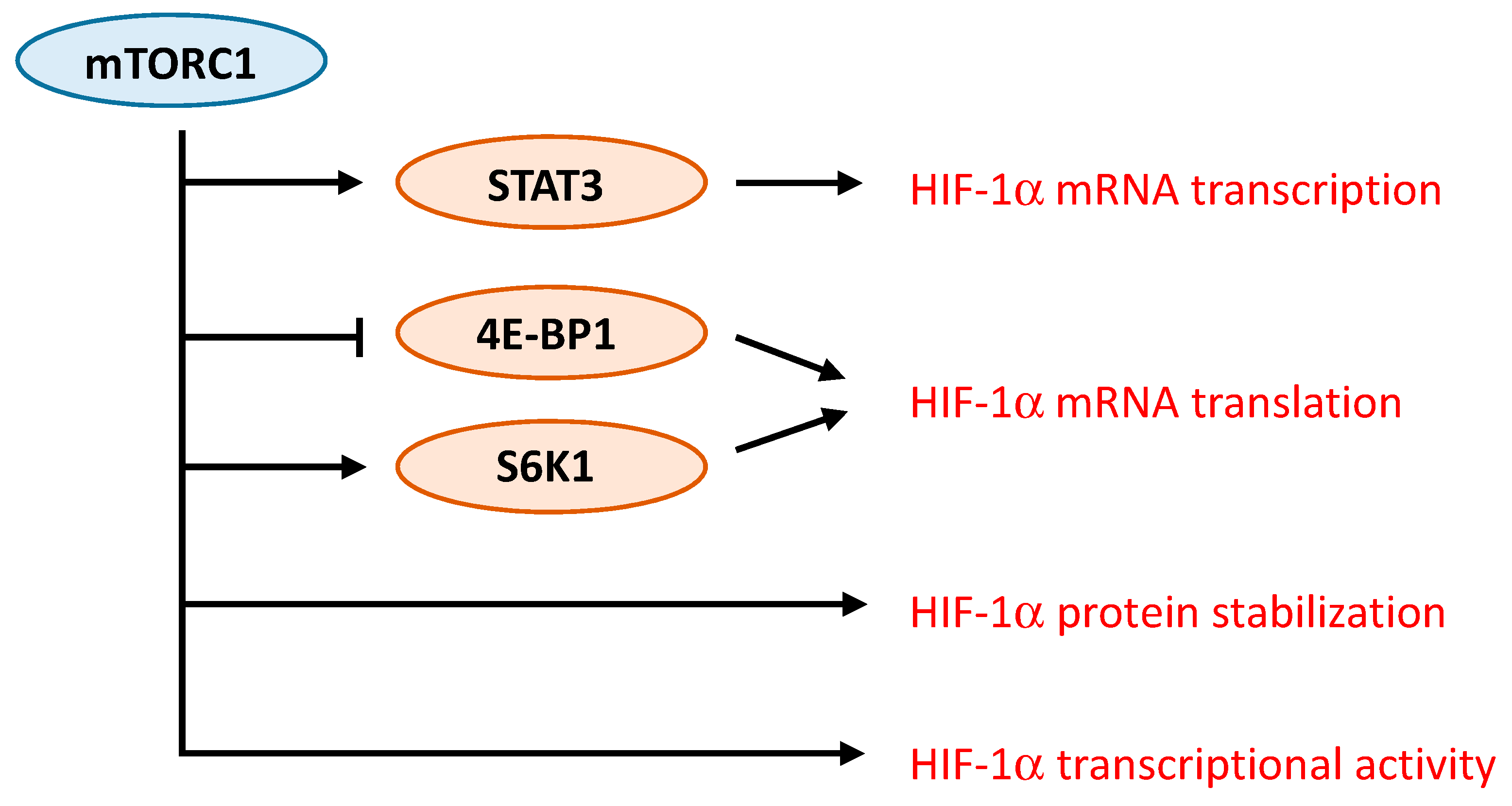
2. mTOR Inhibitors and Tumor Angiogenesis
3. Resistances to the Anti-Angiogenic Effects of mTOR Inhibitors
4. Combined Therapies to Increase the Anti-Angiogenic Efficacy of mTOR Inhibitors
5. mTOR Inhibitors and Normalization of Tumor Vasculature
6. mTOR Inhibitors and Tumor Endothelial Barrier
7. Biomarkers of Efficacy of mTOR Inhibitors
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Saxton, R.A.; Sabatini, D.M. Mtor signaling in growth, metabolism, and disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Shimobayashi, M.; Hall, M.N. Making new contacts: The mtor network in metabolism and signalling crosstalk. Nat. Rev. Mol. Cell Biol. 2014, 15, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. Regulation of mtorc1 and its impact on gene expression at a glance. J. Cell Sci. 2013, 126, 1713–1719. [Google Scholar] [CrossRef] [PubMed]
- Gaubitz, C.; Prouteau, M.; Kusmider, B.; Loewith, R. Torc2 structure and function. Trends Biochem. Sci. 2016, 41, 532–545. [Google Scholar] [CrossRef] [PubMed]
- Oh, W.J.; Jacinto, E. Mtor complex 2 signaling and functions. Cell Cycle 2011, 10, 2305–2316. [Google Scholar] [CrossRef] [PubMed]
- Grabiner, B.C.; Nardi, V.; Birsoy, K.; Possemato, R.; Shen, K.; Sinha, S.; Jordan, A.; Beck, A.H.; Sabatini, D.M. A diverse array of cancer-associated mtor mutations are hyperactivating and can predict rapamycin sensitivity. Cancer Discov. 2014, 4, 554–563. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Montalto, G.; Cervello, M.; Nicoletti, F.; Fagone, P.; Malaponte, G.; Mazzarino, M.C.; et al. Mutations and deregulation of RAS/RAF/MEK/ERK and PI3K/PTEN/AKT/mTOR cascades which alter therapy response. Oncotarget 2012, 3, 954–987. [Google Scholar] [CrossRef] [PubMed]
- Dormond-Meuwly, A.; Dufour, M.; Demartines, N.; Dormond, O. Mtor inhibition and the tumor vasculature. Current Angiogenesis 2012, 1, 11–19. [Google Scholar] [CrossRef]
- Xie, J.; Wang, X.; Proud, C.G. Mtor inhibitors in cancer therapy. F1000Res 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Zaytseva, Y.Y.; Valentino, J.D.; Gulhati, P.; Evers, B.M. Mtor inhibitors in cancer therapy. Cancer Lett. 2012, 319, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Thoreen, C.C.; Kang, S.A.; Chang, J.W.; Liu, Q.; Zhang, J.; Gao, Y.; Reichling, L.J.; Sim, T.; Sabatini, D.M.; Gray, N.S. An atp-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mtorc1. J. Biol. Chem. 2009, 284, 8023–8032. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged rapamycin treatment inhibits mtorc2 assembly and akt/pkb. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, D.; Colombi, M.; Moroni, C.; Hall, M.N. Rapamycin passes the torch: A new generation of mtor inhibitors. Nat. Rev. Drug Discov. 2011, 10, 868–880. [Google Scholar] [CrossRef] [PubMed]
- Rodrik-Outmezguine, V.S.; Okaniwa, M.; Yao, Z.; Novotny, C.J.; McWhirter, C.; Banaji, A.; Won, H.; Wong, W.; Berger, M.; de Stanchina, E.; et al. Overcoming mtor resistance mutations with a new-generation mtor inhibitor. Nature 2016, 534, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Douros, J.; Suffness, M. New antitumor substances of natural origin. Cancer Treat. Rev. 1981, 8, 63–87. [Google Scholar] [CrossRef]
- Populo, H.; Lopes, J.M.; Soares, P. The mtor signalling pathway in human cancer. Int. J. Mol. Sci. 2012, 13, 1886–1918. [Google Scholar] [CrossRef] [PubMed]
- Dufour, M.; Dormond-Meuwly, A.; Demartines, N.; Dormond, O. Targeting the mammalian target of rapamycin (mtor) in cancer therapy: Lessons from past and future perspectives. Cancers 2011, 3, 2478–2500. [Google Scholar] [CrossRef] [PubMed]
- Hudes, G.; Carducci, M.; Tomczak, P.; Dutcher, J.; Figlin, R.; Kapoor, A.; Staroslawska, E.; Sosman, J.; McDermott, D.; Bodrogi, I.; et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 2271–2281. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.C.; Shah, M.H.; Ito, T.; Bohas, C.L.; Wolin, E.M.; Van Cutsem, E.; Hobday, T.J.; Okusaka, T.; Capdevila, J.; de Vries, E.G.; et al. Everolimus for advanced pancreatic neuroendocrine tumors. N. Engl. J. Med. 2011, 364, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Campone, M.; Piccart, M.; Burris, H.A., 3rd; Rugo, H.S.; Sahmoud, T.; Noguchi, S.; Gnant, M.; Pritchard, K.I.; Lebrun, F.; et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N. Engl. J. Med. 2012, 366, 520–529. [Google Scholar] [CrossRef] [PubMed]
- Faes, S.; Demartines, N.; Dormond, O. Resistance to mtorc1 inhibitors in cancer therapy: From kinase mutations to intratumoral heterogeneity of kinase activity. Oxid. Med. Cell. Longev. 2017, 2017, 1726078. [Google Scholar] [CrossRef] [PubMed]
- Potente, M.; Gerhardt, H.; Carmeliet, P. Basic and therapeutic aspects of angiogenesis. Cell 2011, 146, 873–887. [Google Scholar] [CrossRef] [PubMed]
- De Palma, M.; Biziato, D.; Petrova, T.V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 2017, 17, 457–474. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.R.; Hay, M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2011, 11, 393–410. [Google Scholar] [CrossRef] [PubMed]
- Dey, N.; De, P.; Brian, L.J. Evading anti-angiogenic therapy: Resistance to anti-angiogenic therapy in solid tumors. Am. J. Transl. Res. 2015, 7, 1675–1698. [Google Scholar] [CrossRef] [PubMed]
- Weis, S.M.; Cheresh, D.A. Tumor angiogenesis: Molecular pathways and therapeutic targets. Nat. Med. 2011, 17, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Adamis, A.P. Ten years of anti-vascular endothelial growth factor therapy. Nat. Rev. Drug Discov. 2016, 15, 385–403. [Google Scholar] [CrossRef] [PubMed]
- Akselband, Y.; Harding, M.W.; Nelson, P.A. Rapamycin inhibits spontaneous and fibroblast growth factor beta-stimulated proliferation of endothelial cells and fibroblasts. Transplant. Proc. 1991, 23, 2833–2836. [Google Scholar] [PubMed]
- Mohacsi, P.J.; Tuller, D.; Hulliger, B.; Wijngaard, P.L. Different inhibitory effects of immunosuppressive drugs on human and rat aortic smooth muscle and endothelial cell proliferation stimulated by platelet-derived growth factor or endothelial cell growth factor. J. Heart Lung Transplant. 1997, 16, 484–492. [Google Scholar] [PubMed]
- Yu, Y.; Sato, J.D. Map kinases, phosphatidylinositol 3-kinase, and p70 s6 kinase mediate the mitogenic response of human endothelial cells to vascular endothelial growth factor. J. Cell. Physiol. 1999, 178, 235–246. [Google Scholar] [CrossRef]
- Vinals, F.; Chambard, J.C.; Pouyssegur, J. P70 s6 kinase-mediated protein synthesis is a critical step for vascular endothelial cell proliferation. J. Biol. Chem. 1999, 274, 26776–26782. [Google Scholar] [CrossRef] [PubMed]
- Marimpietri, D.; Nico, B.; Vacca, A.; Mangieri, D.; Catarsi, P.; Ponzoni, M.; Ribatti, D. Synergistic inhibition of human neuroblastoma-related angiogenesis by vinblastine and rapamycin. Oncogene 2005, 24, 6785–6795. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Zhao, Y.; Yu, L.; Xu, S.; Fu, G. Microrna-21 mediates the rapamycin-induced suppression of endothelial proliferation and migration. FEBS Lett. 2013, 587, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Humar, R.; Kiefer, F.N.; Berns, H.; Resink, T.J.; Battegay, E.J. Hypoxia enhances vascular cell proliferation and angiogenesis in vitro via rapamycin (mtor)-dependent signaling. FASEB J. 2002, 16, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Bruns, C.J.; Koehl, G.E.; Guba, M.; Yezhelyev, M.; Steinbauer, M.; Seeliger, H.; Schwend, A.; Hoehn, A.; Jauch, K.W.; Geissler, E.K. Rapamycin-induced endothelial cell death and tumor vessel thrombosis potentiate cytotoxic therapy against pancreatic cancer. Clin. Cancer Res. 2004, 10, 2109–2119. [Google Scholar] [CrossRef] [PubMed]
- Barilli, A.; Visigalli, R.; Sala, R.; Gazzola, G.C.; Parolari, A.; Tremoli, E.; Bonomini, S.; Simon, A.; Closs, E.I.; Dall’Asta, V.; et al. In human endothelial cells rapamycin causes mtorc2 inhibition and impairs cell viability and function. Cardiovasc. Res. 2008, 78, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Dormond, O.; Madsen, J.C.; Briscoe, D.M. The effects of mtor-akt interactions on anti-apoptotic signaling in vascular endothelial cells. J. Biol. Chem. 2007, 282, 23679–23686. [Google Scholar] [CrossRef] [PubMed]
- Matter, C.M.; Rozenberg, I.; Jaschko, A.; Greutert, H.; Kurz, D.J.; Wnendt, S.; Kuttler, B.; Joch, H.; Grunenfelder, J.; Zund, G.; et al. Effects of tacrolimus or sirolimus on proliferation of vascular smooth muscle and endothelial cells. J. Cardiovasc. Pharmacol. 2006, 48, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Moss, S.C.; Lightell, D.J., Jr.; Marx, S.O.; Marks, A.R.; Woods, T.C. Rapamycin regulates endothelial cell migration through regulation of the cyclin-dependent kinase inhibitor p27kip1. J. Biol. Chem. 2010, 285, 11991–11997. [Google Scholar] [CrossRef] [PubMed]
- Guba, M.; von Breitenbuch, P.; Steinbauer, M.; Koehl, G.; Flegel, S.; Hornung, M.; Bruns, C.J.; Zuelke, C.; Farkas, S.; Anthuber, M.; et al. Rapamycin inhibits primary and metastatic tumor growth by antiangiogenesis: Involvement of vascular endothelial growth factor. Nat. Med. 2002, 8, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, E.T.; Cao, C.; Niermann, K.; Mu, Y.; Zeng, F.; Hallahan, D.E.; Lu, B. Enhanced radiation damage of tumor vasculature by mtor inhibitors. Oncogene 2005, 24, 5414–5422. [Google Scholar] [CrossRef] [PubMed]
- Dormond-Meuwly, A.; Roulin, D.; Dufour, M.; Benoit, M.; Demartines, N.; Dormond, O. The inhibition of mapk potentiates the anti-angiogenic efficacy of mtor inhibitors. Biochem. Biophys. Res. Commun. 2011, 407, 714–719. [Google Scholar] [CrossRef] [PubMed]
- Del Bufalo, D.; Ciuffreda, L.; Trisciuoglio, D.; Desideri, M.; Cognetti, F.; Zupi, G.; Milella, M. Antiangiogenic potential of the mammalian target of rapamycin inhibitor temsirolimus. Cancer Res. 2006, 66, 5549–5554. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Shen, N.; Mendoza, A.; Khanna, C.; Helman, L.J. Cci-779 inhibits rhabdomyosarcoma xenograft growth by an antiangiogenic mechanism linked to the targeting of mTOR/HIF-1ALPHA/VEGF signaling. Neoplasia 2006, 8, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Mabuchi, S.; Altomare, D.A.; Connolly, D.C.; Klein-Szanto, A.; Litwin, S.; Hoelzle, M.K.; Hensley, H.H.; Hamilton, T.C.; Testa, J.R. Rad001 (everolimus) delays tumor onset and progression in a transgenic mouse model of ovarian cancer. Cancer Res. 2007, 67, 2408–2413. [Google Scholar] [CrossRef] [PubMed]
- Huynh, H.; Chow, K.H.; Soo, K.C.; Toh, H.C.; Choo, S.P.; Foo, K.F.; Poon, D.; Ngo, V.C.; Tran, E. Rad001 (everolimus) inhibits tumour growth in xenograft models of human hepatocellular carcinoma. J. Cell. Mol. Med. 2009, 13, 1371–1380. [Google Scholar] [CrossRef] [PubMed]
- Lane, H.A.; Wood, J.M.; McSheehy, P.M.; Allegrini, P.R.; Boulay, A.; Brueggen, J.; Littlewood-Evans, A.; Maira, S.M.; Martiny-Baron, G.; Schnell, C.R.; et al. Mtor inhibitor rad001 (everolimus) has antiangiogenic/vascular properties distinct from a VEGFR tyrosine kinase inhibitor. Clin. Cancer Res. 2009, 15, 1612–1622. [Google Scholar] [CrossRef] [PubMed]
- Guimaraes, A.R.; Ross, R.; Figuereido, J.L.; Waterman, P.; Weissleder, R. Mri with magnetic nanoparticles monitors downstream anti-angiogenic effects of mtor inhibition. Mol. Imaging Biol. 2011, 13, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Shi, W.Y.; Wu, Z.Y.; Varna, M.; Wang, A.H.; Zhou, L.; Chen, L.; Shen, Z.X.; Lu, H.; Zhao, W.L.; et al. Cytostatic and anti-angiogenic effects of temsirolimus in refractory mantle cell lymphoma. J. Hematol. Oncol. 2010, 3, 30. [Google Scholar] [CrossRef] [PubMed]
- Schnell, C.R.; Stauffer, F.; Allegrini, P.R.; O’Reilly, T.; McSheehy, P.M.; Dartois, C.; Stumm, M.; Cozens, R.; Littlewood-Evans, A.; Garcia-Echeverria, C.; et al. Effects of the dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor nvp-bez235 on the tumor vasculature: Implications for clinical imaging. Cancer Res. 2008, 68, 6598–6607. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.C.; Cohen, M.B.; Panka, D.J.; Collins, M.; Ghebremichael, M.; Atkins, M.B.; Signoretti, S.; Mier, J.W. The efficacy of the novel dual pi3-kinase/mtor inhibitor nvp-bez235 compared with rapamycin in renal cell carcinoma. Clin. Cancer Res. 2010, 16, 3628–3638. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.J.; Koul, D.; LaFortune, T.; Tiao, N.; Shen, R.J.; Maira, S.M.; Garcia-Echevrria, C.; Yung, W.K. Nvp-bez235, a novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor, elicits multifaceted antitumor activities in human gliomas. Mol. Cancer Ther. 2009, 8, 2204–2210. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Mao, J.H.; Qian, L.; Zhu, H.; Gu, D.H.; Pan, X.D.; Yi, F.; Ji, D.M. Pre-clinical evaluation of azd-2014, a novel mtorc1/2 dual inhibitor, against renal cell carcinoma. Cancer Lett. 2015, 357, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Xue, Q.; Hopkins, B.; Perruzzi, C.; Udayakumar, D.; Sherris, D.; Benjamin, L.E. Palomid 529, a novel small-molecule drug, is a torc1/torc2 inhibitor that reduces tumor growth, tumor angiogenesis, and vascular permeability. Cancer Res. 2008, 68, 9551–9557. [Google Scholar] [CrossRef] [PubMed]
- Falcon, B.L.; Barr, S.; Gokhale, P.C.; Chou, J.; Fogarty, J.; Depeille, P.; Miglarese, M.; Epstein, D.M.; McDonald, D.M. Reduced vegf production, angiogenesis, and vascular regrowth contribute to the antitumor properties of dual mtorc1/mtorc2 inhibitors. Cancer Res. 2011, 71, 1573–1583. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, X.; Qin, L.; Xu, T.; Zhu, Z.; Zhong, S.; Zhang, M.; Shen, Z. The dual mtorc1 and mtorc2 inhibitor pp242 shows strong antitumor activity in a pheochromocytoma pc12 cell tumor model. Urology 2015, 85, 273.e1–273.e7. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Qin, Y.; Huang, J.; Qin, J.; Gu, J.; Zhu, H.; Liu, H.; Cai, Y.; Wu, X.; Feng, J. Effect of rapamycin-induced tumor vessel thrombosis combined with docetaxel in non-small-cell lung cancer. Anticancer Drugs 2013, 24, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Chen, S.; Liu, F.; Wu, H.; McHugh, J.; Bergin, I.L.; Gupta, A.; Adams, D.; Guan, J.L. Constitutive activation of mtorc1 in endothelial cells leads to the development and progression of lymphangiosarcoma through vegf autocrine signaling. Cancer Cell 2015, 28, 758–772. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Amato, K.R.; Song, W.; Youngblood, V.; Lee, K.; Boothby, M.; Brantley-Sieders, D.M.; Chen, J. Regulation of endothelial cell proliferation and vascular assembly through distinct mtorc2 signaling pathways. Mol. Cell. Biol. 2015, 35, 1299–1313. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, G.; Yu, K.; Jiang, Z.; Chung, A.; Yao, J.; Ha, C.; Toy, K.; Soriano, R.; Haley, B.; Blackwood, E.; et al. Phosphoproteomic analysis implicates the mtorc2-foxo1 axis in vegf signaling and feedback activation of receptor tyrosine kinases. Sci. Signal. 2013, 6, ra25. [Google Scholar] [CrossRef] [PubMed]
- Dada, S.; Demartines, N.; Dormond, O. Mtorc2 regulates pge2-mediated endothelial cell survival and migration. Biochem. Biophys. Res. Commun. 2008, 372, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Farhan, M.A.; Carmine-Simmen, K.; Lewis, J.D.; Moore, R.B.; Murray, A.G. Endothelial cell mtor complex-2 regulates sprouting angiogenesis. PLoS ONE 2015, 10, e0135245. [Google Scholar] [CrossRef] [PubMed]
- Hudson, C.C.; Liu, M.; Chiang, G.G.; Otterness, D.M.; Loomis, D.C.; Kaper, F.; Giaccia, A.J.; Abraham, R.T. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol. Cell. Biol. 2002, 22, 7004–7014. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Chiles, K.; Feldser, D.; Laughner, E.; Hanrahan, C.; Georgescu, M.M.; Simons, J.W.; Semenza, G.L. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/pten/akt/frap pathway in human prostate cancer cells: Implications for tumor angiogenesis and therapeutics. Cancer Res. 2000, 60, 1541–1545. [Google Scholar] [PubMed]
- Lee, D.F.; Kuo, H.P.; Chen, C.T.; Hsu, J.M.; Chou, C.K.; Wei, Y.; Sun, H.L.; Li, L.Y.; Ping, B.; Huang, W.C.; et al. Ikk beta suppression of tsc1 links inflammation and tumor angiogenesis via the mtor pathway. Cell 2007, 130, 440–455. [Google Scholar] [CrossRef] [PubMed]
- Dormond, O.; Contreras, A.G.; Meijer, E.; Datta, D.; Flynn, E.; Pal, S.; Briscoe, D.M. Cd40-induced signaling in human endothelial cells results in mtorc2- and akt-dependent expression of vascular endothelial growth factor in vitro and in vivo. J. Immunol. 2008, 181, 8088–8095. [Google Scholar] [CrossRef] [PubMed]
- Dey, N.; Sun, Y.; Carlson, J.H.; Wu, H.; Lin, X.; Leyland-Jones, B.; De, P. Anti-tumor efficacy of bez235 is complemented by its anti-angiogenic effects via downregulation of Pi3k-mTOR-HIF1alpha signaling in HER2-defined breast cancers. Am. J. Cancer Res. 2016, 6, 714–746. [Google Scholar] [PubMed]
- Laughner, E.; Taghavi, P.; Chiles, K.; Mahon, P.C.; Semenza, G.L. Her2 (neu) signaling increases the rate of hypoxia-inducible factor 1alpha (hif-1alpha) synthesis: Novel mechanism for hif-1-mediated vascular endothelial growth factor expression. Mol. Cell. Biol. 2001, 21, 3995–4004. [Google Scholar] [CrossRef] [PubMed]
- Duvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a metabolic gene regulatory network downstream of mtor complex 1. Mol. Cell 2010, 39, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Tandon, P.; Gallo, C.A.; Khatri, S.; Barger, J.F.; Yepiskoposyan, H.; Plas, D.R. Requirement for ribosomal protein s6 kinase 1 to mediate glycolysis and apoptosis resistance induced by pten deficiency. Proc. Natl. Acad. Sci. USA 2011, 108, 2361–2365. [Google Scholar] [CrossRef] [PubMed]
- Land, S.C.; Tee, A.R. Hypoxia-inducible factor 1alpha is regulated by the mammalian target of rapamycin (mtor) via an mtor signaling motif. J. Biol. Chem. 2007, 282, 20534–20543. [Google Scholar] [CrossRef] [PubMed]
- Dodd, K.M.; Yang, J.; Shen, M.H.; Sampson, J.R.; Tee, A.R. Mtorc1 drives hif-1alpha and vegf-a signalling via multiple mechanisms involving 4e-bp1, s6k1 and stat3. Oncogene 2015, 34, 2239–2250. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Squadrito, M.L.; De Palma, M. Macrophage regulation of tumor angiogenesis: Implications for cancer therapy. Mol. Aspects Med. 2011, 32, 123–145. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Ma, T.; Shen, X.N.; Xia, X.F.; Xu, G.D.; Bai, X.L.; Liang, T.B. Macrophage-induced tumor angiogenesis is regulated by the tsc2-mtor pathway. Cancer Res. 2012, 72, 1363–1372. [Google Scholar] [CrossRef] [PubMed]
- Mercalli, A.; Calavita, I.; Dugnani, E.; Citro, A.; Cantarelli, E.; Nano, R.; Melzi, R.; Maffi, P.; Secchi, A.; Sordi, V.; et al. Rapamycin unbalances the polarization of human macrophages to m1. Immunology 2013, 140, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Paulis, Y.W.; Soetekouw, P.M.; Verheul, H.M.; Tjan-Heijnen, V.C.; Griffioen, A.W. Signalling pathways in vasculogenic mimicry. Biochim. Biophys. Acta 2010, 1806, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Dunleavey, J.M.; Dudley, A.C. Vascular mimicry: Concepts and implications for anti-angiogenic therapy. Curr. Angiogenes 2012, 1, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Zhang, S.; Zhao, X.; Zhang, W.; Hao, X. Vasculogenic mimicry is associated with poor survival in patients with mesothelial sarcomas and alveolar rhabdomyosarcomas. Int. J. Oncol. 2004, 25, 1609–1614. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Ke, Y.; Sun, X.; Yu, L.; Yang, Z.; Zhang, Y.; Du, M.; Wang, J.; Liu, X.; Huang, S. Mammalian target of rapamycin signaling is involved in the vasculogenic mimicry of glioma via hypoxia-inducible factor-1alpha. Oncol. Rep. 2014, 32, 1973–1980. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Feng, Y.J.; Yao, L.Q.; Cheng, M.J.; Xu, C.J.; Huang, Y.; Zhao, Y.Q.; Jiang, H. Plasticity of ovarian cancer cell skov3ip and vasculogenic mimicry in vivo. Int. J. Gynecol. Cancer 2008, 18, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.M.; Hurwitz, H.I. Understanding and targeting resistance to anti-angiogenic therapies. J. Gastrointest. Oncol. 2013, 4, 253–263. [Google Scholar] [PubMed]
- Bergers, G.; Hanahan, D. Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer 2008, 8, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K.; Carmeliet, P. Snapshot: Tumor angiogenesis. Cell 2012, 149, 1408. [Google Scholar] [CrossRef] [PubMed]
- Birle, D.C.; Hedley, D.W. Signaling interactions of rapamycin combined with erlotinib in cervical carcinoma xenografts. Mol. Cancer Ther. 2006, 5, 2494–2502. [Google Scholar] [CrossRef] [PubMed]
- Mosley, J.D.; Poirier, J.T.; Seachrist, D.D.; Landis, M.D.; Keri, R.A. Rapamycin inhibits multiple stages of c-Neu/Erbb2 induced tumor progression in a transgenic mouse model of HER2-positive breast cancer. Mol. Cancer Ther. 2007, 6, 2188–2197. [Google Scholar] [CrossRef] [PubMed]
- Cao, P.; Maira, S.M.; Garcia-Echeverria, C.; Hedley, D.W. Activity of a novel, dual pi3-kinase/mtor inhibitor nvp-bez235 against primary human pancreatic cancers grown as orthotopic xenografts. Br. J. Cancer 2009, 100, 1267–1276. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Berel, D.; Wang, Y.; Li, P.; Bhowmick, N.A.; Figlin, R.A.; Kim, H.L. A comparison of ku0063794, a dual mtorc1 and mtorc2 inhibitor, and temsirolimus in preclinical renal cell carcinoma models. PLoS ONE 2013, 8, e54918. [Google Scholar] [CrossRef] [PubMed]
- Walpen, T.; Kalus, I.; Schwaller, J.; Peier, M.A.; Battegay, E.J.; Humar, R. Nuclear PIM1 confers resistance to rapamycin-impaired endothelial proliferation. Biochem. Biophys. Res. Commun. 2012, 429, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Semela, D.; Piguet, A.C.; Kolev, M.; Schmitter, K.; Hlushchuk, R.; Djonov, V.; Stoupis, C.; Dufour, J.F. Vascular remodeling and antitumoral effects of mTOR inhibition in a rat model of hepatocellular carcinoma. J. Hepatol. 2007, 46, 840–848. [Google Scholar] [CrossRef] [PubMed]
- Manegold, P.C.; Paringer, C.; Kulka, U.; Krimmel, K.; Eichhorn, M.E.; Wilkowski, R.; Jauch, K.W.; Guba, M.; Bruns, C.J. Antiangiogenic therapy with mammalian target of rapamycin inhibitor rad001 (everolimus) increases radiosensitivity in solid cancer. Clin. Cancer Res. 2008, 14, 892–900. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.D.; Spalding, A.C.; Somnay, Y.R.; Markwart, S.; Ray, M.E.; Hamstra, D.A. Inhibition of mtor radiosensitizes soft tissue sarcoma and tumor vasculature. Clin. Cancer Res. 2009, 15, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.W.; Moretti, L.; Mitchell, L.R.; Jung, D.K.; Lu, B. Combined bcl-2/mammalian target of rapamycin inhibition leads to enhanced radiosensitization via induction of apoptosis and autophagy in non-small cell lung tumor xenograft model. Clin. Cancer Res. 2009, 15, 6096–6105. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, Y.; Mori, M.; Kitahara, S.; Fukumoto, M.; Ezaki, T.; Mori, S.; Echigo, S.; Ohkubo, Y.; Fukumoto, M. Targeting of tumor endothelial cells combining 2 gy/day of x-ray with everolimus is the effective modality for overcoming clinically relevant radioresistant tumors. Cancer Med. 2014, 3, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Fokas, E.; Yoshimura, M.; Prevo, R.; Higgins, G.; Hackl, W.; Maira, S.M.; Bernhard, E.J.; McKenna, W.G.; Muschel, R.J. Nvp-bez235 and nvp-bgt226, dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitors, enhance tumor and endothelial cell radiosensitivity. Radiat. Oncol. 2012, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- Piguet, A.C.; Semela, D.; Keogh, A.; Wilkens, L.; Stroka, D.; Stoupis, C.; St-Pierre, M.V.; Dufour, J.F. Inhibition of mtor in combination with doxorubicin in an experimental model of hepatocellular carcinoma. J. Hepatol. 2008, 49, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Huynh, H.; Chow, P.K.; Palanisamy, N.; Salto-Tellez, M.; Goh, B.C.; Lee, C.K.; Somani, A.; Lee, H.S.; Kalpana, R.; Yu, K.; et al. Bevacizumab and rapamycin induce growth suppression in mouse models of hepatocellular carcinoma. J. Hepatol. 2008, 49, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Hainsworth, J.D.; Spigel, D.R.; Burris, H.A., 3rd; Waterhouse, D.; Clark, B.L.; Whorf, R. Phase ii trial of bevacizumab and everolimus in patients with advanced renal cell carcinoma. J. Clin. Oncol. 2010, 28, 2131–2136. [Google Scholar] [CrossRef] [PubMed]
- Hobday, T.J.; Qin, R.; Reidy-Lagunes, D.; Moore, M.J.; Strosberg, J.; Kaubisch, A.; Shah, M.; Kindler, H.L.; Lenz, H.J.; Chen, H.; et al. Multicenter phase ii trial of temsirolimus and bevacizumab in pancreatic neuroendocrine tumors. J. Clin. Oncol. 2015, 33, 1551–1556. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Manola, J.B.; Pins, M.; McDermott, D.F.; Atkins, M.B.; Dutcher, J.J.; George, D.J.; Margolin, K.A.; DiPaola, R.S. Best: A randomized phase ii study of vascular endothelial growth factor, raf kinase, and mammalian target of rapamycin combination targeted therapy with bevacizumab, sorafenib, and temsirolimus in advanced renal cell carcinoma—A trial of the ecog-acrin cancer research group (e2804). J. Clin. Oncol. 2015, 33, 2384–2391. [Google Scholar] [PubMed]
- Pignochino, Y.; Dell’Aglio, C.; Basirico, M.; Capozzi, F.; Soster, M.; Marchio, S.; Bruno, S.; Gammaitoni, L.; Sangiolo, D.; Torchiaro, E.; et al. The combination of sorafenib and everolimus abrogates mtorc1 and mtorc2 upregulation in osteosarcoma preclinical models. Clin. Cancer Res. 2013, 19, 2117–2131. [Google Scholar] [CrossRef] [PubMed]
- Piguet, A.C.; Saar, B.; Hlushchuk, R.; St-Pierre, M.V.; McSheehy, P.M.; Radojevic, V.; Afthinos, M.; Terracciano, L.; Djonov, V.; Dufour, J.F. Everolimus augments the effects of sorafenib in a syngeneic orthotopic model of hepatocellular carcinoma. Mol. Cancer Ther. 2011, 10, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Tong, L.J.; Ding, J.; Meng, L.H. Systematic combination screening reveals synergism between rapamycin and sunitinib against human lung cancer. Cancer Lett. 2014, 342, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.L.; Wong, C.H.; Lau, C.P.; Zhou, Q.; Hui, C.W.; Lui, V.W.; Ma, B.B.; Chan, A.T.; Yeo, W. Preclinical evaluation of combined tki-258 and rad001 in hepatocellular carcinoma. Cancer Chemother. Pharmacol. 2013, 71, 1417–1425. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.A.; Fox, P.S.; Papadopoulos, N.E.; Bedikian, A.Y.; Hwu, W.J.; Lazar, A.J.; Prieto, V.G.; Culotta, K.S.; Madden, T.L.; Xu, Q.; et al. Phase i study of the combination of sorafenib and temsirolimus in patients with metastatic melanoma. Clin. Cancer Res. 2012, 18, 1120–1128. [Google Scholar] [CrossRef] [PubMed]
- Huynh, H. Azd6244 (arry-142886) enhances the antitumor activity of rapamycin in mouse models of human hepatocellular carcinoma. Cancer 2010, 116, 1315–1325. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, A.W.; Bendell, J.C.; Papadopoulos, K.P.; Burris, H.A., 3rd; Patnaik, A.; Jones, S.F.; Rasco, D.; Cox, D.S.; Durante, M.; Bellew, K.M.; et al. A phase ib trial of the oral mek inhibitor trametinib (gsk1120212) in combination with everolimus in patients with advanced solid tumors. Ann. Oncol. 2015, 26, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Harkavy, B.; Shen, N.; Grohar, P.; Helman, L.J. Rapamycin induces feedback activation of akt signaling through an Igf-1r-dependent mechanism. Oncogene 2007, 26, 1932–1940. [Google Scholar] [CrossRef] [PubMed]
- Kurmasheva, R.T.; Dudkin, L.; Billups, C.; Debelenko, L.V.; Morton, C.L.; Houghton, P.J. The insulin-like growth factor-1 receptor-targeting antibody, cp-751,871, suppresses tumor-derived vegf and synergizes with rapamycin in models of childhood sarcoma. Cancer Res. 2009, 69, 7662–7671. [Google Scholar] [CrossRef] [PubMed]
- Quek, R.; Wang, Q.; Morgan, J.A.; Shapiro, G.I.; Butrynski, J.E.; Ramaiya, N.; Huftalen, T.; Jederlinic, N.; Manola, J.; Wagner, A.J.; et al. Combination mtor and igf-1r inhibition: Phase i trial of everolimus and figitumumab in patients with advanced sarcomas and other solid tumors. Clin. Cancer Res. 2011, 17, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.K.; Tap, W.D.; Qin, L.X.; Livingston, M.B.; Undevia, S.D.; Chmielowski, B.; Agulnik, M.; Schuetze, S.M.; Reed, D.R.; Okuno, S.H.; et al. Cixutumumab and temsirolimus for patients with bone and soft-tissue sarcoma: A multicentre, open-label, phase 2 trial. Lancet Oncol. 2013, 14, 371–382. [Google Scholar] [CrossRef]
- Naing, A.; LoRusso, P.; Fu, S.; Hong, D.S.; Anderson, P.; Benjamin, R.S.; Ludwig, J.; Chen, H.X.; Doyle, L.A.; Kurzrock, R. Insulin growth factor-receptor (IGF-1r) antibody cixutumumab combined with the mtor inhibitor temsirolimus in patients with refractory ewing’s sarcoma family tumors. Clin. Cancer Res. 2012, 18, 2625–2631. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Morales, S.M.; Awada, A.; Blum, J.L.; Tan, A.R.; Ewertz, M.; Cortes, J.; Moy, B.; Ruddy, K.J.; Haddad, T.; et al. A phase ii study of combined ridaforolimus and dalotuzumab compared with exemestane in patients with estrogen receptor-positive breast cancer. Breast Cancer Res. Treat. 2017, 163, 535–544. [Google Scholar] [CrossRef] [PubMed]
- McKeage, M.J.; Baguley, B.C. Disrupting established tumor blood vessels: An emerging therapeutic strategy for cancer. Cancer 2010, 116, 1859–1871. [Google Scholar] [CrossRef] [PubMed]
- Ellis, L.; Shah, P.; Hammers, H.; Lehet, K.; Sotomayor, P.; Azabdaftari, G.; Seshadri, M.; Pili, R. Vascular disruption in combination with mtor inhibition in renal cell carcinoma. Mol. Cancer Ther. 2012, 11, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Kraus, D.; Palasuberniam, P.; Chen, B. Targeting phosphatidylinositol 3-kinase signaling pathway for therapeutic enhancement of vascular-targeted photodynamic therapy. Mol. Cancer Ther. 2017. [Google Scholar] [CrossRef] [PubMed]
- Verheul, H.M.; Salumbides, B.; Van Erp, K.; Hammers, H.; Qian, D.Z.; Sanni, T.; Atadja, P.; Pili, R. Combination strategy targeting the hypoxia inducible factor-1 alpha with mammalian target of rapamycin and histone deacetylase inhibitors. Clin. Cancer Res. 2008, 14, 3589–3597. [Google Scholar] [CrossRef] [PubMed]
- Singleton, P.A.; Mambetsariev, N.; Lennon, F.E.; Mathew, B.; Siegler, J.H.; Moreno-Vinasco, L.; Salgia, R.; Moss, J.; Garcia, J.G. Methylnaltrexone potentiates the anti-angiogenic effects of mtor inhibitors. J. Angiogenes Res. 2010, 2, 5. [Google Scholar] [CrossRef] [PubMed]
- Damiano, V.; Rosa, R.; Formisano, L.; Nappi, L.; Gelardi, T.; Marciano, R.; Cozzolino, I.; Troncone, G.; Agrawal, S.; Veneziani, B.M.; et al. Toll-like receptor 9 agonist imo cooperates with everolimus in renal cell carcinoma by interfering with tumour growth and angiogenesis. Br. J. Cancer 2013, 108, 1616–1623. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, S.; Baluk, P.; Kaidoh, T.; Haskell, A.; Jain, R.K.; McDonald, D.M. Abnormalities in pericytes on blood vessels and endothelial sprouts in tumors. Am. J. Pathol. 2002, 160, 985–1000. [Google Scholar] [CrossRef]
- Baluk, P.; Morikawa, S.; Haskell, A.; Mancuso, M.; McDonald, D.M. Abnormalities of basement membrane on blood vessels and endothelial sprouts in tumors. Am. J. Pathol 2003, 163, 1801–1815. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat. Rev. Drug Discov. 2011, 10, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Normalizing tumor vasculature with anti-angiogenic therapy: A new paradigm for combination therapy. Nat. Med. 2001, 7, 987–989. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Antiangiogenesis strategies revisited: From starving tumors to alleviating hypoxia. Cancer Cell 2014, 26, 605–622. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bindokas, V.; Shen, J.; Fan, H.; Hoffman, R.M.; Xing, H.R. Time-course imaging of therapeutic functional tumor vascular normalization by antiangiogenic agents. Mol. Cancer Ther. 2011, 10, 1173–1184. [Google Scholar] [CrossRef] [PubMed]
- Myers, A.L.; Orr, W.S.; Denbo, J.W.; Ng, C.Y.; Zhou, J.; Spence, Y.; Wu, J.; Davidoff, A.M. Rapamycin-induced tumor vasculature remodeling in rhabdomyosarcoma xenografts increases the effectiveness of adjuvant ionizing radiation. J. Pediatr. Surg. 2012, 47, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Eshleman, J.S.; Carlson, B.L.; Mladek, A.C.; Kastner, B.D.; Shide, K.L.; Sarkaria, J.N. Inhibition of the mammalian target of rapamycin sensitizes u87 xenografts to fractionated radiation therapy. Cancer Res. 2002, 62, 7291–7297. [Google Scholar] [PubMed]
- Wu, L.; Birle, D.C.; Tannock, I.F. Effects of the mammalian target of rapamycin inhibitor cci-779 used alone or with chemotherapy on human prostate cancer cells and xenografts. Cancer Res. 2005, 65, 2825–2831. [Google Scholar] [CrossRef] [PubMed]
- Carman, C.V.; Martinelli, R. T lymphocyte-endothelial interactions: Emerging understanding of trafficking and antigen-specific immunity. Front. Immunol. 2015, 6, 603. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Enis, D.R.; Koh, K.P.; Shiao, S.L.; Pober, J.S. T lymphocyte-endothelial cell interactions. Annu Rev. Immunol. 2004, 22, 683–709. [Google Scholar] [CrossRef] [PubMed]
- Nummer, D.; Suri-Payer, E.; Schmitz-Winnenthal, H.; Bonertz, A.; Galindo, L.; Antolovich, D.; Koch, M.; Buchler, M.; Weitz, J.; Schirrmacher, V.; et al. Role of tumor endothelium in CD4+ CD25+ regulatory T cell infiltration of human pancreatic carcinoma. J. Natl. Cancer Inst. 2007, 99, 1188–1199. [Google Scholar] [CrossRef] [PubMed]
- Motz, G.T.; Santoro, S.P.; Wang, L.P.; Garrabrant, T.; Lastra, R.R.; Hagemann, I.S.; Lal, P.; Feldman, M.D.; Benencia, F.; Coukos, G. Tumor endothelium fasl establishes a selective immune barrier promoting tolerance in tumors. Nat. Med. 2014, 20, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Lanitis, E.; Irving, M.; Coukos, G. Targeting the tumor vasculature to enhance t cell activity. Curr. Opin. Immunol. 2015, 33, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Uldry, E.; Faes, S.; Demartines, N.; Dormond, O. Fine-tuning tumor endothelial cells to selectively kill cancer. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Hendry, S.A.; Farnsworth, R.H.; Solomon, B.; Achen, M.G.; Stacker, S.A.; Fox, S.B. The role of the tumor vasculature in the host immune response: Implications for therapeutic strategies targeting the tumor microenvironment. Front. Immunol. 2016, 7, 621. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yi, T.; Qin, L.; Maldonado, R.A.; von Andrian, U.H.; Kulkarni, S.; Tellides, G.; Pober, J.S. Rapamycin-treated human endothelial cells preferentially activate allogeneic regulatory T cells. J. Clin. Investig. 2013, 123, 1677–1693. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Qin, L.; Manes, T.D.; Kirkiles-Smith, N.C.; Tellides, G.; Pober, J.S. Rapamycin antagonizes tnf induction of vcam-1 on endothelial cells by inhibiting mTORc2. J. Exp. Med. 2014, 211, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Neshat, M.S.; Mellinghoff, I.K.; Tran, C.; Stiles, B.; Thomas, G.; Petersen, R.; Frost, P.; Gibbons, J.J.; Wu, H.; Sawyers, C.L. Enhanced sensitivity of pten-deficient tumors to inhibition of frap/mtor. Proc. Natl. Acad. Sci. USA 2001, 98, 10314–10319. [Google Scholar] [CrossRef] [PubMed]
- Di Nicolantonio, F.; Arena, S.; Tabernero, J.; Grosso, S.; Molinari, F.; Macarulla, T.; Russo, M.; Cancelliere, C.; Zecchin, D.; Mazzucchelli, L.; et al. Deregulation of the pi3k and kras signaling pathways in human cancer cells determines their response to everolimus. J. Clin. Investig. 2010, 120, 2858–2866. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, D.; Boya, P.; Bellet, D.; Faivre, S.; Troalen, F.; Benard, J.; Saulnier, P.; Hopkins-Donaldson, S.; Zangemeister-Wittke, U.; Kroemer, G.; et al. Bcl-2 and CCND1/CDK4 expression levels predict the cellular effects of mtor inhibitors in human ovarian carcinoma. Apoptosis 2004, 9, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Cummings, C.A.; Peters, E.; Lacroix, L.; Andre, F.; Lackner, M.R. The role of next-generation sequencing in enabling personalized oncology therapy. Clin. Transl. Sci. 2016, 9, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Wagle, N.; Grabiner, B.C.; Van Allen, E.M.; Amin-Mansour, A.; Taylor-Weiner, A.; Rosenberg, M.; Gray, N.; Barletta, J.A.; Guo, Y.; Swanson, S.J.; et al. Response and acquired resistance to everolimus in anaplastic thyroid cancer. N. Engl. J. Med. 2014, 371, 1426–1433. [Google Scholar] [CrossRef] [PubMed]
- Wagle, N.; Grabiner, B.C.; Van Allen, E.M.; Hodis, E.; Jacobus, S.; Supko, J.G.; Stewart, M.; Choueiri, T.K.; Gandhi, L.; Cleary, J.M.; et al. Activating mtor mutations in a patient with an extraordinary response on a phase i trial of everolimus and pazopanib. Cancer Discov. 2014, 4, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Phung, T.L.; Eyiah-Mensah, G.; O’Donnell, R.K.; Bieniek, R.; Shechter, S.; Walsh, K.; Kuperwasser, C.; Benjamin, L.E. Endothelial Akt signaling is rate-limiting for rapamycin inhibition of mouse mammary tumor progression. Cancer Res. 2007, 67, 5070–5075. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faes, S.; Santoro, T.; Demartines, N.; Dormond, O. Evolving Significance and Future Relevance of Anti-Angiogenic Activity of mTOR Inhibitors in Cancer Therapy. Cancers 2017, 9, 152. https://doi.org/10.3390/cancers9110152
Faes S, Santoro T, Demartines N, Dormond O. Evolving Significance and Future Relevance of Anti-Angiogenic Activity of mTOR Inhibitors in Cancer Therapy. Cancers. 2017; 9(11):152. https://doi.org/10.3390/cancers9110152
Chicago/Turabian StyleFaes, Seraina, Tania Santoro, Nicolas Demartines, and Olivier Dormond. 2017. "Evolving Significance and Future Relevance of Anti-Angiogenic Activity of mTOR Inhibitors in Cancer Therapy" Cancers 9, no. 11: 152. https://doi.org/10.3390/cancers9110152
APA StyleFaes, S., Santoro, T., Demartines, N., & Dormond, O. (2017). Evolving Significance and Future Relevance of Anti-Angiogenic Activity of mTOR Inhibitors in Cancer Therapy. Cancers, 9(11), 152. https://doi.org/10.3390/cancers9110152