α-Functionalization of Imines via Visible Light Photoredox Catalysis



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
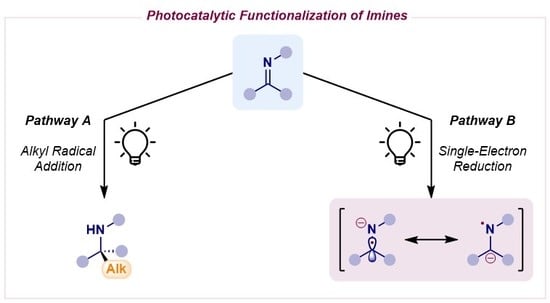
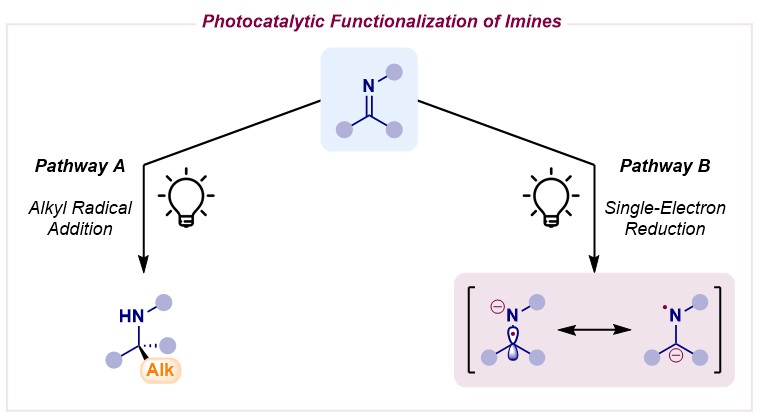
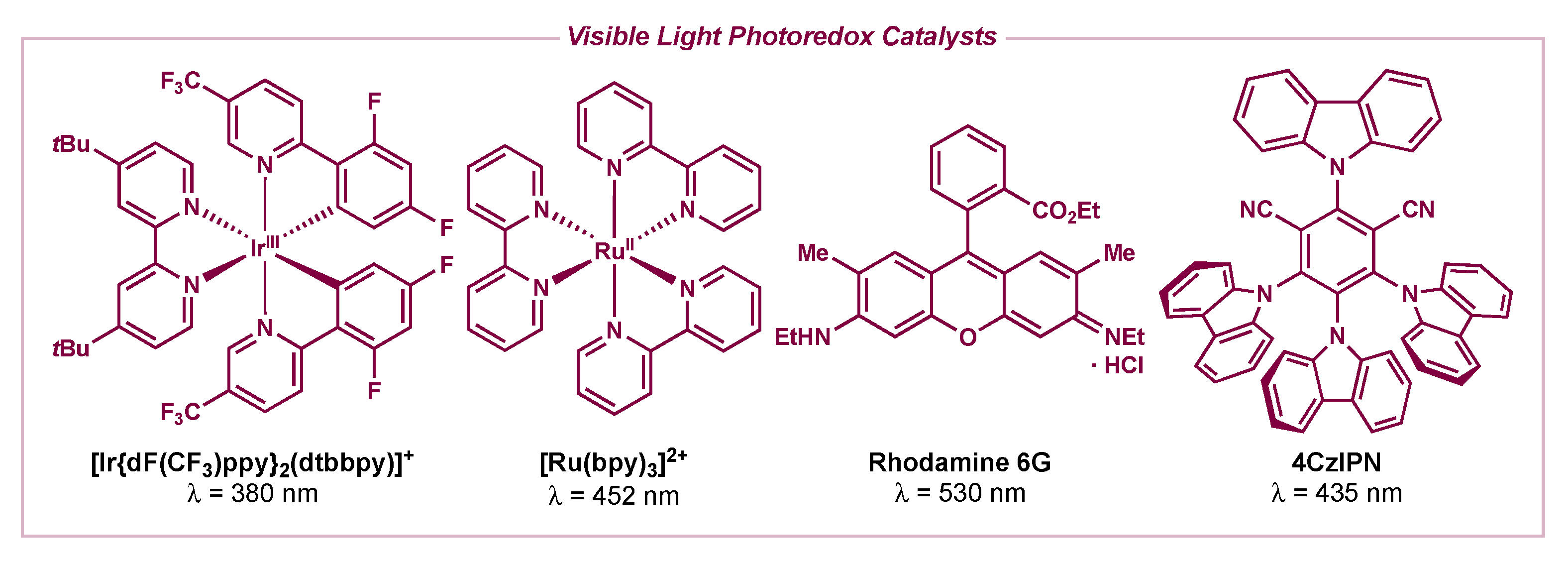
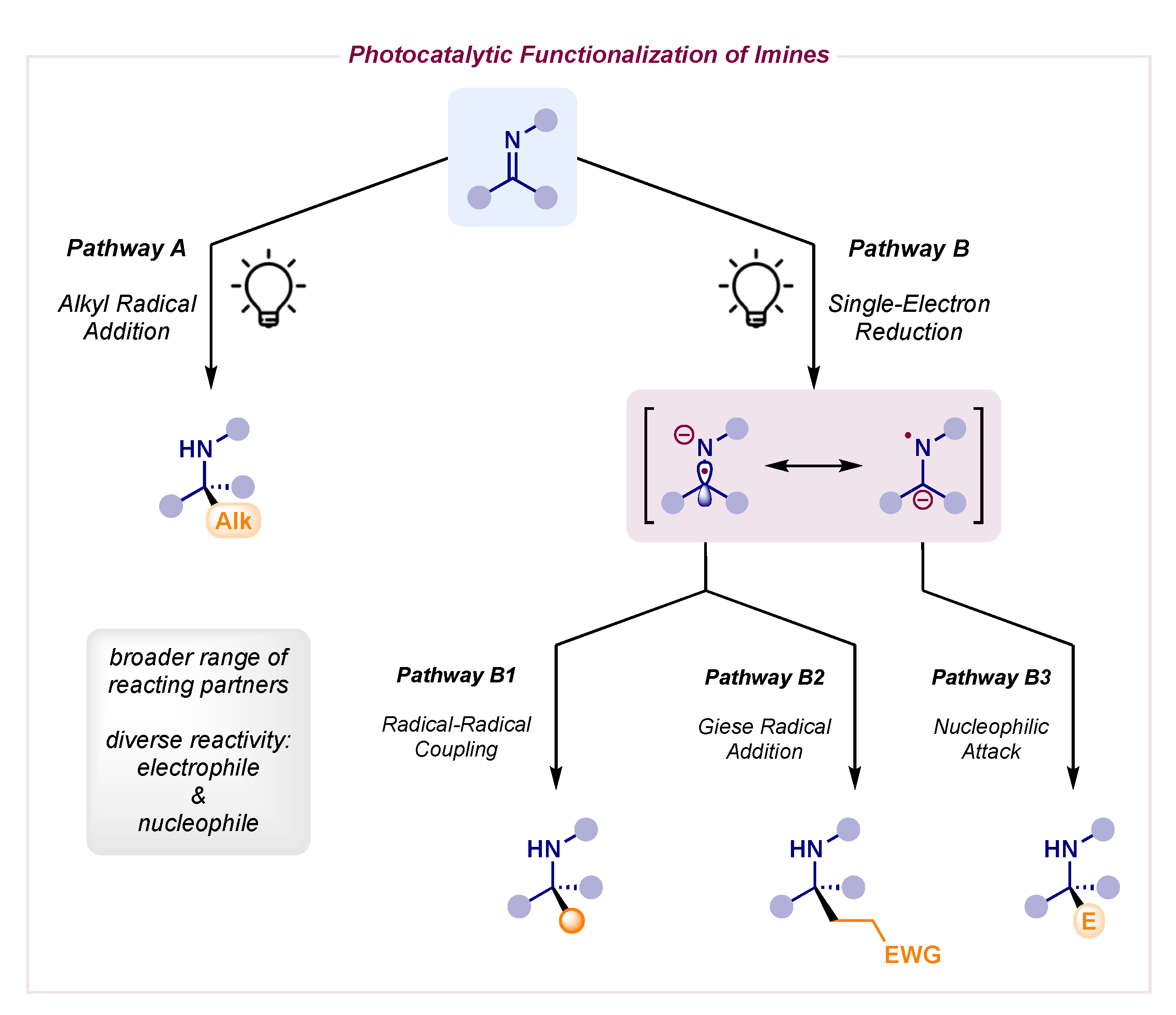
1. Introduction
2. Photocatalytic Radical Additions to Imines—Pathway A
2.1. Racemic Photocatalytic Radical Additions to Imines
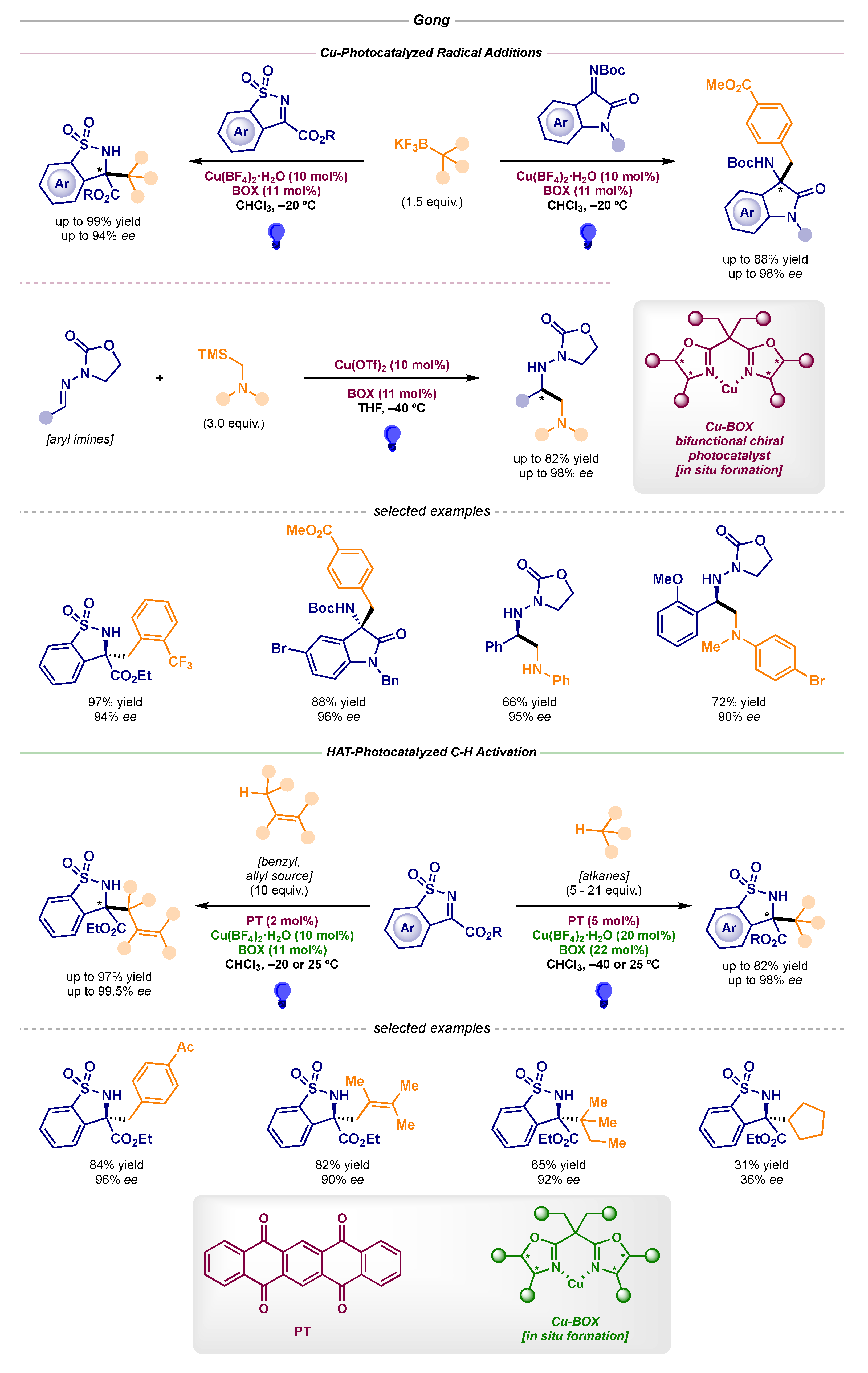
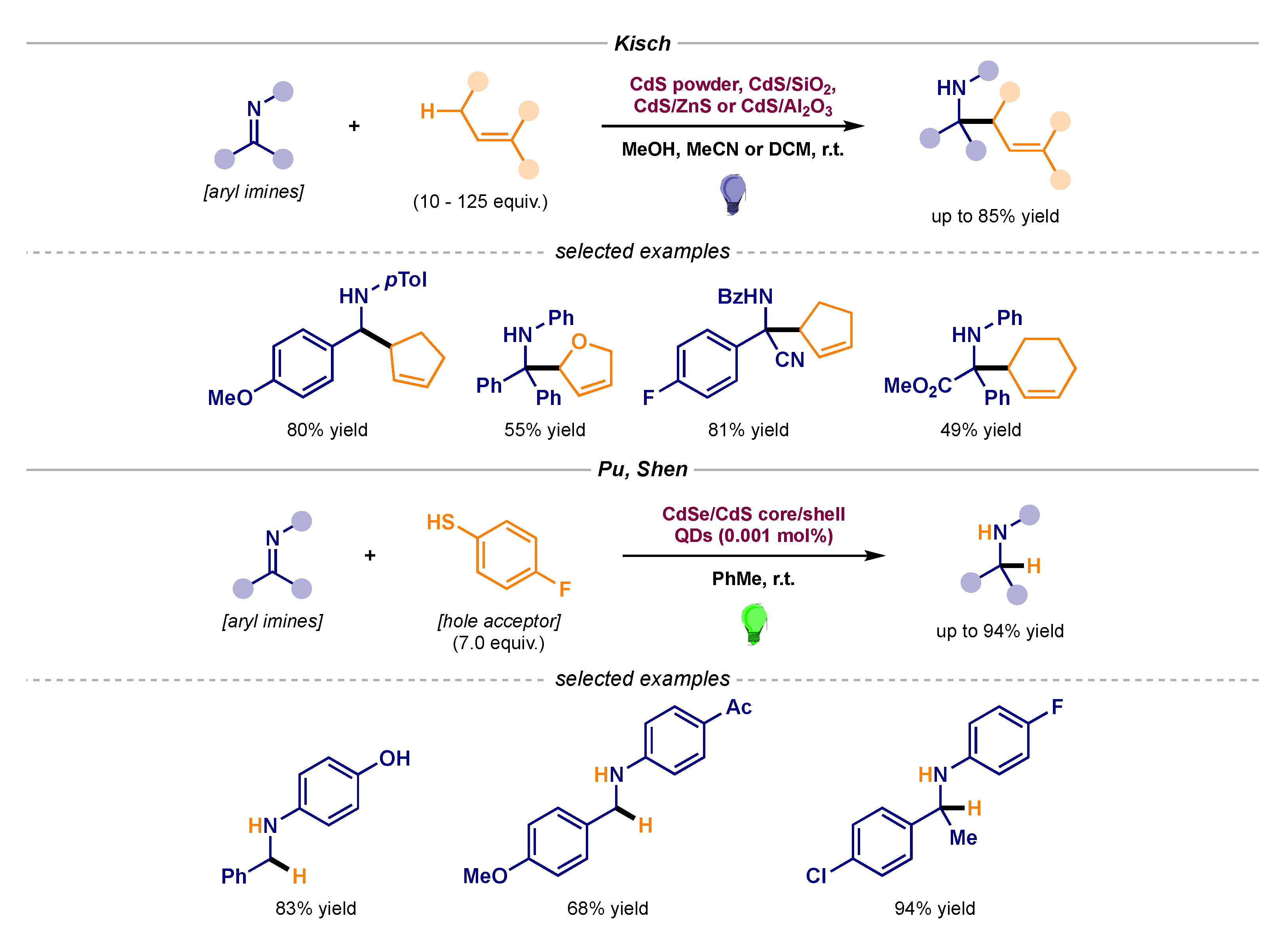
2.2. Stereoselective Photocatalytic Radical Additions to Imines
3. Photocatalytic α-Amino Radical Reactivity via Single-Electron Reduction of Imine Derivatives—Pathway B
3.1. Racemic α-Amino Radical–Radical Couplings—Pathway B1
3.2. Racemic α-Amino Radical Additions to Activated Olefins—Pathway B2
3.3. Racemic α-Amino Carbanion Nucleophilic Attacks—Pathway B3
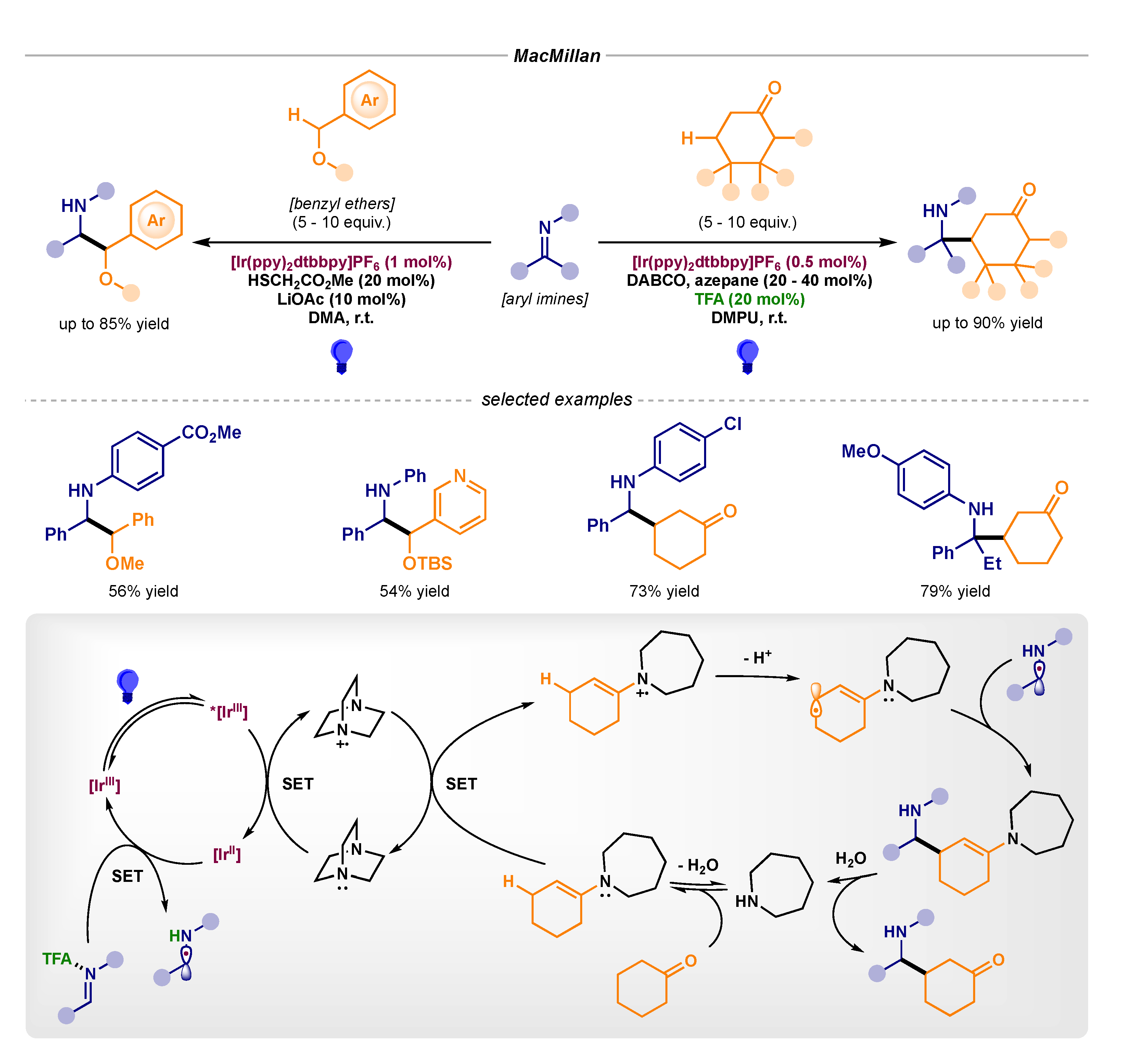
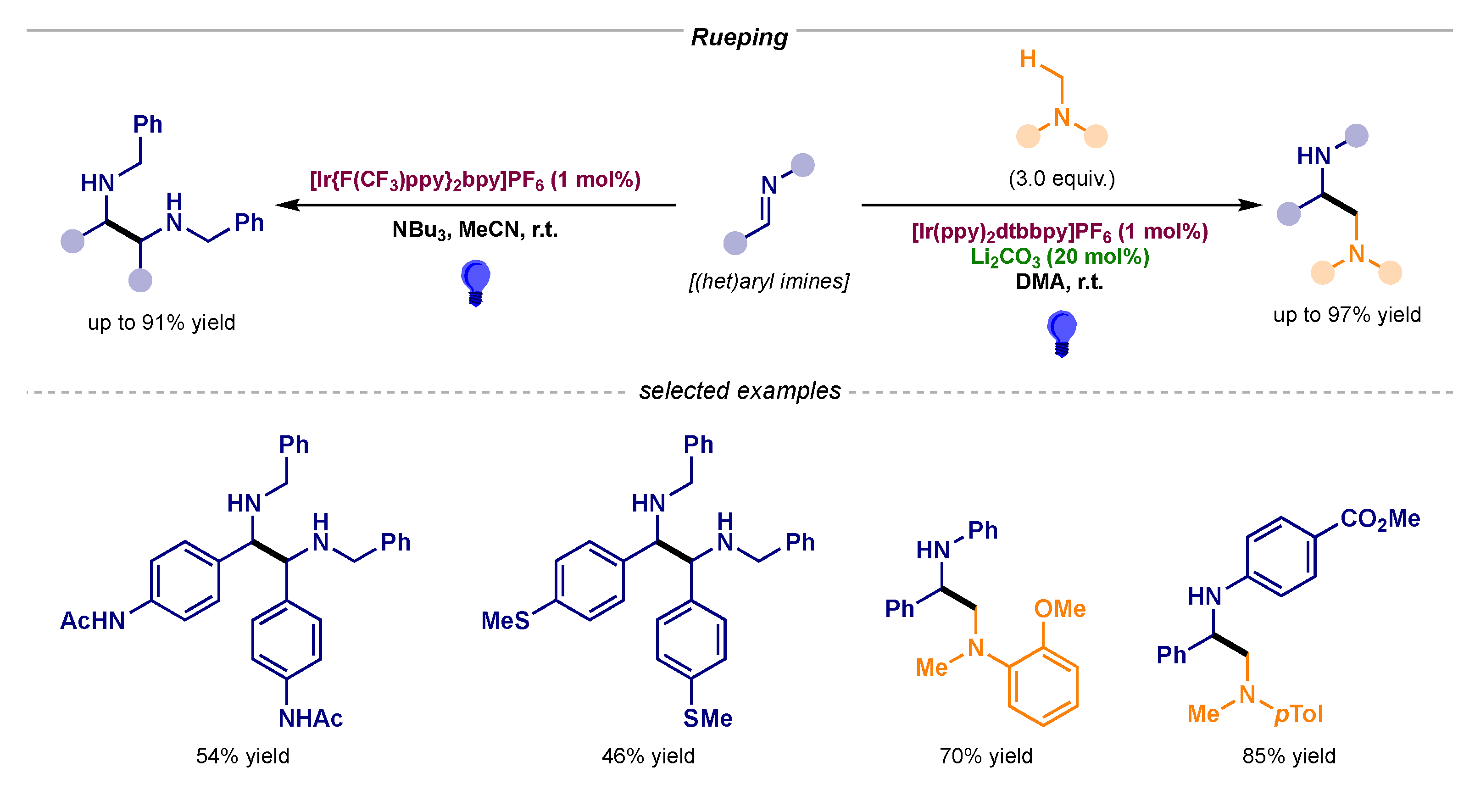
3.4. Stereoselective Photocatalytic α-Amino Radical Reactivity via Single-Electron Reduction of Imine Derivatives—Pathway B
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| abbreviation | full description |
| 4CzIPN | 2,4,5,6-tetra(9H-carbazol-9-yl)isophthalonitrile |
| Ac | acyl |
| Alk | alkyl |
| Anth | anthracenyl |
| Ar | aryl |
| BArF | B[3,5-(CF3)2C6H3]4 |
| BD | 2,3-butanedione |
| Boc | tert-butyloxycarbonyl |
| BOX | bis(oxazoline) |
| bpy | 2,2′-bipyridine |
| Bu | butyl |
| Bz | benzoyl |
| CBA | chiral Brønsted acid |
| CPA | chiral phosphoric acid |
| CPME | cyclopentyl methyl ether |
| Cy | cyclohexyl |
| DABCO | 1,4-diazabicyclo[2.2.2]octane |
| DCM | dichloromethane |
| DFMS | zinc difluoromethanesulfinate |
| DKR | dynamic kinetic resolution |
| DMA | N,N-dimethylacetamide |
| DMF | N,N-dimethylformamide |
| DMPU | N,N’-dimethylpropyleneurea |
| DMSO | dimethylsulfoxide |
| DPZ | 5,6-bis(5-methoxythiophen-2-yl)pyrazine-2,3-dicarbonitrile |
| dr | diastereomeric ratio |
| dtbbpy | 4,4′-di-tert-butyl-2,2′-dipyridyl |
| E | electrophile |
| ee | enantiomeric excess |
| Et | ethyl |
| EWG | electron withdrawing group |
| HAT | hydrogen atom transfer |
| HEH | Hantzsch ester |
| Het | heteroaryl |
| IFET | interfacial electron transfer |
| iPr | iso-propyl |
| MAO | monoamine oxidase |
| Me | methyl |
| Mes | mesityl |
| MS | molecular sieves |
| Naph | naphthyl |
| NHPI | N-hydroxyphthalimide |
| NHS | N-hydroxysuccinimide |
| PCET | proton-coupled electron transfer |
| Ph | phenyl |
| phen | 1,10-phenanthroline |
| ppy | 2-phenylpyridine |
| PT | 5,7,12,14-pentacenetetrone |
| pTol | para-tolyl |
| QD | quantum dot |
| RAE | redox-active ester |
| RPC | radical polar crossover |
| SET | single-electron transfer |
| SLAP | silicon amine protocol |
| SN2 | bimolecular nucleophilic substitution |
| sppy | 3-(pyridin-2-yl)benzenesulfonate |
| TBS | tert-butyldimethylsilyl |
| TCNHPI | N-hydroxytetrachlorophthalimide |
| Tf | trifluoromethanesulfonyl |
| TFA | trifluoroacetic acid |
| TFE | 2,2,2-trifluoroethanol |
| THF | tetrahydrofuran |
| TMS | trimethylsilyl |
| TPP | 2,4,6-triphenylpyrylium tetrafluoroborate |
References
- Visible Light Photocatalysis in Organic Chemistry, 1st ed; Stephenson, C.R.J.; Yoon, T.P.; MacMillan, D.W.C. (Eds.) Wiley-VCH: Weinheim, Germany, 2018. [Google Scholar]
- Matsui, J.K.; Lang, S.B.; Heitz, D.R.; Molander, G.A. Photoredox-Mediated Routes to Radicals: The Value of Catalytic Radical Generation in Synthetic Methods Development. ACS Catal. 2017, 7, 2563–2575. [Google Scholar] [CrossRef]
- Lee, K.N.; Ngai, M.-Y. Recent developments in transition-metal photoredox-catalysed reactions of carbonyl derivatives. Chem. Commun. 2017, 53, 13093–13112. [Google Scholar] [CrossRef] [PubMed]
- Leitch, J.A.; Rossolini, T.; Rogova, T.; Maitland, J.A.P.; Dixon, D.J. α-Amino Radicals via Photocatalytic Single-Electron Reduction of Imine Derivatives. ACS Catal. 2020, 10, 2009–2025. [Google Scholar] [CrossRef]
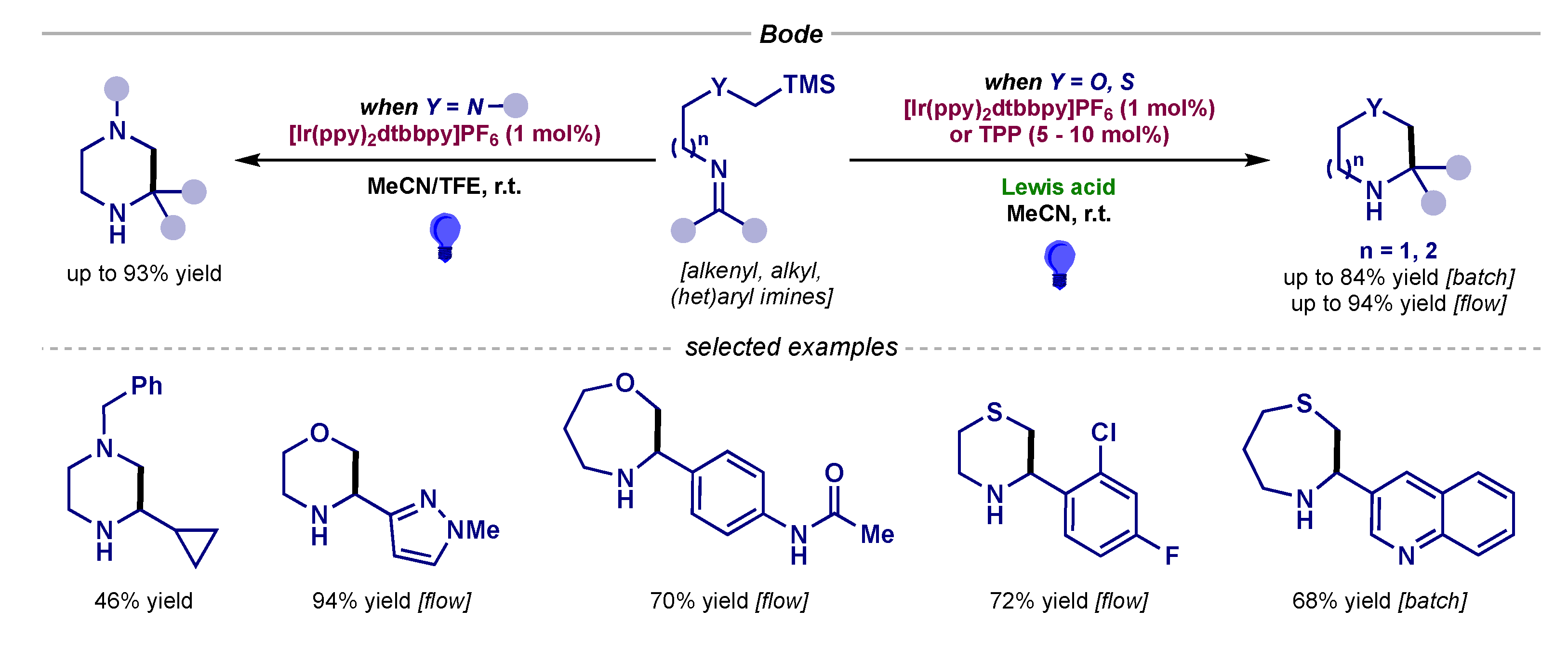
- Hsieh, S.-Y.; Bode, J.W. Silicon Amine Reagents for the Photocatalytic Synthesis of Piperazines from Aldehydes and Ketones. Org. Lett. 2016, 18, 2098–2101. [Google Scholar] [CrossRef] [PubMed]
- Jackl, M.K.; Legnani, L.; Morandi, B.; Bode, J.W. Continuous Flow Synthesis of Morpholines and Oxazepanes with Silicon Amine Protocol (SLAP) Reagents and Lewis Acid Facilitated Photoredox Catalysis. Org. Lett. 2017, 19, 4696–4699. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, S.-Y.; Bode, J.W. Lewis Acid Induced Toggle from Ir(II) to Ir(IV) Pathways in Photocatalytic Reactions: Synthesis of Thiomorpholines and Thiazepanes from Aldehydes and SLAP Reagents. ACS Cent. Sci. 2017, 3, 66–72. [Google Scholar] [CrossRef]
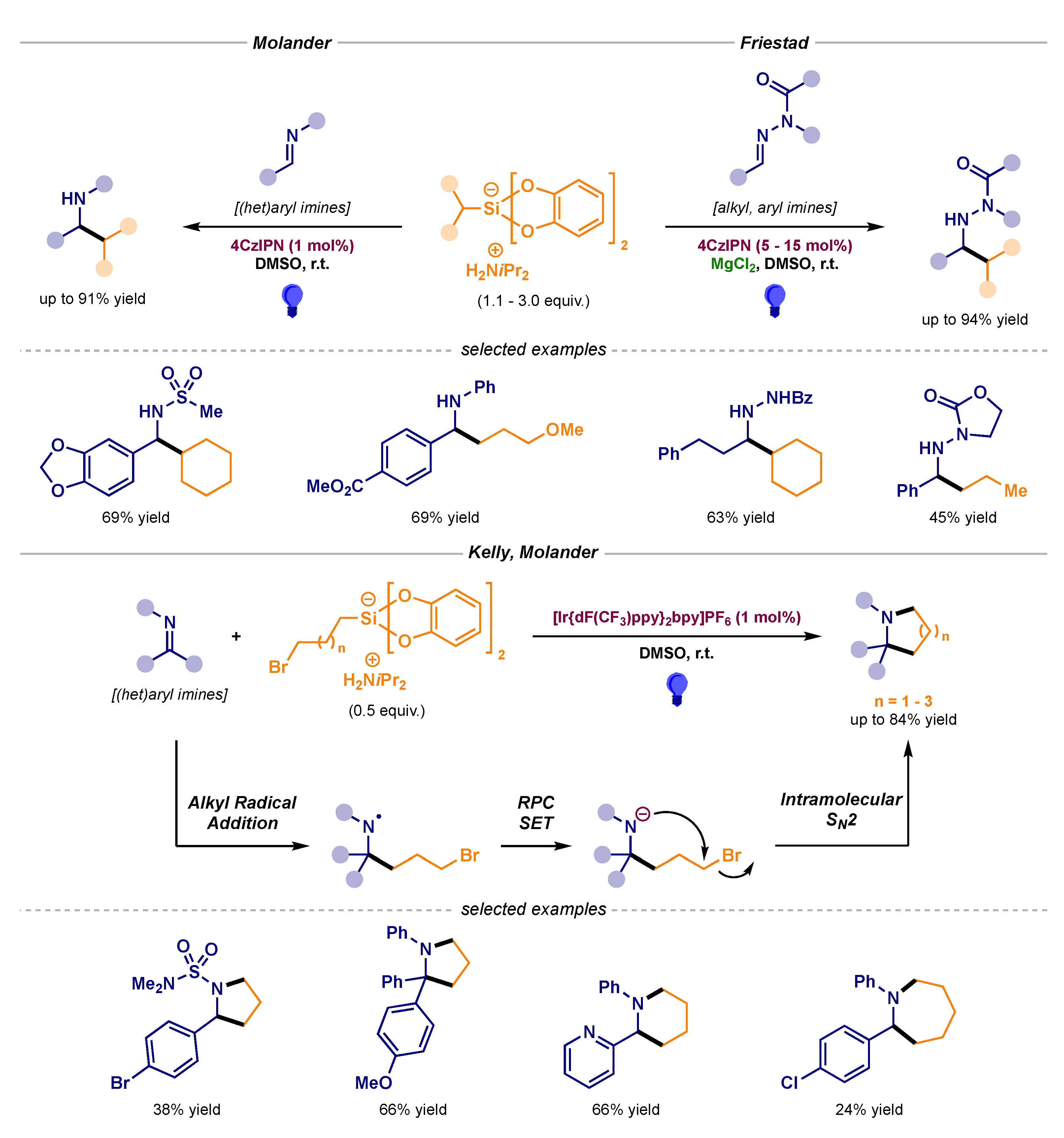
- Patel, N.R.; Kelly, C.B.; Siegenfeld, A.P.; Molander, G.A. Mild, Redox-Neutral Alkylation of Imines Enabled by an Organic Photocatalyst. ACS Catal. 2017, 7, 1766–1770. [Google Scholar] [CrossRef]
- Prieto, A.; Bouyssi, D.; Monteiro, N. Radical-Mediated Formal C(sp2)–H Functionalization of Aldehyde-Derived N,N-Dialkylhydrazones. Eur. J. Org. Chem. 2018, 2378–2393. [Google Scholar] [CrossRef]
- Cullen, S.T.J.; Friestad, G.K. Alkyl Radical Addition to Aliphatic and Aromatic N-Acylhydrazones Using an Organic Photoredox Catalyst. Org. Lett. 2019, 21, 8290–8294. [Google Scholar] [CrossRef]
- Pantaine, L.R.E.; Milligan, J.A.; Matsui, J.K.; Kelly, C.B.; Molander, G.A. Photoredox Radical/Polar Crossover Enables Construction of Saturated Nitrogen Heterocycles. Org. Lett. 2019, 21, 2317–2321. [Google Scholar] [CrossRef]
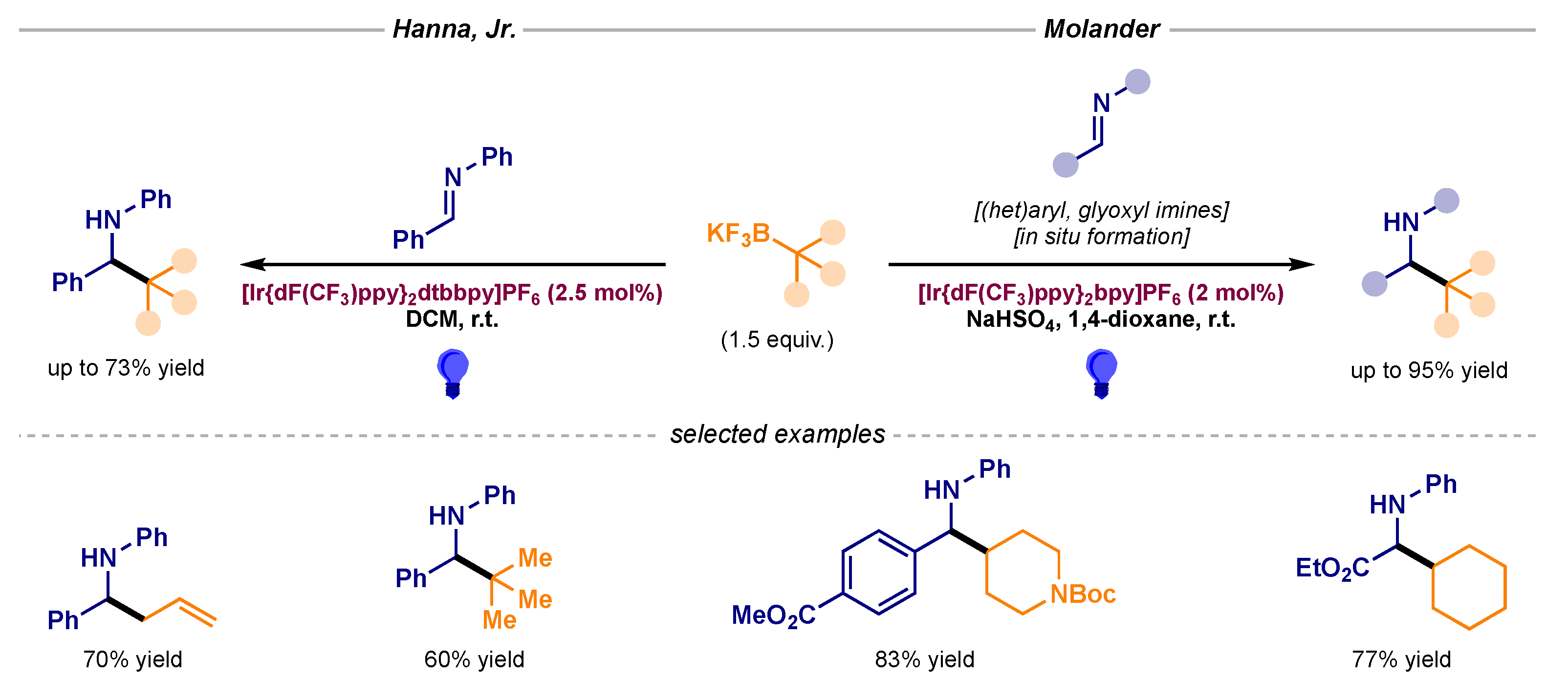
- Plasko, D.P.; Jordan, C.J.; Ciesa, B.E.; Merrill, M.A.; Hanna, J.M., Jr. Visible light-promoted alkylation of imines using potassium organotrifluoroborates. Photochem. Photobiol. Sci. 2018, 17, 534–538. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.; Badir, S.O.; Alam, R.; Molander, G.A. Photoredox-Catalyzed Multicomponent Petasis Reaction with Alkyltrifluoroborates. Org. Lett. 2019, 21, 4853–4858. [Google Scholar] [CrossRef] [PubMed]
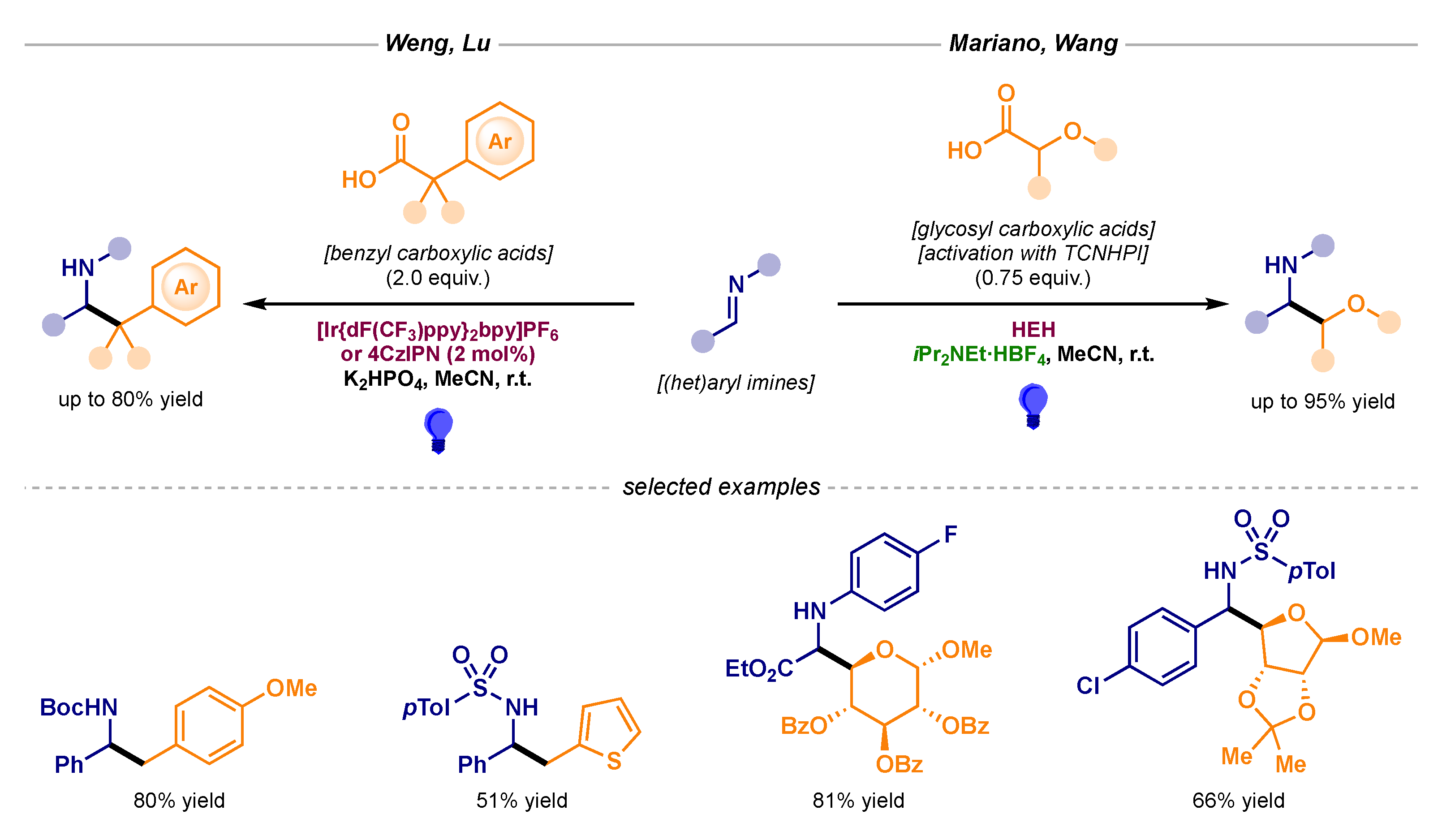
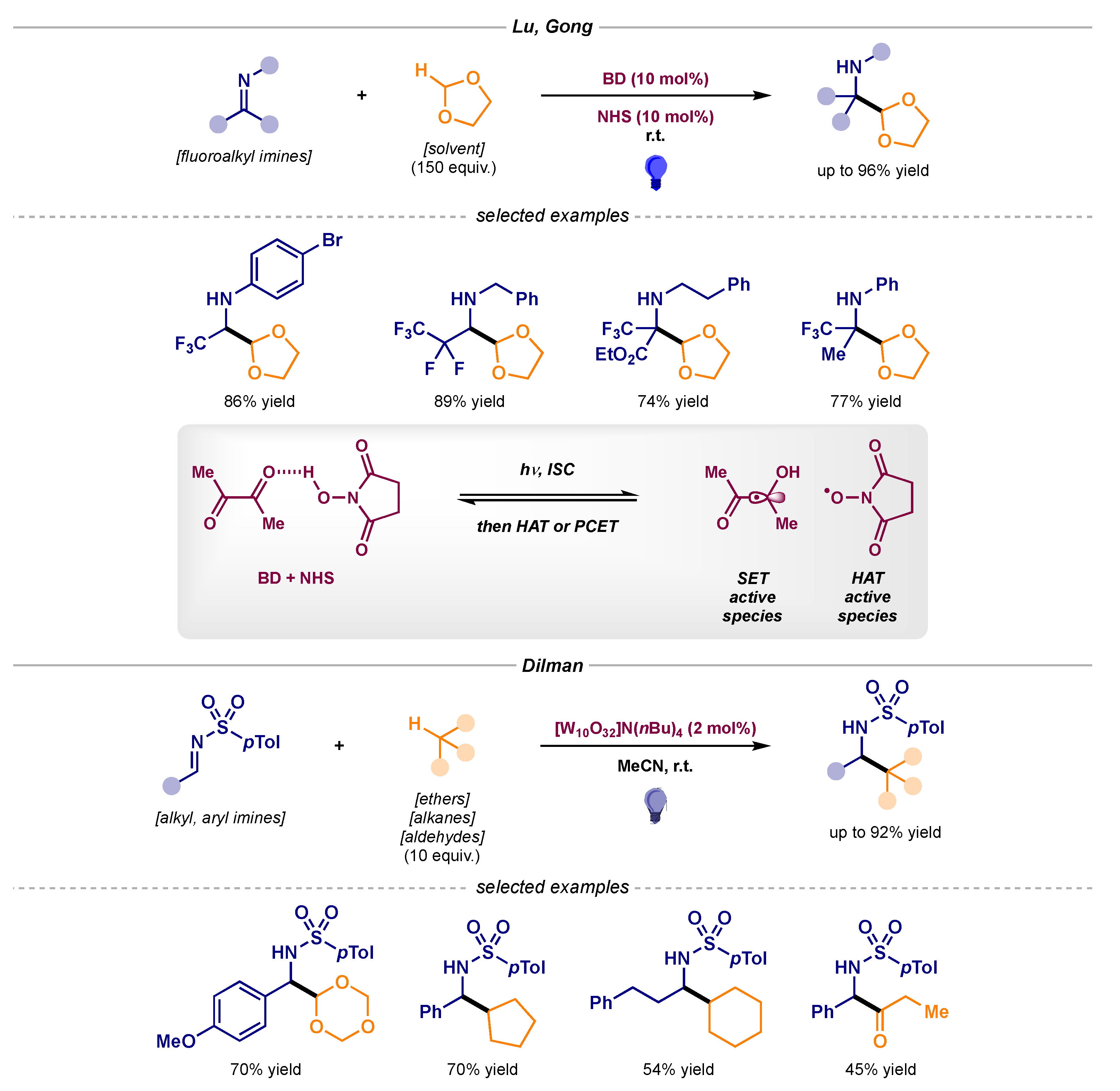
- Guo, J.; Wu, Q.-L.; Xie, Y.; Weng, J.; Lu, G. Visible-Light-Mediated Decarboxylative Benzylation of Imines with Arylacetic Acids. J. Org. Chem. 2018, 83, 12559–12567, The reaction pathway could not be confirmed; both the radical addition and the radical–radical coupling are most likely taking place. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-H.; Yu, S. Radical Alkylation of Imines with 4-Alkyl-1,4-dihydropyridines Enabled by Photoredox/Brønsted Acid Cocatalysis. J. Org. Chem. 2017, 82, 9995–10006, The reaction pathway could not be confirmed; both the radical addition and the radical–radical coupling are most likely taking place. [Google Scholar] [CrossRef]
- Ji, P.; Zhang, Y.; Wei, Y.; Huang, H.; Hu, W.; Mariano, P.A.; Wang, W. Visible-Light-Mediated, Chemo- and Stereoselective Radical Process for the Synthesis of C-Glycoamino Acids. Org. Lett. 2019, 21, 3086–3092. [Google Scholar] [CrossRef]
- Jia, J.; Lefebvre, Q.; Rueping, M. Reductive coupling of imines with redox-active esters by visible light photoredox organocatalysis. Org. Chem. Front. 2020, 7, 602–608. [Google Scholar] [CrossRef]
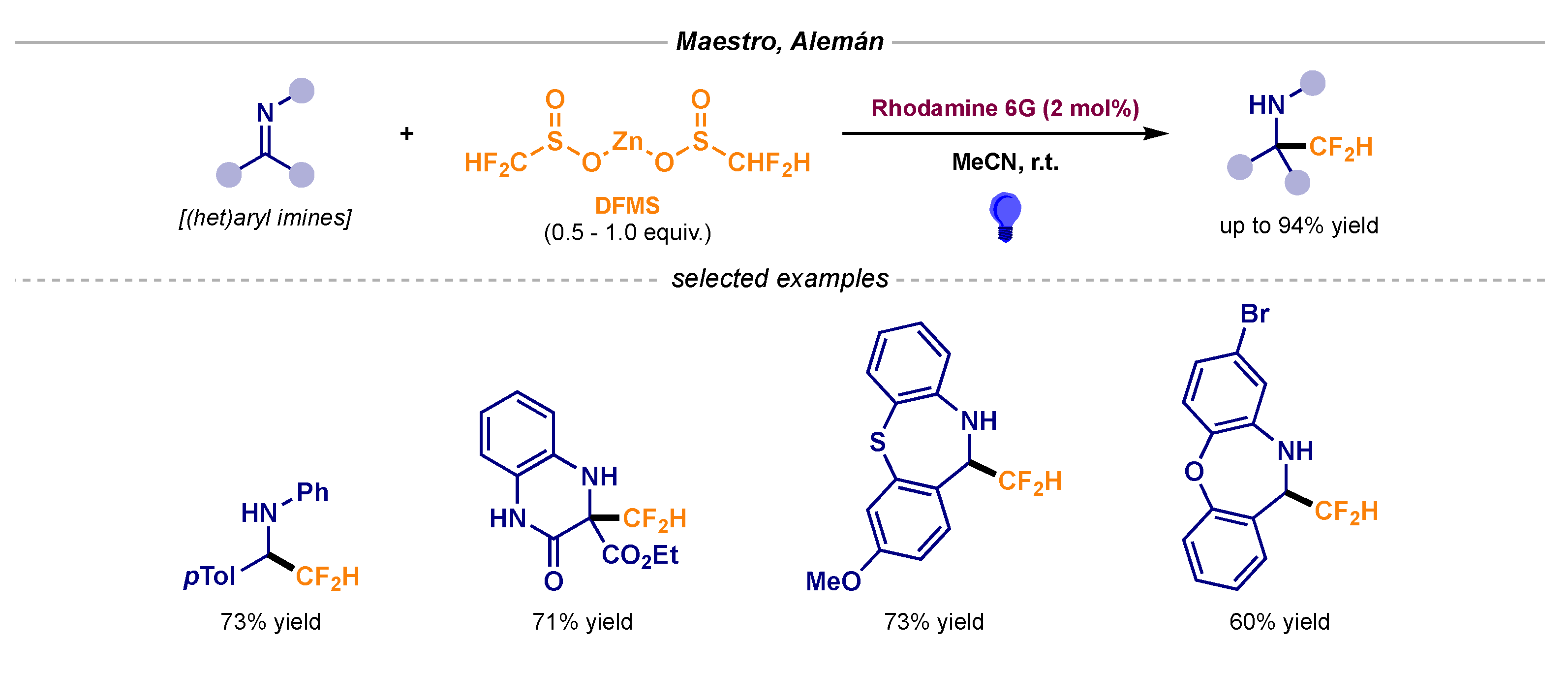
- Garrido-Castro, A.F.; Gini, A.; Maestro, M.C.; Alemán, J. Unlocking the direct photocatalytic difluoromethylation of C=N bonds. Chem. Commun. 2020, 56, 3769–3772. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Zhu, S.; Lu, D.; Gong, Y. Formylation of Fluoroalkyl Imines through Visible-Light-Enabled H-Atom Transfer Catalysis: Access to Fluorinated α-Amino Aldehydes. Org. Lett. 2019, 21, 2019–2024. [Google Scholar] [CrossRef]
- For a photoinduced α-oxyalkylation of imines generated in situ, see: Zhang, L.; Deng, Y.; Shi, F. Light promoted aqueous phase amine synthesis via three-component coupling reactions. Tetrahedron Lett. 2013, 54, 5217–5219. [Google Scholar]
- Supranovich, V.I.; Levin, V.V.; Dilman, A.D. Radical Addition to N-Tosylimines via C−H Activation Induced by Decatungstate Photocatalyst. Org. Lett. 2019, 21, 4271–4274. [Google Scholar] [CrossRef]
- For an inverse hydroboration of imines wherein the boryl radical is generated from N-Heterocyclic Carbene (NHC) boranes via HAT, see: Zhou, N.; Yuan, X.-A.; Zhao, Y.; Xie, J.; Zhu, C. Synergistic Photoredox Catalysis and Organocatalysis for Inverse Hydroboration of Imines. Angew. Chem., Int. Ed. 2018, 57, 3990–3994, The reaction pathway could not be confirmed; both the radical addition and the radical–radical coupling are most likely taking place. [Google Scholar]
- Zhang, H.-H.; Yu, S. Visible-Light-Induced Radical Acylation of Imines with α-Ketoacids Enabled by Electron-Donor−Acceptor Complexes. Org. Lett. 2019, 21, 3711–3715, A radical–radical coupling is most likely taking place. [Google Scholar] [CrossRef] [PubMed]
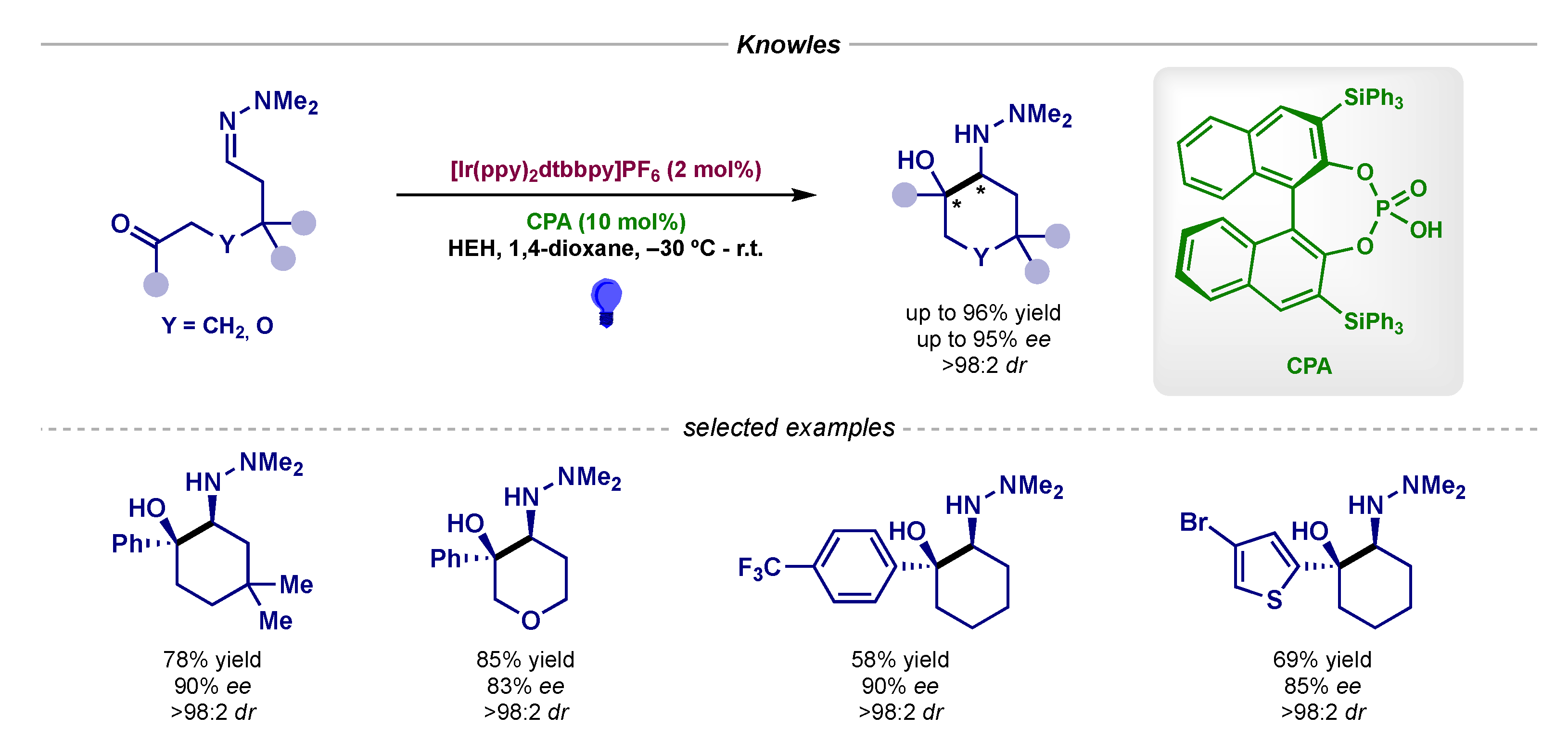
- Rono, L.J.; Yayla, H.G.; Wang, D.Y.; Armstrong, M.F.; Knowles, R.R. Enantioselective Photoredox Catalysis Enabled by Proton-Coupled Electron Transfer: Development of an Asymmetric Aza-Pinacol Cyclization. J. Am. Chem. Soc. 2013, 135, 17735–17738. [Google Scholar] [CrossRef] [PubMed]
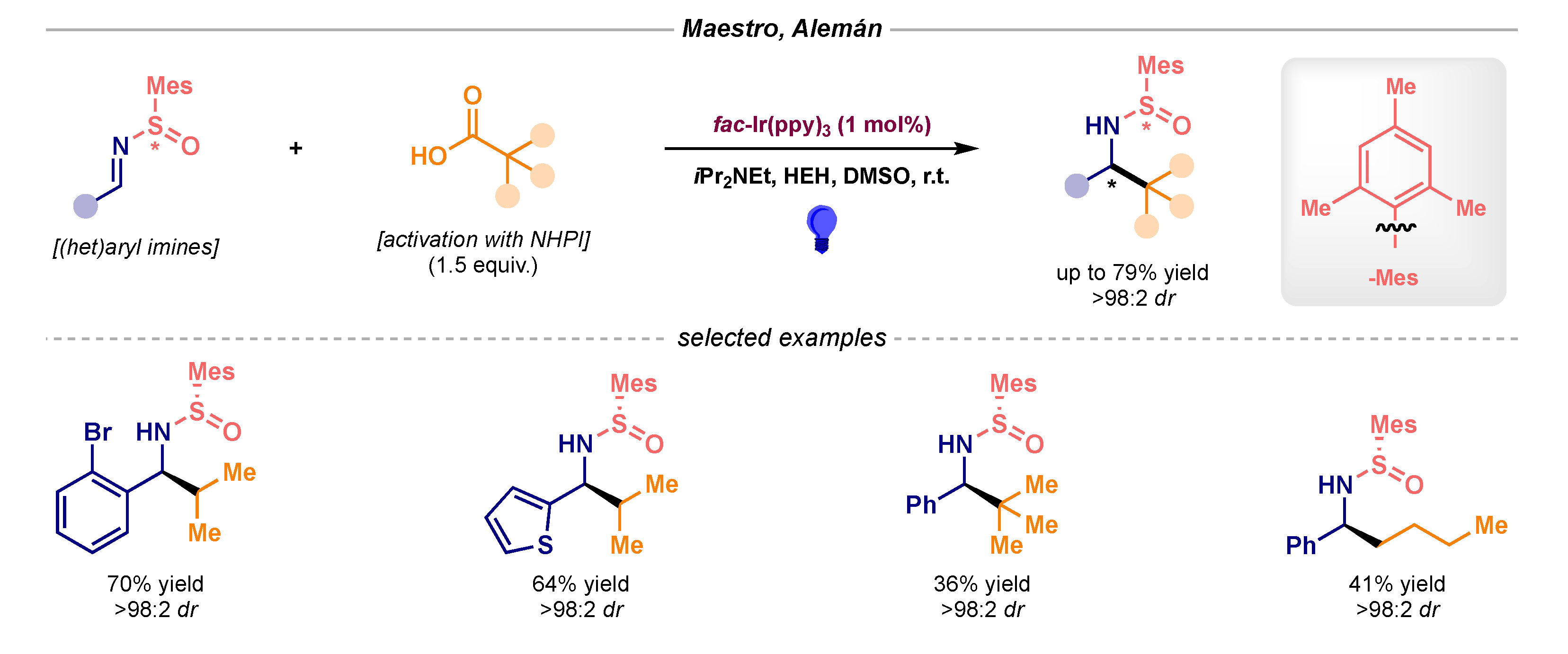
- Garrido-Castro, A.F.; Choubane, H.; Daaou, M.; Maestro, M.C.; Alemán, J. Asymmetric radical alkylation of N-sulfinimines under visible light photocatalytic conditions. Chem. Commun. 2017, 53, 7764–7767. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhou, K.; Wen, Z.; Cao, S.; Shen, X.; Lei, M.; Gong, L. Copper(II)-Catalyzed Asymmetric Photoredox Reactions: Enantioselective Alkylation of Imines Driven by Visible Light. J. Am. Chem. Soc. 2018, 140, 15850–15858. [Google Scholar] [CrossRef]
- Han, B.; Li, Y.; Yu, Y.; Gong, L. Photocatalytic enantioselective α-aminoalkylation of acyclic imine derivatives by a chiral copper catalyst. Nat. Commun. 2019, 10, 3804–3812. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Lei, M.; Gong, L. Photocatalytic regio- and stereoselective C(sp3)–H functionalization of benzylic and allylic hydrocarbons as well as unactivated alkanes. Nat. Catal. 2019, 2, 1016–1026, The reaction pathway could not be confirmed; both the radical addition and the radical–radical coupling are most likely taking place. [Google Scholar] [CrossRef]
- Schindler, W.; Knoch, F.; Kisch, H. Semiconductor-Catalysed Photoaddition: γ,δ-Unsaturated Amines from Cyclopentene and Schiff Bases. Chem. Ber. 1996, 129, 925–932. [Google Scholar] [CrossRef]
- Keck, H.; Schindler, W.; Knoch, F.; Kisch, H. Type B Semiconductor Photocatalysis: The Synthesis of Homoallyl Amines by Cadmium Sulfide-Catalyzed Linear Photoaddition of Olefins and Enol/Allyl Ethers to N-Phenylbenzophenone Imine. Chem. Eur. J. 1997, 3, 1638–1645. [Google Scholar] [CrossRef]
- Kisch, H. Semiconductor Photocatalysis for Chemoselective Radical Coupling Reactions. Acc. Chem. Res. 2017, 50, 1002–1010. [Google Scholar] [CrossRef]
- Xi, Z.-W.; Yang, L.; Wang, D.-Y.; Pu, C.-D.; Shen, Y.-M.; Wu, C.-D.; Peng, X.-G. Visible-Light Photocatalytic Synthesis of Amines from Imines via Transfer Hydrogenation Using Quantum Dots as Catalysts. J. Org. Chem. 2018, 83, 11886–11895. [Google Scholar] [CrossRef] [PubMed]
- Van As, D.J.; Connell, T.U.; Brzozowski, M.; Scully, A.D.; Polyzos, A. Photocatalytic and Chemoselective Transfer Hydrogenation of Diarylimines in Batch and Continuous Flow. Org. Lett. 2018, 20, 905–908. [Google Scholar] [CrossRef] [PubMed]
- Hager, D.; MacMillan, D.W.C. Activation of C−H Bonds via the Merger of Photoredox and Organocatalysis: A Coupling of Benzylic Ethers with Schiff Bases. J. Am. Chem. Soc. 2014, 136, 16986–16989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeffrey, J.L.; Petronijević, F.R.; MacMillan, D.W.C. Selective Radical−Radical Cross-Couplings: Design of a Formal β-Mannich Reaction. J. Am. Chem. Soc. 2015, 137, 8404–8407. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, M.; Fava, E.; Loescher, S.; Jiang, Z.; Rueping, M. Photoredox-Catalyzed Reductive Coupling of Aldehydes, Ketones, and Imines with Visible Light. Angew. Chem., Int. Ed. 2015, 54, 8828–8832. [Google Scholar] [CrossRef]
- Fava, E.; Millet, A.; Nakajima, M.; Loescher, S.; Rueping, M. Reductive Umpolung of Carbonyl Derivatives with Visible-Light Photoredox Catalysis: Direct Access to Vicinal Diamines and Amino Alcohols via α-Amino Radicals and Ketyl Radicals. Angew. Chem., Int. Ed. 2016, 55, 6776–6779. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, S.; Kojiyama, K.; Tsujioka, H.; Sudo, A. Metal-free reductive coupling of C=O and C=N bonds driven by visible light: Use of perylene as a simple photoredox catalyst. Chem. Commun. 2016, 52, 11339–11342. [Google Scholar] [CrossRef]
- Rong, J.; Seeberger, P.H.; Gilmore, K. Chemoselective Photoredox Synthesis of Unprotected Primary Amines Using Ammonia. Org. Lett. 2018, 20, 4081–4085. [Google Scholar] [CrossRef]
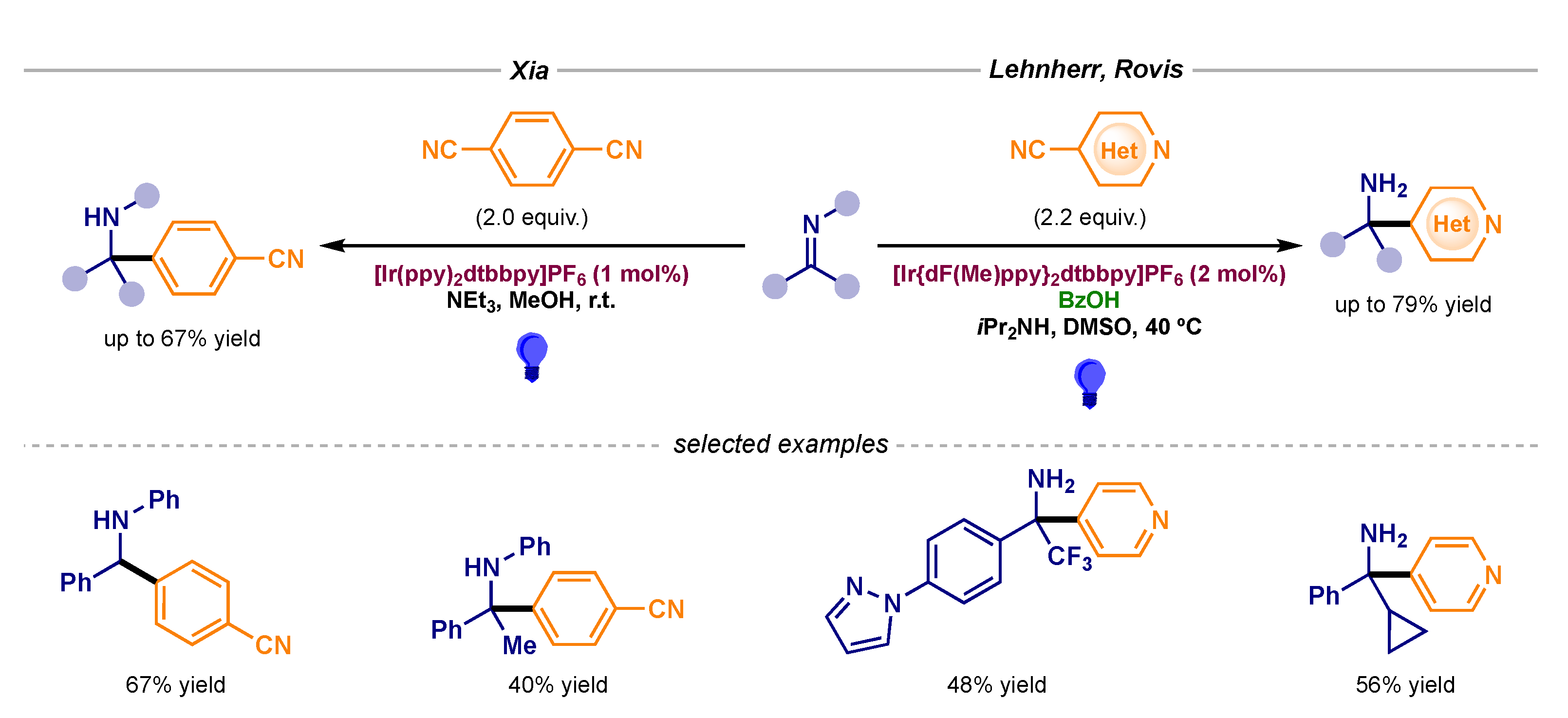
- Xia, Q.; Tian, H.; Dong, J.; Qu, Y.; Li, L.; Song, H.; Liu, Y.; Wang, Q. N-Arylamines Coupled with Aldehydes, Ketones, and Imines by Means of Photocatalytic Proton-Coupled Electron Transfer. Chem. Eur. J. 2018, 24, 9269–9273. [Google Scholar] [CrossRef]
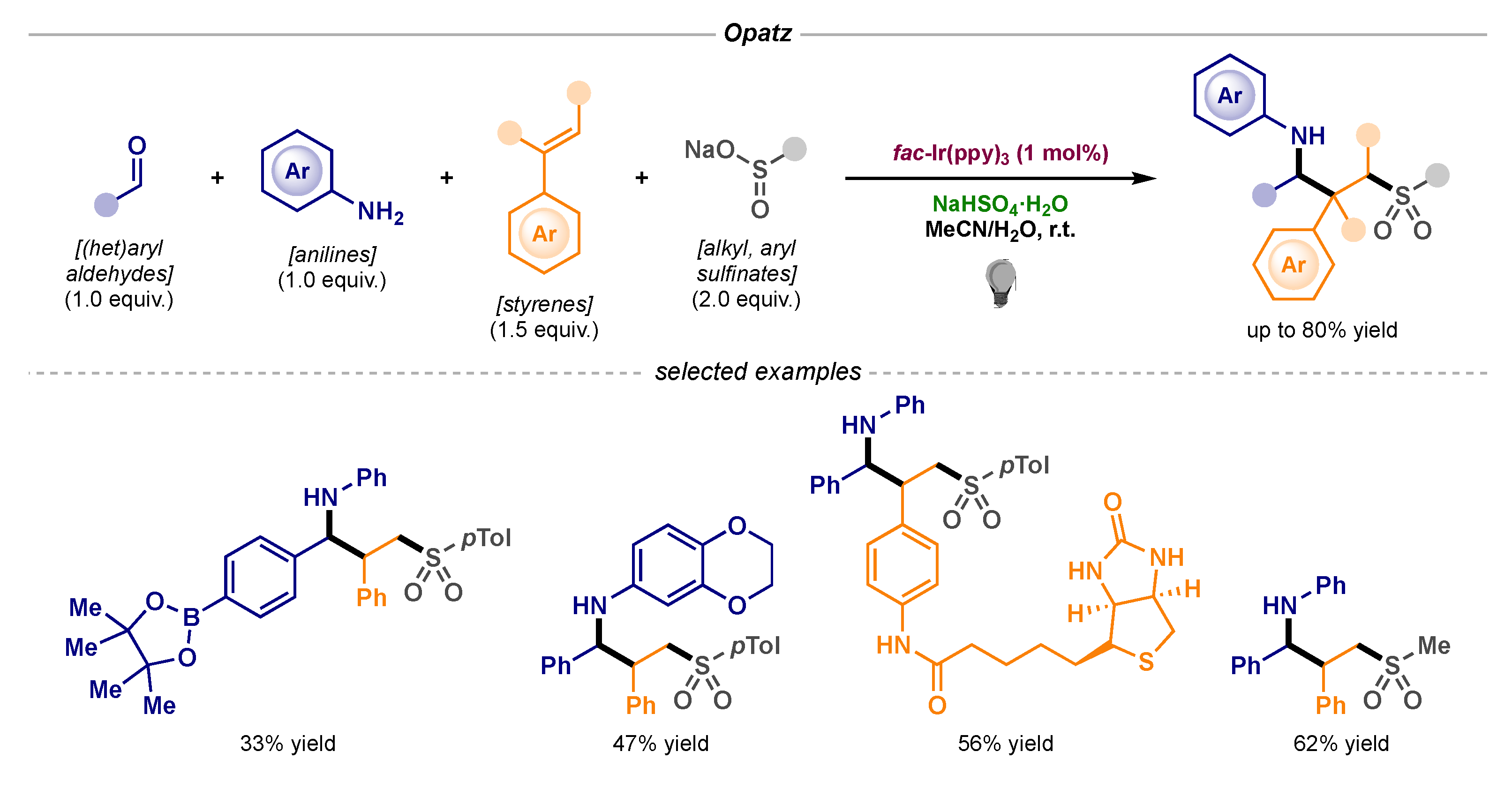
- Kammer, L.M.; Krumb, M.; Spitzbarth, B.; Lipp, B.; Kühlborn, J.; Busold, J.; Mulina, O.M.; Terentev, A.O.; Opatz, T. Photoredox-Catalyzed Four-Component Reaction for the Synthesis of Complex Secondary Amines. Org. Lett. 2020, 22, 3318–3322. [Google Scholar] [CrossRef]
- Chen, M.; Zhao, X.; Yang, C.; Xia, W. Visible-Light-Triggered Directly Reductive Arylation of Carbonyl/Iminyl Derivatives through Photocatalytic PCET. Org. Lett. 2017, 19, 3807–3810. [Google Scholar] [CrossRef] [PubMed]
- Nicastri, M.C.; Lehnherr, D.; Lam, Y.; DiRocco, D.A.; Rovis, T. Synthesis of Sterically Hindered Primary Amines by Concurrent Tandem Photoredox Catalysis. J. Am. Chem. Soc. 2020, 142, 987–998. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Chen, Y. Polarity-Reversed Allylations of Aldehydes, Ketones, and Imines Enabled by Hantzsch Ester in Photoredox Catalysis. Angew. Chem., Int. Ed. 2016, 55, 13312–13315. [Google Scholar] [CrossRef] [PubMed]
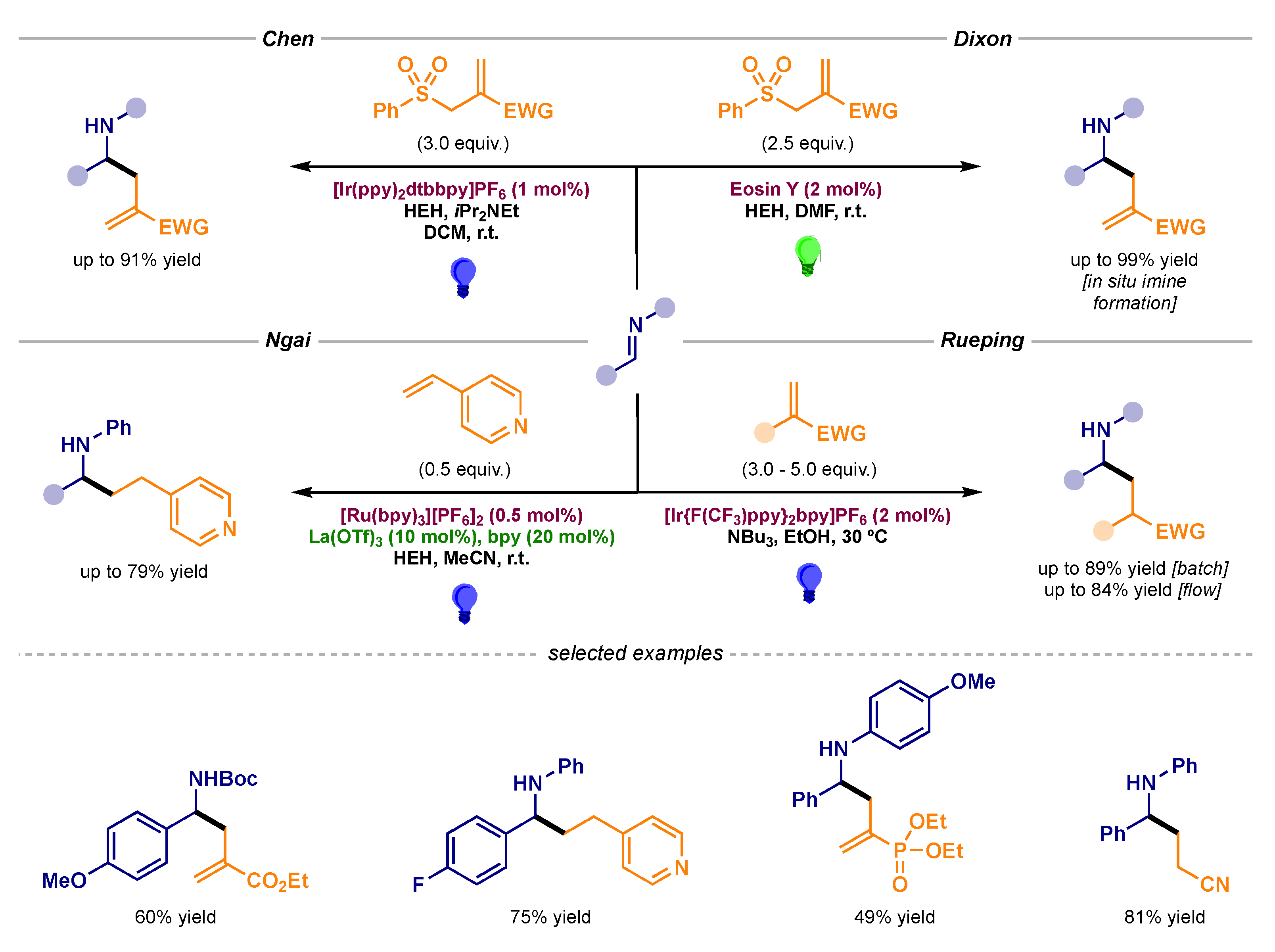
- Fuentes de Arriba, A.L.; Urbitsch, F.; Dixon, D.J. Umpolung synthesis of branched α-functionalized amines from imines via photocatalytic three-component reductive coupling reactions. Chem. Commun. 2016, 52, 14434–14437. [Google Scholar] [CrossRef] [Green Version]
- Rossolini, T.; Leitch, J.A.; Grainger, R.; Dixon, D.J. Photocatalytic Three-Component Umpolung Synthesis of 1,3-Diamines. Org. Lett. 2018, 20, 6794–6798. [Google Scholar] [CrossRef]
- Lee, K.N.; Lei, Z.; Ngai, M.-Y. β-Selective Reductive Coupling of Alkenylpyridines with Aldehydes and Imines via Synergistic Lewis Acid/Photoredox Catalysis. J. Am. Chem. Soc. 2017, 139, 5003–5006. [Google Scholar] [CrossRef]
- Lefebvre, Q.; Porta, R.; Millet, A.; Jia, J.; Rueping, M. One Amine–3 Tasks: Reductive Coupling of Imines with Olefins in Batch and Flow. Chem. Eur. J. 2020, 26, 1363–1367. [Google Scholar] [CrossRef]
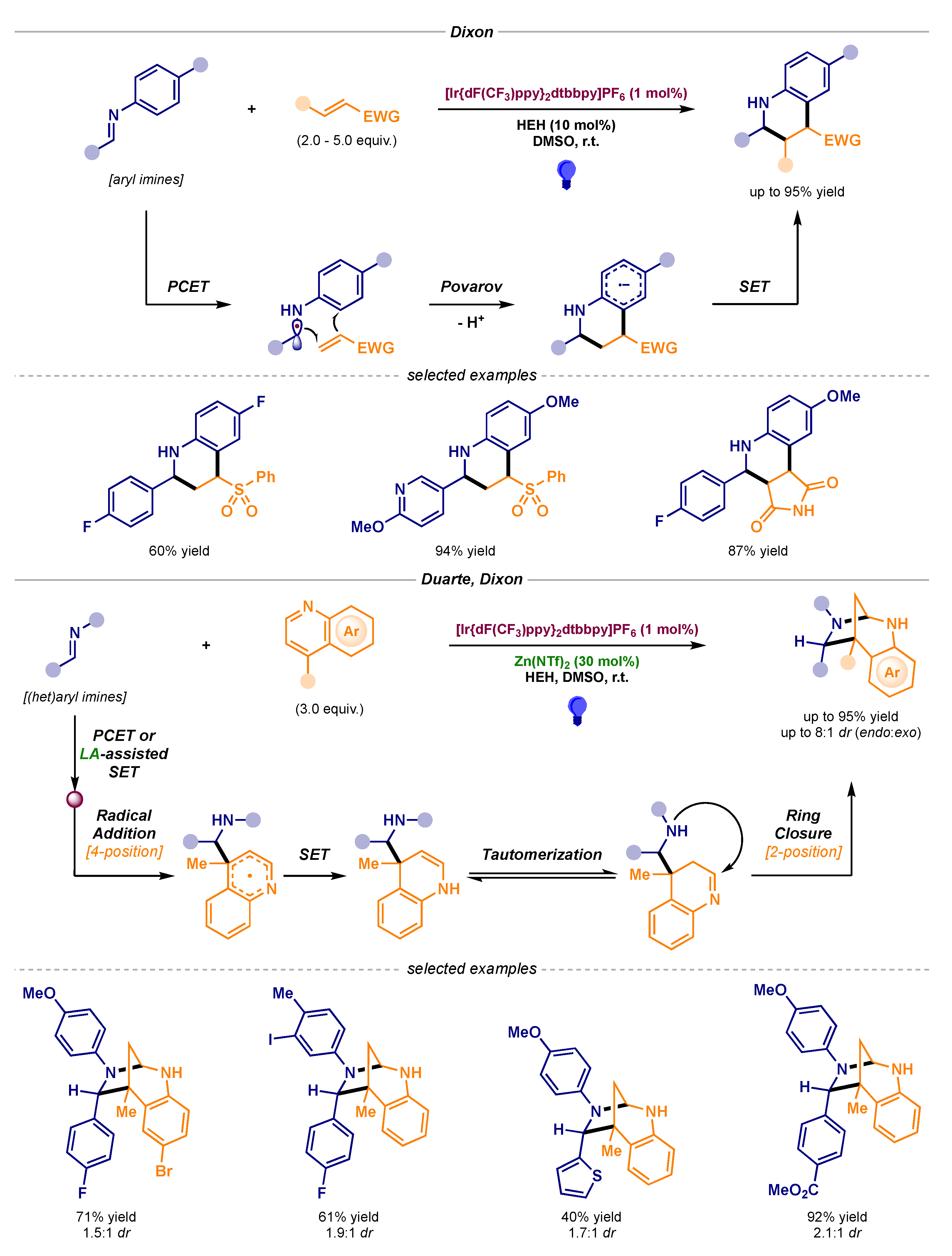
- Leitch, J.A.; Fuentes de Arriba, A.L.; Tan, J.; Hoff, O.; Martínez, C.M.; Dixon, D.J. Photocatalytic reverse polarity Povarov reaction. Chem. Sci. 2018, 9, 6653–6658. [Google Scholar] [CrossRef] [Green Version]
- Leitch, J.A.; Rogova, T.; Duarte, F.; Dixon, D.J. Dearomative Photocatalytic Construction of Bridged 1,3-Diazepanes. Angew. Chem., Int. Ed. 2020, 59, 4121–4130. [Google Scholar] [CrossRef]
- Ju, T.; Fu, Q.; Ye, J.-H.; Zhang, Z.; Liao, L.-L.; Yan, S.-S.; Tian, X.-Y.; Luo, S.-P.; Li, J.; Yu, D.-G. Selective and Catalytic Hydrocarboxylation of Enamides and Imines with CO2 to Generate α,α-Disubstituted α-Amino Acids. Angew. Chem., Int. Ed. 2018, 57, 13897–13901. [Google Scholar] [CrossRef]
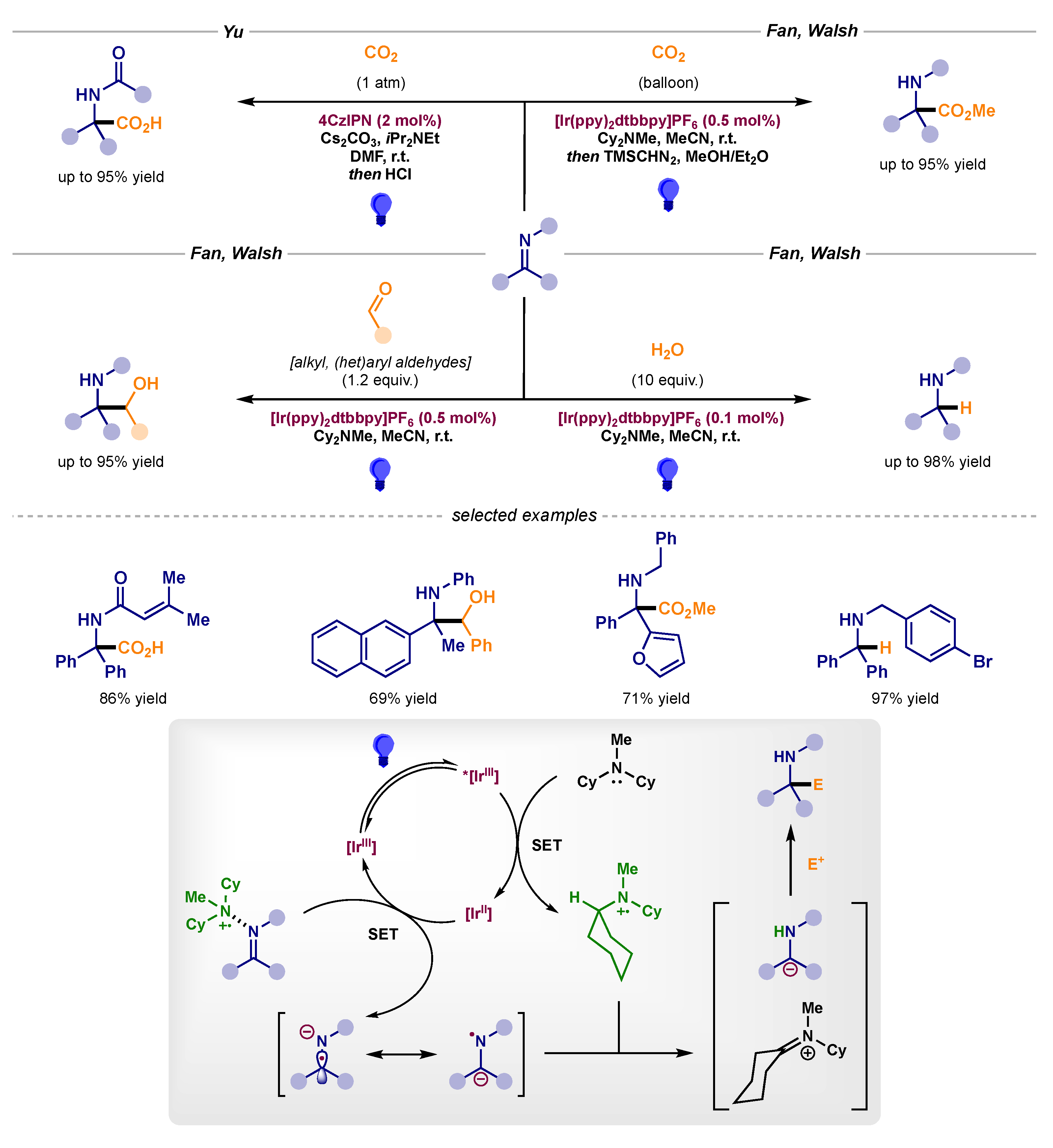
- Fan, X.; Gong, X.; Ma, M.; Wang, R.; Walsh, P.J. Visible light-promoted CO2 fixation with imines to synthesize diaryl α-amino acids. Nat. Commun. 2018, 9, 4936–4943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Ma, M.; Gong, X.; Fan, X.; Walsh, P.J. Reductive Cross-Coupling of Aldehydes and Imines Mediated by Visible Light Photoredox Catalysis. Org. Lett. 2019, 21, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Ma, M.; Gong, X.; Panetti, G.B.; Fan, X.; Walsh, P.J. Visible-Light-Mediated Umpolung Reactivity of Imines: Ketimine Reductions with Cy2NMe and Water. Org. Lett. 2018, 20, 2433–2436. [Google Scholar] [CrossRef] [PubMed]
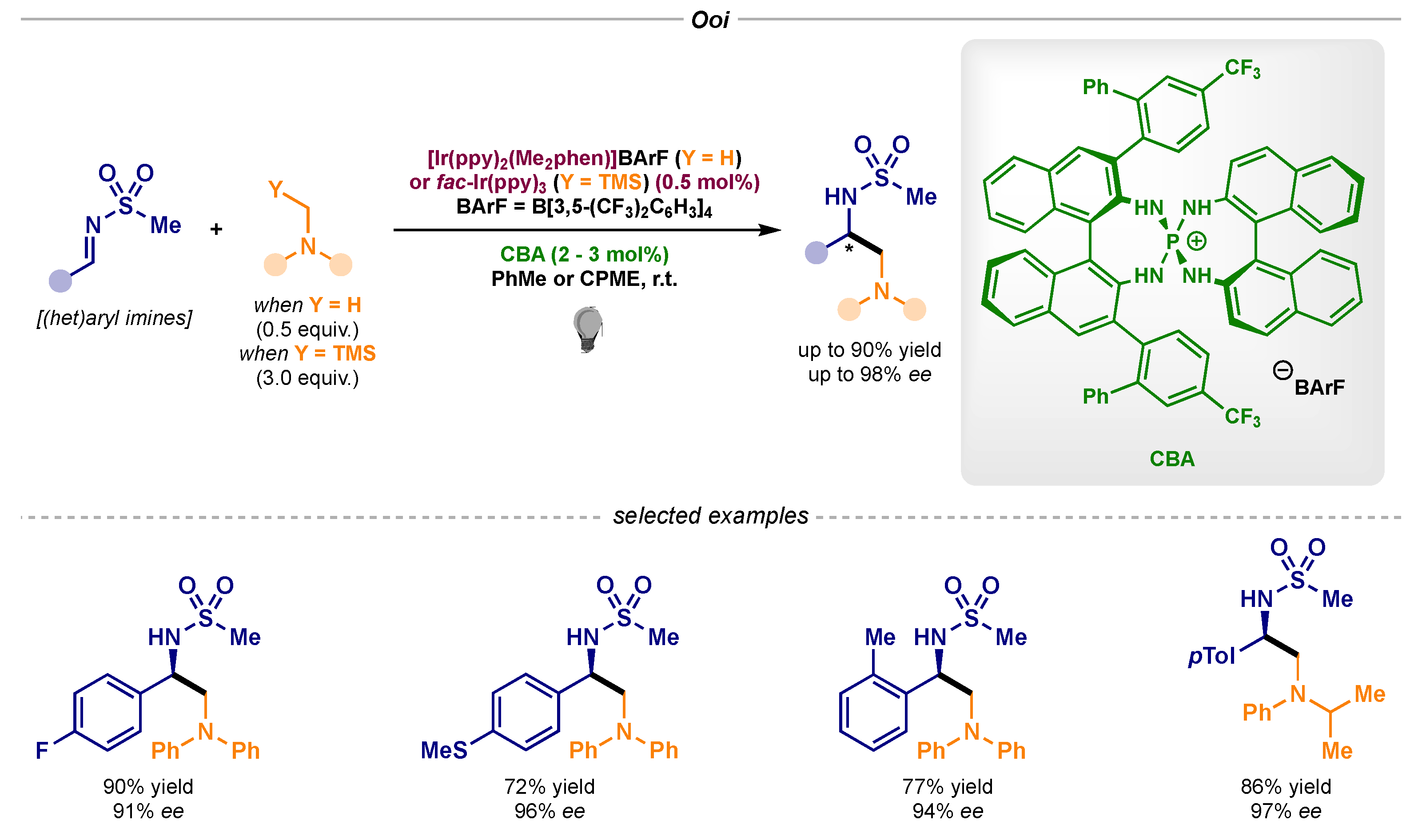
- Uraguchi, D.; Kinoshita, N.; Kizu, T.; Ooi, T. Synergistic Catalysis of Ionic Brønsted Acid and Photosensitizer for a Redox Neutral Asymmetric α-Coupling of N-Arylaminomethanes with Aldimines. J. Am. Chem. Soc. 2015, 137, 13768–13771. [Google Scholar] [CrossRef]
- Kizu, T.; Uraguchi, D.; Ooi, T. Independence from the Sequence of Single-Electron Transfer of Photoredox Process in Redox-Neutral Asymmetric Bond-Forming Reaction. J. Org. Chem. 2016, 81, 6953–6958. [Google Scholar] [CrossRef]
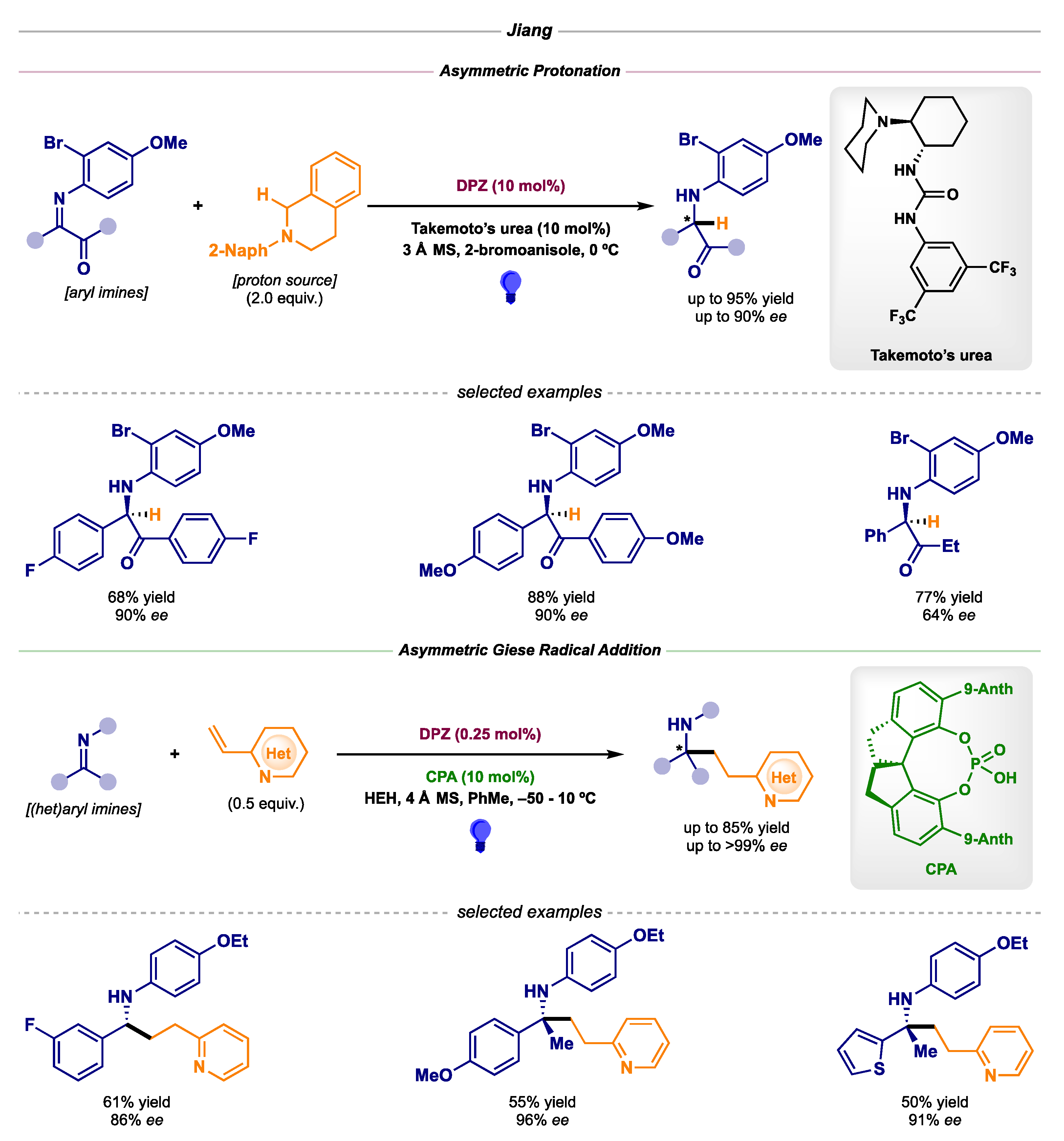
- Lin, L.; Bai, X.; Ye, X.; Zhao, X.; Tan, C.-H.; Jiang, Z. Organocatalytic Enantioselective Protonation for Photoreduction of Activated Ketones and Ketimines Induced by Visible Light. Angew. Chem., Int. Ed. 2017, 56, 13842–13846. [Google Scholar] [CrossRef]
- Cao, K.; Tan, S.M.; Lee, R.; Yang, S.; Jia, H.; Zhao, X.; Qiao, B.; Jiang, Z. Catalytic Enantioselective Addition of Prochiral Radicals to Vinylpyridines. J. Am. Chem. Soc. 2019, 141, 5437–5443. [Google Scholar] [CrossRef]
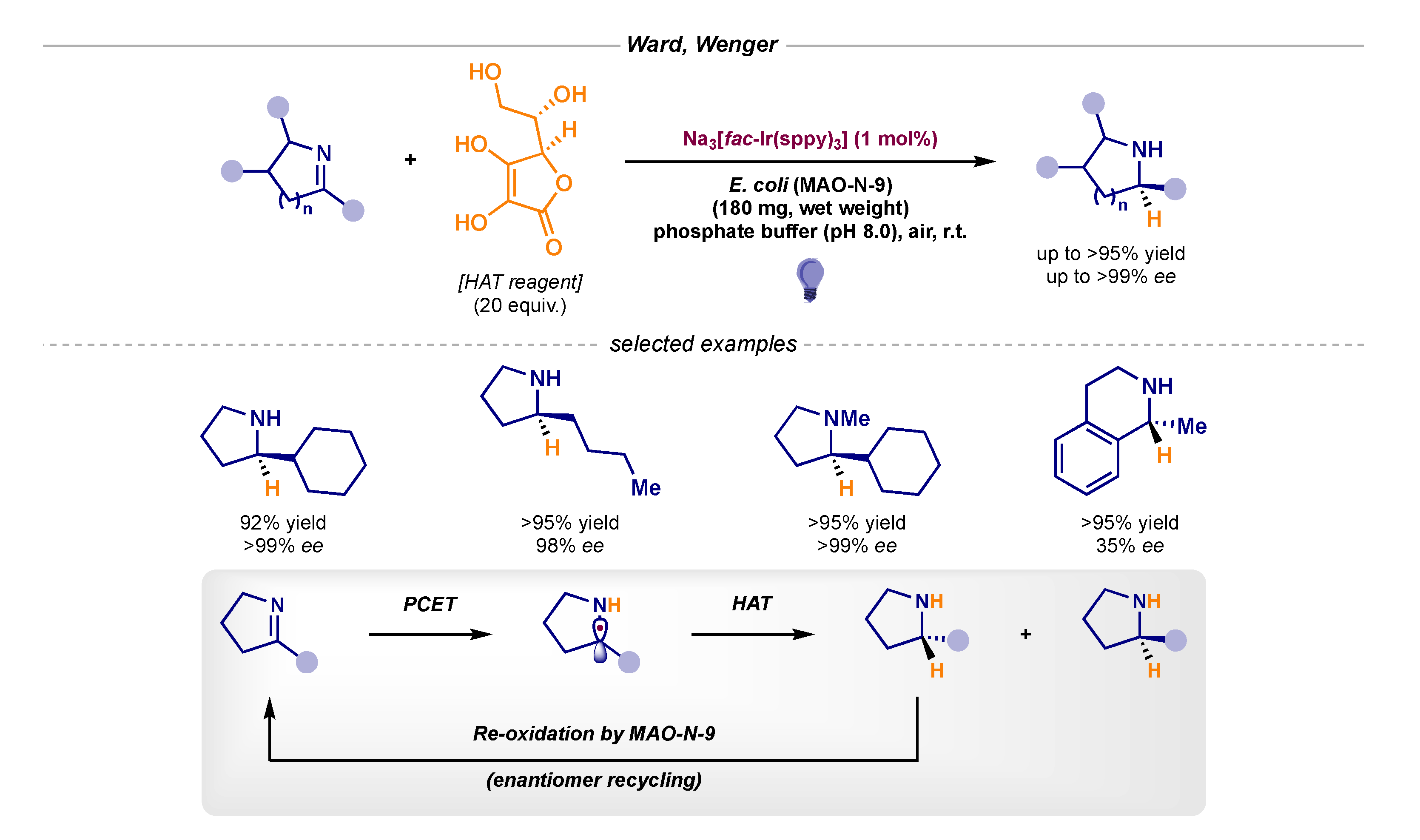
- Guo, X.; Okamoto, Y.; Schreier, M.R.; Ward, T.R.; Wenger, O.S. Enantioselective synthesis of amines by combining photoredox and enzymatic catalysis in a cyclic reaction network. Chem. Sci. 2018, 9, 5052–5056. [Google Scholar] [CrossRef] [Green Version]






















© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garrido-Castro, A.F.; Maestro, M.C.; Alemán, J. α-Functionalization of Imines via Visible Light Photoredox Catalysis. Catalysts 2020, 10, 562. https://doi.org/10.3390/catal10050562
Garrido-Castro AF, Maestro MC, Alemán J. α-Functionalization of Imines via Visible Light Photoredox Catalysis. Catalysts. 2020; 10(5):562. https://doi.org/10.3390/catal10050562
Chicago/Turabian StyleGarrido-Castro, Alberto F., M. Carmen Maestro, and José Alemán. 2020. "α-Functionalization of Imines via Visible Light Photoredox Catalysis" Catalysts 10, no. 5: 562. https://doi.org/10.3390/catal10050562
APA StyleGarrido-Castro, A. F., Maestro, M. C., & Alemán, J. (2020). α-Functionalization of Imines via Visible Light Photoredox Catalysis. Catalysts, 10(5), 562. https://doi.org/10.3390/catal10050562