2.1. Catalysts Characterization
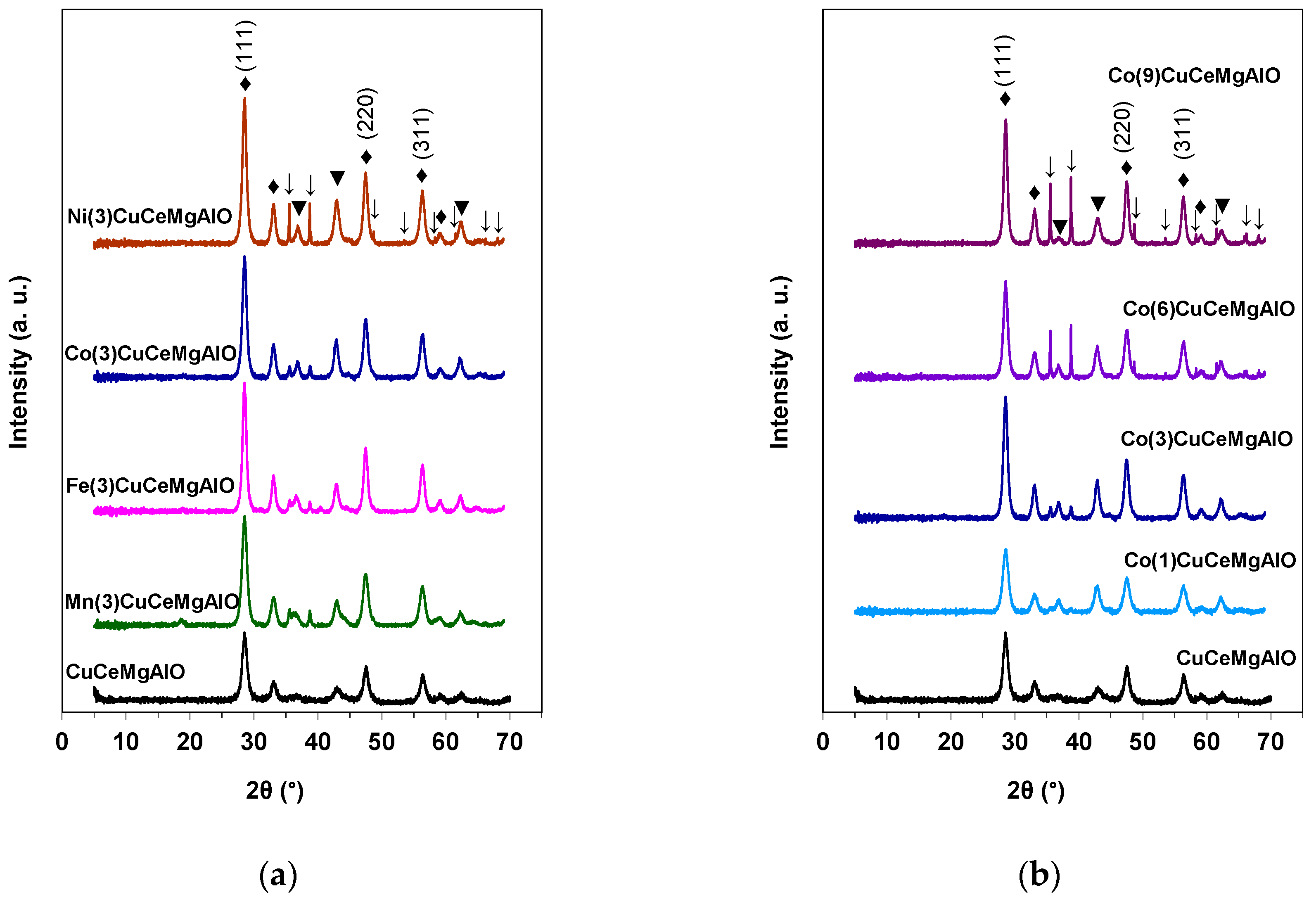
The XRD patterns of the as-prepared M(3)CuCeMgAl and Co(x)CuCeMgAl LDH precursors are displayed in
Figure 1. All the M(3)CuCeMgAl LDH precursors consisted of well-crystallized LDH phase (Powder Diffraction File (PDF) 37-0630) together with poorly crystallized boehmite AlOOH phase (PDF 83-2384) (
Figure 1a). The M-free CuCeMgAl LDH precursor consisted of a mixture of both LDH and boehmite poorly crystallized phases (
Figure 1a). The absence of diffraction lines corresponding to cerium- or transition metal-containing additional phases can be noted, suggesting that these cations were well dispersed in the M(3)CuCeMgAl LDH precursor samples, as already observed in CuCeMgAl LDH materials [
27]. The Co(x)CuCeMgAl LDH precursors also consisted of a mixture of well-crystallized LDH and poorly crystallized boehmite AlOOH phases (
Figure 1b). However, diffraction lines corresponding to CuO tenorite phase (PDF 41-0254) could be observed at higher Co contents, i.e.,
x = 6 and 9 at.%. This could be explained as follows: at high Co contents the M
2+/M
3+ atomic ratio became higher than the upper limit corresponding to the LDH phase formation, i.e., M
2+/M
3+ = 4, and, because of its pronounced Jahn–Teller effect, Cu
2+ left the brucite-like layers, forming a separate phase to the benefit of Co
2+ species, which entered the LDH layers [
9].
The (110) and (003) reflections were used to calculate the lattice parameters
a (
a = 2 ×
d110) and
c (
c = 3 ×
d003), respectively, assuming a hexagonal symmetry for the LDH structure (
Table 1). In both M(3)CuCeMgAl and Co(x)CuCeMgAl LDH series the values of the cell parameter
a were close together, accounting for similar mean intermetallic distances in the layers. This suggests that the different cations were homogeneously distributed in the hydroxide layers. The cell parameter
c, which is a function of different factors, like the nature of the interlayer anion, the average charge of the LDH layers, and the amount of water in the interlayer space, accounted for the presence of mainly nitrate as compensating anion [
28], originating from the precursor salts used for the LDH synthesis.
The mean crystallite size in the
c direction (
Table 1) was estimated from the full-width at half-maximum of the LDH (003) reflection by using the Debye-Scherrer equation. For both M(3)CuCeMgAl and Co(x)CuCeMgAl LDH precursors, nanometric crystallite sizes within the range 9.7–12.2 nm were observed.
The XRD patterns of the calcined M(3)CuCeMgAlO and Co(x)CuCeMgAlO mixed oxides are displayed in
Figure 2. The periclase-like Mg(Al)O mixed oxide phase (PDF 04-0829) together with CeO
2 fluorite phase (PDF 75-0076) were identified in all the calcined mixed oxides. Except for CuCeMgAlO system, more or less intense diffraction lines attributed to CuO tenorite phase were observed in both M(3)CuCeMgAlO (
Figure 2a) and Co(x)CuCeMgAlO (
Figure 2b) mixed oxides, and, for the latter, their intensity increased with increasing the Co content. Obviously, adding transition metals M (M = Mn, Fe, Co, and Ni) to CuCeMgAlO composition and increasing their content determined the segregation of CuO tenorite phase.
The lattice parameters of the ceria fluorite structure were calculated using the Bragg’s law from the three most intense (111), (220), and (311) reflections, and are presented in
Table 2. It can be observed that they were all close together, i.e.,
a = 0.5416 ± 0.0002 nm, and close to that corresponding to the M-free CuCeMgAlO sample (0.5415 nm). On the other hand, the lattice parameters of the periclase-like phase calculated from the (200) reflection at ca. 43° 2θ (
Table 2) were close together for both M(3)CuCeMgAlO and Co(x)CuCeMgAlO mixed oxides, i.e.,
a = 0.4215 ± 0.0004 nm, but were significantly higher than that of the M-free CuCeMgAlO sample (0.4201 nm). These data suggest that the cations M in both M(3)CuCeMgAlO and Co(x)CuCeMgAlO mixed oxides were homogeneously dispersed in the periclase-like Mg(Al)O phase rather than in the ceria phase. However, the existence of small quantities of poorly crystallized transition metal oxide-based side phases in the transition metal-promoted CuCeMgAlO mixed oxides could not be totally ruled out.
The full-width at half-maximum (FWHM) of the three most intense (111), (220), and (311) diffraction lines of CeO
2 phase were used to calculate the mean crystallite size with the Debye–Scherrer equation:
where
D is the crystallite size,
λ is the wavelength of the Cu K-alpha radiation (0.15406 nm), and
θ is the Bragg diffraction angle. They are presented in
Table 2. It can be observed that the crystallite size of ceria varied in the range of 13.7 ± 2.9 nm. The Debye–Scherrer equation was also used to calculate the mean crystallite size of the periclase-like phase from the two most intense reflections at ca. 43 and 62° 2
θ (
Table 2). The crystallite size of the solid solution periclase-like phase was similar for all the transition metal-promoted CuCeMgAlO mixed oxides, i.e.,
D = 13.8 ± 1.5 nm, being higher than that observed for the M-free CuCeMgAlO system.
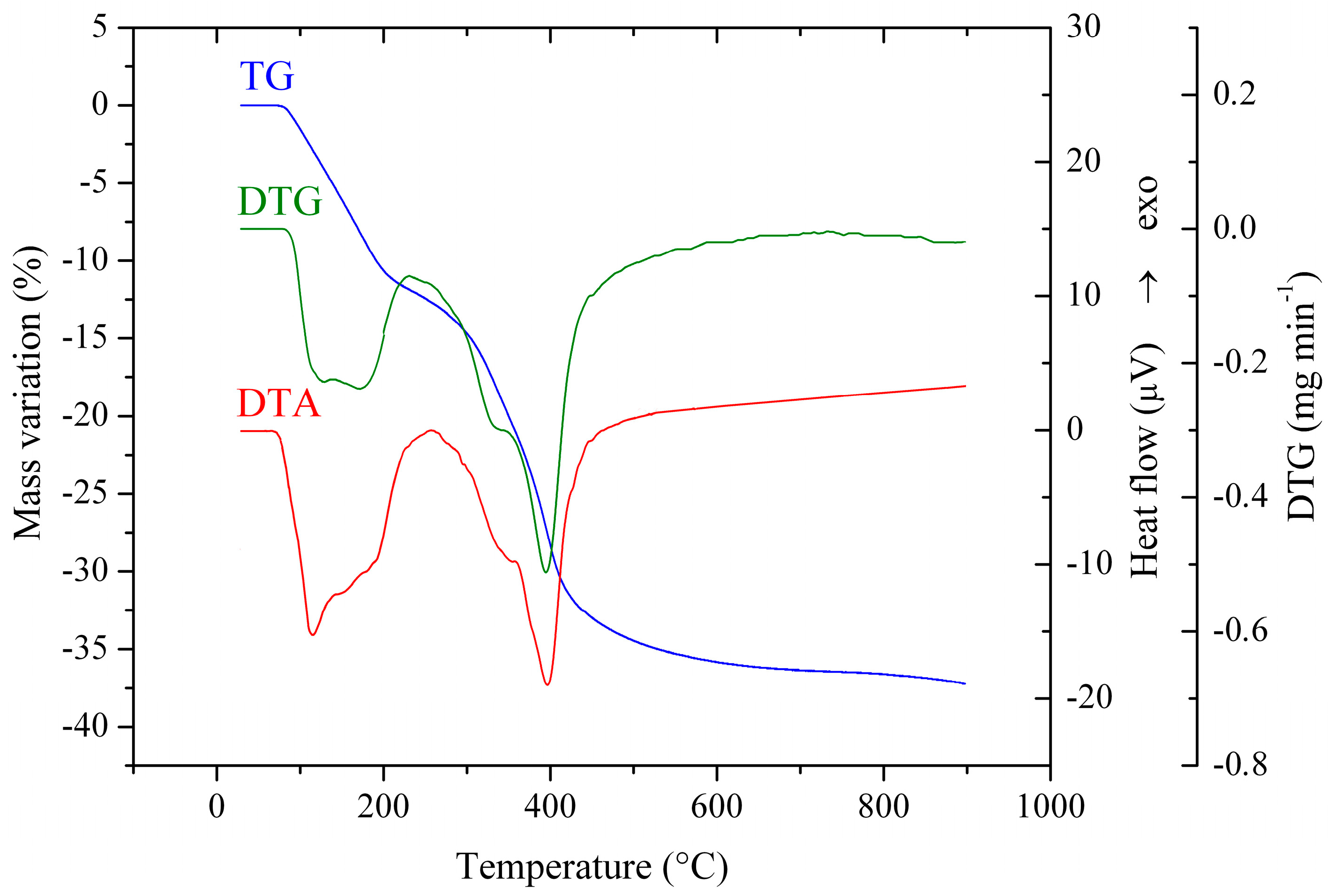
The decomposition of the LDH precursors into their corresponding mixed oxides has been followed by thermogravimetric and differential thermal analyses. The TG curves of selected LDH precursors are presented in
Figure S1, while the TG-DTG-DTA curves of the as-prepared Co(3)CuCeMgAl LDH precursor, representative of all samples, are presented in
Figure 3. They are characteristic for the LDH materials [
29] with two weight losses. The first weight loss of 9–13% (with DTG-DTA peaks at 110 and 170 °C for the Co(3)CuCeMgAl LDH precursor) was ascribed to the removal of weakly bounded and interlayered water molecules, while the second weight loss in the range of 20–24% (with DTG-DTA peaks at 350 and 395 °C for Co(3)CuCeMgAl LDH) was attributed to the dehydroxylation of the LDH layers and the decomposition of the anions from the interlayer space.
The structure and morphology of the LDH-derived mixed oxides were further studied via transmission electron microscopy (TEM) and scanning electron microscopy (SEM). The TEM images of selected mixed oxides (
Figure S2) revealed that they have a scale-like structure with nanoparticle sizes of ca. 5–15 nm. Regular and smooth lattice fringes could also be observed, confirming the good crystallinity of the materials. The SEM images of all the mixed oxides are displayed in
Figure S3. All the images show agglomerated powders with micro- and nanometric features that generated a large surface area. Notably, in powders with a higher Co content, crystalline, rectangular, parallel-piped shapes of different sizes were observed. These corresponded to copper oxides with tenorite structure [
30], in good agreement with the conclusions drawn from the XRD analysis.
The cationic composition of the calcined mixed oxide catalysts was determined by EDX spectroscopy and it is tabulated in
Table 3. It can be observed that, for all the samples, the cationic content was close to the calculated values within the limits of experimental error of the method used, except for Mn(3)- and Fe(3)CuCeMgAlO samples, for which it was slightly more different. The Mg/Al and Cu/Ce atomic ratios varied in the ranges 3.0–3.3 and 1.5–1.7, respectively, being thus close to the fixed values of 3 and 1.5, respectively, except for Mn(3)CuCeMgAlO sample, for which they were slightly higher, i.e., 3.5 and 2.1, respectively.
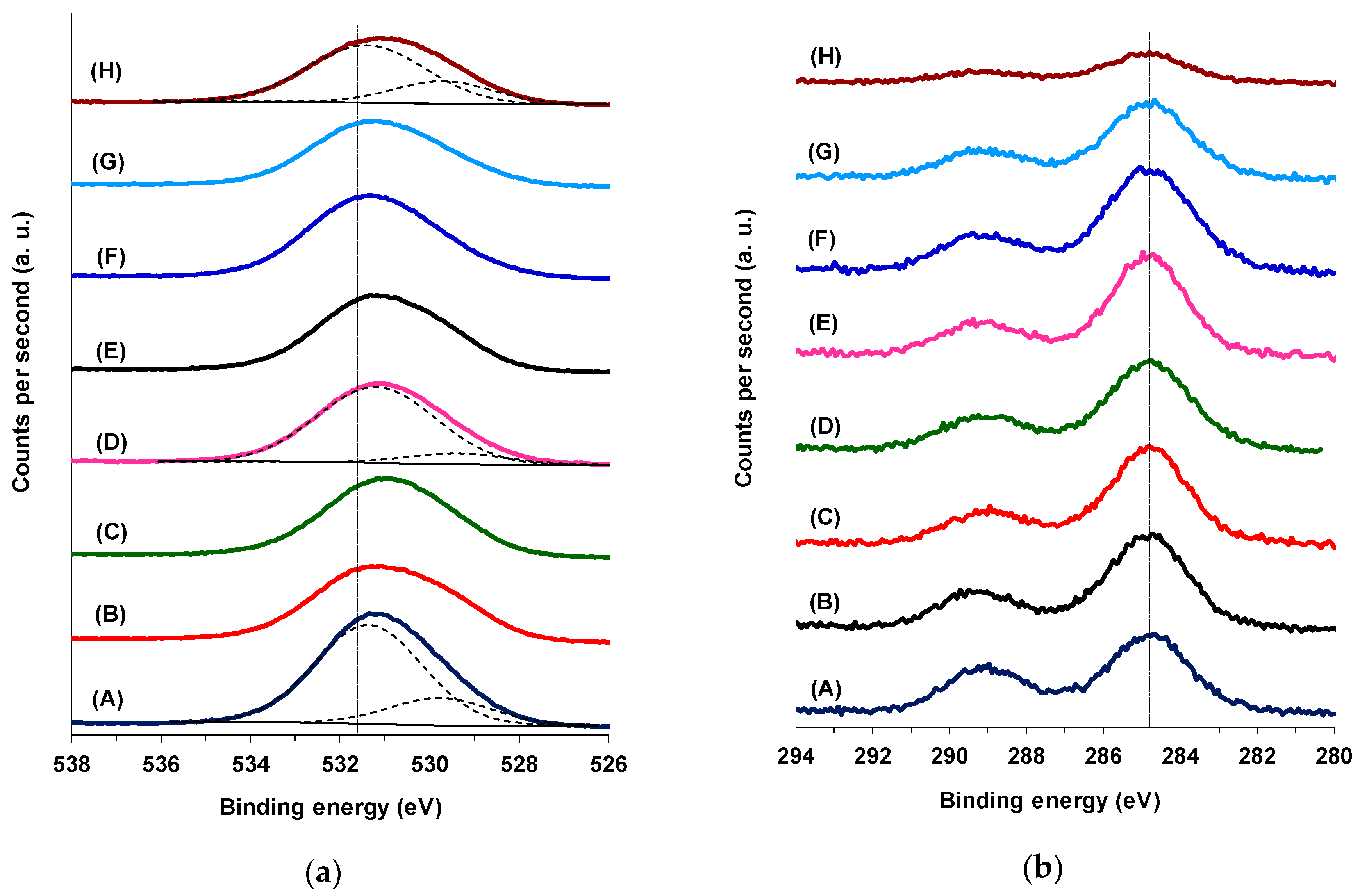
The surface composition of both M(3)CuCeMgAlO and Co(x)CuCeMgAlO mixed oxides together with the oxidation states of the different elements were studied by X-ray photoelectron spectroscopy (XPS). All the expected cations together with oxygen and carbon were evidenced on the surface of the different oxides, their respective content being tabulated in
Table S1. For all the mixed oxides the O 1s core level XPS spectra (
Figure 4a) consisted of a peak deconvoluted into two components at ca. 529.7 and 531.6 eV, which were ascribed to lattice oxygen in oxide and oxygen in the lateral structure, respectively [
31]. The oxygen in the lateral structure was associated to hydroxyl and/or carbonate species [
32] and also to subsurface oxygen ions with particular coordination and lower electron density than the lattice oxygen probably located at the interface of the crystalline CuO and CeO
2 phases [
21,
31].
The C 1s core level XPS spectra (
Figure 4b) showed two main contributions at 284.8 and ca. 289.2 eV attributed to adventitious hydrocarbon species and carbon in carbonate, respectively [
32]. This confirmed the need of a thermal treatment of the LDH-derived catalysts in the reactor before each activity test in order to clean their surface [
33].
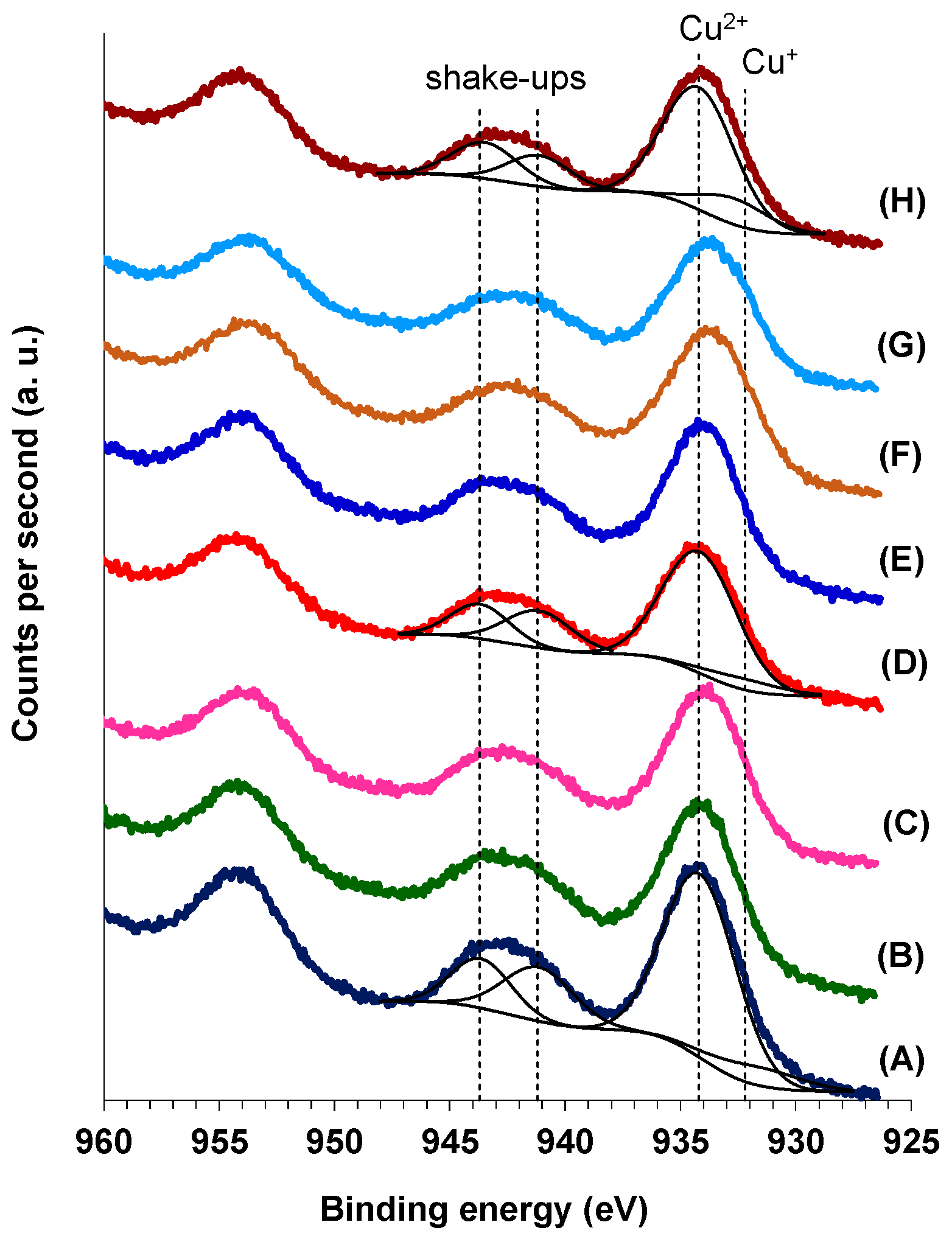
Figure 5 shows the photoelectron profile of Cu 2p region for both M(3)CuCeMgAlO and Co(x)CuCeMgAlO mixed oxides, while in
Table 4 are presented the surface concentrations of copper together with Cu
2+/Cu atomic ratios. Cu 2p
3/2 emission with two components centered at 932.2 and 934.2 eV was observed in all the mixed oxides, indicating the presence of both Cu
+ and Cu
2+ surface species, respectively [
34,
35]. The shake-ups of Cu 2p
3/2 emission line, centered at 941.2 and 943.7 eV, confirmed the presence of surface Cu
2+ as well [
35]. The presence of Cu
+ surface species in the mixed oxides suggests that, at least at the interface, CuO phase could be doped with higher valence cations, such as M
3+, Al
3+, and/or Ce
4+, existing in the mixed oxide.
The data in
Table 4 show that, except for Mn(3)- and Fe(3)CuCeMgAlO, the surface of the catalysts was enriched in Cu compared to the bulk composition (
Table 3). The Cu
2+/Cu surface atomic ratio was calculated by the method developed in [
35] using the following equation:
where
A is the total area of the main Cu 2p
3/2 emission line,
B is the area of the shake-up peak, and
A1
s/
Bs is a factor representing the ratio between the area of the main peak and that of the shake-up peak for a sample containing only Cu
2+ which is equal to 1.89 ± 0.08 for 20 eV pass energy [
35]. The surface Cu
2+/Cu atomic ratio varied in the range from 0.76 to 0.92, being lower for both M(3)CuCeMgAlO and Co(x)CuCeMgAlO mixed oxides compared to the unpromoted CuCeMgAlO, except for Co(3)CuCeMgAlO. This shows that, except for the last, the amount of surface Cu
+ species was higher in the promoted catalysts compared to the unpromoted one.
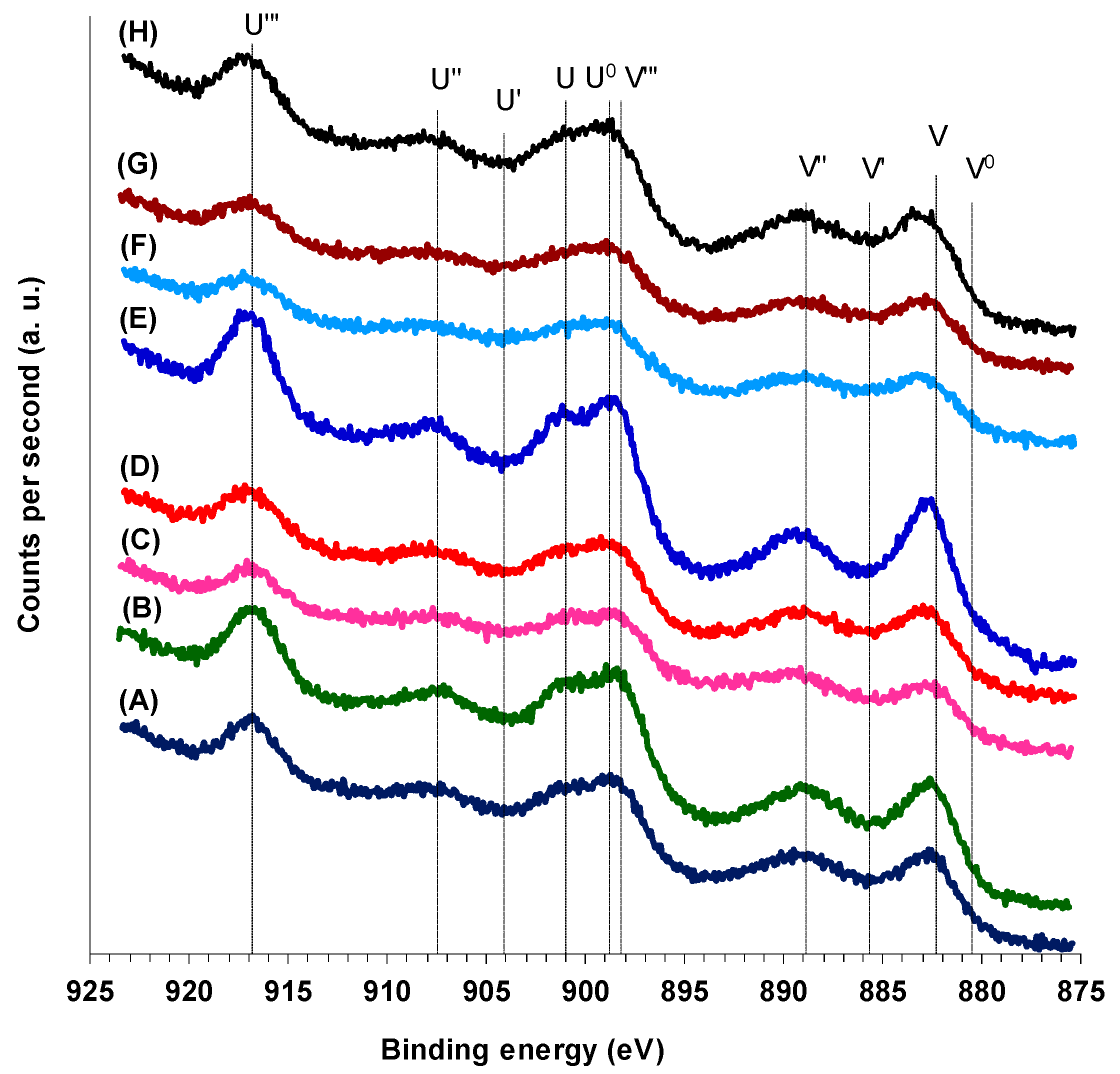
The X-ray photoelectron spectra of the Ce 3d
3/2 and Ce 3d
5/2 core levels of both M(3)CuCeMgAlO and Co(x)CuCeMgAlO mixed oxides are presented in
Figure 6. It can be observed that all the XPS spectra show 10 peaks, suggesting that both Ce
3+ and Ce
4+ species were present on the surface of all the mixed oxides. Indeed, six peaks corresponding to three pairs of spin-orbit doublets, conventionally labeled in order of decreasing energy U‴, U″, U (for Ce 3d
3/2) and V‴, V″, V (for Ce 3d
5/2), were attributed to Ce
4+ species, while four peaks corresponding to two pairs of spin-orbit doublets, labeled U′, U
0 (for Ce 3d
3/2) and V′, V
0 (for Ce 3d
5/2), were attributed to Ce
3+ species [
36]. Their characteristic binding energies were taken from [
37]. The U‴ peak was not observed in the Ce 3d spectrum of a pure Ce
3+ oxide, being exclusively attributed to Ce
4+ species. Therefore, it was used as a quantitative measure of the amount of Ce
4+ [
38]. Thus, taking into consideration that for pure CeO
2 the U‴ peak represented ca. 14% of total integral intensity [
38], the surface Ce
4+ content was calculated using the following equation:
where %
U‴ represents the percentage of
U‴ peak area with respect to the total Ce 3d area. The Ce
4+/Ce surface atomic ratio thus calculated is tabulated in
Table 4. It varied in the range from 0.79 to 0.93, being lower for both M(3)CuCeMgAlO and Co(x)CuCeMgAlO mixed oxides compared to the unpromoted CuCeMgAlO. This shows that the amount of surface Ce
3+ species was higher in the promoted catalysts compared to the unpromoted one, in agreement with previously reported results for co-doped CeO
2 [
23]. Moreover, the Ce
4+/Ce surface atomic ratio roughly parallels the Cu
2+/Cu surface atomic ratio (
Figure S4) for both M(3)CuCeMgAlO and Co(x)CuCeMgAlO series of mixed oxides, suggesting that the promoter equally favored the reduction of both Ce
4+ and Cu
2+ surface species in the promoted catalysts and, hence, improved the synergistic effect between Cu and Ce [
23]. It is worth noting that responsible for the presence of surface Ce
3+ species was, at least partly, the reduction of Ce
4+ under the X-ray beam during the XPS analysis. The data in
Table 4 also show that the surface concentration of cerium was lower than its bulk concentration (
Table 3) for all the mixed oxides and, except for the Mn(3)CuCeMgAlO sample, the Cu/Ce surface atomic ratio (
Table 4) was significantly higher than the bulk ratio (
Table 3).
The Al 2p and Mg 2p X-ray photoelectron spectra of all the mixed oxides are shown in
Figures S5 and S6, respectively.
Figure S6 also shows their Auger Mg KLL spectra. Although the Al 2p peak appeared at almost the same binding energy as Cu 3p, the deconvolution of the XPS signal allowed us to unambiguously identify and quantify it. The relative peak positions of both Al and Mg were very stable (
Table S2) and accounted for Al
3+ and Mg
2+ in their corresponding oxides. This was confirmed for Mg by calculating the modified Auger parameter (
m-AP) using the following formula:
where
BEMg2p is the binding energy of Mg 2p and
KEMgKLL, the kinetic energy of the Mg KLL peak. Indeed, the values obtained for
m-AP (
Table S2) were specific for MgO [
39] in all the mixed oxide samples. The Mg/Al surface atomic ratio (
Table 4) was lower than the bulk ratio for all the mixed oxides studied, indicating an Al enrichment of the surface.
The photoelectron profiles of Mn 2p, Fe 2p, and Ni 2p of the Mn(3)-, Fe(3)-, and Ni(3)CuCeMgAlO mixed oxides, respectively, are shown in
Figure S7, while their corresponding surface concentrations are tabulated in
Table 4. Each emission line (2p
3/2 and 2p
1/2) of the X-ray photoelectron spectrum of Mn 2p was fitted with three peaks corresponding to Mn
3+, Mn
4+, and a shake-up [
40]. The 2p
3/2 components at 641.7 and 643.3 eV attributed to Mn
3+ and Mn
4+, respectively, were used to calculate the Mn
4+/Mn
3+ surface ratio, which was equal to 0.67. The photoelectron spectrum of Fe 2p showed the presence of both Fe
2+ and Fe
3+ species on the surface of the Fe(3)CuCeMgAlO mixed oxide. The Fe
3+/Fe
2+ surface ratio was found to be 0.46. The observed Ni 2p photoelectron spectrum corresponded to hydroxylated nickel oxide [
41]. The Ni 2p
3/2 emission line and its shake-up peak centered at 856 and 861.9 eV, respectively, were attributed to high spin Ni
2+ species [
42,
43]. The surface concentrations of Mn and Ni were higher, while that of Fe was lower (
Table 4) compared to their corresponding bulk concentrations (
Table 3).
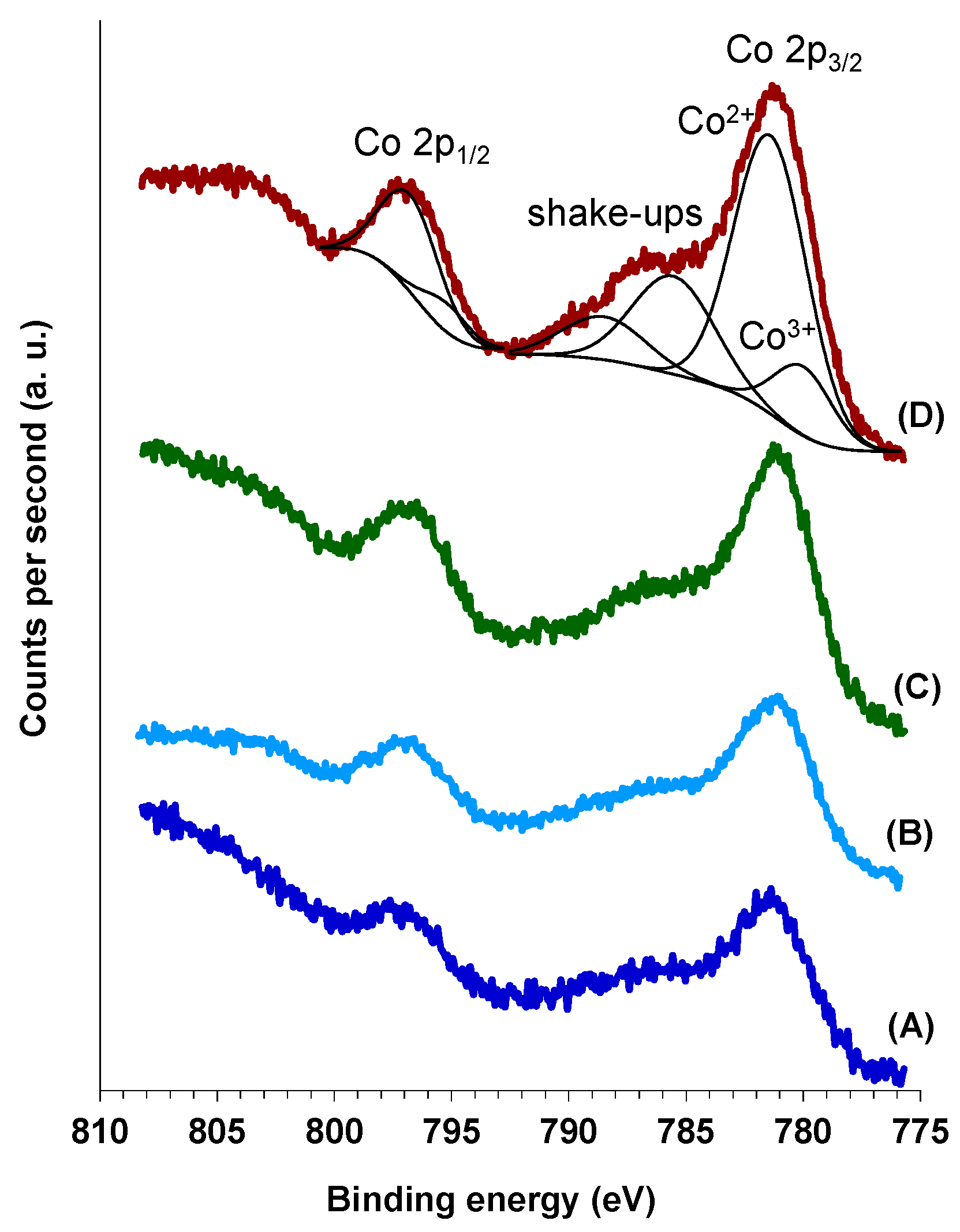
Figure 7 shows the photoelectron profiles of Co 2p region for the Co(x)CuCeMgAlO mixed oxides, while in
Table 4 are presented the surface concentrations of cobalt. The photoelectron spectra were deconvoluted into two spin-orbit doublets corresponding to Co
3+ and Co
2+ species, respectively, and two shake-up peaks [
44]. Thus, the Co 2p
3/2 component at 780.0 eV and a 2p
3/2–2p
1/2 splitting of ca. 15.3 eV was attributed to Co
3+ species, while that at 781.4 eV and a 2p
3/2–2p
1/2 splitting of ca. 15.4 eV was attributed to Co
2+ species. The surface Co
3+/Co
2+ atomic ratios are presented in
Table 4. It can be observed that the Co
3+/Co
2+ surface ratio passes through a maximum with increasing the Co content in the Co(x)CuCeMgAlO series, which corresponded to the Co(3)CuCeMgAlO system. The data in
Table 4 also show that the surface of all the Co-promoted mixed oxides was enriched in Co compared to the bulk composition (
Table 3).
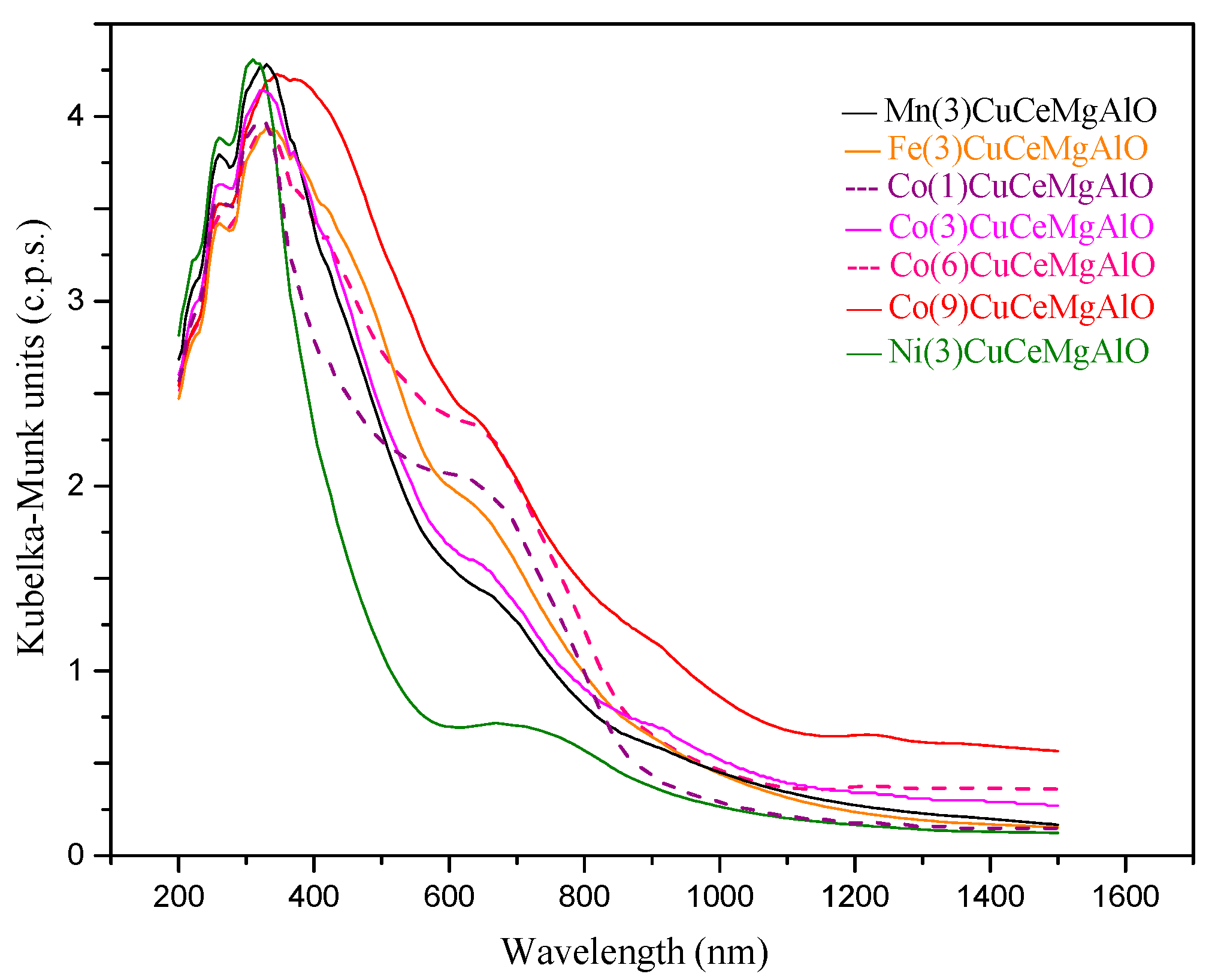
The electronic spectra of all the transition metal-promoted CuCeMgAlO mixed oxides (
Figure 8) contained both charge transfer (CT) bands and d-d transition bands. The strong bands in the ultraviolet range are characteristic to charge transfer transitions between copper and oxygen ions [
45]. The spectra of all the investigated materials showed two absorption maxima in this region at ca. 260 and 320 nm, which were assigned to CT bands characteristic for mononuclear Cu
2+ centers and oligomeric species (Cu
2+–O
2−–Cu
2+)
n2+, respectively [
21,
46]. The weak bands from the visible range (ca. 625–670 nm) were attributed to d
z2 → d
x2−y2 transitions characteristic for Cu
2+ ion in an octahedral stereochemistry [
45]. Moreover, for cobalt-containing oxides, new bands appeared in the near-infrared region of the spectra as the cobalt content increased. These bands were assigned to
4T
1g →
4A
2g (910 nm) and
4T
1g →
4T
2g (1220 nm) transitions, which are characteristic for Co
2+ ion in an octahedral stereochemistry [
45]. This was in line with the dispersion of cobalt species in the periclase-like Mg(Al)O phase, as suggested by the XRD analysis.
The adsorption-desorption isotherms of the unpromoted and transition metal-promoted CuCeMgAlO mixed oxides are shown in
Figure S8. All the solids revealed type IV isotherms with either H3-type or a combination of H3 with H2b types hysteresis loops specific for mesoporous materials with either slit-shaped pores or more complex pore structures [
47]. The specific surface areas (
Table 5) of all the transition metal-promoted CuCeMgAlO samples were close together and, for the Co(x)CuCeMgAlO mixed oxides, varied irrespective of the Co content, in line with their similar particle sizes of the periclase-like phase (
D = 13.8 ± 1.5 nm). However, their specific surface area was lower than that of the unpromoted CuCeMgAlO mixed oxide, in line with its lower particle size of the periclase-like phase (
D = 10.1 nm).
The pore volume of all mixed oxides varied in the range of 0.17 cm
3 g
−1 for Fe(3)CuCeMgAlO to 0.35 cm
3 g
−1 for Ni(3)CuCeMgAlO (
Table 5). The pore size distributions of the mixed oxides (
Figure S9) obtained from the desorption branch of isotherms indicated unimodal pore structures extending from 2 to ca. 20 nm, with well-defined maxima ranging from 7.4 nm for Ni(3)CuCeMgAlO to 10.8 nm for Co(6)CuCeMgAlO (
Table 5). A shoulder at 15.2 nm was also noticed in the pore size distribution of the Co(1)CuCeMgAlO sample. For the M-free CuCeMgAlO sample a well-defined bimodal pore size distribution with maxima at 3.7 and 11.4 could be observed.
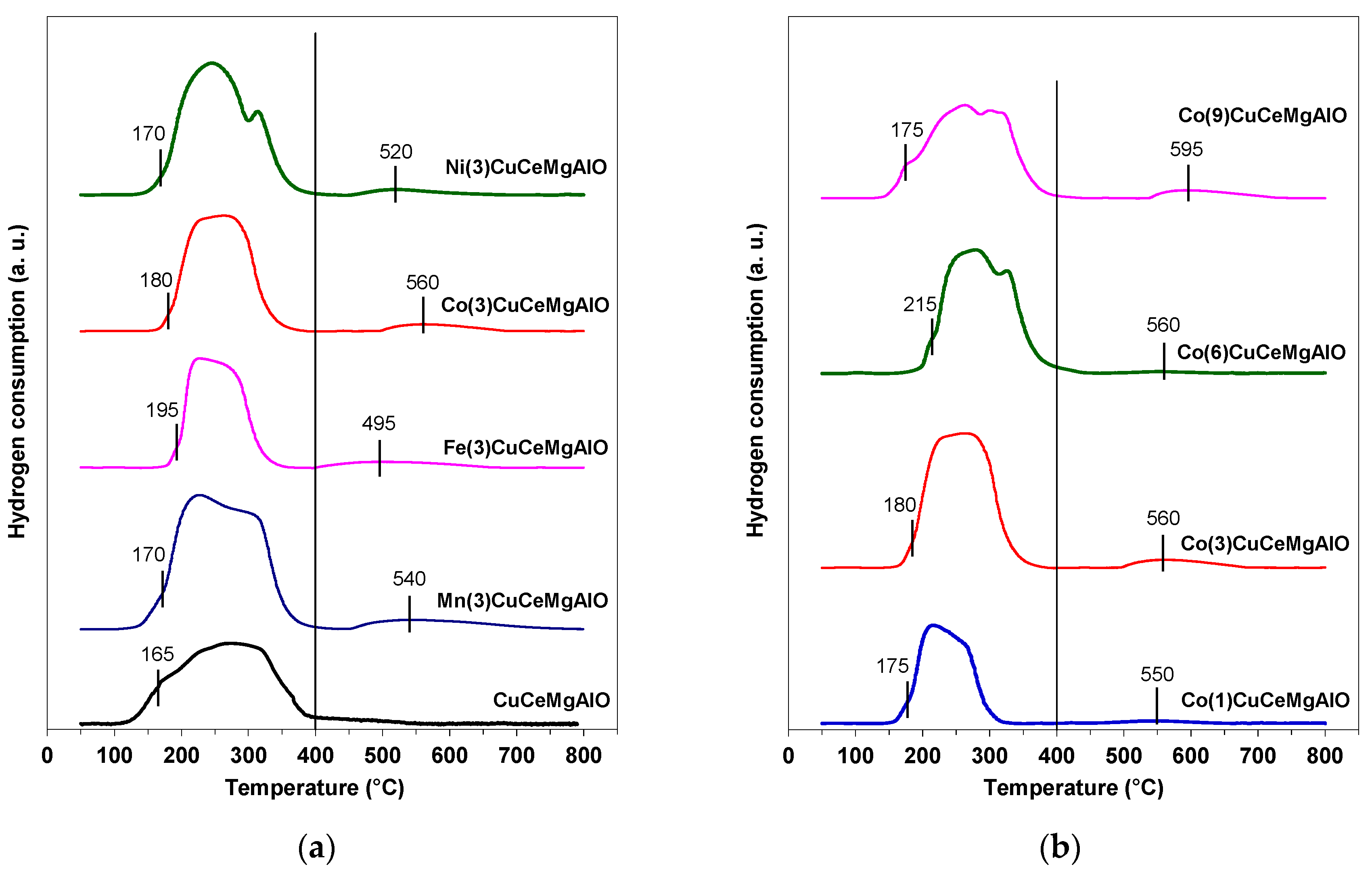
The reduction behavior of the different mixed oxides was investigated by H
2-TPR measurements, their reduction profiles being displayed in
Figure 9 with the corresponding H
2 consumptions tabulated in
Table 6. For the unpromoted CuCeMgAlO system, only one broad and intense reduction peak was observed in the temperature range 125–390 °C, with a tail extending up to 550 °C. This TPR profile accounted for the reduction of both Cu
2+ and Ce
4+ species, as described elsewhere [
21]. Thus, the broad and intense reduction peak with several maxima below 390 °C was attributed to the successive reduction of Cu
2+ species doped in the ceria particles, from well-dispersed and sintered CuO particles interacting more or less strongly with the CeMgAlO support and to the reduction of Ce
4+ species from highly reducible smaller ceria particles, while the tail was attributed to the reduction of Ce
4+ species from less reducible larger ceria particles [
21]. The hydrogen spillover on the metallic Cu particles was likely involved in the reduction of surface ceria lowering its reduction temperature [
48]. It is worth noting that the reduction model of small and large ceria particles is equivalent with that of surface and bulk ceria, often used to explain the reduction profile of Ce-containing materials, as small and large particles are mostly surface and mostly bulk, respectively [
48].
Due to the different nature of the transition-metal cations and the degree of crystallinity of the phases present, different reducibilities were expected for the M(3)CuCeMgAlO mixed oxides. Their H
2-TPR patterns showed an intense and broad peak in the low-temperature region, which was narrower compared to that of the M-free CuCeMgAlO mixed oxide and clearly ended at a temperature lower than 400 °C. A weak reduction peak at higher temperatures was also observed. The first reduction peak was due to the same reduction processes as observed for the M-free CuCeMgAlO system combined with the reduction of the transition-metal cations M. The latter promoted the redox ability of the material, resulting in sharp and overlapping peaks, in agreement with previously reported results [
22]. The high-temperature weak peak can be attributed to the reduction of either Ce
4+ species from less reducible larger ceria particles [
48] or M
n+ transition-metal ions from spinel-like phases with different stoichiometries [
11]. Although the latter were not evidenced by X-ray diffraction, their presence could not be totally ruled out. The first maximum reduction temperature for the M(3)CuCeMgAlO systems was higher than that observed for the M-free CuCeMgAlO mixed oxide. This was obviously due to the significantly higher surface area of the latter [
49]. Based on their first maximum reduction temperature, the reducibility of the M(3)CuCeMgAlO mixed oxides, in terms of easiness of reduction, increased as follows: Fe(3)CuCeMgAlO < Co(3)CuCeMgAlO < Mn(3)CuCeMgAlO ≈ Ni(3)CuCeMgAlO. This order corresponds to the decrease of the crystallite size of the Mg(Al)O periclase-like phase (
Table 2), suggesting that the surface transition-metal species dispersed in the Mg(Al)O matrix determined the easiness of reduction of the studied mixed oxides. In terms of hydrogen consumption, their reducibility followed the order: Fe(3)CuCeMgAlO < Co(3)CuCeMgAlO < Ni(3)CuCeMgAlO < Mn(3)CuCeMgAlO. Notably, the hydrogen consumption for the Fe(3)CuCeMgAlO mixed oxide was lower than that necessary for the reduction of all the Cu species calculated based on the oxide composition (
Table 6), suggesting that, for this mixed oxide, Cu was not quantitatively reduced, likely due to a hindered access of hydrogen to the reducible species, as reported earlier [
11].
In the Co(x)CuCeMgAlO series, the low-temperature intense peak represented the overlap of the reduction of copper and highly reducible cerium species, as described above, with the reduction of cobalt species, its broadness going increasingly with increasing the Co content. Interestingly, the temperature maximum of the weak high-temperature reduction peak increased with increasing the CeO
2 crystallite size (
Table 2), suggesting that it could be attributed to the reduction of Ce
4+ species from less reducible larger ceria particles rather than to the reduction of M
n+ transition-metal ions from spinel-like phases. Indeed, it was shown that the high-temperature reduction of ceria strongly depended on its crystallinity: the higher the crystallite size, the higher the reduction temperature [
46]. In terms of easiness of reduction, the reducibility of the Co(x)CuCeMgAlO mixed oxides increased in the order Co(6)CuCeMgAlO < Co(3)CuCeMgAlO < Co(1)CuCeMgAlO ≈ Co(9)CuCeMgAlO, which corresponded to the decrease of the crystallite size of the Mg(Al)O periclase-like phase (
Table 2), as observed for the M(3)CuCeMgAlO series. In terms of hydrogen consumption, the reducibility followed the order: Co(1)CuCeMgAlO < Co(9)CuCeMgAlO ≤ Co(6)CuCeMgAlO < Co(3)CuCeMgAlO. Unexpectedly, the hydrogen consumption did not increase with increasing the Co content, suggesting that the transition-metal species were decorated with non-reducible Mg(Al)O mixed oxide, which diminished the accessibility of hydrogen [
11]. This was in line with both the XRD analysis and the UV-VIS spectroscopy data showing that the cobalt cations were homogeneously dispersed in the Mg(Al)O periclase-like phase. This was also supported by the XRD analysis of the Co(x)CuCeMgAl LDH precursors, showing that Co replaced Cu in the brucite-like layers for Co contents higher than 3 at.%, obviously leading to Mg(Al,Co)O periclase-like particles and segregated CuO after calcination. Thus, together with the CuO particles, only the cobalt species on the surface of the Mg(Al,Co)O periclase-like particles were reduced (the smaller the particle size, the easier the reduction), while those located in the bulk were not. Notably, in agreement with this, the hydrogen consumption for the Co(1)CuCeMgAlO mixed oxide was lower than that needed for the reduction of all the Cu species calculated based on the oxide composition (
Table 6). Indeed, at low Co content, when Cu remained in the brucite-like layers of the LDH precursor, both Co and Cu species were dispersed in the Mg(Al)O matrix of the calcined oxide and, hence, the reduction of the transition-metal species located in the bulk was prevented.
2.2. Catalytic Properties
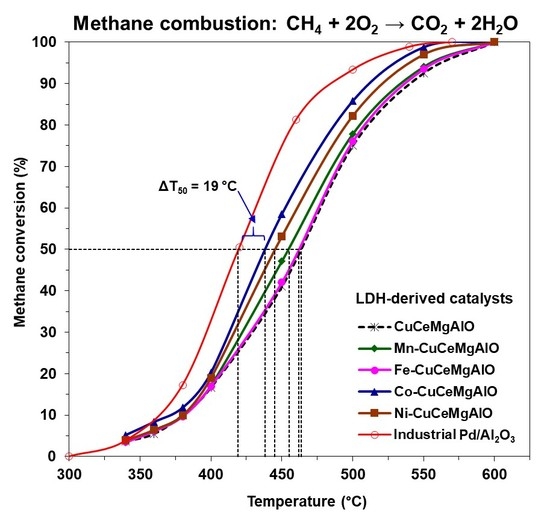
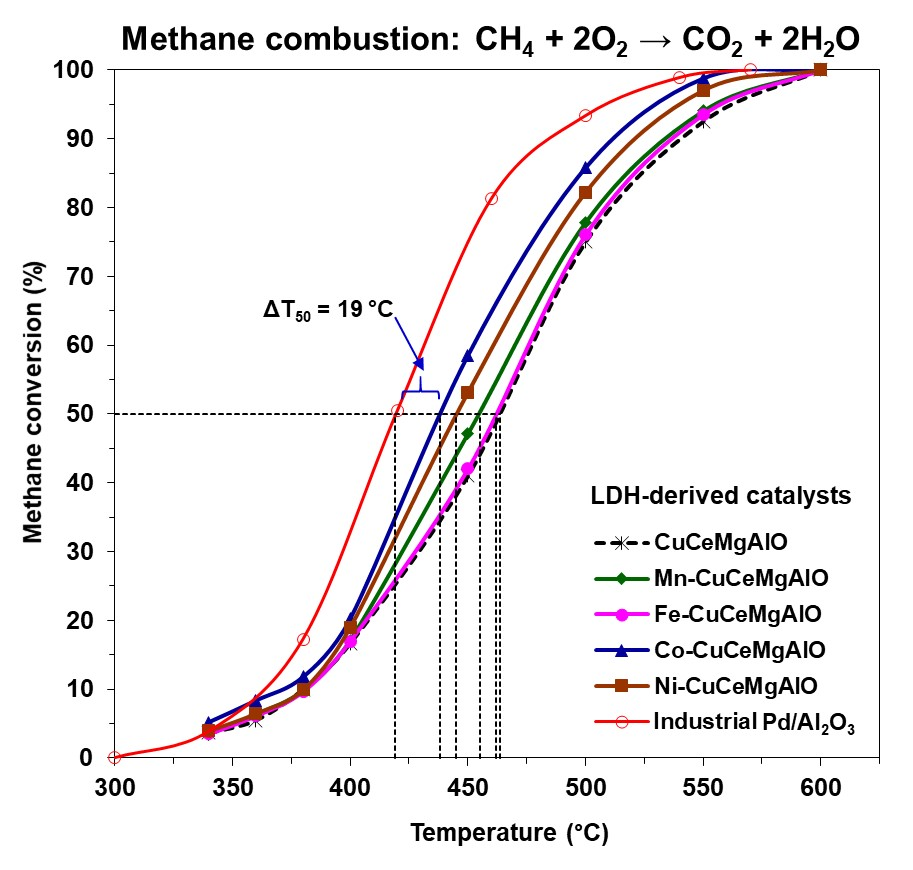
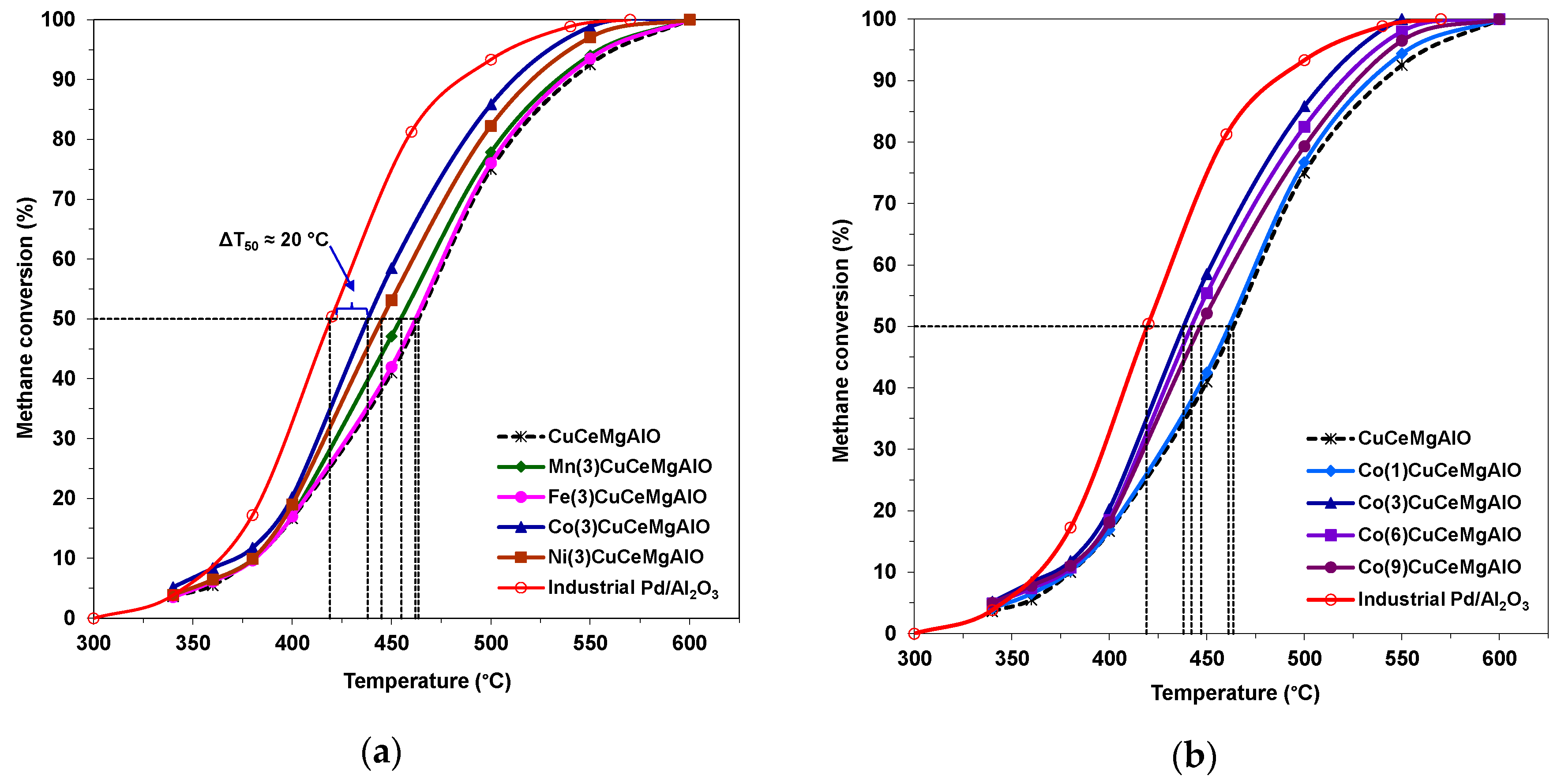
The catalytic performance of the mixed oxide catalysts was evaluated in the complete oxidation of methane as a model molecule and compared to that of a reference Pd/Al
2O
3 catalyst previously reported in [
14], with the light-off curves obtained being presented in
Figure 10. As a measure of the catalytic activity, the temperatures corresponding to 10, 50, and 90% methane conversion (T
10, T
50, and T
90, respectively), together with the intrinsic and specific activities at 400 °C, are listed in
Table 7. The light-off curve for Pd/Al
2O
3 catalyst, characterized by relatively low T
50 and T
90 values of 419 and 484 °C, respectively, accounted for its high activity in methane complete oxidation.
The light-off curves of both M(3)CuCeMgAlO and Co(x)CuCeMgAlO catalysts were between those of Pd/Al
2O
3 and M-free CuCeMgAlO, indicating, on one hand, that all the transition-metal M (M = Mn, Fe, Co, Ni) cations had a promoting effect on the CuCeMgAlO system, and, on the other hand, that this promoting effect depended on the transition-metal M content for M = Co. Obviously, the nature of the transition-metal M strongly influenced the catalytic activity of the CuCeMgAlO system. Indeed, in terms of both T
50 and T
90, the catalytic activity followed the order: CuCeMgAlO < Fe(3)CuCeMgAlO < Mn(3)CuCeMgAlO < Ni(3)CuCeMgAlO < Co(3)CuCeMgAlO. The catalytic behavior of the CuCeMgAlO system was attributed to both the excellent dispersion of copper and the synergy effect between Cu and Ce [
21]. According to the XRD analysis, the copper dispersion in the M(3)CuCeMgAlO mixed oxides seemed to be lower compared to CuCeMgAlO, as the segregation of CuO tenorite phase was evidenced. Therefore, to explain their increased activity, an enhanced synergy effect between Cu and Ce in the presence of transition-metal cations M should be taken into consideration, as suggested by the XPS analysis (
Figure S4) and in line with previously reported results [
22,
23,
24,
25,
26]. Indeed, the synergistic interaction between M, Cu, and Ce seemed to be a more important factor affecting the catalytic performance than the degree of crystallinity and surface area of the M(3)CuCeMgAlO materials, in agreement with previously reported results [
25]. It is noteworthy that the order of activity observed did not follow the easiness of reduction for the M(3)CuCeMgAlO mixed oxides, suggesting that the most reducible species in the H
2-TPR experiments were not necessarily the most active in the reaction conditions. On the other hand, the lowest activity of the Fe(3)CuCeMgAlO catalyst in the M(3)CuCeMgAlO series could be correlated to the partial reduction of Cu observed in H
2-TPR experiments. Keeping this in mind, the superior activity of the Fe(3)CuCeMgAlO compared to the unpromoted CuCeMgAlO system, mainly in terms of intrinsic reaction rate, can only be explained by a synergistic interaction between Fe, Cu, and Ce.
For the most active Co(3)CuCeMgAlO catalyst the T
50 value was 25 °C lower than that of the M-free CuCeMgAlO catalyst and only 19 °C higher than that of the reference Pd/Al
2O
3 catalyst. Moreover, the complete conversion of methane was achieved over the Co(3)CuCeMgAlO system at a temperature close to that corresponding to the Pd/Al
2O
3 catalyst (T
100 ≈ 570 °C). Notably, both specific and intrinsic activities of the Co(3)CuCeMgAlO catalyst calculated at 400 °C (
Table 7) showed the superiority of this system, and, hence, the effect of Co content on its catalytic performance was studied. It appeared that the cobalt content strongly influenced, in a complex manner, the catalytic activity of the Co(x)CuCeMgAlO catalysts (
Figure 10b), which, in terms of both T
50 and T
90 and specific and intrinsic rates, followed the order: CuCeMgAlO < Co(1)CuCeMgAlO < Co(9)CuCeMgAlO < Co(6)CuCeMgAlO < Co(3)CuCeMgAlO. In other words, the catalytic activity passed through a maximum, corresponding to the Co(3)CuCeMgAlO system with increasing the Co content from 1 to 9 at.%. This evolution could be explained taking into consideration an interplay between the Cu-Co-Ce synergistic interaction and the segregation of CuO tenorite phase, the latter going increasingly with increasing the Co content, as evidenced by the X-ray diffraction (
Figure 2b) and SEM analyses (
Figure S3). Indeed, for Co contents lower than 3 at.%, the amount of promoter involved in the synergistic interaction with Cu and Ce was too low, the copper itself being partially not accessible, as shown in the H
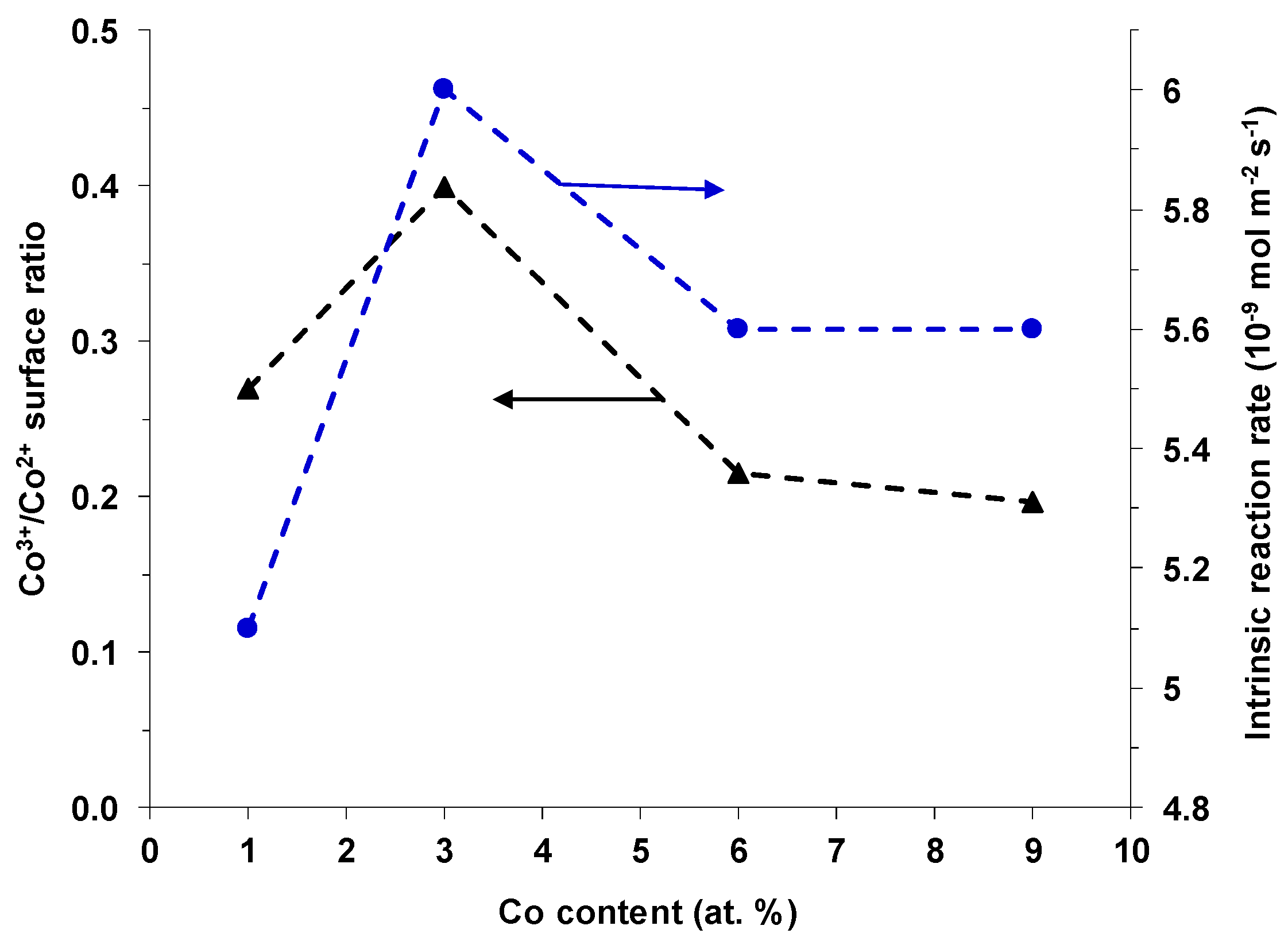
2-TPR experiments, and, hence, the density of the most active Cu-Co-Ce catalytic sites was lower. For Co contents higher than 3 at.%, the strong segregation of CuO phase took place, resulting again in a decrease of the amount of the most active Cu-Co-Ce catalytic sites compared to the Co(3)CuCeMgAlO system, which was the most active catalyst in this series. Moreover, it was observed that both the rate of methane transformation and the Co
3+/Co
2+ surface atomic ratio roughly followed the same trend when plotted as a function of the Co content
x in the Co(x)CuCeMgAlO catalysts (
Figure 11). This suggests that the Co
3+/Co
2+ surface atomic ratio is another factor determining the activity of these catalysts in methane combustion, in agreement with the literature [
8].
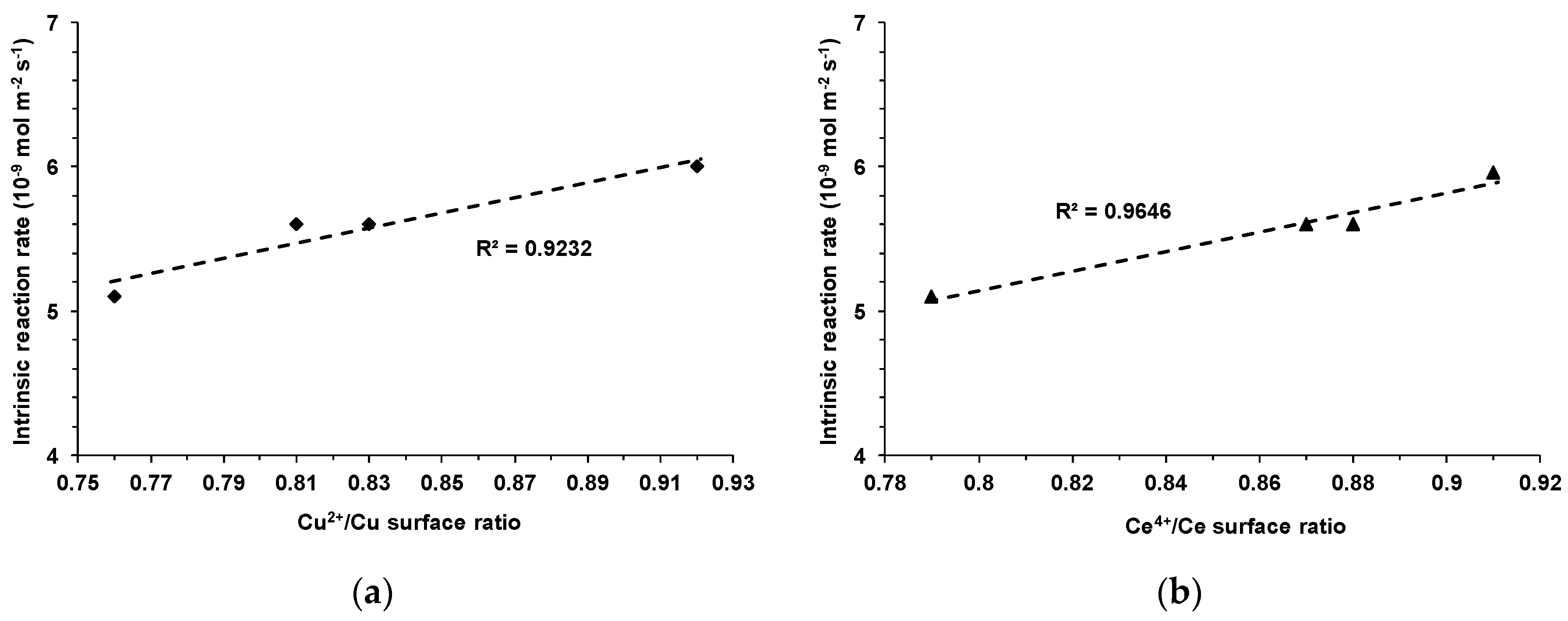
Interestingly, pretty good linear correlations were observed between both Cu
2+/Cu and Ce
4+/Ce surface atomic ratios and the rate of methane transformation over Co(x)CuCeMgAlO catalysts (
Figure 12), clearly suggesting that Cu
2+ and Ce
4+ surface species were also involved in the catalytic process. Notably, all these correlations showing the increase of the reaction rate with increasing Co
3+, Cu
2+, and Ce
4+ surface concentrations unambiguously demonstrated that the synergistic interaction between these species was a key factor controlling the activity of the Co(x)CuCeMgAlO catalysts in the complete oxidation of methane. The absence of these correlations for the M(3)CuCeMgAlO series does not necessarily minimize the role of the M-Cu-Ce synergistic interaction, but rather shows that the nature of the cation M influenced the physicochemical characteristics and, hence, the catalytic properties of these materials in a complex manner. Thus, for example, while the surface concentration of Fe is by ca. 30% lower compared with its bulk concentration, the surface concentrations of Mn, Co, and Ni are by ca. 73, 74, and 178%, respectively, higher. Also, for Co(3)- and Ni(3)CuCeMgAlO, the surface of the catalysts is enriched in Cu compared to the bulk composition, while for Mn(3)- and Fe(3)CuCeMgAlO it is poorer. Moreover, for Fe(3)-, Co(3)-, and Ni(3)CuCeMgAlO, the Cu/Ce surface atomic ratio is significantly higher than the bulk ratio, while for Mn(3)CuCeMgAlO it is lower. All these differences, which were not observed in the Co(x)CuCeMgAlO series, could have been at the origin of the different behavior of the M(3)CuCeMgAlO series.
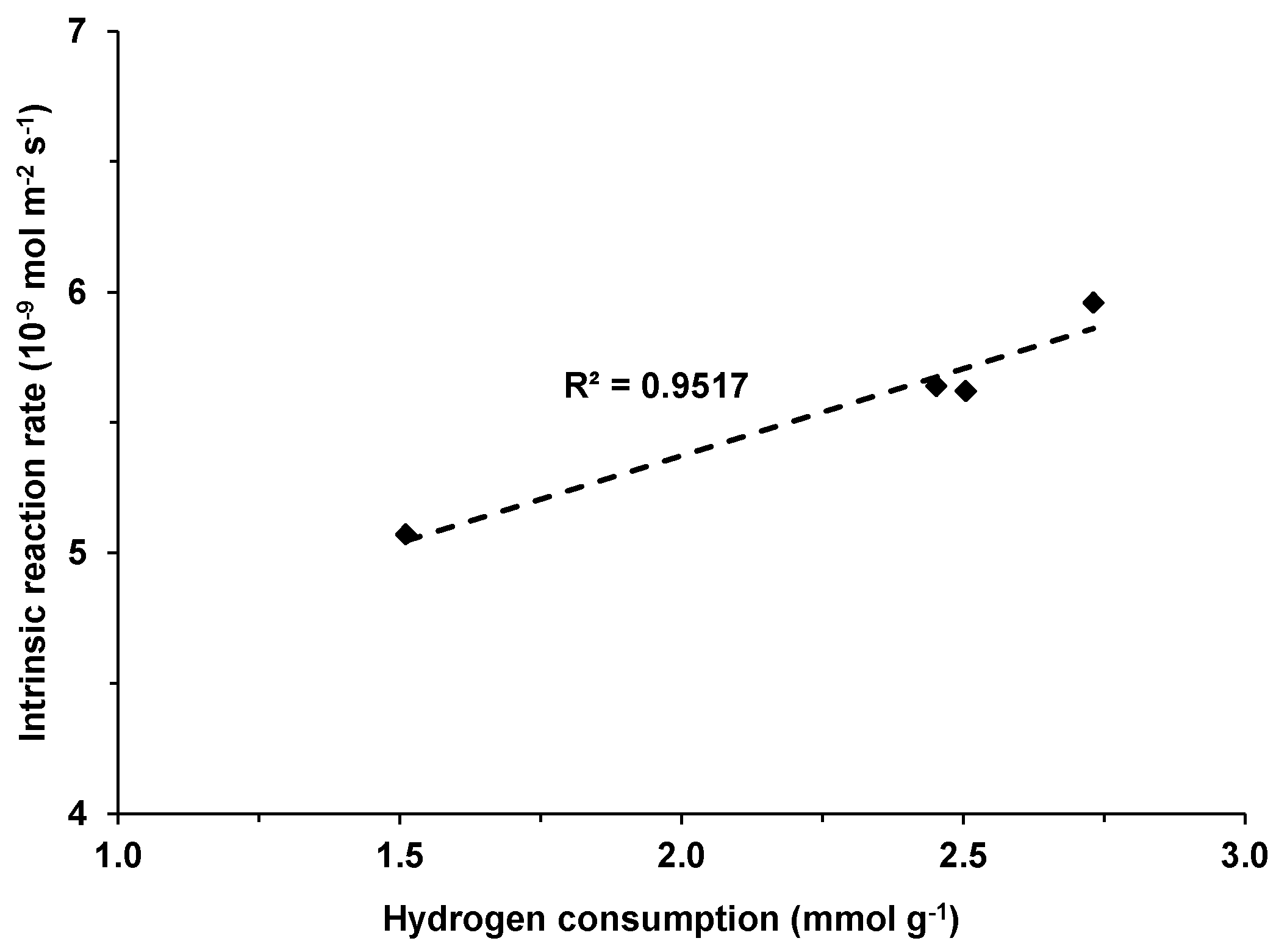
No correlation could be observed between the catalytic performance and the reducibility of the mixed oxide in the M(3)CuCeMgAlO series. However, for the Co(x)CuCeMgAlO series, a good linear correlation between the rate of methane conversion and the hydrogen consumption in the H
2-TPR experiments was noticed (
Figure 13). On one hand, this suggested that all the reducible species evidenced in the H
2-TPR experiments were involved in the catalytic oxidation of methane over the Co-promoted CuCeMgAlO catalysts, in line with a Cu-Co-Ce synergistic interaction, and, on the other hand, confirmed the heterogeneous redox mechanism for methane combustion over these catalytic materials. Notably, the correlation reaction rate – reducibility was previously reported for several mixed oxides catalysts obtained from LDH precursors containing Cu [
12,
14,
50] and Ce [
33] for the complete oxidation of methane.
The apparent activation energies (E
a) for the transformation of methane on the transition metal-promoted CuCeMgAlO catalysts were calculated from the slope of the low-conversion linear part of the ln
r vs. 10
3/
T Arrhenius plots (
Figure S10) and are presented in
Table 7. It can be observed that the activation energy values for the M(3)CuCeMgAlO catalysts were slightly higher than that corresponding to M-free CuCeMgAlO. This difference can be attributed to the segregation of CuO in the former. However, their enhanced catalytic activity was obviously due to a greater density of highly active sites consisting of CuO synergistically interacting with both M and Ce cations, the different nature of the cation M accounting for the differences observed in the activation energies of the M(3)CuCeMgAlO series. Although the lowest activation energy in this series corresponded to the most active Co(3)CuCeMgAlO catalyst, it did not parallel the catalytic activity likely due to different densities of surface active sites. Regarding the activation energy for the Co(x)CuCeMgAlO series, it was slightly lower for Co(1)CuCeMgAlO system compared to CuCeMgAlO, in line with their relative activities, while it was higher for the other Co-containing systems and varied within a narrow range irrespective of the Co content. Also, it did not parallel the catalytic activity, as different densities of active sites may exist on the surface of Co(x)CuCeMgAlO mixed oxides.
Although similar LDH-derived Cu- and/or Ce-containing mixed oxides [
12,
21,
33,
50] catalysts were shown to display good stabilities during the total oxidation of methane, the stability on stream of the most active Co(3)CuCeMgAlO catalyst was tested at 520 °C for 60 h. No change of the catalytic performance could be observed (
Figure S11), suggesting that Co(3)CuCeMgAlO system is stable on stream, at least for the reaction conditions and the reaction time chosen.


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}