
Understanding Resistance Mechanisms and Expanding the Therapeutic Utility of PARP Inhibitors

Abstract
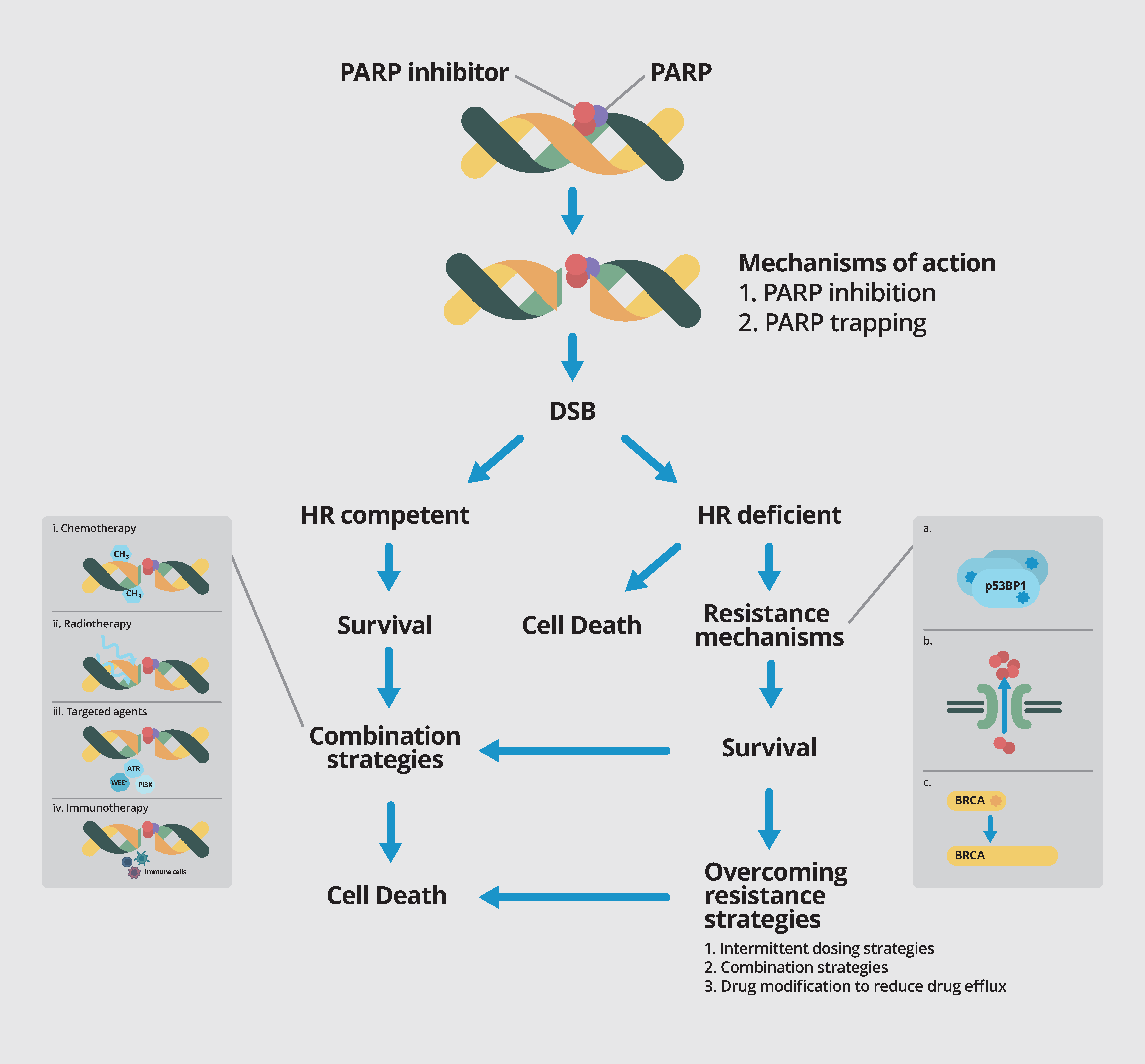
:1. Introduction
2. DNA Damage Repair (DDR) and Mechanisms of Action of PARP Inhibition
3. Currently Available PARP Inhibitors
4. Understanding the Mechanisms of Resistance to PARP Inhibitors
5. Overcoming PARP Resistance
6. Expanding the Therapeutic Utility of PARP Inhibitors beyond BRCA Mutant Cancers
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Durkacz, B.W.; Omidiji, O.; Gray, D.A.; Shall, S. (ADP-ribose)n participates in DNA excision repair. Nature 1980, 283, 593–596. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-repair defects and olaparib in metastatic prostate cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef] [PubMed]
- Robson, M.; Im, S.A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N. Engl. J. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Gelmon, K.A.; Tischkowitz, M.; Mackay, H.; Swenerton, K.; Robidoux, A.; Tonkin, K.; Hirte, H.; Huntsman, D.; Clemons, M.; Gilks, B.; et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: A phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011, 12, 852–861. [Google Scholar] [CrossRef]
- Satoh, M.S.; Lindahl, T. Role of poly(ADP-ribose) formation in DNA repair. Nature 1992, 356, 356–358. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G., Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, T.A.; Shi, Y.; Rodriguez, L.E.; Solomon, L.R.; Donawho, C.K.; DiGiammarino, E.L.; Panchal, S.C.; Wilsbacher, J.L.; Gao, W.; Olson, A.M.; et al. Mechanistic dissection of PARP1 trapping and the impact on in vivo tolerability and efficacy of PARP inhibitors. Mol. Cancer Res. 2015, 13, 1465–1477. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.Y.; Renaud, A.; Zhang, Y.; Ji, J.; Takeda, S.; Morris, J.; Teicher, B.; Doroshow, J.H.; Pommier, Y. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol. Cancer Ther. 2014, 13, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Fong, P.C.; Yap, T.A.; Boss, D.S.; Carden, C.P.; Mergui-Roelvink, M.; Gourley, C.; De Greve, J.; Lubinski, J.; Shanley, S.; Messiou, C.; et al. Poly(ADP)-ribose polymerase inhibition: Frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J. Clin. Oncol. 2010, 28, 2512–2519. [Google Scholar] [CrossRef] [PubMed]
- Tutt, A.; Robson, M.; Garber, J.E.; Domchek, S.M.; Audeh, M.W.; Weitzel, J.N.; Friedlander, M.; Arun, B.; Loman, N.; Schmutzler, R.K.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: A proof-of-concept trial. Lancet 2010, 376, 235–244. [Google Scholar] [CrossRef]
- Audeh, M.W.; Carmichael, J.; Penson, R.T.; Friedlander, M.; Powell, B.; Bell-McGuinn, K.M.; Scott, C.; Weitzel, J.N.; Oaknin, A.; Loman, N.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: A proof-of-concept trial. Lancet 2010, 376, 245–251. [Google Scholar] [CrossRef]
- Kaye, S.B.; Lubinski, J.; Matulonis, U.; Ang, J.E.; Gourley, C.; Karlan, B.Y.; Amnon, A.; Bell-McGuinn, K.M.; Chen, L.M.; Friedlander, M.; et al. Phase ii, open-label, randomized, multicenter study comparing the efficacy and safety of olaparib, a poly (ADP-ribose) polymerase inhibitor, and pegylated liposomal doxorubicin in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer. J. Clin. Oncol. 2012, 30, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N. Engl. J. Med. 2012, 366, 1382–1392. [Google Scholar] [CrossRef] [PubMed]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.L.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: A preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014, 15, 852–861. [Google Scholar] [CrossRef]
- Ledermann, J.A.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Overall survival in patients with platinum-sensitive recurrent serous ovarian cancer receiving olaparib maintenance monotherapy: An updated analysis from a randomised, placebo-controlled, double-blind, phase 2 trial. Lancet Oncol. 2016, 17, 1579–1589. [Google Scholar] [CrossRef]
- Bang, Y.J.; Im, S.A.; Lee, K.W.; Cho, J.Y.; Song, E.K.; Lee, K.H.; Kim, Y.H.; Park, J.O.; Chun, H.G.; Zang, D.Y.; et al. Randomized, double-blind phase ii trial with prospective classification by ATM protein level to evaluate the efficacy and tolerability of olaparib plus paclitaxel in patients with recurrent or metastatic gastric cancer. J. Clin. Oncol. 2015, 33, 3858–3865. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Myzak, M.C.; Johnson Iii, B.A.; De Jesus-Acosta, A.; Le, D.T.; Jaffee, E.M.; Azad, N.S.; Donehower, R.C.; Zheng, L.; Oberstein, P.E.; et al. Olaparib in combination with irinotecan, cisplatin, and mitomycin C in patients with advanced pancreatic cancer. Oncotarget 2017. [Google Scholar] [CrossRef] [PubMed]
- Drew, Y.; Ledermann, J.; Hall, G.; Rea, D.; Glasspool, R.; Highley, M.; Jayson, G.; Sludden, J.; Murray, J.; Jamieson, D.; et al. Phase 2 multicentre trial investigating intermittent and continuous dosing schedules of the poly(ADP-ribose) polymerase inhibitor rucaparib in germline BRCA mutation carriers with advanced ovarian and breast cancer. Br. J. Cancer 2016, 114, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M.; et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 part 1): An international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef]
- Coleman, R.L.; Swisher, E.M.; Oza, A.M.; Scott, C.L.; Giordano, H.; Lin, K.K.; Konecny, G.E.; Tinker, A.; O’Malley, D.M.; Kristeleit, R.S.; et al. Refinement of prespecified cutoff for genomic loss of heterozygosity (LOH) in ARIEL2 part 1: A phase II study of rucaparib in patients (PTS) with high grade ovarian carcinoma (HGOC). J. Clin. Oncol. 2016, 34, 5540. [Google Scholar]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Telli, M.L.; Timms, K.M.; Reid, J.; Hennessy, B.; Mills, G.B.; Jensen, K.C.; Szallasi, Z.; Barry, W.T.; Winer, E.P.; Tung, N.M.; et al. Homologous recombination deficiency (HRD) score predicts response to platinum-containing neoadjuvant chemotherapy in patients with triple-negative breast cancer. Clin. Cancer Res. 2016, 22, 3764–3773. [Google Scholar] [CrossRef] [PubMed]
- Middleton, M.R.; Friedlander, P.; Hamid, O.; Daud, A.; Plummer, R.; Falotico, N.; Chyla, B.; Jiang, F.; McKeegan, E.; Mostafa, N.M.; et al. Randomized phase II study evaluating veliparib (ABT-888) with temozolomide in patients with metastatic melanoma. Ann. Oncol. 2015, 26, 2173–2179. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.S.; Blais, N.; Mazieres, J.; Reck, M.; Jones, C.M.; Juhasz, E.; Urban, L.; Orlov, S.; Barlesi, F.; Kio, E.; et al. Randomized, placebo-controlled, phase ii study of veliparib in combination with carboplatin and paclitaxel for advanced/metastatic non-small cell lung cancer. Clin. Cancer Res. 2017, 23, 1937–1944. [Google Scholar] [CrossRef] [PubMed]
- De Bono, J.; Ramanathan, R.K.; Mina, L.; Chugh, R.; Glaspy, J.; Rafii, S.; Kaye, S.; Sachdev, J.; Heymach, J.; Smith, D.C.; et al. Phase I, dose-escalation, two-part trial of the PARP inhibitor talazoparib in patients with advanced germline BRCA1/2 mutations and selected sporadic cancers. Cancer Discov. 2017, 7, 620–629. [Google Scholar] [CrossRef] [PubMed]
- Edwards, S.L.; Brough, R.; Lord, C.J.; Natrajan, R.; Vatcheva, R.; Levine, D.A.; Boyd, J.; Reis-Filho, J.S.; Ashworth, A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature 2008, 451, 1111–1115. [Google Scholar] [CrossRef] [PubMed]
- Sakai, W.; Swisher, E.M.; Karlan, B.Y.; Agarwal, M.K.; Higgins, J.; Friedman, C.; Villegas, E.; Jacquemont, C.; Farrugia, D.J.; Couch, F.J.; et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 2008, 451, 1116–1120. [Google Scholar] [CrossRef] [PubMed]
- Norquist, B.; Wurz, K.A.; Pennil, C.C.; Garcia, R.; Gross, J.; Sakai, W.; Karlan, B.Y.; Taniguchi, T.; Swisher, E.M. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J. Clin. Oncol. 2011, 29, 3008–3015. [Google Scholar] [CrossRef] [PubMed]
- Jaspers, J.E.; Kersbergen, A.; Boon, U.; Sol, W.; van Deemter, L.; Zander, S.A.; Drost, R.; Wientjens, E.; Ji, J.; Aly, A.; et al. Loss of 53bp1 causes PARP inhibitor resistance in BRCA1-mutated mouse mammary tumors. Cancer Discov. 2013, 3, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Xu, X.; Bunting, S.F.; Liu, J.; Wang, R.H.; Cao, L.L.; Wu, J.J.; Peng, T.N.; Chen, J.; Nussenzweig, A.; et al. A selective requirement for 53bp1 in the biological response to genomic instability induced by BRCA1 deficiency. Mol. Cell 2009, 35, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Bunting, S.F.; Callen, E.; Kozak, M.L.; Kim, J.M.; Wong, N.; Lopez-Contreras, A.J.; Ludwig, T.; Baer, R.; Faryabi, R.B.; Malhowski, A.; et al. Brca1 functions independently of homologous recombination in DNA interstrand crosslink repair. Mol. Cell 2012, 46, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.; Lottersberger, F.; Buonomo, S.B.; Sfeir, A.; de Lange, T. 53BP1 regulates DSB repair using RIF1 to control 5′ end resection. Science 2013, 339, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Di Virgilio, M.; Callen, E.; Yamane, A.; Zhang, W.; Jankovic, M.; Gitlin, A.D.; Feldhahn, N.; Resch, W.; Oliveira, T.Y.; Chait, B.T.; et al. RIF1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science 2013, 339, 711–715. [Google Scholar] [CrossRef] [PubMed]
- Nakada, S.; Yonamine, R.M.; Matsuo, K. RNF8 regulates assembly of RAD51 at DNA double-strand breaks in the absence of BRCA1 and 53BP1. Cancer Res. 2012, 72, 4974–4983. [Google Scholar] [CrossRef] [PubMed]
- Binkhathlan, Z.; Lavasanifar, A. P-glycoprotein inhibition as a therapeutic approach for overcoming multidrug resistance in cancer: Current status and future perspectives. Curr. Cancer Drug Targets 2013, 13, 326–346. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.H.; Yu, A.M. ABC transporters in multidrug resistance and pharmacokinetics, and strategies for drug development. Curr. Pharm. Des. 2014, 20, 793–807. [Google Scholar] [CrossRef] [PubMed]
- Basanta, D.; Anderson, A.R. Exploiting ecological principles to better understand cancer progression and treatment. Interface Focus 2013. [Google Scholar] [CrossRef] [PubMed]
- Korolev, K.S.; Xavier, J.B.; Gore, J. Turning ecology and evolution against cancer. Nat. Rev. Cancer 2014, 14, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.H.; Evans, T.J.; Middleton, M.R.; Molife, L.R.; Spicer, J.; Dieras, V.; Roxburgh, P.; Giordano, H.; Jaw-Tsai, S.; Goble, S.; et al. A phase I study of intravenous and oral rucaparib in combination with chemotherapy in patients with advanced solid tumours. Br. J. Cancer 2017, 116, 884–892. [Google Scholar] [CrossRef] [PubMed]
- Balmana, J.; Tung, N.; Isakoff, S.; Grana, B.; Ryan, P.; Saura, C.; Lowe, E.; Frewer, P.; Winer, E.; Baselga, J. Phase I trial of olaparib in combination with cisplatin for the treatment of patients with advanced breast, ovarian and other solid tumors. Ann. Oncol. 2014, 25, 1656–1663. [Google Scholar] [CrossRef] [PubMed]
- Henneman, L.; van Miltenburg, M.H.; Michalak, E.M.; Braumuller, T.M.; Jaspers, J.E.; Drenth, A.P.; de Korte-Grimmerink, R.; Gogola, E.; Szuhai, K.; Schlicker, A.; et al. Selective resistance to the PARP inhibitor olaparib in a mouse model for BRCA1-deficient metaplastic breast cancer. Proc. Natl. Acad. Sci. USA 2015, 112, 8409–8414. [Google Scholar] [CrossRef] [PubMed]
- Garnett, M.J.; Edelman, E.J.; Heidorn, S.J.; Greenman, C.D.; Dastur, A.; Lau, K.W.; Greninger, P.; Thompson, I.R.; Luo, X.; Soares, J.; et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 2012, 483, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Choy, E.; Butrynski, J.E.; Harmon, D.C.; Morgan, J.A.; George, S.; Wagner, A.J.; D’Adamo, D.; Cote, G.M.; Flamand, Y.; Benes, C.H.; et al. Phase II study of olaparib in patients with refractory ewing sarcoma following failure of standard chemotherapy. BMC Cancer 2014, 14, 813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ordonez, J.L.; Amaral, A.T.; Carcaboso, A.M.; Herrero-Martin, D.; del Carmen Garcia-Macias, M.; Sevillano, V.; Alonso, D.; Pascual-Pasto, G.; San-Segundo, L.; Vila-Ubach, M.; et al. The PARP inhibitor olaparib enhances the sensitivity of ewing sarcoma to trabectedin. Oncotarget 2015, 6, 18875–18890. [Google Scholar] [CrossRef] [PubMed]
- Oza, A.M.; Cibula, D.; Benzaquen, A.O.; Poole, C.; Mathijssen, R.H.; Sonke, G.S.; Colombo, N.; Spacek, J.; Vuylsteke, P.; Hirte, H.; et al. Olaparib combined with chemotherapy for recurrent platinum-sensitive ovarian cancer: A randomised phase 2 trial. Lancet Oncol. 2015, 16, 87–97. [Google Scholar] [CrossRef]
- Robins, H.I.; Zhang, P.; Gilbert, M.R.; Chakravarti, A.; de Groot, J.F.; Grimm, S.A.; Wang, F.; Lieberman, F.S.; Krauze, A.; Trotti, A.M.; et al. A randomized phase I/II study of ABT-888 in combination with temozolomide in recurrent temozolomide resistant glioblastoma: An nrg oncology rtog group study. J. Neurooncol. 2016, 126, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Farago, A.F.; Drapkin, B.J.; Charles, A.; Yeap, B.; Heist, R.S.; Azzoli, C.G.; Jackman, D.M.; Barbie, D.A.; Choy, E.; Sequist, L.V.; et al. Abstract ct048: Phase 1/2 study of olaparib tablets and temozolomide in patients with small cell lung cancer (SCLC) following failure of prior chemotherapy. Cancer Res. 2017, 77, CT048. [Google Scholar] [CrossRef]
- Hirai, T.; Shirai, H.; Fujimori, H.; Okayasu, R.; Sasai, K.; Masutani, M. Radiosensitization effect of poly(ADP-ribose) polymerase inhibition in cells exposed to low and high liner energy transfer radiation. Cancer Sci. 2012, 103, 1045–1050. [Google Scholar] [CrossRef] [PubMed]
- Wurster, S.; Hennes, F.; Parplys, A.C.; Seelbach, J.I.; Mansour, W.Y.; Zielinski, A.; Petersen, C.; Clauditz, T.S.; Munscher, A.; Friedl, A.A.; et al. PARP1 inhibition radiosensitizes HNSCC cells deficient in homologous recombination by disabling the DNA replication fork elongation response. Oncotarget 2016, 7, 9732–9741. [Google Scholar] [CrossRef] [PubMed]
- Nowsheen, S.; Bonner, J.A.; Yang, E.S. The poly(ADP-ribose) polymerase inhibitor ABT-888 reduces radiation-induced nuclear EGFR and augments head and neck tumor response to radiotherapy. Radiother. Oncol. 2011, 99, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Barazzuol, L.; Jena, R.; Burnet, N.G.; Meira, L.B.; Jeynes, J.C.; Kirkby, K.J.; Kirkby, N.F. Evaluation of poly (ADP-ribose) polymerase inhibitor ABT-888 combined with radiotherapy and temozolomide in glioblastoma. Radiat. Oncol. 2013, 8, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemasson, B.; Wang, H.; Galban, S.; Li, Y.; Zhu, Y.; Heist, K.A.; Tsein, C.; Chenevert, T.L.; Rehemtulla, A.; Galban, C.J.; et al. Evaluation of concurrent radiation, temozolomide and ABT-888 treatment followed by maintenance therapy with temozolomide and ABT-888 in a genetically engineered glioblastoma mouse model. Neoplasia 2016, 18, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Foiani, M. Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell Biol. 2008, 9, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.S.; O’Carrigan, B.; Jackson, S.P.; Yap, T.A. Targeting DNA repair in cancer: Beyond PARP inhibitors. Cancer Discov. 2017, 7, 20–37. [Google Scholar] [CrossRef] [PubMed]
- Karnak, D.; Engelke, C.G.; Parsels, L.A.; Kausar, T.; Wei, D.; Robertson, J.R.; Marsh, K.B.; Davis, M.A.; Zhao, L.; Maybaum, J.; et al. Combined inhibition of wee1 and PARP1/2 for radiosensitization in pancreatic cancer. Clin. Cancer Res. 2014, 20, 5085–5096. [Google Scholar] [CrossRef] [PubMed]
- Snedeker, J.C.; Burleson, T.M.; Tibes, R.; Porter, C.C. Combined inhibition of wee1 and poly(ADP ribose) polymerase1/2 synergistically inhibits acute myeloid leukemia cells by enhancing DNA damage and the induction of apoptosis. Blood 2014, 124, 3621. [Google Scholar]
- Kim, H.; George, E.; Ragland, R.L.; Rafail, S.; Zhang, R.; Krepler, C.; Morgan, M.A.; Herlyn, M.; Brown, E.J.; Simpkins, F. Targeting the ATR/CHK1 axis with PARP inhibition results in tumor regression in BRCA-mutant ovarian cancer models. Clin. Cancer Res. 2017, 23, 3097–3108. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Shen, Q.; Zhang, P.; Tao, R.; Chang, W.; Li, R.; Xie, G.; Liu, W.; Zhang, L.; Kapoor, P.; et al. CHK1 inhibition potentiates the therapeutic efficacy of PARP inhibitor BMN673 in gastric cancer. Am. J. Cancer Res. 2017, 7, 473–483. [Google Scholar] [PubMed]
- Sen, T.; Tong, P.; Stewart, C.A.; Cristea, S.; Valliani, A.; Shames, D.S.; Redwood, A.; Fan, Y.; Li, L.; Glisson, B.S.; et al. CHK1 inhibition in small cell lung cancer produces single-agent activity in biomarker-defined disease subsets and combination activity with cisplatin or olaparib. Cancer Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, Y.H.; Garcia-Garcia, C.; Serra, V.; He, L.; Torres-Lockhart, K.; Prat, A.; Anton, P.; Cozar, P.; Guzman, M.; Grueso, J.; et al. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discov. 2012, 2, 1036–1047. [Google Scholar] [CrossRef] [PubMed]
- Juvekar, A.; Burga, L.N.; Hu, H.; Lunsford, E.P.; Ibrahim, Y.H.; Balmana, J.; Rajendran, A.; Papa, A.; Spencer, K.; Lyssiotis, C.A.; et al. Combining a PI3K inhibitor with a PARP inhibitor provides an effective therapy for brca1-related breast cancer. Cancer Discov. 2012, 2, 1048–1063. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Billalabeitia, E.; Seitzer, N.; Song, S.J.; Song, M.S.; Patnaik, A.; Liu, X.S.; Epping, M.T.; Papa, A.; Hobbs, R.M.; Chen, M.; et al. Vulnerabilities of PTEN-TP53-deficient prostate cancers to compound PARP-PI3K inhibition. Cancer Discov. 2014, 4, 896–904. [Google Scholar] [CrossRef] [PubMed]
- De, P.; Sun, Y.; Carlson, J.H.; Friedman, L.S.; Leyland-Jones, B.R.; Dey, N. Doubling down on the PI3K-AKT-mTOR pathway enhances the antitumor efficacy of PARP inhibitor in triple negative breast cancer model beyond BRCA-ness. Neoplasia 2014, 16, 43–72. [Google Scholar] [CrossRef] [PubMed]
- Mo, W.; Liu, Q.; Lin, C.C.; Dai, H.; Peng, Y.; Liang, Y.; Peng, G.; Meric-Bernstam, F.; Mills, G.B.; Li, K.; et al. mTOR inhibitors suppress homologous recombination repair and synergize with PARP inhibitors via regulating SUV39H1 in BRCA-proficient triple-negative breast cancer. Clin. Cancer Res. 2016, 22, 1699–1712. [Google Scholar] [CrossRef] [PubMed]
- Chan, N.; Bristow, R.G. “Contextual” synthetic lethality and/or loss of heterozygosity: Tumor hypoxia and modification of DNA repair. Clin. Cancer Res. 2010, 16, 4553–4560. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.F.; Barry, W.T.; Birrer, M.; Lee, J.M.; Buckanovich, R.J.; Fleming, G.F.; Rimel, B.; Buss, M.K.; Nattam, S.; Hurteau, J.; et al. Combination cediranib and olaparib versus olaparib alone for women with recurrent platinum-sensitive ovarian cancer: A randomised phase 2 study. Lancet Oncol. 2014, 15, 1207–1214. [Google Scholar] [CrossRef]
- Neri, P.; Ren, L.; Gratton, K.; Stebner, E.; Johnson, J.; Klimowicz, A.; Duggan, P.; Tassone, P.; Mansoor, A.; Stewart, D.A.; et al. Bortezomib-induced “brcaness” sensitizes multiple myeloma cells to PARP inhibitors. Blood 2011, 118, 6368–6379. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, T.; Flies, D.B.; Marjon, N.A.; Mantia-Smaldone, G.; Ronner, L.; Gimotty, P.A.; Adams, S.F. Ctla-4 blockade synergizes therapeutically with PARP inhibition in BRCA1-deficient ovarian cancer. Cancer Immunol. Res. 2015, 3, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.K.; Hsu, J.M.; Hsu, J.L.; Yu, W.H.; Du, Y.; Lee, H.H.; et al. PARP inhibitor upregulates PD-l1 expression and enhances cancer-associated immunosuppression. Clin. Cancer Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Peng, Y.; Wei, L.; Zhang, W.; Yang, L.; Lan, L.; Kapoor, P.; Ju, Z.; Mo, Q.; Shih Ie, M.; et al. ARID1a deficiency impairs the DNA damage checkpoint and sensitizes cells to PARP inhibitors. Cancer Discov. 2015, 5, 752–767. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Omlin, A.; de Bono, J.S. Development of therapeutic combinations targeting major cancer signaling pathways. J. Clin. Oncol. 2013, 31, 1592–1605. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Drug | Company | IC50/nM | Relative PARP Trapping Potency [9,11] | Predominant Toxicities |
|---|---|---|---|---|
| Olaparib | Astra Zeneca | 6 | 1 | GI toxicities, fatigue, anemia |
| Rucaparib | Clovis | 21 | 1 | GI toxicities, fatigue, anemia, liver dysfunction |
| Niraparib | Tesaro | 60 | ~2 | Myelosuppression, GI toxicities, fatigue |
| Veliparib | AbbVie | 30 | <0.2 | Fatigue, alopecia, GI toxicities, myelosuppression |
| Talazoparib | Pfizer | 4 | ~100 | GI toxicities, fatigue, lymphopenia |
| Drug | PARP Inhibitor | Phase | Tumor Type | NCT |
|---|---|---|---|---|
| Cytotoxic | ||||
| Platinums | ||||
| Carboplatin | Olaparib | I | Solid tumors | 02418624 |
| Carboplatin + paclitaxel | Talazoparib | I | Solid tumors | 02317874 |
| Carboplatin + paclitaxel | Veliparib | III | Breast | 02163694 |
| Carboplatin + etoposide | Veliparib | II | SCLC | 02289690 |
| Carboplatin + gemcitabine | Veliparib | II | Germ cell | 02860819 |
| Carboplatin + paclitaxel + avastin | Olaparib | III | Ovarian | 02477644 |
| Cisplatin | Veliparib | II | Breast | 02595905 |
| Cisplatin + gemcitabine | Talazoparib | I | Solid tumors | 02537561 |
| Temozolomide-based | ||||
| Irinotecan +/− temozolomide | Talazoparib | I | Paediatric tumors | 02392793 |
| Temozolomide or irinotecan | Niraparib | I | Ewing’s sarcoma | 02044120 |
| Temozolomide + capecitabine | Veliparib | I | PNET | 02831179 |
| 5FU-based | ||||
| FOLFOX | Veliparib | I/II | Pancreas | 0149865 |
| FOLFIRI | Veliparib | II | Pancreas | 02890355 |
| Others | ||||
| Liposomal irinotecan | Veliparib | I | Solid tumors | 02631733 |
| Decitabine | Talazoparib | I | AML | 02878785 |
| Radiation | ||||
| RT | Olaparib | I | HNSCC | 02229656 |
| RT | Olaparib | I | Breast | 02227082 |
| RT | Olaparib | I | Esophagus | 01460888 |
| RT | Olaparib | I | Sarcoma | 02787642 |
| RT +/− cisplatin | Olaparib | I | NSCLC | 01562210 |
| RT + carboplatin + paclitaxel | Veliparib | I/II | NSCLC | 01386385 |
| Rd223 | Niraparib | I | Prostate | 03076203 |
| Targeted therapy | ||||
| Cell cycle check point inhibitors | ||||
| AZD1775 (Wee1) | Olaparib | I | Solid tumors | 02511795 |
| Prexasertib (CHK1) | Olaparib | I | Solid tumors | 03057145 |
| VX-970 (ATR) + cisplatin | Veliparib | I | Solid tumors | 02723864 |
| Dinaciclib (CDK) | Veliparib | I | Solid tumors | 01434316 |
| Anti-angiogenics | ||||
| Cediranib (VEGF) | Olaparib | II | Ovarian; GBM; solid tumors; | 02345265; 02974621; 02498613 |
| Ramucirumab (VEGF) | Olaparib | I/II | Gastric | 03008278 |
| Bevacizumab (VEGF) | Niraparib | I/II | Ovarian | 02354131 |
| PI3K/AKT/mTOR pathway | ||||
| AZD5363 (PI3K) | Olaparib | I | Solid tumors | 02338622 |
| Everolimus (mTOR) | Niraparib | I | Breast, ovarian | 03154281 |
| Other targeted therapies | ||||
| Selumetinib | Olaparib | I | Solid tumors | 03162627 |
| AT13387 (Hsp90) | Olaparib | I | Ovarian and breast | 02898207 |
| Lapatanib (HER2) | Veliparib | I | Breast | 02158507 |
| Hormonal therapy | ||||
| Abiraterone | Olaparib | II | Prostate | 01972217 |
| Enzalutamide | Niraparib | I | Prostate | 02500901 |
| Immunotherapy | ||||
| Anti-PD1 | ||||
| Nivolumab | Veliparib | I | Solid tumors, lymphoma | 03061188 |
| Pembrolizumab | Niraparib | I | Breast, ovarian | 02657889 |
| Nivolumab + platinum doublet | Veliparib | II | NSCLC | 02944396 |
| Anti-PDL1 | ||||
| Durvalumab | Olaparib | II | Breast | 03167619 |
| Durvalumab + tremelimumab | Olaparib | I | Ovarian | 02953457 |
| Atezolizumab | Rucaparib | I | Gynaecological | 03101280 |
| Atezolizumab | Veliparib | II | Breast | 02849496 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, J.S.J.; Tan, D.S.P. Understanding Resistance Mechanisms and Expanding the Therapeutic Utility of PARP Inhibitors. Cancers 2017, 9, 109. https://doi.org/10.3390/cancers9080109
Lim JSJ, Tan DSP. Understanding Resistance Mechanisms and Expanding the Therapeutic Utility of PARP Inhibitors. Cancers. 2017; 9(8):109. https://doi.org/10.3390/cancers9080109
Chicago/Turabian StyleLim, Joline S. J., and David S. P. Tan. 2017. "Understanding Resistance Mechanisms and Expanding the Therapeutic Utility of PARP Inhibitors" Cancers 9, no. 8: 109. https://doi.org/10.3390/cancers9080109
APA StyleLim, J. S. J., & Tan, D. S. P. (2017). Understanding Resistance Mechanisms and Expanding the Therapeutic Utility of PARP Inhibitors. Cancers, 9(8), 109. https://doi.org/10.3390/cancers9080109