Hydrogenation of Aqueous Acetic Acid over Ru-Sn/TiO2 Catalyst in a Flow-Type Reactor, Governed by Reverse Reaction




Abstract
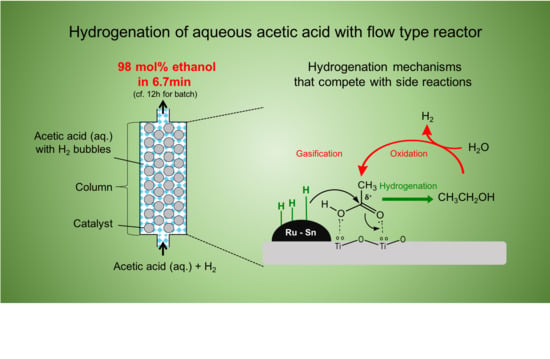
:
1. Introduction
2. Results and Discussion
2.1. Hydrogenation of Aqueous Acetic Acid to Ethanol in Flow Type Reactor
2.2. Side Reactions Competing with Ethanol Production
2.3. Influences of Catalyst Column Size and Reaction Mechanism
2.4. Roles of Flow and Batch Reactors
2.5. Hydrogenation of Lactic Acid to Propane-1,2-diol
3. Experimental
3.1. Materials and Catalyst Preparation
3.2. Hydrogenation with Flow Type Reactor
3.3. Product Determination
4. Conclusions
- Selectivity of the acetic acid formation against gasification was markedly improved in the flow reactor, which enabled the reaction at higher temperatures. For this reason, ethanol was obtained in 78 and 98 mol % yields at 280 and 200 °C for short residence times of 0.5 and 6.7 min (LHSV: 15.1−1 and 1.23 h−1), respectively, (batch type: 12 h).
- Oxidation of the product ethanol to acetic acid (reverse reaction) with water as an oxidant occurred as a side reaction, which decreased the apparent rate of ethanol production.
- The hydrogenation of aqueous acetic acid was governed by equilibrium reactions, and hence, the ethanol/acetic acid molar ratio did not change for the prolonged reaction. This limited the reaction temperature to less than 240 °C, for which the ethanol/acetic acid molar ratio at equilibrium was 49 thus giving complete conversion of acetic acid to ethanol.
- The ethanol/acetic acid molar ratio at equilibrium varied from 238:1 to 3.0:1 depending on the reaction temperature (from 200 to 300 °C).
- Prolonged reactions above 240 °C gave gaseous product because gasification is irreversible. Conversely, the amount of acetic acid converted to ethanol was determined by an equilibrium process.
- The use of a flow reactor is advantageous for the efficient activation of hydrogen and increases the rate of hydrogenation of acetic acid to ethanol rather than the reverse reaction. Thus, conversion to ethanol is completed before gasification reactions start to affect the yield.
- Lactic acid was also reduced selectively to propane-1,2-diol in an 87 mol % yield with a residence time less than 0.5 min.
- A hydrogenation mechanism is proposed, providing insights into the development of efficient hydrogenation catalysts and reaction systems.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Urry, J. The problem of energy. Theory Cult. Soc. 2014, 31, 3–20. [Google Scholar] [CrossRef]
- Kim, S.; Dale, B.E. Global potential bioethanol production from wasted crops and crop residues. Biomass Bioenergy 2004, 26, 361–375. [Google Scholar] [CrossRef]
- Dodić, S.N.; Popov, S.D.; Dodić, J.M.; Ranković, J.A.; Zavargo, Z.Z. Potential contribution of bioethanol fuel to the transport sector of Vojvodina. Renew. Sustain. Energy Rev. 2009, 13, 2197–2200. [Google Scholar] [CrossRef]
- Owusu, P.A.; Asumadu-Sarkodie, S. A review of renewable energy sources, sustainability issues and climate change mitigation. Cogent Eng. 2016, 3, 1–14. [Google Scholar] [CrossRef]
- Yang, Y.; Bae, J.; Kim, J.; Suh, S. Replacing gasoline with corn ethanol results in significant environmental problem-shifting. Environ. Sci. Technol. 2012, 46, 3671–3678. [Google Scholar] [CrossRef]
- Takagi, M.; Abe, S.; Suzuki, S.; Emert, G.; Yata, N. A method for production of alcohol directly from cellulose using cellulase and yeast. Chem. Microb. Protein 1977, 551–571. [Google Scholar]
- Schell, D.J.; Hinman, N.D.; Wyman, C.E.; Werdene, P.J. Whole broth cellulase production for use in simultaneous saccharification and fermentation. Appl. Biochem. Biotechnol. 1990, 24, 287–297. [Google Scholar] [CrossRef]
- Saka, S.; Rabemanolontsoa, H.; Minami, E.; Kawamoto, H. Advanced ethanol production with acetic acid fermentation from lignocellulosics. J. Jpn. Pet. Inst. 2019, 62, 199–204. [Google Scholar] [CrossRef] [Green Version]
- Le Berre, C. Acetic acid. Eur. Chem. News. 2001, 74, 20. [Google Scholar]
- Dimian, A.C.; Kiss, A.A. Novel energy efficient process for acetic acid production by methanol carbonylation. Chem. Eng. Res. Des. 2020, 159, 1–12. [Google Scholar] [CrossRef]
- Besson, M.; Gallezot, P.; Pinel, C. Conversion of biomass into chemicals over metal catalysts. Chem. Rev. 2014, 114, 1827–1870. [Google Scholar] [CrossRef]
- Cheah, K.Y.; Tang, T.S.; Mizukami, F.; Niwa, S.I.; Toba, M.; Choo, Y.M. Selective hydrogenation of oleic acid to 9-octadecen-1-ol: Catalyst preparation and optimum reaction conditions. J. Am. Oil Chem. Soc. 1992, 69, 410–416. [Google Scholar] [CrossRef]
- Chen, L.; Li, Y.; Zhang, X.; Zhang, Q.; Wang, T.; Ma, L. Mechanistic insights into the effects of support on the reaction pathway for aqueous-phase hydrogenation of carboxylic acid over the supported Ru catalysts. Appl. Catal. A Gen. 2014, 478, 117–128. [Google Scholar] [CrossRef]
- Mendes, M.J.; Santos, O.A.A.; Jordão, E.; Silva, A.M. Hydrogenation of oleic acid over ruthenium catalysts. Appl. Catal. A Gen. 2001, 217, 253–262. [Google Scholar] [CrossRef]
- Zhang, S.; Duan, X.; Ye, L.; Lin, H.; Xie, Z.; Yuan, Y. Production of ethanol by gas phase hydrogenation of acetic acid over carbon nanotube-supported Pt-Sn nanoparticles. Proc. Catal. Today 2013, 215, 260–266. [Google Scholar] [CrossRef]
- Tahara, K.; Nagahara, E.; Itoi, Y.; Nishiyama, S.; Tsuruya, S.; Masai, M. Liquid-phase hydrogenation of carboxylic acid on supported bimetallic Ru-Sn-alumina catalysts. Appl. Catal. A Gen. 1997, 154, 75–86. [Google Scholar] [CrossRef]
- Wan, H.; Chaudhari, R.V.; Subramaniam, B. Aqueous phase hydrogenation of acetic acid and its promotional effect on p-cresol hydrodeoxygenation. Energy Fuels 2013, 27, 487–493. [Google Scholar] [CrossRef]
- Kawamoto, H.; Fujii, T.; Ito, Y.; Saka, S. Effects of different solvents on hydrogenation of acetic acid over Pt/TiO2 for bioethanol production. J. Jpn. Inst. Energy 2016, 95, 162–166. [Google Scholar] [CrossRef]
- Ito, Y.; Kawamoto, H.; Saka, S. Efficient and selective hydrogenation of aqueous acetic acid on Ru-Sn/TiO2 for bioethanol production from lignocellulosics. Fuel 2016, 178, 118–123. [Google Scholar] [CrossRef] [Green Version]
- Hessel, V.; Löb, P.; Krtschil, U.; Löwe, H. Microstructured reactors for development and production in pharmaceutical and fine chemistry. In Synthesis New Avenues to Efficient Chemical Synthesis; Seeberger, P.H., Blume, T., Eds.; Springer: Berlin/Heidelberg, Germany, 2007; pp. 205–240. [Google Scholar]
- Roberge, D.M.; Zimmermann, B.; Rainone, F.; Gottsponer, M.; Eyholzer, M.; Kockmann, N. Microreactor technology and continuous processes in the fine chemical and pharmaceutical industry: Is the revolution underway? Org. Process. Res. Dev. 2008, 12, 905–910. [Google Scholar] [CrossRef]
- Roberge, D.M.; Bieler, N.; Mathier, M.; Eyholzer, M.; Zimmermann, B.; Barthe, P.; Guermeur, C.; Lobet, O.; Moreno, M.; Woehl, P. Development of an industrial multi-injection microreactor for fast and exothermic reactions—Part II. Chem. Eng. Technol. 2008, 31, 1155–1161. [Google Scholar] [CrossRef]
- Hessel, V. Novel process windows-gates to maximizing process intensification via flow chemistry. Chem. Eng. Technol. 2009, 32, 1641. [Google Scholar] [CrossRef]
- Wiles, C.; Watts, P. Continuous flow reactors: A perspective. Green Chem. 2012, 14, 38–54. [Google Scholar] [CrossRef]
- Hartman, R.L.; McMullen, J.P.; Jensen, K.F. Deciding whether to go with the flow: Evaluating the merits of flow reactors for synthesis. Angew. Chem. Int. Ed. 2011, 50, 7502–7519. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Jiao, J.; Song, P.; Nie, W.; Yi, C.; Zhang, Q.; Li, P. Recent progress in continuous-flow hydrogenation. ChemSusChem 2020, 13, 2876–2893. [Google Scholar] [CrossRef]
- Durndell, L.J.; Wilson, K.; Lee, A.F. Platinum-catalysed cinnamaldehyde hydrogenation in continuous flow. RSC Adv. 2015, 5, 80022–80026. [Google Scholar] [CrossRef] [Green Version]
- Numwong, N.; Luengnaruemitchai, A.; Chollacoop, N.; Yoshimura, Y. Partial hydrogenation of polyunsaturated fatty acid methyl esters over Pd/activated carbon: Effect of type of reactor. Chem. Eng. J. 2012, 210, 173–181. [Google Scholar] [CrossRef]
- Gómez-Quero, S.; Cárdenas-Lizana, F.; Keane, M.A. Liquid phase catalytic hydrodechlorination of 2,4-dichlorophenol over Pd/Al2O3: Batch vs. continuous operation. Chem. Eng. J. 2011, 166, 1044–1051. [Google Scholar] [CrossRef]
- Wang, Y.; Prinsen, P.; Triantafyllidis, K.S.; Karakoulia, S.A.; Yepez, A.; Len, C.; Luque, R. Batch versus continuous flow performance of supported mono- and bimetallic nickel catalysts for catalytic transfer hydrogenation of furfural in isopropanol. ChemCatChem 2018, 10, 3459–3468. [Google Scholar] [CrossRef]
- Osako, T.; Torii, K.; Hirata, S.; Uozumi, Y. Chemoselective continuous-flow hydrogenation of aldehydes catalyzed by platinum nanoparticles dispersed in an amphiphilic resin. ACS Catal. 2017, 7, 7371–7377. [Google Scholar] [CrossRef]
- Olcay, H.; Xu, Y.; Huber, G.W. Effects of hydrogen and water on the activity and selectivity of acetic acid hydrogenation on ruthenium. Green Chem. 2014, 16, 911–924. [Google Scholar] [CrossRef]
- Sloan, E.D.; Koh, C.A. Molecular structures and similarities to ice. In Clathrate Hydrates of Natural Gases, 3rd ed.; Sloan, E.D., Ed.; CRC Press: Boca Raton, FL, USA, 2007; pp. 45–102. [Google Scholar]
- Mao, W.L.; Mao, H.K. Hydrogen storage in molecular compounds. Proc. Natl. Acad. Sci. USA 2004, 101, 708–710. [Google Scholar] [CrossRef] [Green Version]
- Ziparo, C.; Giannasi, A.; Ulivi, L.; Zoppi, M. Raman spectroscopy study of molecular hydrogen solubility in water at high pressure. Int. J. Hydrog. Energy 2011, 36, 7951–7955. [Google Scholar] [CrossRef]
- Diagne, C.; Idriss, H.; Kiennemann, A. Hydrogen production by ethanol reforming over Rh/CeO2-ZrO2 catalysts. Catal. Commun. 2002, 3, 565–571. [Google Scholar] [CrossRef]
- Xiong, H.; DeLaRiva, A.; Wang, Y.; Datye, A.K. Low-temperature aqueous-phase reforming of ethanol on bimetallic PdZn catalysts. Catal. Sci. Technol. 2015, 5, 254–263. [Google Scholar] [CrossRef]
- Nozawa, T.; Yoshida, A.; Hikichi, S.; Naito, S. Effects of Re addition upon aqueous phase reforming of ethanol over TiO2 supported Rh and Ir catalysts. Int. J. Hydrog. Energy 2015, 40, 4129–4140. [Google Scholar] [CrossRef]
- Nozawa, T.; Mizukoshi, Y.; Yoshida, A.; Naito, S. Aqueous phase reforming of ethanol and acetic acid over TiO2 supported Ru catalysts. Appl. Catal. B Environ. 2014, 146, 221–226. [Google Scholar] [CrossRef]
- Sauer, M.; Porro, D.; Mattanovich, D.; Branduardi, P. Microbial production of organic acids: Expanding the markets. Trends Biotechnol. 2008, 26, 100–108. [Google Scholar] [CrossRef]
- Vaidya, U.R.; Nadkarni, V.M. Unsaturated polyester resins from poly (ethylene terephthalate) waste. 1. synthesis and characterization. Ind. Eng. Chem. Res. 1987, 26, 194–198. [Google Scholar] [CrossRef]
- Luo, G.; Yan, S.; Qiao, M.; Zhuang, J.; Fan, K. Effect of tin on Ru-B/γ-Al2O3 catalyst for the hydrogenation of ethyl lactate to 1,2-propanediol. Appl. Catal. A Gen. 2004, 275, 95–102. [Google Scholar] [CrossRef]
- Fiume, M.M.; Bergfeld, W.F.; Belsito, D.V.; Hill, R.A.; Klaassen, C.D.; Liebler, D.; Marks, J.G.; Shank, R.C.; Slaga, T.J.; Snyder, P.W.; et al. Safety assessment of propylene glycol, tripropylene glycol, and PPGs as used in cosmetics. Int. J. Toxicol. 2012, 31, 245S–260S. [Google Scholar] [CrossRef]
- Luo, G.; Yan, S.; Qiao, M.; Fan, K. Effect of promoters on the structures and properties of the RuB/γ-Al2O3 catalyst. J. Mol. Catal. A Chem. 2005, 230, 69–77. [Google Scholar] [CrossRef]
- Zhang, Z.; Jackson, J.E.; Miller, D.J. Aqueous-phase hydrogenation of lactic acid to propylene glycol. Appl. Catal. A Gen. 2001, 219, 89–98. [Google Scholar] [CrossRef]
- Broadbent, H.S.; Campbell, G.C.; Bartley, W.J.; Johnson, J.H. Rhenium and its compounds as hydrogenation catalysts. III. rhenium heptoxide. J. Org. Chem. 1959, 24, 1847–1854. [Google Scholar] [CrossRef]
- Mao, B.W.; Cai, Z.Z.; Huang, M.Y.; Jiang, Y.Y. Hydrogenation of carboxylic acids catalyzed by magnesia-supported poly-γ-aminopropylsiloxane-Ru complex. Polym. Adv. Technol. 2003, 14, 278–281. [Google Scholar] [CrossRef]
- Jones, R.V.; Godorhazy, L.; Varga, N.; Szalay, D.; Urge, L.; Darvas, F. Continuous-flow high pressure hydrogenation reactor for optimization and high-throughput synthesis. J. Comb. Chem. 2006, 8, 110–116. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction Temperature (°C) | ||||||
|---|---|---|---|---|---|---|
| 200 | 220 | 240 | 260 | 280 | 300 | |
| Wide column | 238 | 120 | 49 | 12 | 6.1 | 3.0 |
| Narrow column | 3.9 | 4.6 | 2.4 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Y.; Konishi, K.; Minami, E.; Saka, S.; Kawamoto, H. Hydrogenation of Aqueous Acetic Acid over Ru-Sn/TiO2 Catalyst in a Flow-Type Reactor, Governed by Reverse Reaction. Catalysts 2020, 10, 1270. https://doi.org/10.3390/catal10111270
Zhao Y, Konishi K, Minami E, Saka S, Kawamoto H. Hydrogenation of Aqueous Acetic Acid over Ru-Sn/TiO2 Catalyst in a Flow-Type Reactor, Governed by Reverse Reaction. Catalysts. 2020; 10(11):1270. https://doi.org/10.3390/catal10111270
Chicago/Turabian StyleZhao, Yuanyuan, Kansei Konishi, Eiji Minami, Shiro Saka, and Haruo Kawamoto. 2020. "Hydrogenation of Aqueous Acetic Acid over Ru-Sn/TiO2 Catalyst in a Flow-Type Reactor, Governed by Reverse Reaction" Catalysts 10, no. 11: 1270. https://doi.org/10.3390/catal10111270
APA StyleZhao, Y., Konishi, K., Minami, E., Saka, S., & Kawamoto, H. (2020). Hydrogenation of Aqueous Acetic Acid over Ru-Sn/TiO2 Catalyst in a Flow-Type Reactor, Governed by Reverse Reaction. Catalysts, 10(11), 1270. https://doi.org/10.3390/catal10111270