Identification of Main Active Sites and the Role of NO2 on NOx Reduction with CH4 over In/BEA Catalyst: A Computational Study

Abstract
:1. Introduction
2. Results and Discussion
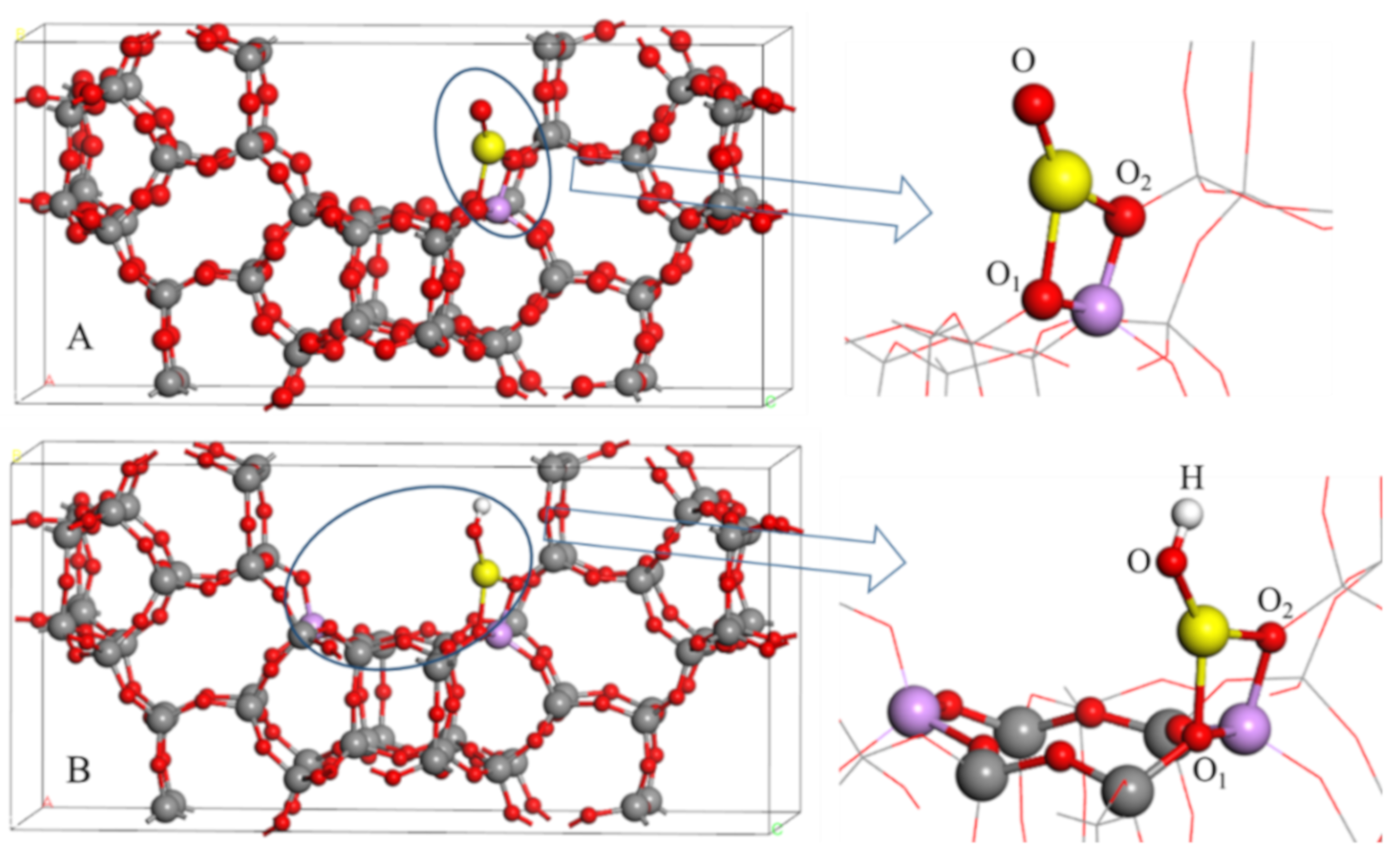
2.1. The Optimized Structures of BEA Zeolite and In/BEA Model
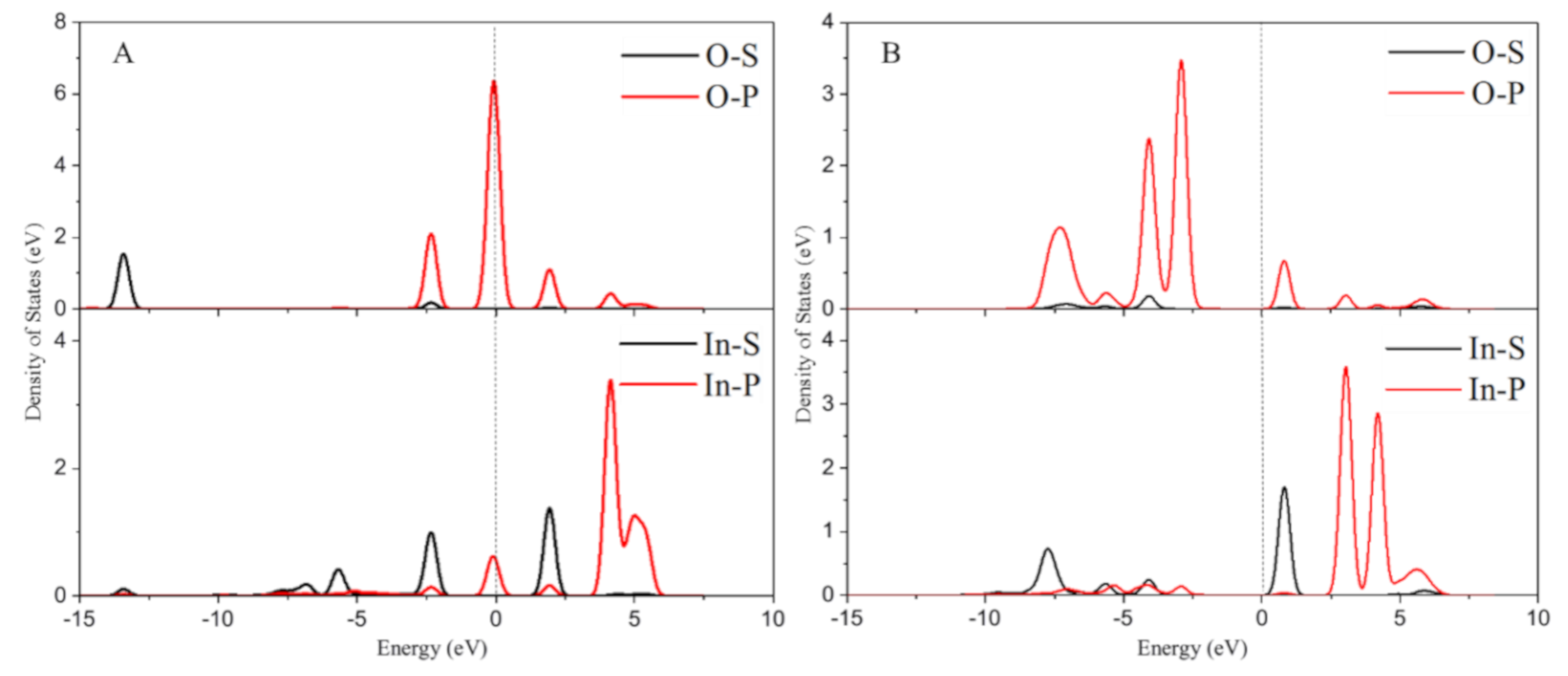
2.2. The Electronic Structure Analysis for In/BEA Model
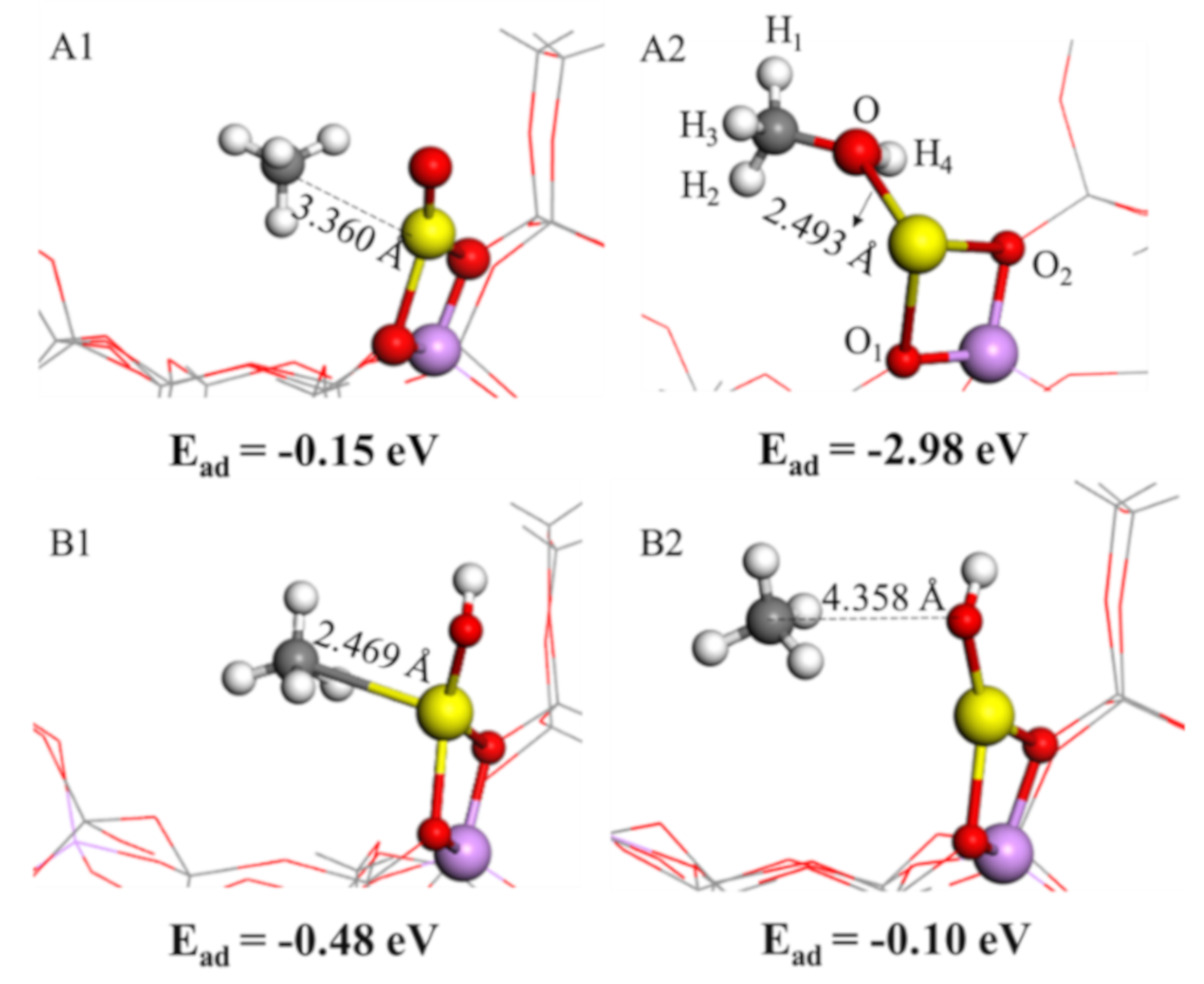
2.3. CH4 Adsorption Characteristics
2.4. NO and NO2 Adsorption Characteristics
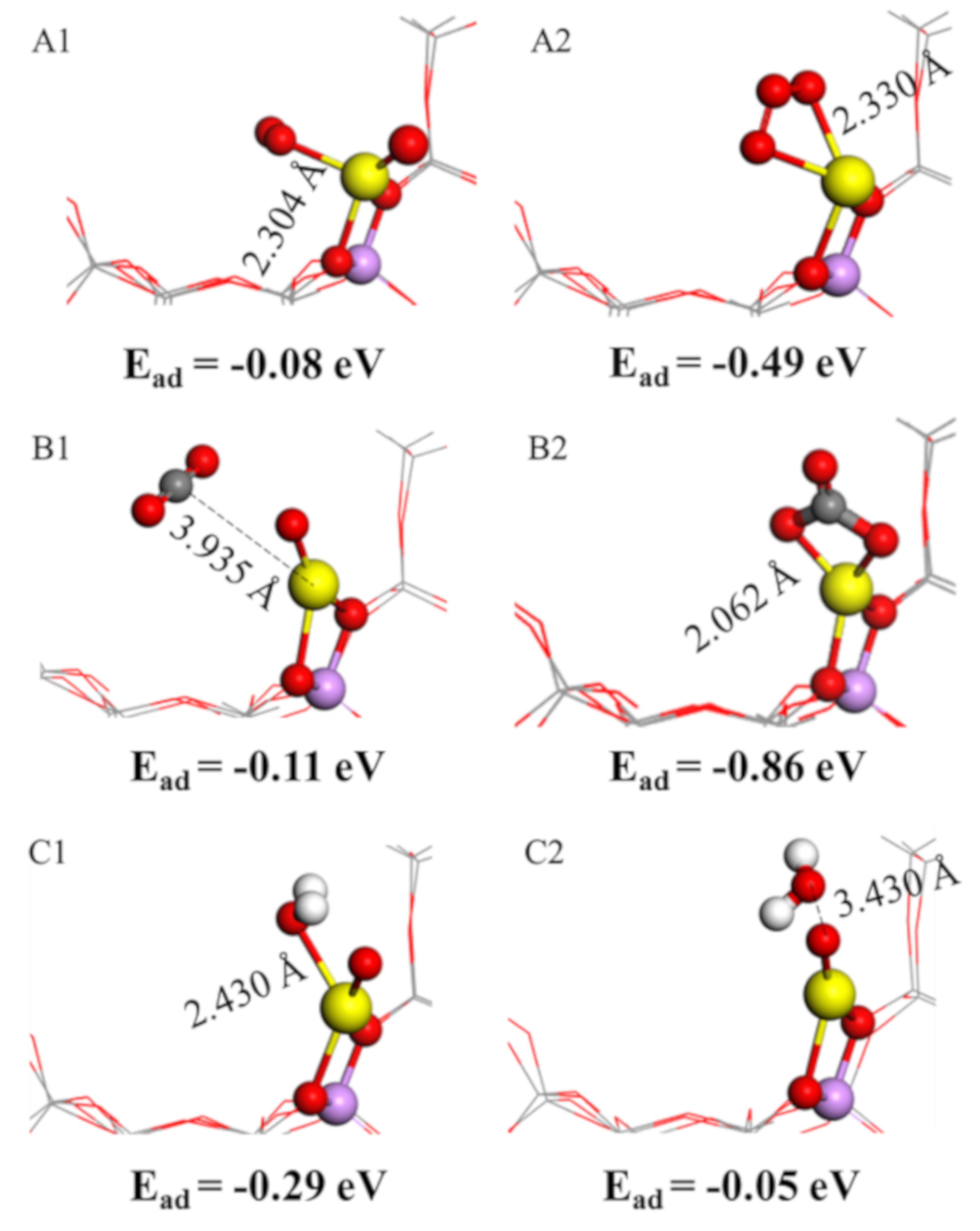
2.5. O2, H2O and CO2 Adsorption Characteristics
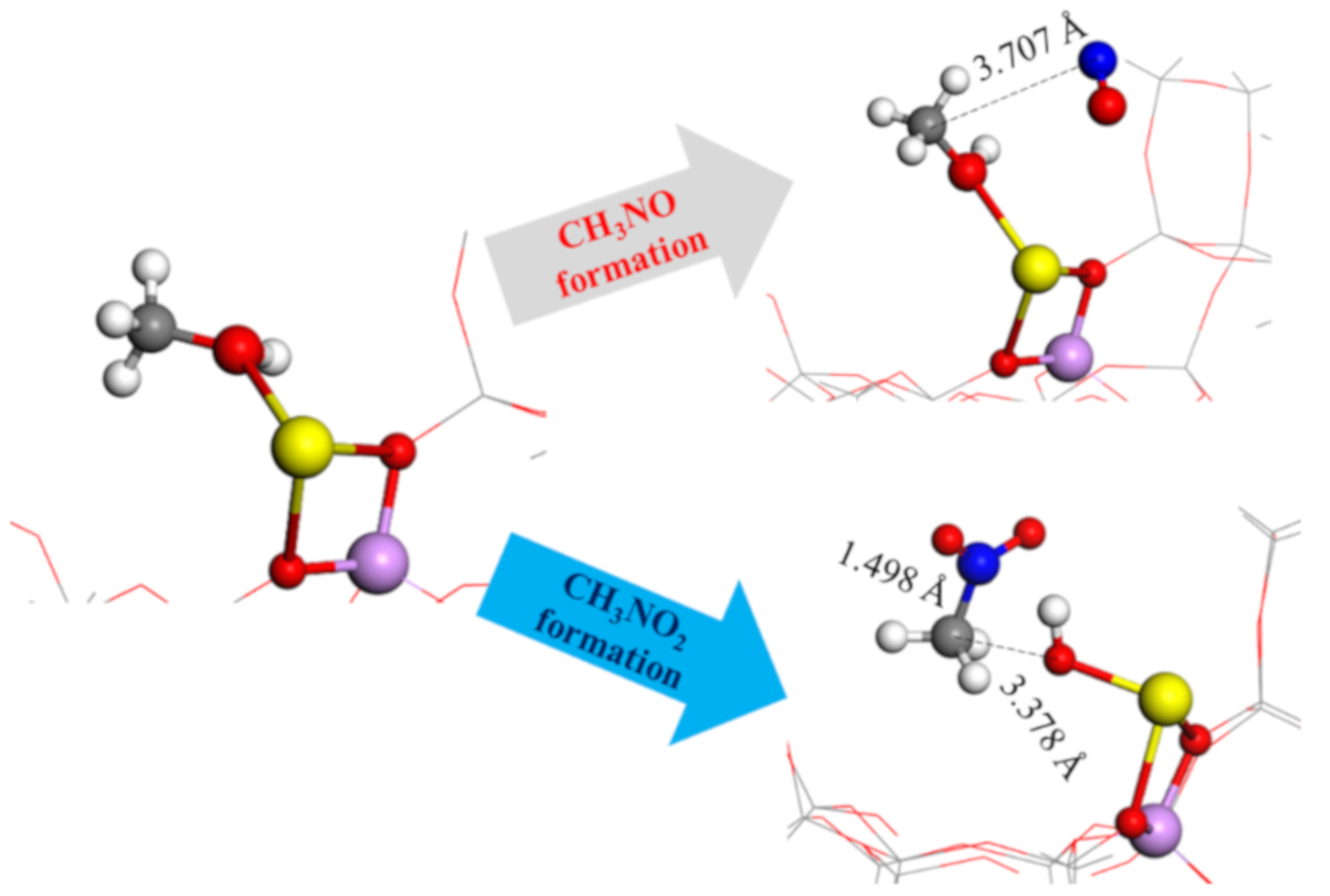
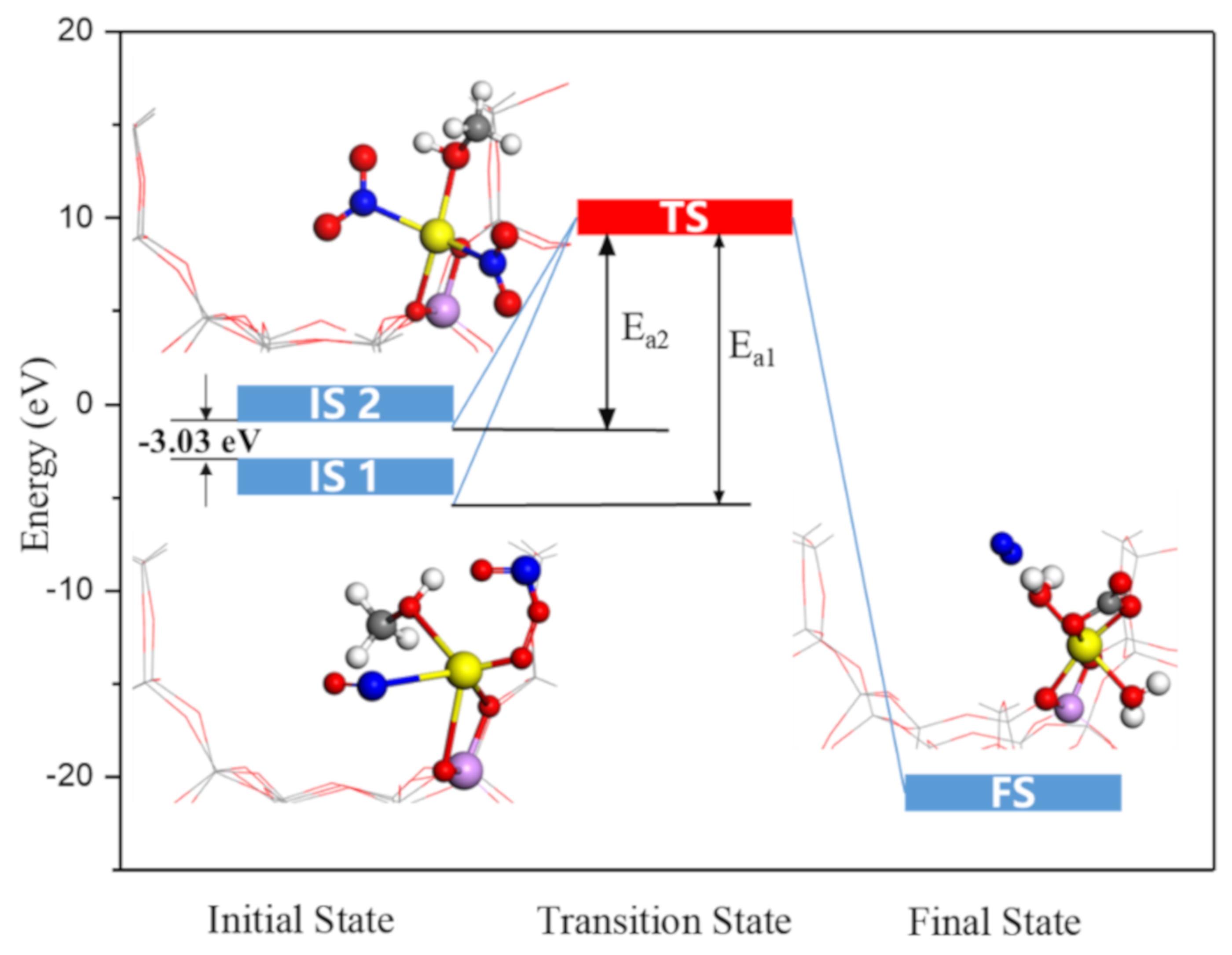
2.6. Nitromethanes’ Formation and Reaction Enthalpy
3. Computational Methods
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Li, G.; Wang, X.; Jia, C.; Liu, Z. An in situ Fourier transform infrared study on the mechanism of NO reduction by acetylene over mordenite-based catalysts. J. Catal. 2008, 257, 291–296. [Google Scholar] [CrossRef]
- Liu, J.; Li, X.; Zhao, Q.; Hao, C.; Zhang, D. Insight into the mechanism of selective catalytic reduction of NOx by propene over the Cu/Ti0.7Zr0.3O2 catalyst by fourier transform infrared spectroscopy and density functional theory calculations. Environ. Sci. Technol. 2013, 47, 4528–4535. [Google Scholar] [CrossRef]
- Li, Y.; Armor, J.N. Catalytic reduction of nitrogen oxides with methane in the presence of excess oxygen. Appl. Catal. B-Environ. 1992, 1, L31–L40. [Google Scholar] [CrossRef]
- Kikuchi, E.; Yogo, K. Selective catalytic reduction of nitrogen monoxide by methane on zeolite catalysts in an oxygen-rich atmosphere. Catal. Today 1994, 22, 73–86. [Google Scholar] [CrossRef]
- Gao, E.; Sun, G.; Zhang, W.; Bernards, M.T.; He, Y.; Pan, H.; Shi, Y. Surface lattice oxygen activation via Zr4+ cations substituting on A2+ sites of MnCr2O4 forming ZrxMn1-xCr2O4 catalysts for enhanced NH3-SCR performance. Chem. Eng. J. 2020, 380, 122397. [Google Scholar] [CrossRef]
- Lónyi, F.; Valyon, J.; Gutierrez, L.; Ulla, M.A.; Lombardo, E.A. The SCR of NO with CH4 over Co-, Co,Pt-, and H-mordenite catalysts. Appl. Catal. B-Environ. 2007, 73, 1–10. [Google Scholar] [CrossRef]
- Lukyanov, D.B.; Lombardo, E.A.; Sill, G.A.; D’Itri, J.L.; Hall, W.K. Selective Catalytic Reduction (SCR) of NO with Methane over CoZSM-5 and HZSM-5 Zeolites: On the Role of Free Radicals and Competitive Oxidation Reactions. J. Catal. 1996, 163, 447–456. [Google Scholar] [CrossRef]
- Wang, Y.; Lei, Z.; Chen, B.; Guo, Q.; Liu, N. Adsorption of NO and N2O on Fe-BEA and H-BEA zeolites. Appl. Surf. Sci. 2010, 256, 4042–4047. [Google Scholar] [CrossRef]
- Yan, Z.; Zuo, Z.; Li, Z.; Zhang, J. A cluster DFT study of NH3 and NO adsorption on the (MoO2)2+/HZSM-5 surface: Lewis versus Brønsted acid sites. Appl. Surf. Sci. 2014, 321, 339–347. [Google Scholar] [CrossRef]
- Lónyi, F.; Solt, H.E.; Valyon, J.; Boix, A.; Gutierrez, L.B. The SCR of NO with methane over In H- and Co,In,H-ZSM-5 catalysts: The promotional effect of cobalt. Appl. Catal. B-Environ. 2012, 117, 212–223. [Google Scholar] [CrossRef]
- Meng, D.; Zhan, W.; Guo, Y.; Guo, Y.; Wang, L.; Lu, G. A highly effective catalyst of Sm-MnOx for the NH3-SCR of NOx at low temperature: Promotional role of Sm and its catalytic performance. ACS Catal. 2015, 5, 5973–5983. [Google Scholar] [CrossRef]
- Lónyi, F.; Solt, H.E.; Valyon, J.; Boix, A.; Gutierrez, L.B. The activation of NO and CH4 for NO-SCR reaction over In- and Co-containing H-ZSM-5 catalysts. J. Mol. Catal. A-Chem. 2011, 345, 75–80. [Google Scholar] [CrossRef]
- Kikuchi, E.; Ogura, M.; Terasaki, I.; Goto, Y. Selective reduction of nitric oxide with methane on gallium and indium containing H-ZSM-5 catalysts: Formation of active sites by solid-state ion exchange. J. Catal. 1996, 161, 465–470. [Google Scholar] [CrossRef]
- Kubacka, A.; Janas, J.; Sulikowski, B. In/Co-ferrierite: A highly active catalyst for the CH4-SCR NO process under presence of steam. Appl. Catal. B-Environ. 2006, 69, 43–48. [Google Scholar] [CrossRef]
- Marnellos, G.E.; Efthimiadis, E.A.; Vasalos, I.A. Mechanistic and kinetic analysis of the NOx selective catalytic reduction by hydrocarbons in excess O2 over In/Al2O3 in the presence of SO2 and H2O. Appl. Catal. B-Environ. 2004, 48, 1–15. [Google Scholar] [CrossRef]
- Maunula, T.; Ahola, J.; Hamada, H. Reaction mechanism and kinetics of NOx reduction by methane on In/ZSM-5 under lean conditions. Appl. Catal. B-Environ. 2006, 64, 13–24. [Google Scholar] [CrossRef]
- Lónyi, F.; Solt, H.E.; Valyon, J.; Decolatti, H.; Gutierrez, L.B.; Miró, E. An operando DRIFTS study of the active sites and the active intermediates of the NO-SCR reaction by methane over In,H- and In,Pd,H-zeolite catalysts. Appl. Catal. B-Environ. 2010, 100, 133–142. [Google Scholar] [CrossRef]
- Pan, H.; Jian, Y.; Yu, Y.; Chen, N.; He, C.; He, C. Promotional mechanism of propane on selective catalytic reduction of NOx by methane over In/H-BEA at low temperature. Appl. Surf. Sci. 2016, 390, 608–616. [Google Scholar] [CrossRef]
- Zhou, D.; Zhang, Y.; Zhu, H.; Ma, D.; Bao, X. The structure, stability, and reactivity of Mo-oxo species in H-ZSM5 zeolites: Density functional theory study. J. Phys. Chem. C 2007, 111, 2081–2091. [Google Scholar] [CrossRef]
- Pietrzyk, P.; Podolska, K.; Sojka, Z. Role of NOδ+ intermediates in NO reduction with propene over NiZSM-5 zeolite revealed by EPR and IR spectroscopic investigations and DFT modeling. J. Phys. Chem. C 2011, 115, 13008–13015. [Google Scholar] [CrossRef]
- Cowan, A.D.; Dumpelmann, R.; Cant, N.W. The rate-determining step in the selective reduction of nitric-oxide by methane over a Co-ZSM5 catalyst in the presence of oxygen. J. Catal. 1995, 151, 356–363. [Google Scholar] [CrossRef]
- Cant, N.W.; Liu, I.O.Y. The mechanism of the selective reduction of nitrogen oxides by hydrocarbons on zeolite catalysts. Catal. Today 2000, 63, 133–146. [Google Scholar] [CrossRef]
- Kikuyama, S.; Matsukuma, I.; Kikuchi, R.; Sasaki, K.; Eguchi, K. A role of components in Pt-ZrO2/Al2O3 as a sorbent for removal of NO and NO2. Appl. Catal. A-Gen. 2002, 226, 23–30. [Google Scholar] [CrossRef]
- Newsam, J.M.; Treacy, M.M.J.; Koetsier, W.T.; Gruyter, C.B.D.; Thomas, J.M. Structural characterization of zeolite beta. Proc. R. Soc. Lond. A Math. Phys. Sci. 1988, 420, 375–405. [Google Scholar]
- Lide, D.R. CRC Handbook of Chemistry and Physics; CRC Press: Boca Raton, FL, USA, 2004. [Google Scholar]
- Yang, W.; Parr, R.G. Hardness, softness, and the fukui function in the electronic theory of metals and catalysis. Proc. Natl. Acad. Sci. USA 1985, 82, 6723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, D.; Zhang, L.; Wan, L.; Zhang, Y.; Bian, Y.; Jiang, J. Conformational effects, molecular orbitals, and reaction activities of bis(phthalocyaninato) lanthanum double-deckers: Density functional theory calculations. Phys. Chem. Chem. Phys. 2011, 13, 13277. [Google Scholar] [CrossRef]
- Sinev, M.Y.; Margolis, L.Y.; Korchak, V.N. Heterogeneous free-radical reactions in oxidation processes. Russ. Chem. Rev. 1995, 64, 349–364. [Google Scholar] [CrossRef]
- Córdoba, L.F.; Sachtler, W.M.H.; Montes De Correa, C. NO reduction by CH4 over Pd/Co-sulfated zirconia catalysts. Appl. Catal. B-Environ. 2005, 56, 269–277. [Google Scholar] [CrossRef]
- Lei Huang, K.Z.S.N. Promotional effect of the TiO2 (001) facet in the selective catalytic reduction of NO with NH3: In situ DRIFTS and DFT studies. Catal. Sci. Technol. 2016, 6, 8516–8524. [Google Scholar] [CrossRef]
- Li, Y.J.; Armor, J.N. Selective reduction of NOx by methane on Co-Ferrierites: I. reaction and kinetic studies. J. Catal. 1994, 150, 376–387. [Google Scholar] [CrossRef]
- Li, Y.J.; Slager, T.L.; Armor, J.N. Selective reduction of NOx by methane on Co-Ferrierites: II. catalyst characterization. J. Catal. 1994, 150, 388–399. [Google Scholar] [CrossRef]
- Yokoyama, C.; Misono, M. Catalytic reduction of nitrogen oxides by propene in the presence of oxygen over cerium ion-exchanged zeolites: III. effects of carriers. J. Catal. 1996, 160, 95–105. [Google Scholar] [CrossRef]
- Ren, L.; Zhang, T.; Liang, D.; Xu, C.; Tang, J.; Lin, L. Effect of addition of Zn on the catalytic activity of a Co/HZSM-5 catalyst for the SCR of NOx with CH4. Appl. Catal. B-Environ. 2002, 35, 317–321. [Google Scholar] [CrossRef]
- Pietrogiacomi, D.; Campa, M.C.; Ardemani, L.; Occhiuzzi, M. Operando FTIR study of Fe-MOR, Co-MOR, and Ni-MOR as catalysts for simultaneous abatement of NOx and N2O with CH4 in the presence of O2. An insight on reaction pathway. Catal. Today 2019, 336, 131–138. [Google Scholar] [CrossRef]
- Fujita, H.; Kanougi, T.; Atoguchi, T. Distribution of Brønsted acid sites on beta zeolite H-BEA: A periodic density functional theory calculation. Appl. Catal. A-Gen. 2006, 313, 160–166. [Google Scholar] [CrossRef]
- Delley, B. From molecules to solids with the DMol3 approach. J. Chem. Phys. 2000, 113, 7756–7764. [Google Scholar] [CrossRef]
- Hammer, B. Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functionals. Phys. Rev. B 1999, 59, 7413–7421. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Wang, Y. Generalized gradient approximation for the exchange-correlation hole of a many-electron system. Phys. Rev. B 1996, 54, 16533–16539. [Google Scholar] [CrossRef] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Models | dAl–O1 | dAl–O2 | dIn–O | dIn–O1 | dIn–O2 | dO–H | qAl | qIn | qO | qH | qO1 | qO2 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| L-model | 1.822 | 1.827 | 1.872 | 2.249 | 2.229 | - | 1.445 | 0.978 | −0.608 | - | −0.933 | −0.933 |
| B-model | 1.870 | 1.872 | 1.954 | 2.072 | 2.106 | 0.981 | 1.465 | 1.273 | −0.553 | 0.329 | −0.941 | −0.950 |
| Models | In | O | H | |||
|---|---|---|---|---|---|---|
| L-model | 0.425 | 0.411 | 0.155 | 0.310 | - | - |
| B-model | 0.391 | 0.003 | 0.120 | 0.001 | 0.059 | 0.003 |
| Bonds | L-model | B-model | ||
|---|---|---|---|---|
| In site | O site | In site | O site | |
| In–O | 1.873 | 2.493 | 1.986 | 1.954 |
| In–O1 | 2.247 | 2.596 | 2.118 | 2.072 |
| In–O2 | 2.241 | 3.246 | 2.124 | 2.106 |
| Al–O1 | 1.824 | 1.794 | 1.854 | 1.870 |
| Al–O2 | 1.824 | 1.786 | 1.860 | 1.873 |
| O–H | - | 0.975 | 0.980 | 0.981 |
| C–O | - | 1.445 | - | 4.358 |
| C–H1 | 1.098 | 1.096 | 1.096 | 1.097 |
| C–H2 | 1.098 | 1.098 | 1.105 | 1.097 |
| C–H3 | 1.097 | 1.100 | 1.109 | 1.098 |
| C–H4 | 1.099 | - | 1.102 | 1.098 |
| qCH4 | 0.034 | 0.305 | 0.052 | −0.002 |
| Eads | −0.15 | −2.98 | −0.48 | −0.10 |
| L-Model | B-Model | ||||
|---|---|---|---|---|---|
| NO–In and O site | NO2-In and O site | NO–In and O site | NO2-In site | NO2-O site | |
| dIn–O | 2.169 | 2.330 | 1.995 | 1.990 | 2.183 |
| dIn–O1 | 2.332 | 2.324 | 2.158 | 2.159 | 2.375 |
| dIn–O2 | 2.345 | 2.298 | 2.204 | 2.187 | 2.453 |
| dAl–O1 | 1.813 | 1.818 | 1.840 | 1.833 | 1.811 |
| dAl–O2 | 1.814 | 1.817 | 1.832 | 1.846 | 1.811 |
| dIn-N | - | - | 2.635 | 2.322 | - |
| dN-O | 1.387 | 1.310 | - | - | 1.395 |
| dN-O3 | 1.197 | 1.210 | 1.171 | 1.224 | 1.209 |
| dN-O4 | - | 1.304 | - | 1.220 | 1.234 |
| dO–H | - | - | 0.976 | 1.008 | |
| qIn | 0.859 | 0.917 | 1.072 | 1.132 | 0.816 |
| qO | −0.422 | −0.407 | −0.609 | −0.613 | −0.333 |
| qNO/NO2 | −0.267 | −0.248 | 0.120 | 0.122 | 0.037 |
| Eads | −1.84 | −1.93 | −1.07 | −0.56 | −0.42 |
| O2 | CO2 | H2O | ||||
|---|---|---|---|---|---|---|
| In site | O site | In site | O site | In site | O site | |
| dIn–O | 1.933 | 2.330 | 1.874 | 2.058 | 1.897 | 1.894 |
| dIn–O1 | 2.235 | 2.336 | 2.255 | 2.178 | 2.279 | 2.335 |
| dIn–O2 | 2.234 | 2.368 | 2.200 | 2.166 | 2.244 | 2.196 |
| dAl–O1 | 1.828 | 1.813 | 1.822 | 1.840 | 1.817 | 1.801 |
| dAl–O2 | 1.824 | 1.811 | 1.835 | 1.845 | 1.823 | 1.836 |
| dO2/H2O/CO2 | 1.269 | 1.346 | 1.174, 1.175 | 1.380, 1.209 | 0.995, 0.980 | 0.964, 0.967 |
| qO2/H2O/CO2 | 0.092 | 0.094 | 0.077 | 0.215 | 0.027 | 0.016 |
| Eads | −0.08 | −0.49 | −0.11 | −0.86 | −0.29 | −0.05 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, E.; Pan, H.; Wang, L.; Shi, Y.; Chen, J. Identification of Main Active Sites and the Role of NO2 on NOx Reduction with CH4 over In/BEA Catalyst: A Computational Study. Catalysts 2020, 10, 572. https://doi.org/10.3390/catal10050572
Gao E, Pan H, Wang L, Shi Y, Chen J. Identification of Main Active Sites and the Role of NO2 on NOx Reduction with CH4 over In/BEA Catalyst: A Computational Study. Catalysts. 2020; 10(5):572. https://doi.org/10.3390/catal10050572
Chicago/Turabian StyleGao, Erhao, Hua Pan, Li Wang, Yao Shi, and Jun Chen. 2020. "Identification of Main Active Sites and the Role of NO2 on NOx Reduction with CH4 over In/BEA Catalyst: A Computational Study" Catalysts 10, no. 5: 572. https://doi.org/10.3390/catal10050572
APA StyleGao, E., Pan, H., Wang, L., Shi, Y., & Chen, J. (2020). Identification of Main Active Sites and the Role of NO2 on NOx Reduction with CH4 over In/BEA Catalyst: A Computational Study. Catalysts, 10(5), 572. https://doi.org/10.3390/catal10050572