Targeting Autophagy in ALK-Associated Cancers

Abstract
:1. Introduction
2. Autophagy: Definition and Measurements
2.1. History of Autophagy
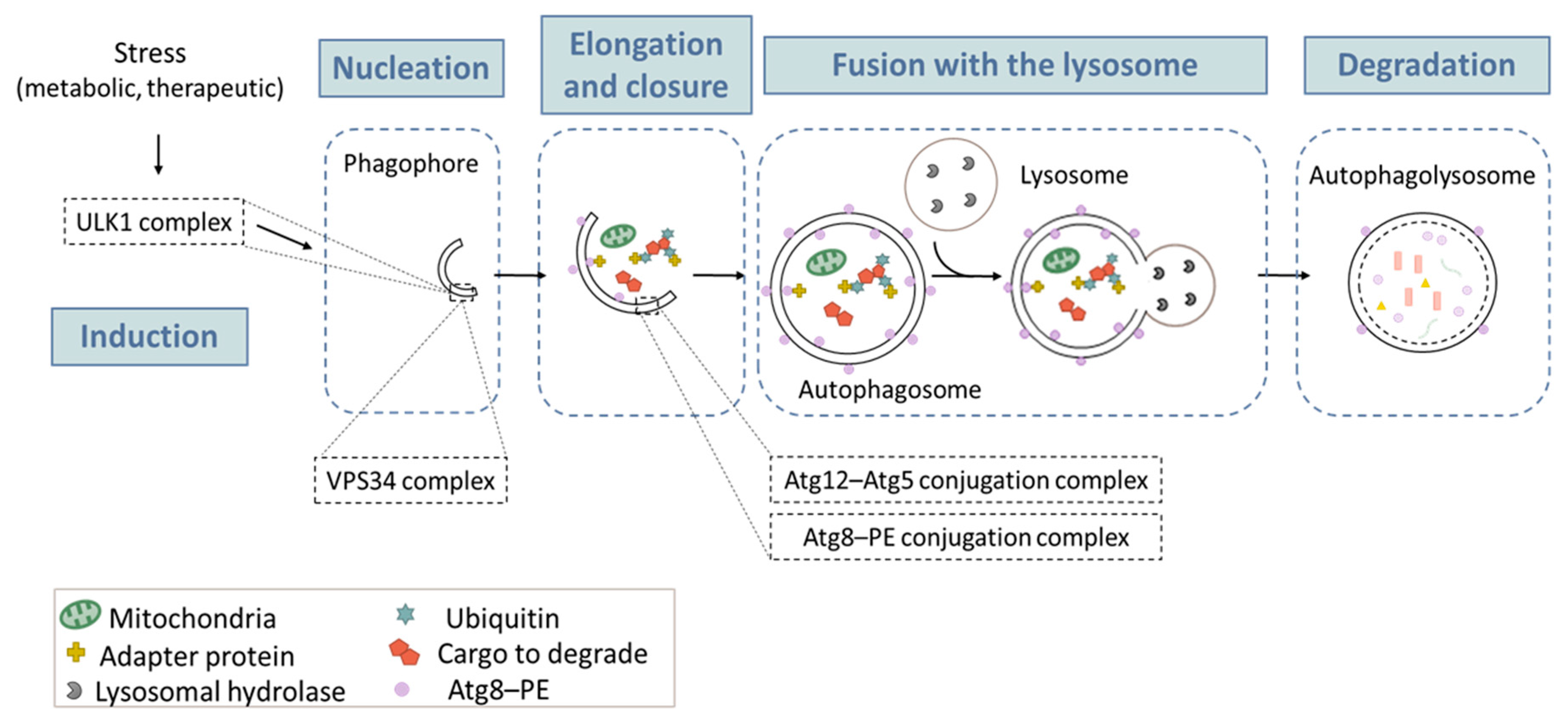
2.2. The Autophagic Process
2.3. Classical Measure of Autophagy
2.3.1. Transmission Electron Microscopy (TEM)
2.3.2. Labelling of Acidic Vesicules
2.3.3. Turnover of PE-Conjugated ATG8 Proteins
2.3.4. Fluorescent LC3B Probes
2.3.5. Autophagic Cargo Sequestration Assay
3. Autophagy in Cancer Therapy
3.1. Cytoprotective or Cytotoxic Functions of Autophagy Following Cancer therapy
3.2. Autophagic Switch
3.3. Other Therapeutic Use of Autophagy And Autophagosomes
3.3.1. Oncogene Degradation
3.3.2. Autophagosomes, As Carriers for Vaccination
4. ALK-associated Cancers
4.1. Neuroblastoma
4.2. Rhabdomyosarcoma
4.3. Glioblastoma
4.4. Anaplastic Large Cell Lymphoma
4.5. Non-Small Cell Lung Carcinoma
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Iwahara, T.; Fujimoto, J.; Wen, D.; Cupples, R.; Bucay, N.; Arakawa, T.; Mori, S.; Ratzkin, B.; Yamamoto, T. Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene 1997, 14, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Hallberg, B.; Palmer, R.H. Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat. Rev. Cancer 2013, 13, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Anaplastic lymphoma kinase (ALK): Structure, oncogenic activation, and pharmacological inhibition. Pharmacol. Res. 2013, 68, 68–94. [Google Scholar] [CrossRef] [PubMed]
- Chiarle, R.; Voena, C.; Ambrogio, C.; Piva, R.; Inghirami, G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat. Rev. Cancer 2008, 8, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Gambacorti-Passerini, C.; le Coutre, P.; Mologni, L.; Fanelli, M.; Bertazzoli, C.; Marchesi, E.; Di Nicola, M.; Biondi, A.; Corneo, G.M.; Belotti, D.; et al. Inhibition of the ABL kinase activity blocks the proliferation of BCR/ABL+ leukemic cells and induces apoptosis. Blood Cells. Mol. Dis. 1997, 23, 380–394. [Google Scholar] [CrossRef] [PubMed]
- Sawyers, C.L.; Hochhaus, A.; Feldman, E.; Goldman, J.M.; Miller, C.B.; Ottmann, O.G.; Schiffer, C.A.; Talpaz, M.; Guilhot, F.; Deininger, M.W.; et al. Imatinib induces hematologic and cytogenetic responses in patients with chronic myelogenous leukemia in myeloid blast crisis: results of a phase II study. Blood 2002, 99, 3530–3539. [Google Scholar] [CrossRef] [PubMed]
- Crescenzo, R.; Inghirami, G. Anaplastic lymphoma kinase inhibitors. Curr. Opin. Pharmacol. 2015, 23, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Mologni, L. Inhibitors of the anaplastic lymphoma kinase. Expert. Opin. Investig. Drugs 2012, 21, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Christensen, J.G.; Zou, H.Y.; Arango, M.E.; Li, Q.; Lee, J.H.; McDonnell, S.R.; Yamazaki, S.; Alton, G.R.; Mroczkowski, B.; Los, G. Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma. Mol. Cancer Ther. 2007, 6, 3314–3322. [Google Scholar] [CrossRef] [PubMed]
- Gambacorti-Passerini, C.; Messa, C.; Pogliani, E.M. Crizotinib in anaplastic large-cell lymphoma. N. Engl. J. Med. 2010, 364, 775–776. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.H.; Bazhenova, L.; Camidge, D.R.; Solomon, B.J.; Herman, J.; Kain, T.; Bang, Y.J.; Kwak, E.L.; Shaw, A.T.; Salgia, R.; et al. Rapid and dramatic radiographic and clinical response to an ALK inhibitor (crizotinib, PF02341066) in an ALK translocation-positive patient with non-small cell lung cancer. J. Thorac. Oncol. 2010, 5, 2044–2046. [Google Scholar] [CrossRef] [PubMed]
- Butrynski, J.E.; D’Adamo, D.R.; Hornick, J.L.; Dal Cin, P.; Antonescu, C.R.; Jhanwar, S.C.; Ladanyi, M.; Capelletti, M.; Rodig, S.J.; Ramaiya, N.; et al. Crizotinib in ALK-rearranged inflammatory myofibroblastic tumor. N. Engl. J. Med. 2010, 363, 1727–1733. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.L.; Soda, M.; Yamashita, Y.; Ueno, T.; Takashima, J.; Nakajima, T.; Yatabe, Y.; Takeuchi, K.; Hamada, T.; Haruta, H.; et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N. Engl. J. Med. 2010, 363, 1734–1739. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Okuda, K.; Zheng, W.; Butrynski, J.; Capelletti, M.; Wang, L.; Gray, N.S.; Wilner, K.; Christensen, J.G.; Demetri, G.; et al. The neuroblastoma-associated F1174L ALK mutation causes resistance to an ALK kinase inhibitor in ALK-translocated cancers. Cancer Res. 2010, 70, 10038–10043. [Google Scholar] [CrossRef] [PubMed]
- Mosse, Y.P.; Lim, M.S.; Voss, S.D.; Wilner, K.; Ruffner, K.; Laliberte, J.; Rolland, D.; Balis, F.M.; Maris, J.M.; Weigel, B.J.; et al. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: a Children’s Oncology Group phase 1 consortium study. Lancet. Oncol. 2013, 14, 472–480. [Google Scholar] [CrossRef]
- Gambacorti, P.C.; Farina, F.; Stasia, A.; Redaelli, S.; Ceccon, M.; Mologni, L.; Messa, C.; Guerra, L.; Giudici, G.; Sala, E.; et al. Crizotinib in advanced, chemoresistant anaplastic lymphoma kinase-positive lymphoma patients. J. Natl. Cancer Inst. 2014, 106, djt378. [Google Scholar] [CrossRef] [PubMed]
- Mologni, L. Current and future treatment of anaplastic lymphoma kinase-rearranged cancer. World J. Clin. Oncol. 2015, 6, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Werner, M.T.; Zhao, C.; Zhang, Q.; Wasik, M.A. Nucleophosmin-Anaplastic lymphoma kinase: The ultimate oncogene and therapeutic target. Blood 2017, 129, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Pietrocola, F.; Pedro, J.M.B.; Amaravadi, R.K.; Baehrecke, E.H.; Cecconi, F.; Codogno, P.; Debnath, J.; Gewirtz, D.A.; Karantza, V.; et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015, 34, e201490784. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeldt, M.T.; Ryan, K.M. The role of autophagy in tumour development and cancer therapy. Expert. Rev. Mol. Med. 2009, 11, e36. [Google Scholar] [CrossRef] [PubMed]
- Joffre, C.; Djavaheri-Mergny, M.; Pattingre, S.; Giuriato, S. L’autophagie: Le yin et le yang des cancers. Medecine/Sciences 2017, 33, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Deter, R.L.; Baudhuin, P.; De Duve, C. Participation of lysosomes in cellular autophagy induced in rat liver by glucagon. J. Cell Biol. 1967, 35. [Google Scholar] [CrossRef]
- Mortimore, G.E.; Schworer, C.M. Induction of autophagy by amino-acid deprivation in perfused rat liver. Nature 1977, 270, 174–176. [Google Scholar] [CrossRef] [PubMed]
- Takeshige, K.; Baba, M.; Tsuboi, S.; Noda, T.; Ohsumi, Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J. Cell Biol. 1992, 119, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Ohsumi, Y.; Yoshimori, T. Autophagosome formation in mammalian cells. Cell Struct. Funct. 2002, 27, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Ohsumi, Y. Historical landmarks of autophagy research. Cell Res. 2014, 24, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.L.; Cuervo, A.M. Autophagy and human disease: Emerging themes. Curr. Opin. Genet. Dev. 2014, 26, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Klionsky, D.J. Mammalian autophagy: Core molecular machinery and signaling regulation. Curr Opin. Cell Biol. 2010, 22, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef] [PubMed]
- Abada, A.; Elazar, Z. Getting ready for building: signaling and autophagosome biogenesis. EMBO Rep. 2014, 15, 839–852. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakrama, F.Z.; Seguin-Py, S.; Le Grand, J.N.; Fraichard, A.; Delage-Mourroux, R.; Despouy, G.; Perez, V.; Jouvenot, M.; Boyer-Guittaut, M. GABARAPL1 (GEC1) associates with autophagic vesicles. Autophagy 2010, 6, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Szalai, P.; Hagen, L.K.; Sætre, F.; Luhr, M.; Sponheim, M.; Øverbye, A.; Mills, I.G.; Seglen, P.O.; Engedal, N. Autophagic bulk sequestration of cytosolic cargo is independent of LC3, but requires GABARAPs. Exp. Cell Res. 2015, 333, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Kimura, S.; Noda, T.; Yoshimori, T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2007, 3, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Gump, J.M.; Thorburn, A. Sorting cells for basal and induced autophagic flux by quantitative ratiometric flow cytometry. Autophagy 2014, 10, 1327–1334. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Zhong, W.; Zhou, J.; Sheng, F.; Fang, Z.; Wei, Y.; Chen, Y.; Deng, X.; Xia, B.; Lin, J. Monitoring autophagic flux by an improved tandem fluorescent-tagged LC3 (mTagRFP-mWasabi-LC3) reveals that high-dose rapamycin impairs autophagic flux in cancer cells. Autophagy 2012, 8, 1215–1226. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Ueno, T.; Uchiyama, Y. Chapter Six-Use of pHlurorin-mKate2-human LC3 to Monitor Autophagic Responses. Methods Enzymol. 2017, 587, 87–96. [Google Scholar] [PubMed]
- Kaizuka, T.; Morishita, H.; Hama, Y.; Tsukamoto, S.; Matsui, T.; Toyota, Y.; Kodama, A.; Ishihara, T.; Mizushima, T.; Mizushima, N. An Autophagic Flux Probe that Releases an Internal Control. Mol. Cell 2016, 64, 835–849. [Google Scholar] [CrossRef] [PubMed]
- Morishita, H.; Kaizuka, T.; Hama, Y.; Mizushima, N. A new probe to measure autophagic flux in vitro and in vivo. Autophagy 2017, 13, 757–758. [Google Scholar] [CrossRef] [PubMed]
- Seglen, P.O.; Luhr, M.; Mills, I.G.; Sætre, F.; Szalai, P.; Engedal, N. Macroautophagic cargo sequestration assays. Methods 2015, 75, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Kopitz, J.; Kisen, G.; Gordon, P.B.; Bohley, P.; Seglen, P.O. Nonselective autophagy of cytosolic enzymes by isolated rat hepatocytes. J. Cell Biol. 1990, 111, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Luhr, M.; Szalai, P.; Sætre, F.; Gerner, L.; Seglen, P.O.; Engedal, N. A Simple Cargo Sequestration Assay for Quantitative Measurement of Nonselective Autophagy in Cultured Cells. Methods Enzymol. 2017, 587, 351–364. [Google Scholar] [PubMed]
- Maycotte, P.; Thorburn, A. Autophagy and cancer therapy. Cancer Biol. Ther. 2011, 11, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Rebecca, V.W.; Amaravadi, R.K. Emerging strategies to effectively target autophagy in cancer. Oncogene 2015. [Google Scholar] [CrossRef] [PubMed]
- Gewirtz, D.A. The four faces of autophagy: implications for cancer therapy. Cancer Res. 2014, 74, 647–651. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Goehe, R.W.; Di, X.; Hicks, M.A.; Torti, S.V.; Torti, F.M.; Harada, H.; Gewirtz, D.A. A novel cytostatic form of autophagy in sensitization of non-small cell lung cancer cells to radiation by vitamin D and the vitamin D analog, EB 1089. Autophagy 2014, 10, 2346–2361. [Google Scholar] [CrossRef] [PubMed]
- Dou, Q.; Chen, H.-N.; Wang, K.; Yuan, K.; Lei, Y.; Li, K.; Lan, J.; Chen, Y.; Huang, Z.; Xie, N.; et al. Ivermectin Induces Cytostatic Autophagy by Blocking the PAK1/Akt Axis in Breast Cancer. Cancer Res. 2016, 76, 4457–4469. [Google Scholar] [CrossRef] [PubMed]
- Heydt, Q.; Larrue, C.; Saland, E.; Bertoli, S.; Sarry, J.-E.; Besson, A.; Manenti, S.; Joffre, C.; Mansat-De Mas, V. Oncogenic FLT3-ITD supports autophagy via ATF4 in acute myeloid leukemia. Oncogene 2017. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.B.; Hui, B.; Shi, Y.H.; Zhou, J.; Peng, Y.F.; Gu, C.Y.; Yang, H.; Shi, G.M.; Ke, A.W.; Wang, X.Y.; et al. Autophagy activation in hepatocellular carcinoma contributes to the tolerance of oxaliplatin via reactive oxygen species modulation. Clin. Cancer Res. 2011, 17, 6229–6238. [Google Scholar] [CrossRef] [PubMed]
- Thorburn, J.; Andrysik, Z.; Staskiewicz, L.; Gump, J.; Maycotte, P.; Oberst, A.; Green, D.R.; Espinosa, J.M.; Thorburn, A. Autophagy controls the kinetics and extent of mitochondrial apoptosis by regulating PUMA levels. Cell Rep. 2014, 7, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Luo, R.Z.; Lu, Y.; Zhang, X.; Yu, Q.; Khare, S.; Kondo, S.; Kondo, Y.; Yu, Y.; Mills, G.B.; et al. The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cancer cells. J. Clin. Investig. 2008, 118, 3917–3929. [Google Scholar] [CrossRef] [PubMed]
- Viale, A.; Pettazzoni, P.; Lyssiotis, C.A.; Ying, H.; Sánchez, N.; Marchesini, M.; Carugo, A.; Green, T.; Seth, S.; Giuliani, V.; et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014, 514, 628–632. [Google Scholar] [CrossRef] [PubMed]
- Laussmann, M.A.; Passante, E.; Düssmann, H.; Rauen, J.A.; Würstle, M.L.; Delgado, M.E.; Devocelle, M.; Prehn, J.H.M.; Rehm, M. Proteasome inhibition can induce an autophagy-dependent apical activation of caspase-8. Cell Death Differ. 2011, 18, 1584–1597. [Google Scholar] [CrossRef] [PubMed]
- Young, M.M.; Takahashi, Y.; Khan, O.; Park, S.; Hori, T.; Yun, J.; Sharma, A.K.; Amin, S.; Hu, C.-D.; Zhang, J.; et al. Autophagosomal membrane serves as platform for intracellular death-inducing signaling complex (iDISC)-mediated caspase-8 activation and apoptosis. J. Biol. Chem. 2012, 287, 12455–12468. [Google Scholar] [CrossRef] [PubMed]
- Nezis, I.P.; Shravage, B.V.; Sagona, A.P.; Johansen, T.; Baehrecke, E.H.; Stenmark, H. Autophagy as a trigger for cell death: Autophagic degradation of inhibitor of apoptosis dBruce controls DNA fragmentation during late oogenesis in Drosophila. Autophagy 2010, 6, 1214–1215. [Google Scholar] [CrossRef] [PubMed]
- Gump, J.M.; Staskiewicz, L.; Morgan, M.J.; Bamberg, A.; Riches, D.W.H.; Thorburn, A. Autophagy variation within a cell population determines cell fate through selective degradation of Fap-1. Nat. Cell Biol. 2014, 16, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Bonapace, L.; Bornhauser, B.C.; Schmitz, M.; Cario, G.; Ziegler, U.; Niggli, F.K.; Schafer, B.W.; Schrappe, M.; Stanulla, M.; Bourquin, J.-P. Induction of autophagy-dependent necroptosis is required for childhood acute lymphoblastic leukemia cells to overcome glucocorticoid resistance. J. Clin. Investig. 2010, 120, 1310–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Z.; Sanchez-Lopez, E.; Karin, M. Autophagy, Inflammation, and Immunity: A Troika Governing Cancer and Its Treatment. Cell 2016, 166, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Pietrocola, F.; Bravo-San Pedro, J.M.; Galluzzi, L.; Kroemer, G. Autophagy in natural and therapy-driven anticancer immunosurveillance. Autophagy 2017, 8627. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Kögel, D. Cell death by autophagy: Emerging molecular mechanisms and implications for cancer therapy. Oncogene 2015, 34, 5105–5113. [Google Scholar] [CrossRef] [PubMed]
- DeMasters, G.; Di, X.; Newsham, I.; Shiu, R.; Gewirtz, D.A. Potentiation of radiation sensitivity in breast tumor cells by the vitamin D3 analogue, EB 1089, through promotion of autophagy and interference with proliferative recovery. Mol. Cancer Ther. 2006, 5, 2786–2797. [Google Scholar] [CrossRef] [PubMed]
- Wilson, E.N.; Bristol, M.L.; Di, X.; Maltese, W.A.; Koterba, K.; Beckman, M.J.; Gewirtz, D.A. A switch between cytoprotective and cytotoxic autophagy in the radiosensitization of breast tumor cells by chloroquine and vitamin D. Horm. Cancer 2011, 2, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Høyer-Hansen, M.; Bastholm, L.; Mathiasen, I.S.; Elling, F.; Jäättelä, M. Vitamin D analog EB1089 triggers dramatic lysosomal changes and Beclin 1-mediated autophagic cell death. Cell Death Differ. 2005, 12, 1297–1309. [Google Scholar] [CrossRef] [PubMed]
- Shen, P.; Chen, M.; He, M.; Chen, L.; Song, Y.; Xiao, P.; Wan, X.; Dai, F.; Pan, T.; Wang, Q. Inhibition of ERα/ERK/P62 cascades induces “autophagic switch” in the estrogen receptor-positive breast cancer cells exposed to gemcitabine. Oncotarget 2016, 7, 48501–48516. [Google Scholar] [CrossRef] [PubMed]
- Fung, C.; Chen, X.; Grandis, J.R.; Duvvuri, U. EGFR tyrosine kinase inhibition induces autophagy in cancer cells. Cancer Biol. Ther. 2012, 13, 1417–1424. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Henson, E.S.; Xiao, W.; Huang, D.; McMillan-Ward, E.M.; Israels, S.J.; Gibson, S.B. Tyrosine kinase receptor EGFR regulates the switch in cancer cells between cell survival and cell death induced by autophagy in hypoxia. Autophagy 2016, 12, 1029–1046. [Google Scholar] [CrossRef] [PubMed]
- Henson, E.; Chen, Y.; Gibson, S. EGFR family members’ regulation of autophagy is at a crossroads of cell survival and death in cancer. Cancers 2017, 9, 27. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, S.; Kolukula, V.K.; Preet, A.; Albanese, C.; Avantaggiati, M.L. Dissecting the pathways that destabilize mutant p53: The proteasome or autophagy? Cell Cycle 2013, 12, 1022–1029. [Google Scholar] [CrossRef] [PubMed]
- Li, D.D.; Sun, T.; Wu, X.Q.; Chen, S.P.; Deng, R.; Jiang, S.; Feng, G.K.; Pan, J.X.; Zhang, X.S.; Zeng, Y.X.; et al. The Inhibition of Autophagy Sensitises Colon Cancer Cells with Wild-Type p53 but Not Mutant p53 to Topotecan Treatment. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- De Vries, A.; Flores, E.R.; Miranda, B.; Hsieh, H.-M.; van Oostrom, C.T.M.; Sage, J.; Jacks, T. Targeted point mutations of p53 lead to dominant-negative inhibition of wild-type p53 function. Proc. Natl. Acad. Sci. USA 2002, 99, 2948–2953. [Google Scholar] [CrossRef] [PubMed]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Xiao, H.L.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Le Toumelin, G.; Criollo, A.; Rain, J.-C.; Gautier, F.; Juin, P.; Tasdemir, E.; Pierron, G.; Troulinaki, K.; Tavernarakis, N.; et al. Functional and physical interaction between Bcl-XL and a BH3-like domain in Beclin-1. EMBO J. 2007, 26, 2527–2539. [Google Scholar] [CrossRef] [PubMed]
- Akar, U.; Chaves-Reyez, A.; Barria, M.; Tari, A.; Sanguino, A.; Kondo, Y.; Kondo, S.; Arun, B.; Lopez-Berestein, G.; Ozpolat, B. Silencing of Bcl-2 expression by small interfering RNA induces autophagic cell death in MCF-7 breast cancer cells. Autophagy 2008, 4, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.D.; Wu, D.; Gu, J.H.; Ge, J.B.; Wu, J.C.; Han, R.; Liang, Z.Q.; Qin, Z.H. The Pro-Survival Role of Autophagy Depends on Bcl-2 Under Nutrition Stress Conditions. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Lamy, L.; Ngo, V.N.; Emre, N.C.T.; Shaffer, A.L.; Yang, Y.; Tian, E.; Nair, V.; Kruhlak, M.J.; Zingone, A.; Landgren, O.; et al. Control of autophagic cell death by caspase-10 in multiple myeloma. Cancer Cell 2013, 23, 435–449. [Google Scholar] [CrossRef] [PubMed]
- Lavieu, G.; Scarlatti, F.; Sala, G.; Levade, T.; Ghidoni, R.; Botti, J.; Codogno, P. Is autophagy the key mechanism by which the sphingolipid rheostat controls the cell fate decision? Autophagy 2007, 3, 45–47. [Google Scholar] [CrossRef] [PubMed]
- Newton, J.; Lima, S.; Maceyka, M.; Spiegel, S. Revisiting the sphingolipid rheostat: Evolving concepts in cancer therapy. Exp. Cell Res. 2015, 333, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Scarlatti, F.; Bauvy, C.; Ventruti, A.; Sala, G.; Cluzeaud, F.; Vandewalle, A.; Ghidoni, R.; Codogno, P. Ceramide-mediated macroautophagy involves inhibition of protein kinase B and up-regulation of beclin 1. J. Biol. Chem. 2004, 279, 18384–18391. [Google Scholar] [CrossRef] [PubMed]
- Lavieu, G.; Scarlatti, F.; Sala, G.; Carpentier, S.; Levade, T.; Ghidoni, R.; Botti, J.; Codogno, P. Regulation of autophagy by sphingosine kinase 1 and its role in cell survival during nutrient starvation. J. Biol. Chem. 2006, 281, 8518–8527. [Google Scholar] [CrossRef] [PubMed]
- Willems, L.; Chapuis, N.; Puissant, A.; Maciel, T.T.; Green, A.S.; Jacque, N.; Vignon, C.; Park, S.; Guichard, S.; Herault, O.; et al. The dual mTORC1 and mTORC2 inhibitor AZD8055 has anti-tumor activity in acute myeloid leukemia. Leukemia 2012, 26, 1195–1202. [Google Scholar] [CrossRef] [PubMed]
- Isakson, P.; Bjoras, M.; Boe, S.O.; Simonsen, A. Autophagy contributes to therapy-induced degradation of the PML/RARA oncoprotein. Blood 2010, 116, 2324–2331. [Google Scholar] [CrossRef] [PubMed]
- Goussetis, D.J.; Gounaris, E.; Wu, E.J.; Vakana, E.; Sharma, B.; Bogyo, M.; Altman, J.K.; Platanias, L.C. Autophagic degradation of the BCR-ABL oncoprotein and generation of antileukemic responses by arsenic trioxide. Blood 2012, 120, 3555–3562. [Google Scholar] [CrossRef] [PubMed]
- Larrue, C.; Saland, E.; Boutzen, H.; Vergez, F.; David, M.; Joffre, C.; Hospital, M.A.; Tamburini, J.; Delabesse, E.; Manenti, S.; et al. Proteasome inhibitors induce FLT3-ITD degradation through autophagy in AML cells. Blood 2016, 127, 882–892. [Google Scholar] [CrossRef] [PubMed]
- Torgersen, M.L.; Engedal, N.; Boe, S.O.; Hokland, P.; Simonsen, A. Targeting autophagy potentiates the apoptotic effect of histone deacetylase inhibitors in t(8;21) AML cells. Blood 2013, 122, 2467–2476. [Google Scholar] [CrossRef] [PubMed]
- Shibutani, S.T.; Saitoh, T.; Nowag, H.; Münz, C.; Yoshimori, T. Autophagy and autophagy-related proteins in the immune system. Nat. Immunol. 2015, 16, 1014–1024. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Adjemian, S.; Yang, H.; Catani, J.P.P.; Hannani, D.; Martins, I.; Michaud, M.; Kepp, O.; Sukkurwala, A.Q.; Vacchelli, E.; et al. ATP-dependent recruitment, survival and differentiation of dendritic cell precursors in the tumor bed after anticancer chemotherapy. Oncoimmunology 2013, 2, e24568. [Google Scholar] [CrossRef] [PubMed]
- Michaud, M.; Martins, I.; Sukkurwala, A.Q.; Adjemian, S.; Ma, Y.; Pellegatti, P.; Shen, S.; Kepp, O.; Scoazec, M.; Mignot, G.; et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 2011, 334, 1573–1577. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.; Tortola, L.; Perlot, T.; Wirnsberger, G.; Novatchkova, M.; Nitsch, R.; Sykacek, P.; Frank, L.; Schramek, D.; Komnenovic, V.; et al. A dual role for autophagy in a murine model of lung cancer. Nat. Commun. 2014, 5, 3056. [Google Scholar] [CrossRef] [PubMed]
- Chemali, M.; Radtke, K.; Desjardins, M.; English, L. Alternative pathways for MHC class I presentation: A new function for autophagy. Cell. Mol. Life Sci. 2011, 68, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Schmid, D.; Pypaert, M.; Münz, C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity 2007, 26, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Palucka, A.K. Dendritic cells as therapeutic vaccines against cancer. Nat. Rev. Immunol. 2005, 5, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Bloy, N.; Pol, J.; Aranda, F.; Eggermont, A.; Cremer, I.; Fridman, W.H.; Fučíková, J.; Galon, J.; Tartour, E.; Spisek, R.; et al. Trial watch: Dendritic cell-based anticancer therapy. Oncoimmunology 2014, 3, e963424. [Google Scholar] [CrossRef] [PubMed]
- Carreno, B.M.; Magrini, V.; Becker-Hapak, M.; Kaabinejadian, S.; Hundal, J.; Petti, A.A.; Ly, A.; Lie, W.-R.; Hildebrand, W.H.; Mardis, E.R.; et al. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 2015, 348, 803–808. [Google Scholar] [CrossRef] [PubMed]
- van der Bruggen, P.; Van den Eynde, B.J. Processing and presentation of tumor antigens and vaccination strategies. Curr. Opin. Immunol. 2006, 18, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, L.-X.; Pang, P.; Cui, Z.; Aung, S.; Haley, D.; Fox, B.A.; Urba, W.J.; Hu, H.-M. Tumor-derived autophagosome vaccine: mechanism of cross-presentation and therapeutic efficacy. Clin. Cancer Res. 2011, 17, 7047–7057. [Google Scholar] [CrossRef] [PubMed]
- Münz, C. Of LAP, CUPS, and DRibbles—Unconventional Use of Autophagy Proteins for MHC Restricted Antigen Presentation. Front. Immunol. 2015, 6, 200. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.; Zhou, Z.; Shu, S.; Fang, Y.; Twitty, C.; Hilton, T.L.; Aung, S.; Urba, W.J.; Fox, B.A.; Hu, H.-M.; et al. Autophagy-assisted antigen cross-presentation: Autophagosome as the argo of shared tumor-specific antigens and DAMPs. Oncoimmunology 2012, 1, 976–978. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Luo, Q.; Xie, H.; Huang, X.; Ni, Y.; Mou, Y.; Hu, Q. Therapeutic antitumor efficacy of tumor-derived autophagosome (DRibble) vaccine on head and neck cancer. Int. J. Nanomedicine 2015, 10, 1921–1930. [Google Scholar] [CrossRef] [PubMed]
- Page, D.B.; Hulett, T.W.; Hilton, T.L.; Hu, H.-M.; Urba, W.J.; Fox, B.A. Glimpse into the future: harnessing autophagy to promote anti-tumor immunity with the DRibbles vaccine. J. Immunother. Cancer 2016, 4, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Mologni, L. Expanding the portfolio of anti-ALK weapons. Transl. Lung Cancer Res. 2015, 4, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.; Soda, M.; Togashi, Y.; Ota, Y.; Sekiguchi, Y.; Hatano, S.; Asaka, R.; Noguchi, M.; Mano, H. Identification of a novel fusion, SQSTM1-ALK, in ALK-positive large B-cell lymphoma. Haematologica 2011, 96, 464–467. [Google Scholar] [CrossRef] [PubMed]
- Mitou, G.; Frentzel, J.; Desquesnes, A.; Le Gonidec, S.; Alsaati, T.; Beau, I.; Lamant, L.; Meggetto, F.; Espinos, E.; Codogno, P.; et al. Targeting autophagy enhances the anti-tumoral action of crizotinib in ALK-positive anaplastic large cell lymphoma. Oncotarget 2015, 6, 30149–30164. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Zhang, L.; Cheng, Y.; Patel, R.; Wu, H.; Zhang, Y.; Wang, M.; Ji, S.; Belani, C.P.; Yang, J.M.; et al. Induction of autophagy contributes to crizotinib resistance in ALK-positive lung cancer. Cancer Biol. Ther. 2014, 15, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Aveic, S.; Pantile, M.; Seydel, A.; Esposito, M.R.; Zanon, C.; Li, G.; Tonini, G.P. Combating autophagy is a strategy to increase cytotoxic effects of novel ALK inhibitor entrectinib in neuroblastoma cells. Oncotarget 2016, 7, 5646–5663. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, L.; Guan, S.; Cao, W.; Wang, H.; Chen, Z.; Zhao, Y.; Yu, Y.; Zhang, H.; Pang, J.C.; et al. Novel ALK inhibitor AZD3463 inhibits neuroblastoma growth by overcoming crizotinib resistance and inducing apoptosis. Sci. Rep. 2016, 6, 19423. [Google Scholar] [CrossRef] [PubMed]
- Megiorni, F.; McDowell, H.P.; Camero, S.; Mannarino, O.; Ceccarelli, S.; Paiano, M.; Losty, P.D.; Pizer, B.; Shukla, R.; Pizzuti, A.; et al. Crizotinib-induced antitumour activity in human alveolar rhabdomyosarcoma cells is not solely dependent on ALK and MET inhibition. J. Exp. Clin. Cancer Res. 2015, 34, 112. [Google Scholar] [CrossRef] [PubMed]
- Lorente, M.; Torres, S.; Salazar, M.; Carracedo, A.; Hernandez-Tiedra, S.; Rodriguez-Fornes, F.; Garcia-Taboada, E.; Melendez, B.; Mollejo, M.; Campos-Martin, Y.; et al. Stimulation of the midkine/ALK axis renders glioma cells resistant to cannabinoid antitumoral action. Cell Death Differ. 2011, 18, 959–973. [Google Scholar] [CrossRef] [PubMed]
- Lorente, M.; Torres, S.; Salazar, M.; Carracedo, A.; Hernandez-Tiedra, S.; Rodriguez-Fornes, F.; Garcia-Taboada, E.; Melendez, B.; Mollejo, M.; Campos-Martin, Y.; et al. Stimulation of ALK by the growth factor midkine renders glioma cells resistant to autophagy-mediated cell death. Autophagy 2011, 7, 1071–1073. [Google Scholar] [CrossRef] [PubMed]
- Park, J.R.; Eggert, A.; Caron, H. Neuroblastoma: Biology, Prognosis, and Treatment. Hematol. Oncol. Clin. North. Am. 2010, 24, 65–86. [Google Scholar] [CrossRef] [PubMed]
- Brodeur, G.; Seeger, R.; Schwab, M.; Varmus, H.; Bishop, J. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science 1984, 224, 1121–1124. [Google Scholar] [CrossRef] [PubMed]
- Deyell, R.J.; Attiyeh, E.F. Advances in the understanding of constitutional and somatic genomic alterations in neuroblastoma. Cancer Genet. 2011, 204, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Janoueix-Lerosey, I.; Lequin, D.; Brugières, L.; Ribeiro, A.; de Pontual, L.; Combaret, V.; Raynal, V.; Puisieux, A.; Schleiermacher, G.; Pierron, G.; et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature 2008, 455, 967–970. [Google Scholar] [CrossRef] [PubMed]
- Pugh, T.J.; Morozova, O.; Attiyeh, E.F.; Asgharzadeh, S.; Wei, J.S.; Auclair, D.; Carter, S.L.; Cibulskis, K.; Hanna, M.; Kiezun, A.; et al. The genetic landscape of high-risk neuroblastoma. Nat. Genet. 2013, 45, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Lamant, L.; Pulford, K.; Bischof, D.; Morris, S.W.; Mason, D.Y.; Delsol, G.; Mariame, B. Expression of the ALK tyrosine kinase gene in neuroblastoma. Am. J. Pathol. 2000, 156, 1711–1721. [Google Scholar] [CrossRef]
- Mossé, Y.P.; Laudenslager, M.; Longo, L.; Cole, K.A.; Wood, A.; Attiyeh, E.F.; Laquaglia, M.J.; Sennett, R.; Lynch, J.E.; Perri, P.; et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 2008, 455, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Fix, A.; Lucchesi, C.; Ribeiro, A.; Lequin, D.; Pierron, G.; Schleiermacher, G.; Delattre, O.; Janoueix-Lerosey, I. Characterization of amplicons in neuroblastoma: High-resolution mapping using DNA microarrays, relationship with outcome, and identification of overexpressed genes. Genes Chromosom. Cancer 2008, 4, 819–834. [Google Scholar] [CrossRef] [PubMed]
- Krytska, K.; Ryles, H.T.; Sano, R.; Raman, P.; Infarinato, N.R.; Hansel, T.D.; Makena, M.R.; Song, M.M.; Reynolds, C.P.; Mossé, Y.P. Crizotinib synergizes with chemotherapy in preclinical models of neuroblastoma. Clin. Cancer Res. 2016, 22, 948–960. [Google Scholar] [CrossRef] [PubMed]
- Cheung, N.-K.V.; Dyer, M.A. Neuroblastoma: Developmental biology, cancer genomics and immunotherapy. Nat. Rev. Cancer 2013, 13, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Moore, N.F.; Azarova, A.M.; Bhatnagar, N.; Ross, K.N.; Drake, L.E.; Frumm, S.; Liu, Q.S.; Christie, A.L.; Sanda, T.; Chesler, L.; et al. Molecular rationale for the use of PI3K/AKT/mTOR pathway inhibitors in combination with crizotinib in ALK-mutated neuroblastoma. Oncotarget 2014, 5, 8737–8749. [Google Scholar] [CrossRef] [PubMed]
- McDowell, H.P. Update on childhood rhabdomyosarcoma. Arch. Dis. Child. 2003, 88, 354–357. [Google Scholar] [CrossRef] [PubMed]
- Kuttesch, J.F.J. Multidrug resistance in pediatric oncology. Investig. New Drugs 1996, 14, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Van Gaal, J.C.; Flucke, U.E.; Roeffen, M.H.S.; de Bont, E.S.J.M.; Sleijfer, S.; Mavinkurve-Groothuis, A.M.C.; Suurmeijer, A.J.H.; van der Graaf, W.T.A.; Versleijen-Jonkers, Y.M.H. Anaplastic lymphoma kinase aberrations in rhabdomyosarcoma: clinical and prognostic implications. J. Clin. Oncol. 2012, 30, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Bonvini, P.; Zin, A.; Alaggio, R.; Pawel, B.; Bisogno, G.; Rosolen, A. High ALK mRNA expression has a negative prognostic significance in rhabdomyosarcoma. Br. J. Cancer 2013, 109, 3084–3091. [Google Scholar] [CrossRef] [PubMed]
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant astrocytic glioma: Genetics, biology, and paths to treatment. Genes Dev. 2007, 21, 2683–2710. [Google Scholar] [CrossRef] [PubMed]
- Jawhari, S.; Ratinaud, M.-H.; Verdier, M. Glioblastoma, hypoxia and autophagy: A survival-prone “ménage-à-trois”. Cell Death Dis. 2016, 7, e2434. [Google Scholar] [CrossRef] [PubMed]
- Karagkounis, G.; Stranjalis, G.; Argyrakos, T.; Pantelaion, V.; Mastoris, K.; Rontogianni, D.; Komaitis, S.; Kalamatianos, T.; Sakas, D.; Tiniakos, D. Anaplastic lymphoma kinase expression and gene alterations in glioblastoma: Correlations with clinical outcome. J. Clin. Pathol. 2017, 70, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Junca, A.; Villalva, C.; Tachon, G.; Rivet, P.; Cortes, U.; Guilloteau, K.; Balbous, A.; Godet, J.; Wager, M.; Karayan-Tapon, L. Crizotinib targets in glioblastoma stem cells. Cancer Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Koyama-Nasu, R.; Haruta, R.; Nasu-Nishimura, Y.; Taniue, K.; Katou, Y.; Shirahige, K.; Todo, T.; Ino, Y.; Mukasa, A.; Saito, N.; et al. The pleiotrophin-ALK axis is required for tumorigenicity of glioblastoma stem cells. Oncogene 2014, 33, 2236–2244. [Google Scholar] [CrossRef] [PubMed]
- Chiba, R.; Akiya, M.; Hashimura, M.; Oguri, Y.; Inukai, M.; Hara, A.; Saegusa, M. ALK signaling cascade confers multiple advantages to glioblastoma cells through neovascularization and cell proliferation. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- Kanzawa, T.; Germano, I.M.; Komata, T.; Ito, H.; Kondo, Y.; Kondo, S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ. 2004, 11, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Yuan, G.; Yan, S.F.; Xue, H.; Zhang, P.; Sun, J.T.; Li, G. Cucurbitacin i induces protective autophagy in glioblastoma in Vitro and in Vivo. J. Biol. Chem. 2014, 289, 10607–10619. [Google Scholar] [CrossRef] [PubMed]
- Mulay, S.R.; Desai, J.; Kumar, S.V.; Eberhard, J.N.; Thomasova, D.; Romoli, S.; Grigorescu, M.; Kulkarni, O.P.; Popper, B.; Vielhauer, V.; et al. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Nat. Commun. 2016, 7, 10274. [Google Scholar] [CrossRef] [PubMed]
- Turner, S.D.; Lamant, L.; Kenner, L.; Brugières, L. Anaplastic large cell lymphoma in paediatric and young adult patients. Br. J. Haematol. 2016, 560–572. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.W.; Kirstein, M.N.; Valentine, M.B.; Dittmer, K.G.; Shapiro, D.N.; Saltman, D.L.; Look, A.T. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 1994, 263, 1281. [Google Scholar] [CrossRef] [PubMed]
- Lamant, L.; Dastugue, N.; Pulford, K.; Delsol, G.; Mariame, B. A new fusion gene TPM3-ALK in anaplastic large cell lymphoma created by a (1;2)(q25;p23) translocation. Blood 1999, 93, 3088–3095. [Google Scholar] [PubMed]
- Lowe, E.J.; Gross, T.G. Anaplastic large cell lymphoma in children and adolescents. Pediatr. Hematol. Oncol. 2013, 30, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Torossian, A.; Frentzel, J.; Daugrois, C.; Broin, N.; Gandarillas, S.; Al Saati, T.; Lamant, L.; Brousset, P.; Giuriato, S.; Espinos, E. Inserm UMR1037, Toulouse, France. Unpublished data. 2017. [Google Scholar]
- Frentzel, J.; Sorrentino, D.; Torossian, A.; Mitou, G.; Hoareau-Allera, C.; Blasco, R.B.; Meggetto, F.; Espinos, E.; Chiarle, R.; Giuriato, S. Inserm UMR1037, Toulouse, France. Unpublished data. 2017. [Google Scholar]
- Rikova, K.; Guo, A.; Zeng, Q.; Possemato, A.; Yu, J.; Haack, H.; Nardone, J.; Lee, K.; Reeves, C.; Li, Y.; et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 2007, 131, 1190–1203. [Google Scholar] [CrossRef] [PubMed]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Camidge, D.R.; Doebele, R.C. Treating ALK-positive lung cancer--early successes and future challenges. Nat. Rev. Clin. Oncol. 2012, 9, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Solomon, B.J.; Mok, T.; Kim, D.W.; Wu, Y.L.; Nakagawa, K.; Mekhail, T.; Felip, E.; Cappuzzo, F.; Paolini, J.; Usari, T.; et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. [Erratum appears in N Engl J Med. 2015 Oct 15;373(16):1582; PMID: 26466011]. N. Engl. J. Med. 2014, 371, 2167–2177. [Google Scholar] [CrossRef] [PubMed]
- Doebele, R.C.; Pilling, A.B.; Aisner, D.L.; Kutateladze, T.G.; Le, A.T.; Weickhardt, A.J.; Kondo, K.L.; Linderman, D.J.; Heasley, L.E.; Franklin, W.A.; et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin. Cancer Res. 2012, 18, 1472–1482. [Google Scholar] [CrossRef] [PubMed]
- Van der Wekken, A.J.; Saber, A.; Hiltermann, T.J.N.; Kok, K.; van den Berg, A.; Groen, H.J.M. Resistance mechanisms after tyrosine kinase inhibitors afatinib and crizotinib in non-small cell lung cancer, a review of the literature. Crit. Rev. Oncol. Hematol. 2016, 100, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Morel, E.; Mehrpour, M.; Botti, J.; Dupont, N.; Hamaï, A.; Nascimbeni, A.C.; Codogno, P. Autophagy: A Druggable Process. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 375–398. [Google Scholar] [CrossRef] [PubMed]
- Schläfli, A.M.; Berezowska, S.; Adams, O.; Langer, R.; Tschan, M.P. Reliable LC3 and p62 autophagy marker detection in formalin fixed paraffin embedded human tissue by immunohistochemistry. Eur. J. Histochem. 2015, 59, 2481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraya, A.A.; Piao, S.; Xu, X.; Zhang, G.; Herlyn, M.; Gimotty, P.; Levine, B.; Amaravadi, R.K.; Speicher, D.W. Identification of secreted proteins that reflect autophagy dynamics within tumor cells. Autophagy 2015, 11, 60–74. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, W.; Levi, E.; Petrogiannis-Haliotis, T.; Lehmann, L.; Wang, Z.; Kadin, M.E. A murine xenograft model for human CD30+ anaplastic large cell lymphoma. Successful growth inhibition with an anti-CD30 antibody (HeFi-1). Am. J. Pathol. 1999, 155, 1353–1359. [Google Scholar] [CrossRef]
- Cheng, M.; Quail, M.R.; Gingrich, D.E.; Ott, G.R.; Lu, L.; Wan, W.; Albom, M.S.; Angeles, T.S.; Aimone, L.D.; Cristofani, F.; et al. CEP-28122, a highly potent and selective orally active inhibitor of anaplastic lymphoma kinase with antitumor activity in experimental models of human cancers. Mol. Cancer Ther. 2012, 11, 670–679. [Google Scholar] [CrossRef] [PubMed]
- Abate, F.; Todaro, M.; van der Krogt, J.-A.; Boi, M.; Landra, I.; Machiorlatti, R.; Tabbò, F.; Messana, K.; Abele, C.; Barreca, A.; et al. A novel patient-derived tumorgraft model with TRAF1-ALK anaplastic large-cell lymphoma translocation. Leukemia 2015, 29, 1390–1401. [Google Scholar] [CrossRef] [PubMed]
- Fang, D.D.; Zhang, B.; Gu, Q.; Lira, M.; Xu, Q.; Sun, H.; Qian, M.; Sheng, W.; Ozeck, M.; Wang, Z.; et al. HIP1–ALK, A Novel ALK Fusion Variant that Responds to Crizotinib. J. Thorac. Oncol. 2014, 9, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Hooper, J.E.; Cantor, E.L.; Ehlen, M.S.; Banerjee, A.; Malempati, S.; Stenzel, P.; Woltjer, R.L.; Gandour-Edwards, R.; Goodwin, N.C.; Yang, Y.; et al. A patient-derived xenograft model of parameningeal embryonal rhabdomyosarcoma for preclinical studies. Sarcoma 2015. [Google Scholar] [CrossRef] [PubMed]
- Braekeveldt, N.; Bexell, D. Patient-derived xenografts as preclinical neuroblastoma models. Cell Tissue Res. 2017. [Google Scholar] [CrossRef] [PubMed]


| Cancer model | Condition 1 | Condition 2 | Reference |
|---|---|---|---|
| Breast cancer | Radiotherapy | Radiotherapy + vitamin B3 | [62,63] |
| Breast cancer | Gemcitabine | Gemcitabine + Estrogen receptor signaling | [65] |
| Non Small Cell Lung cancer | Low inhibition of EGFR signaling | High inhibition of EGFR signaling | [66,67] |
| Colon cancer | Topotecan | Topotecan + mutated p53 status | [70] |
| Breast cancer Neuroblastoma Multiple myeloma | Beclin 1/Bcl-2 complex | Disruption of Beclin 1/Bcl-2 complex | [74] [75] [76] |
| Breast cancer Colon cancer | Sphingosine 1 phosphate signaling | Ceramide signaling | [79] [80] |
| Acute myeloid leukemia | Strong mTOR inhibition | Low mTOR inhibition | [81] |

{kind=link}
{kind=link}
| Cancer Type | ALK Gene Aberration | Treatment | Method(s) Used to Monitor Autophagy | Role of Autophagy | Signaling Pathway Involved | Refs |
|---|---|---|---|---|---|---|
| ALK+ALCL | Translocation (mainly NPM-ALK) | Crizotinib | Electron microscopy Western Blotting Immunohistochemistry Autophagy array (qPCR) Acridine Orange | Cytoprotective | Akt-mTOR suggested | [103] |
| ALK+NSCLC | Translocation (mainly EML4-ALK) | Crizotinib | Western blotting Electron microscopy | Cytoprotective | Akt-mTOR | [104] |
| NB | Mutations Amplification | Entrectinib AZD3463 | Western blotting | Cytoprotective Cytotoxic role suggested | Not studied PI3K/Akt/mTOR | [105] [106] |
| ARMS | Gain in copy number | Crizotinib | Acridine Orange Western blotting | Cytotoxic role suggested | PI3K/Akt/mTOR suggested | [107] |
| GBM | No aberration reported | THC | Immunohistochemistry Western blotting | Cytotoxic | Midkine through Akt/mTORC1 | [108] [109] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frentzel, J.; Sorrentino, D.; Giuriato, S. Targeting Autophagy in ALK-Associated Cancers. Cancers 2017, 9, 161. https://doi.org/10.3390/cancers9120161
Frentzel J, Sorrentino D, Giuriato S. Targeting Autophagy in ALK-Associated Cancers. Cancers. 2017; 9(12):161. https://doi.org/10.3390/cancers9120161
Chicago/Turabian StyleFrentzel, Julie, Domenico Sorrentino, and Sylvie Giuriato. 2017. "Targeting Autophagy in ALK-Associated Cancers" Cancers 9, no. 12: 161. https://doi.org/10.3390/cancers9120161
APA StyleFrentzel, J., Sorrentino, D., & Giuriato, S. (2017). Targeting Autophagy in ALK-Associated Cancers. Cancers, 9(12), 161. https://doi.org/10.3390/cancers9120161