Exploring the Mechanism of Catalysis with the Unified Reaction Valley Approach (URVA)—A Review




Abstract
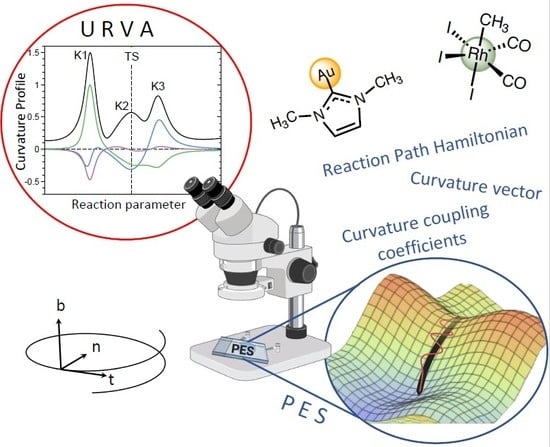
:
1. Introduction
2. The Unified Reaction Valley Approach (URVA)
2.1. Background: The Reaction Path Hamiltonian
2.2. Basic Methodology of URVA
3. Computational Methods
4. Results
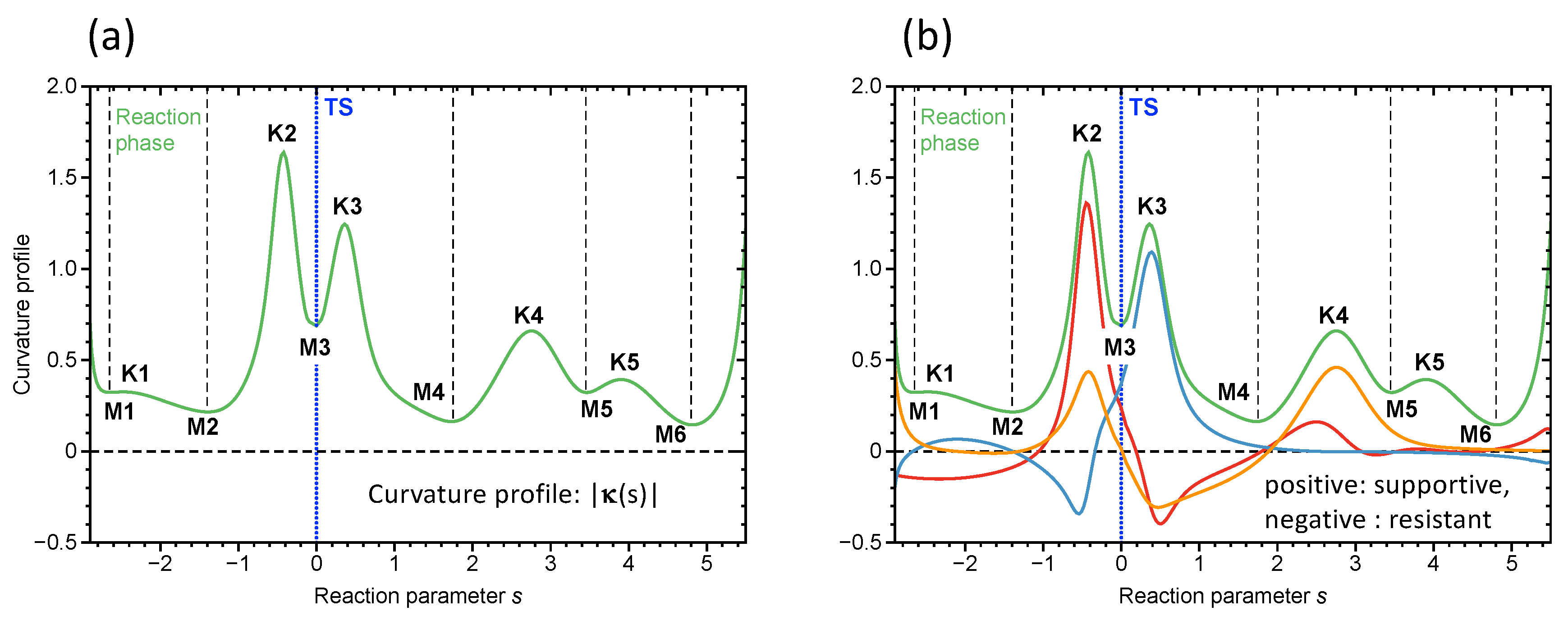
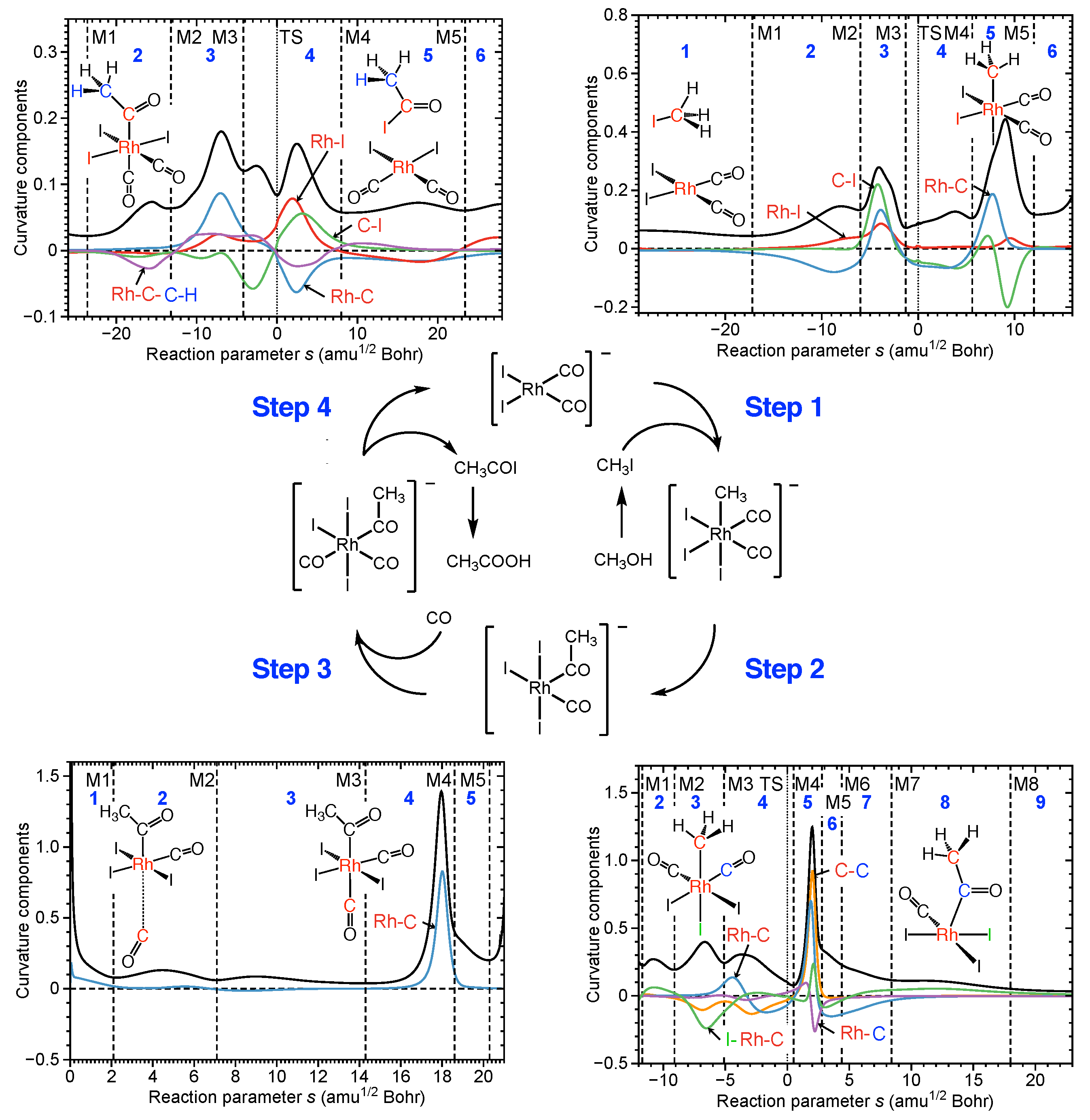
4.1. Rh Catalyzed Methanol Carbonylation—The Monsanto Process
4.2. Sharpless Epoxidation of Allylic Alcohols—Transition to Heterogenous Catalysis
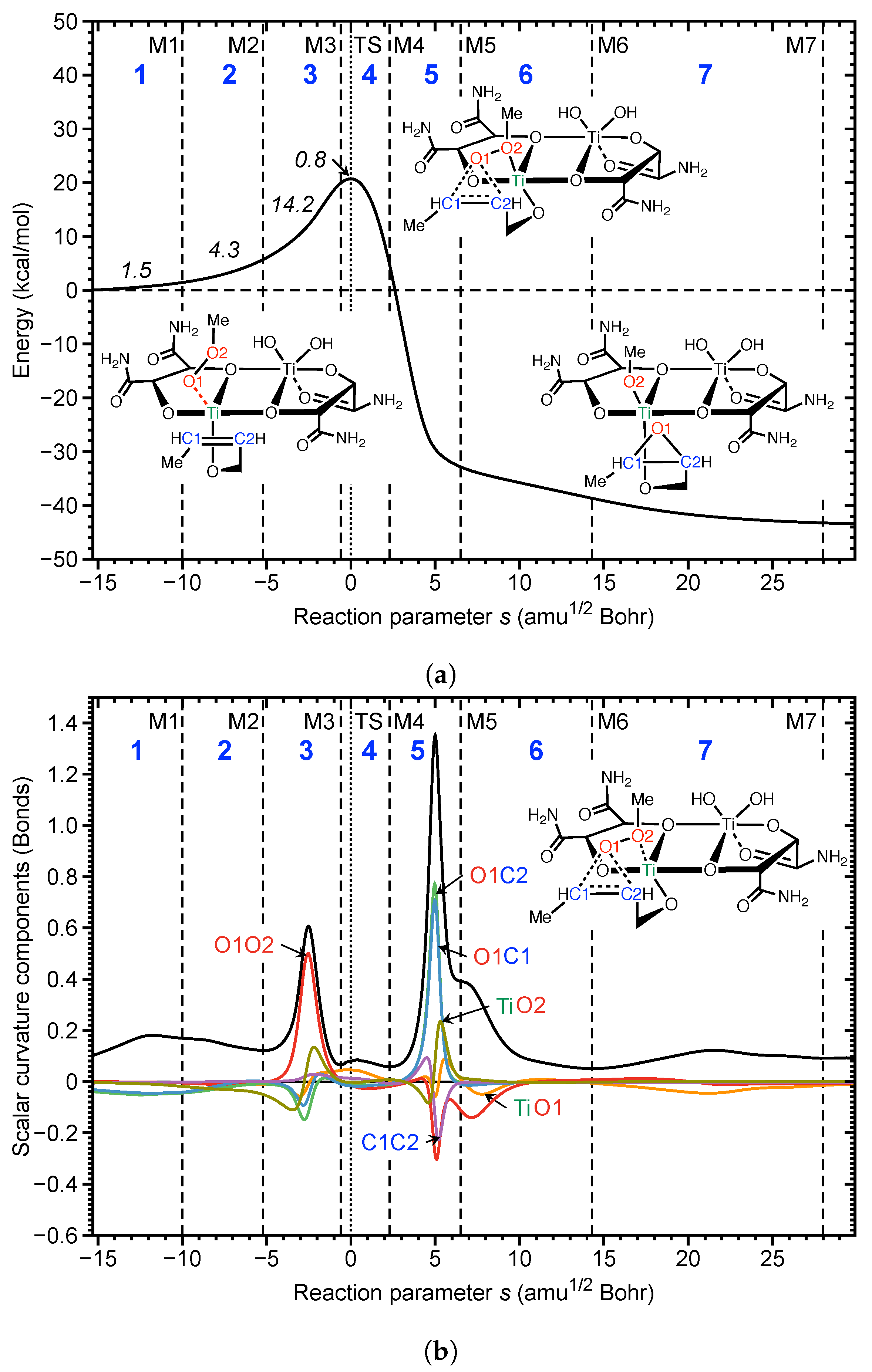
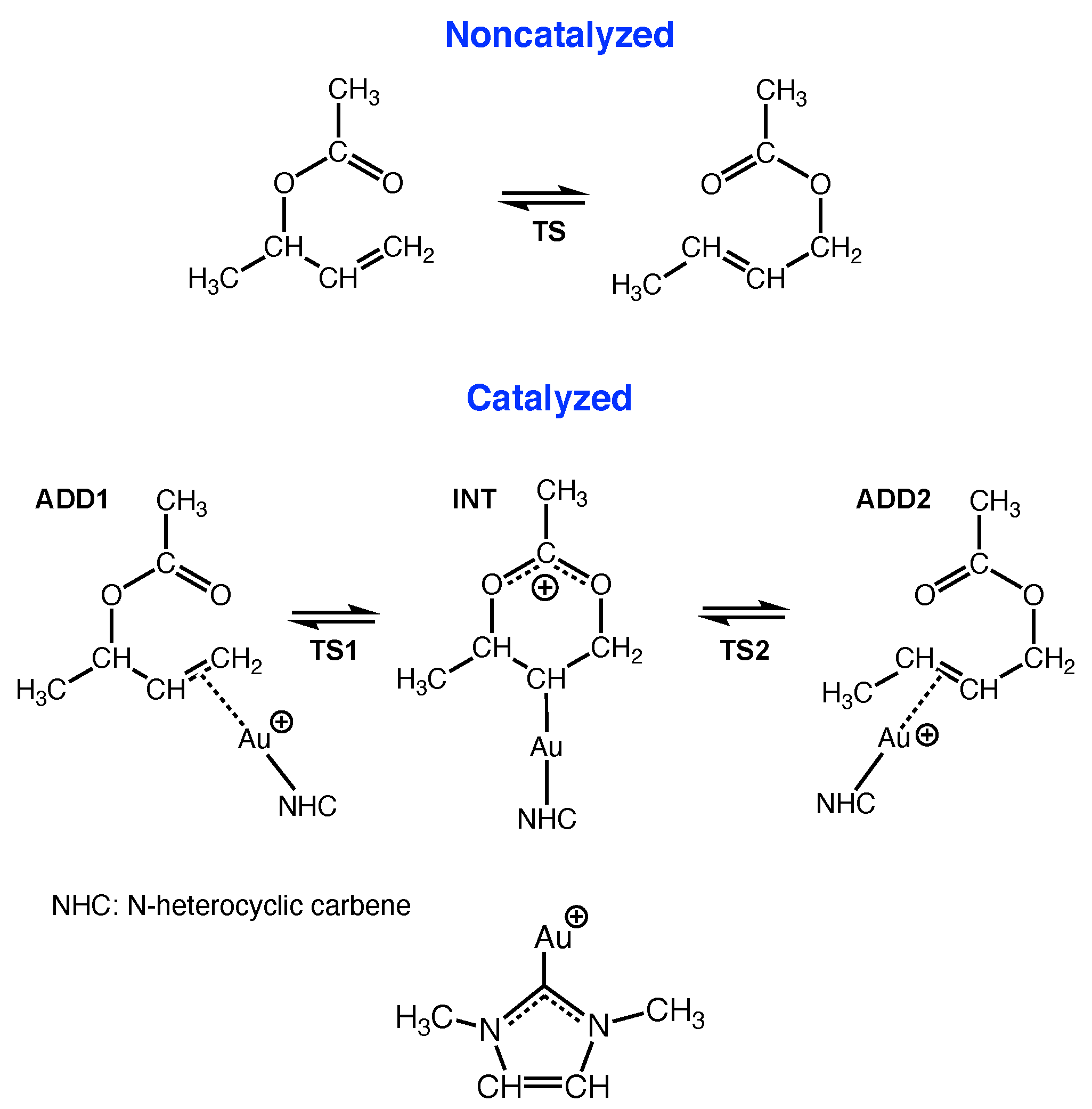
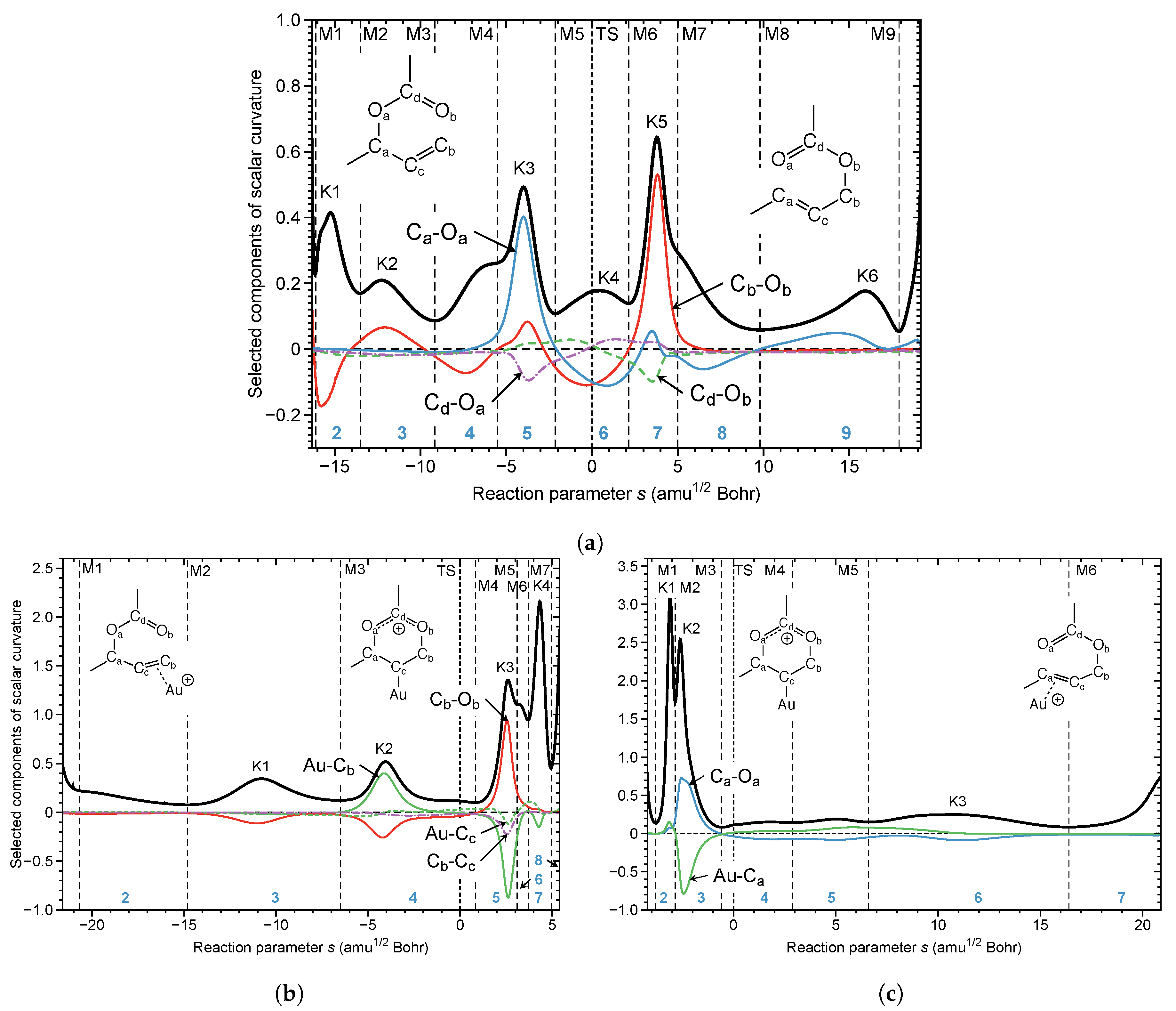
4.3. Au(I) Assisted [3,3]-Sigmatropic Rearrangement of Allyl Acetate
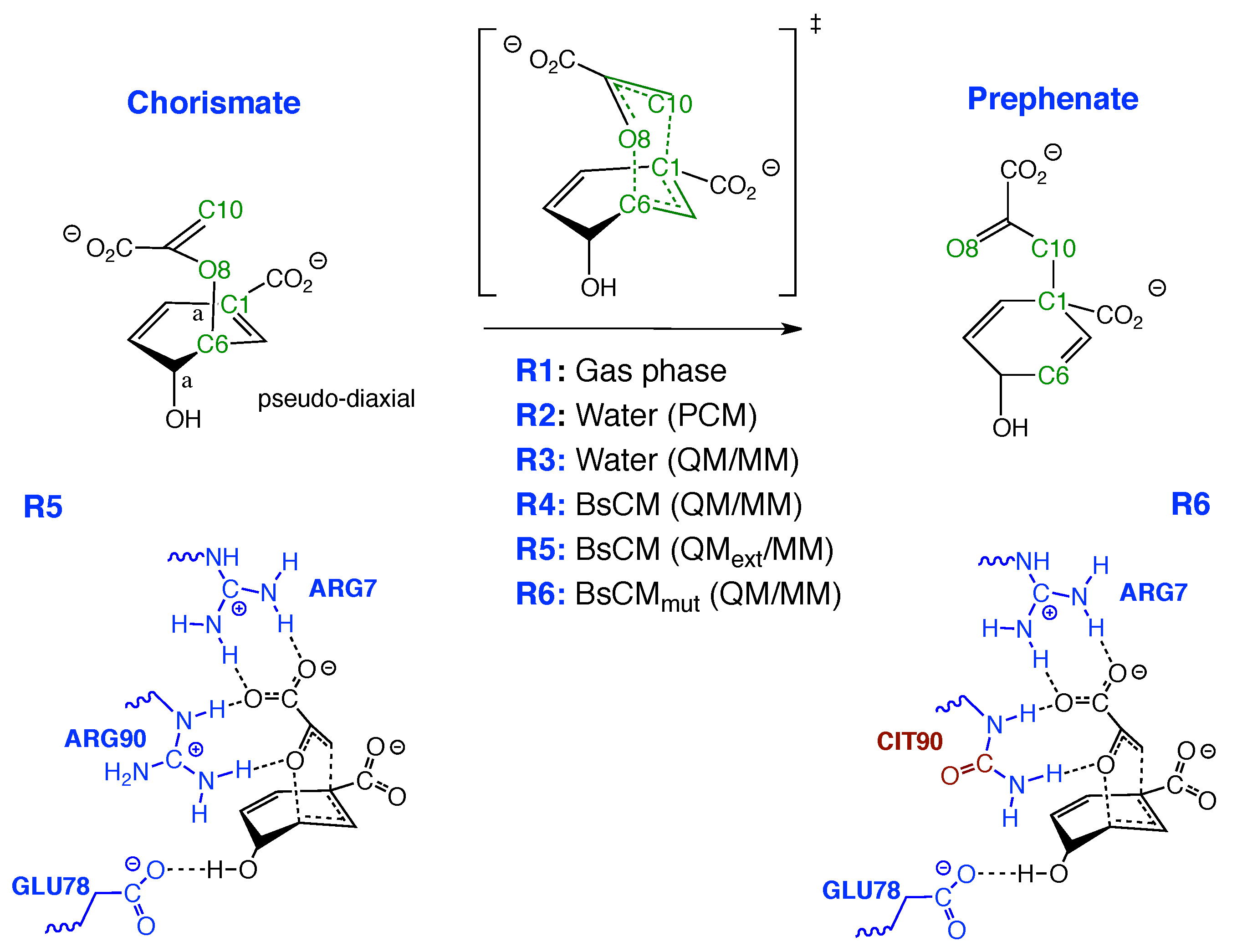
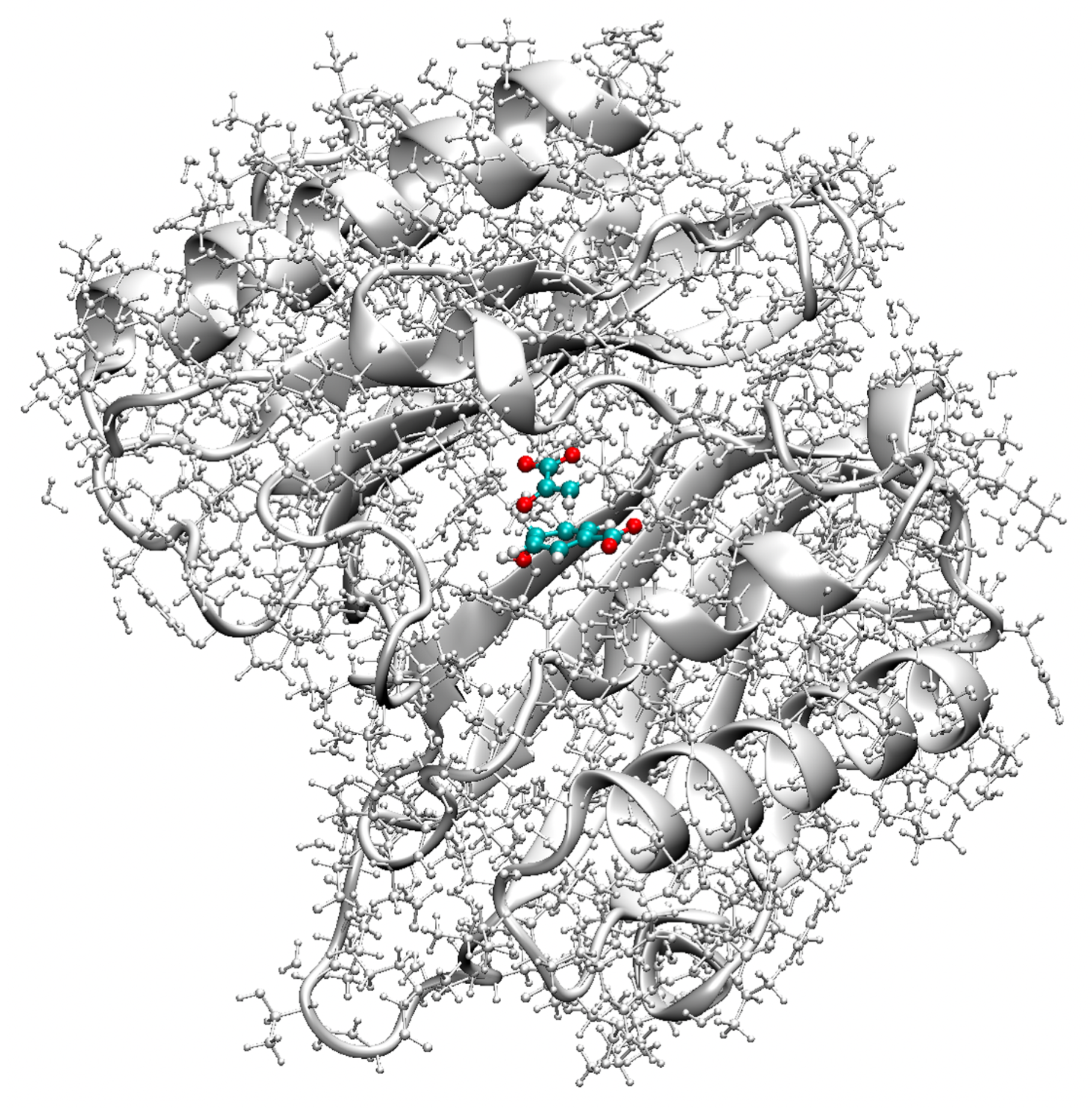
4.4. Bacillus Subtilis Chorismate Mutase Catalyzed Claisen Rearrangement
5. Conclusions
- The Rh catalyzed methanol carbonylation is an example for a coordination-sphere-driven catalysis catalyzing a chemical reaction by changing the coordination sphere of the transition metal to facilitate bond forming/bond breaking processes. The Rh coordination number changes from 4 to 6 in step 1, from 6 to 5 in step 2 from 5 to 6 in step 3 and back to 4 in step 4. Step 1 with an activation energy of 45.0 kcal/mol is the cause of the harsh reaction conditions. URVA identifies the approach of the reactants and the cleavage of the C–I bond as the most energy demanding processes of this step. These findings provide valuable information for catalyst modification aiming at milder reaction conditions. The long approach phase can be shortened by chelating the reactant and C–I bond breakage can be supported via polarization of the C–I bond, which will lower the overall activation energy. Work is in progress along these lines.
- In the Sharpless epoxidation of allylic alcohols the dimeric Ti catalyst mimics a surface typical of heterogenous catalysis, thus facilitating a stereospecific collision of the reaction partners, where the O–O bond of the oxidizing peroxide glides over the Ti atom. During this process the metal atom polarizes the peroxide oxygen atoms facilitating O–O bond breakage. Another important feature of the Sharpless reaction revealed by URVA is that both new C–O epoxide bonds are synchronously finalized after the TS, i.e., without energy consumption. Therefore, catalyst optimization should predominantly focus on further support of O–O breakage.
- The URVA analysis of Au(I) assisted [3,3]-sigmatropic rearrangement of allyl acetate shows how the Au(I) catalyst breaks up the non-catalyzed rearrangement into two energy saving steps by switching between Au(I)- and Au(I)- complexation. The unfavorably high activation energy of non-catalyzed reaction is caused the fact that the migrating CO bond is broken before the TS. In contrast, the -acidic cationic Au(I) catalyst forms a Au(I)--complex via the ethylene unit in the first step, supports the formation of the new CO bond while conserving the CO bond to be broken, and transforms at the end of this step into an intermediate Au(I)--complex resembling the TS of the non-catalyzed reaction. In the second step, the Au(I)--complex transforms back into a more stable Au(I)--complex including the energy conserving breakage of the migrating CO bond.
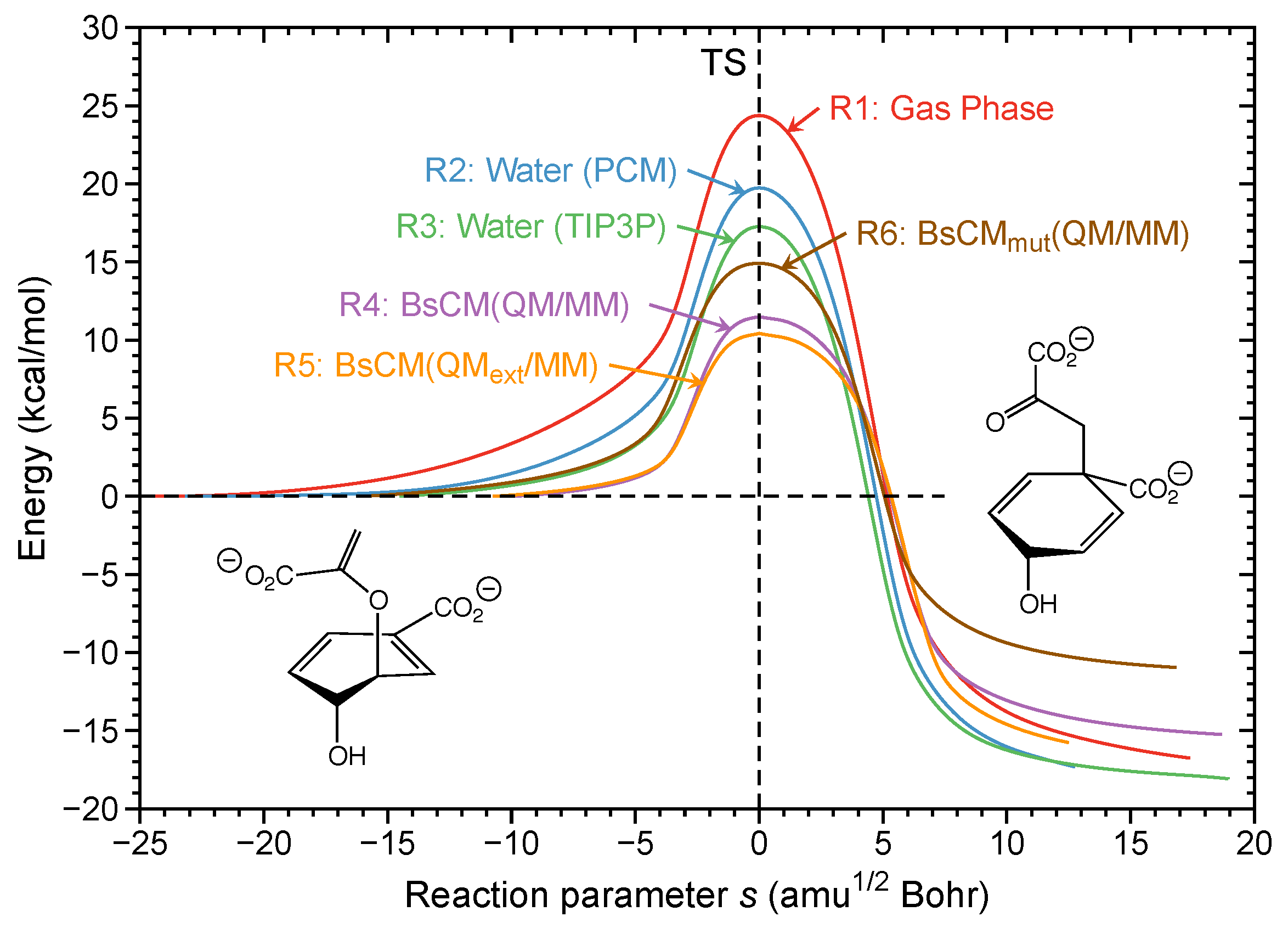
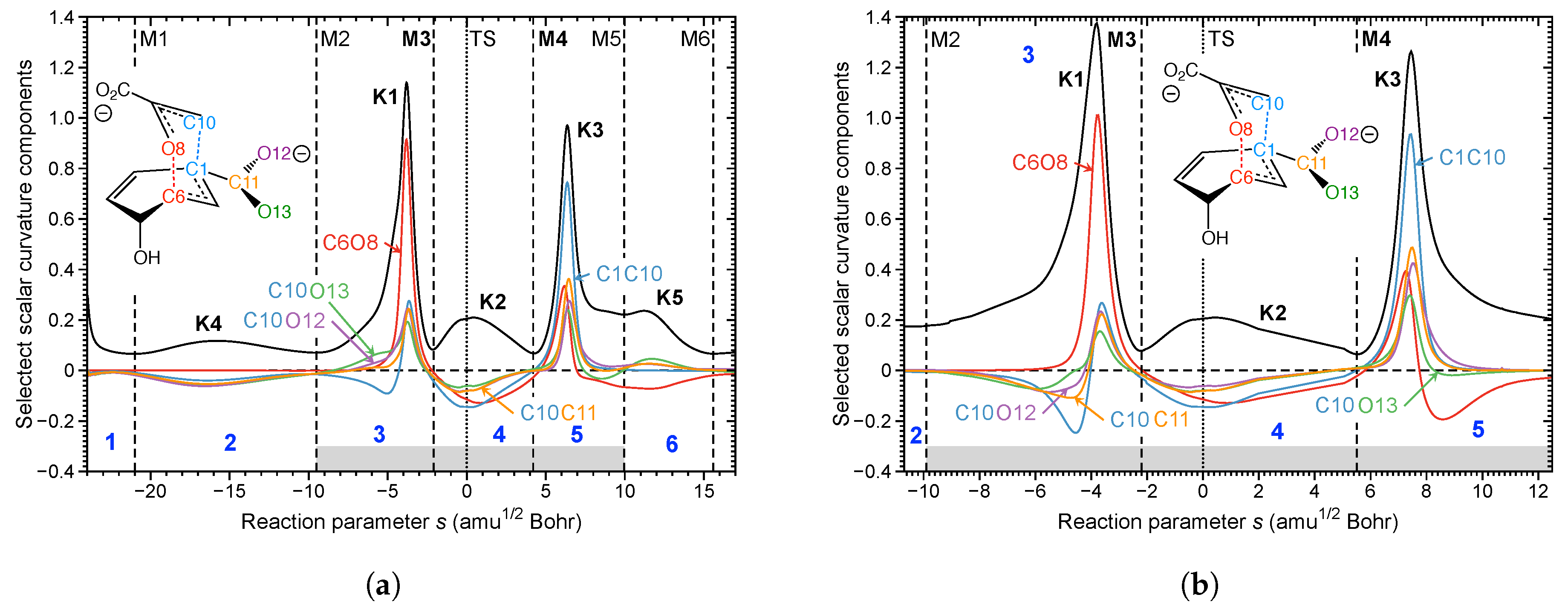
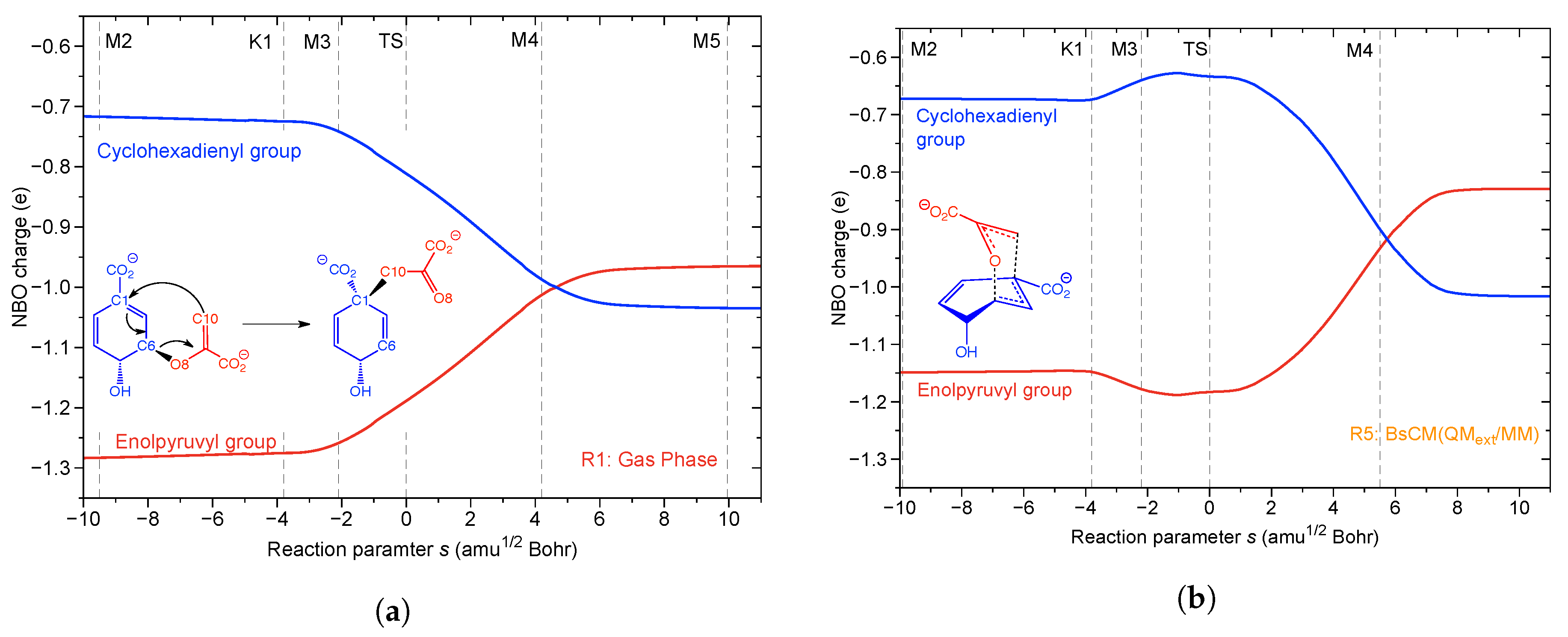
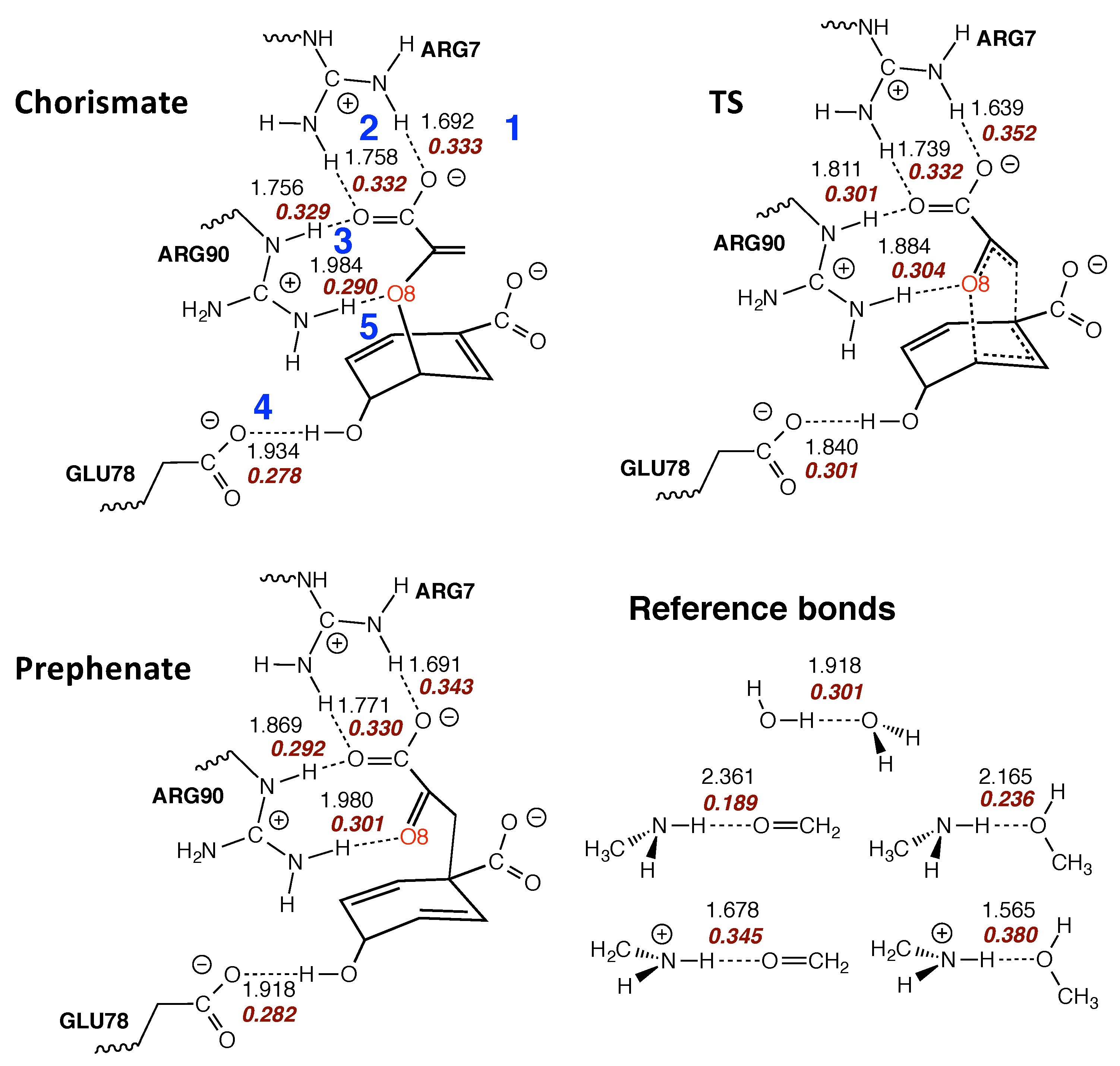
- The Bacillus subtilis chorismate mutase catalyzed Claisen rearrangement is an example for space-confinement-driven catalysis, perfectly designed by nature. URVA indisputably proves that the actual mechanism of the chorismate rearrangement is the same in the gas phase, solution and in the enzyme. The process of CO bond cleavage starts before the TS and the new CC bond formation is finalized after the TS. There are subtitle differences in the pre-chemical phases, which are a result of the different environments. The pre-chemical phases become shorter in aqueous solution and disappear for the reaction in the enzyme, where the chemical process of CO bond cleavage starts directly in the entrance channel. The local mode analysis reveals that the inter-molecular H-bond network between chorismate and BsCM does not change during the whole rearrangement, which eliminates suggestions that the enzyme lowers the barrier by stabilizing the TS through specific H-bonding.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| URVA | unified reaction valley approach |
| pURVA | standalone program of the unified reaction valley approach, written in python |
| RC | reaction complex |
| RP | reaction path |
| TS | transition state |
| RPH | reaction path Hamiltonian |
| PES | potential energy surface |
| IRC | intrinsic reaction coordinate |
| DFT | density functional theory |
| B3LYP | Becke 3–parameter Lee–Yang–Parr functional |
| SDD | Stuttgart–Dresden effective core potential |
| DLPNO–CCSD(T) | domain based local pair natural orbital coupled cluster |
| PCM | polarizable continuum solvent model |
| TIP3P | three–site transferrable intermolecular potential |
| QM/MM | quantum mechanics and molecular mechanics |
| ONIOM | Own N–layer integrated molecular orbital and molecular mechanics |
| ARG | arginine |
| GLU | glutamic acid |
| H-bond | hydrogen bond |
| NHC | N–heterocyclic carbene |
| BsCM | Bacillus subtilis chorismate mutase |
| NBO | natural bond orbital |
| BSO | bond strength order |
Appendix A. pURVA

References
- Beckman, J. The Publication Strategies of Jöns Jacob Berzelius (1779–1848): Negotiating National and Linguistic Boundaries in Chemistry. Ann. Sci. 2016, 73, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Wisniak, J. The History of Catalysis. From the Beginning to Nobel Prizes. Educ. Quim. 2010, 21, 60–69. [Google Scholar] [CrossRef]
- Paul, C.J.K. (Ed.) Contemporary Catalysis: Science, Technology, and Applications; Royal Society of Chemistry: London, UK, 2017. [Google Scholar]
- Ruiz, J.C.S. (Ed.) Applied Industrial Catalysis; Arcler Press LLC: New York, NY, USA, 2017. [Google Scholar]
- Ludwig, J.R.; Schindler, C.S. Catalyst: Sustainable Catalysis. Chem 2017, 2, 313–316. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.L. Transition-Metal Catalysis and Organocatalysis: Where Can Progress Be Expected? Angew. Chem. Int. Ed. 2016, 55, 5352–5353. [Google Scholar] [CrossRef] [Green Version]
- Chelucci, G.; Baldino, S.; Baratta, W. Recent Advances in Osmium–Catalyzed Hydrogenation and Dehydrogenation Reactions. Acc. Chem. Res. 2015, 48, 363–379. [Google Scholar] [CrossRef]
- Do, J.L.; Mottillo, C.; Tan, D.; Štrukil, V.; Friščić, T. Mechanochemical Ruthenium–Catalyzed Olefin Metathesis. J. Am. Chem. Soc. 2015, 137, 2476–2479. [Google Scholar] [CrossRef]
- Biffis, A.; Centomo, P.; Del Zotto, A.; Zecca, M. Pd Metal Catalysts for Cross–Couplings and Related Reactions in the 21st Century: A Critical Review. Chem. Rev. 2018, 118, 2249–2295. [Google Scholar] [CrossRef]
- Jones, J.W. Another Nobel Prize for Catalysis: Frances Arnold in 2018. ACS Catal. 2018, 8, 10913. [Google Scholar] [CrossRef] [Green Version]
- Grand View Research, Inc. Catalyst Market Size; Grand View Research, Inc.: San Francisco, CA, USA, 2019. [Google Scholar]
- Olveira, S.; Forster, S.P.; Seeger, S. Nanocatalysis: Academic Discipline and Industrial Realities. J. Nanotech. 2014, 2014, 324089. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.; Zhou, X.; Andoy, N.M.; Han, K.S.; Choudhary, E.; Zou, N.; Chen, G.; Shen, H. Spatiotemporal Catalytic Dynamics within Single Nanocatalysts Revealed by Single–Molecule Microscopy. Chem. Soc. Rev. 2014, 43, 1107–1117. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Chen, T.; Song, P.; Zhang, Y.; Xu, W. Single–Molecule Nanocatalysis of Pt Nanoparticles. J. Phys. Chem. C 2018, 122, 1746–1752. [Google Scholar] [CrossRef]
- Tripathi, P.; Sinha, S. Industrial Biocatalysis: An Insight into Trends and Future Directions. Curr. Sustain. Renew. Energy Rep. 2020, 64, 1–7. [Google Scholar] [CrossRef]
- Sandoval, B.A.; Hyster, T.K. Emerging Strategies for Expanding the Toolbox of Enzymes in Biocatalysis. Curr. Opin. Chem. Biol. 2020, 55, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.P.; Brown, M.J.B.; Diaz-Rodriguez, A.; Lloyd, R.C.; Roiban, G.D. Biocatalysis: A Pharma Perspective. Adv. Synth. Catal. 2019, 361, 2421–2432. [Google Scholar] [CrossRef] [Green Version]
- Ahn, S.; Hong, M.; Sundararajan, M.; Ess, D.H.; Baik, M.H. Design and Optimization of Catalysts Based on Mechanistic Insights Derived from Quantum Chemical Reaction Modeling. Chem. Rev. 2019, 119, 6509–6560. [Google Scholar] [CrossRef]
- Durand, D.J.; Fey, N. Computational Ligand Descriptors for Catalyst Design. Chem. Rev. 2019, 119, 6561–6594. [Google Scholar] [CrossRef] [Green Version]
- Vogiatzis, K.D.; Polynski, M.V.; Kirkland, J.K.; Townsend, J.; Hashemi, A.; Liu, C.; Pidko, E.A. Computational Approach to Molecular Catalysis by 3d Transition Metals: Challenges and Opportunities. Chem. Rev. 2019, 119, 2453–2523. [Google Scholar] [CrossRef] [Green Version]
- Quesne, M.G.; Silveri, F.; de Leeuw, N.H.; Catlow, C.R.A. Advances in Sustainable Catalysis: A Computational Perspective. Front. Chem. 2019, 7, 182–205. [Google Scholar] [CrossRef] [Green Version]
- Harvey, J.N. Mechanism and Kinetics in Homogeneous Catalysis: A Computational Viewpoint. In Transition Metals in Coordination Environments. Challenges and Advances in Computational Chemistry and Physics; Broclawik, E., Borowski, T., Radoń, M., Eds.; Springer: New York, NY, USA, 2019; Volume 29, pp. 289–314. [Google Scholar]
- Grajciar, L.; Heard, C.J.; Bondarenko, A.A.; Polynski, M.V.; Nachtigall, P. Towards Operando Computational Modeling in Heterogeneous Catalysis. Chem. Soc. Rev. 2018, 47, 8307–8348. [Google Scholar] [CrossRef] [Green Version]
- Lam, Y.; Grayson, M.N.; Holland, M.C.; Simon, A.; Houk, K.N. Theory and Modeling of Asymmetric Catalytic Reactions. Acc. Chem. Res. 2016, 49, 750–762. [Google Scholar] [CrossRef]
- Foscato, M.; Jensen, V.R. Automated in Silico Design of Homogeneous Catalysts. ACS Catal. 2020, 10, 2354–2377. [Google Scholar] [CrossRef]
- Toyao, T.; Maeno, Z.; Takakusagi, S.; Kamachi, T.; Takigawa, I.; Shimizu, K. Machine Learning for Catalysis Informatics: Recent Applications and Prospects. ACS Catal. 2020, 10, 2260–2297. [Google Scholar] [CrossRef]
- Espinosa-Garcia, J.; Monge-Palacios, M.; Corchado, J.C. Constructing Potential Energy Surfaces for Polyatomic Systems: Recent Progress and New Problems. Adv. Phys. Chem. 2012, 2012, 164752. [Google Scholar] [CrossRef] [Green Version]
- Rybkin, V.V. Sampling Potential Energy Surfaces in the Condensed Phase with Many—Body Electronic Structure Methods. Chemistry 2020, 26, 362–368. [Google Scholar] [CrossRef]
- Quintas-Sánchez, E.; Dawes, R. AUTOSURF: A Freely Available Program To Construct Potential Energy Surfaces. J. Chem. Inf. Model. 2019, 59, 262–271. [Google Scholar] [CrossRef]
- Dewyer, A.L.; Argüelles, A.J.; Zimmerman, P.M. Methods for Exploring Reaction Space in Molecular Systems. WIREs 2018, 8, e1354. [Google Scholar] [CrossRef]
- Unke, O.T.; Koner, D.; Patra, S.; Käser, S.; Meuwly, M. High–dimensional Potential Energy Surfaces for Molecular Simulations: From Empiricism to Machine Learning. Mach. Learn. Sci. Technol. 2020, 1, 013001. [Google Scholar] [CrossRef]
- Noé, F.; Tkatchenko, A.; Müller, K.R.; Clementi, C. Machine Learning for Molecular Simulation. Ann. Rev. Phys. Chem. 2020, 71, 361–390. [Google Scholar] [CrossRef] [Green Version]
- Abbott, A.S.; Turney, J.M.; Zhang, B.; Smith, D.A.; Altarawy, D.; Schaefer, H.F. PES-Learn: An Open-Source Software Package for the Automated Generation of Machine Learning Models of Molecular Potential Energy Surfaces. J. Chem. Theory Comput. 2019, 15, 4386–4398. [Google Scholar] [CrossRef]
- Quapp, W.; Hirsch, M.; Heidrich, D. Following the Streambed Reaction on Potential–energy Surfaces: A New Robust Method. Theor. Chem. Acc. 2000, 105, 145–155. [Google Scholar] [CrossRef]
- Bofill, J.M.; Anglada, J.M. Finding Transition States using Reduced Potential-energy Surfaces. Theor. Chem. Acc. 2001, 105, 463–472. [Google Scholar] [CrossRef]
- Schlegel, H.B. Exploring potential energy surfaces for chemical reactions: An overview of some practical methods. J. Comput. Chem. 2003, 24, 1514–1527. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, M.; Quapp, W. The reaction pathway of a potential energy surface as curve with induced tangent. Chem. Phys. Lett. 2004, 395, 150–156. [Google Scholar] [CrossRef]
- Aguilar-Mogas, A.; Giménez, X.; Bofill, J.M. Finding reaction paths using the potential energy as reaction coordinate. J. Chem. Phys. 2008, 128, 104102. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kim, J.W.; Kim, Z.; Kim, W.Y. Efficient prediction of reaction paths through molecular graph and reaction network analysis. Chem. Sci. 2018, 9, 825–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, S.; Harabuchi, Y.; Takagi, M.; Saita, K.; Suzuki, K.; Ichino, T.; Sumiya, Y.; Sugiyama, K.; Ono, Y. Implementation and Performance of the Artificial Force Induced Reaction Method in the GRRM17 Program. J. Comput. Chem. 2018, 39, 233–250. [Google Scholar] [CrossRef] [Green Version]
- Maeda, S.; Harabuchi, Y.; Ono, Y.; Taketsugu, T.; Morokuma, K. Intrinsic reaction coordinate: Calculation, bifurcation, and automated search. Int. J. Quant. Chem. 2015, 115, 258–269. [Google Scholar] [CrossRef]
- Tsutsumi, T.; Ono, Y.; Arai, Z.; Taketsugu, T. Visualization of the Intrinsic Reaction Coordinate and Global Reaction Route Map by Classical Multidimensional Scaling. J. Chem. Theory Comput. 2018, 14, 4263–4270. [Google Scholar] [CrossRef]
- Quapp, W.; Bofill, J.M. Some Mathematical Reasoning on the Artifical Force Induced Reaction Method. J. Comput. Chem. 2020, 41, 629–634. [Google Scholar] [CrossRef] [Green Version]
- Hare, S.R.; Bratholm, L.A.; Glowacki, D.R.; Carpenter, B.K. Low dimensional representations along intrinsic reaction coordinates and molecular dynamics trajectories using interatomic distance matrices. Chem. Sci. 2019, 10, 9954–9968. [Google Scholar] [CrossRef]
- Geerlings, P.; Chamorro, E.; Chattaraj, P.; De Proft, F.; Gázquez, J.; Liu, S.; Morell, C.; Toro-Labbé, A.; Vela, A.; Ayers, P. Conceptual density functional theory: Status, prospects, issues. Theoret. Chem. Acc. 2020, 139, 36-1–36-18. [Google Scholar] [CrossRef]
- Stuyver, T.; Proft, F.D.; Geerlings, P.; Shaik, S. How Do Local Reactivity Descriptors Shape the Potential Energy Surface Associated with Chemical Reactions? The Valence Bond Delocalization Perspective. J. Am. Chem. Soc. 2020, 142, 10102–10113. [Google Scholar] [CrossRef]
- Toro-Labbé, A.; Gutieŕrez-Oliva, S.; Murray, J.; Politzer, P. A new perspective on chemical and physical processes: The reaction force. Mol. Phys. 2007, 105, 2619–2625. [Google Scholar] [CrossRef]
- Urcelay, F.; Toro-Labbé, A.; Gutieŕrez-Oliva, S. Spectral Decomposition of the Reaction Force Constant. J. Phys. Chem. A 2020, 124, 2372–2379. [Google Scholar] [CrossRef]
- Rincon, L.; Torres, F.J.; Mora, J.R.; Zambrano, C.H.; Rodriguez, V. A valence bond perspective of the reaction force formalism. Theor. Chem. Acc. 2020, 139, 13. [Google Scholar] [CrossRef]
- Bader, R. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1994. [Google Scholar]
- Bader, R. Atoms in Molecules. Chem. Rev. 1998, 1, 64–86. [Google Scholar]
- Savin, A.; Nesper, R.; Wengert, S.; Fässler, T.F. ELF: The Electron Localization Function. Angew. Chem. Int. Ed. 1997, 36, 1808–1832. [Google Scholar] [CrossRef]
- Andrés, J.; Gracia, L.; Gonzalez-Navarrete, P.; Safont, V. Chemical structure and reactivity by means of quantum chemical topology analysis. Comput. Theor. Chem. 2015, 1053, 17–30. [Google Scholar] [CrossRef] [Green Version]
- Andrés, J.; Gonzalez-Navarrete, P.; Safont, V.S. Unraveling Reaction Mechanisms by Means of Quantum Chemical Topology Analysis. Int. J. Quantum. Chem. 2014, 114, 1239–1252. [Google Scholar] [CrossRef] [Green Version]
- Polo, V.; Andres, J.; Berski, S.; Domingo, L.R.; Silvi, B. Understanding Reaction Mechanisms in Organic Chemistry from Catastrophe Theory Applied to the Electron Localization Function Topology. J. Phys. Chem. A 2008, 112, 7128–7136. [Google Scholar] [CrossRef]
- Martino, M.; Salvadori, A.; Lazzari, F.; Paoloni, L.; Nandi, S.; Mancini, G.; Barone, V.; Rampino, S. Chemical promenades: Exploring potential-energy surfaces with immersive virtual reality. J. Comput. Chem. 2020, 41, 1310–1323. [Google Scholar] [CrossRef]
- Yang, Z.; Houk, K.N. The Dynamics of Chemical Reactions: Atomistic Visualizations of Organic Reactions, and Homage to vant Hoff. Chemistry 2018, 24, 3916–3924. [Google Scholar] [CrossRef] [PubMed]
- Black, K.; Liu, P.; Xu, L.; Doubleday, C.; Houk, K.N. Dynamics, transition states, and timing of bond formation in Diels–Alder reactions. Proc. Natl. Acad. Sci. USA 2012, 109, 12860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meuwly, M. Reactive molecular dynamics: From small molecules to proteins. WIREs Comput. Mol. Sci. 2019, 9, e1386. [Google Scholar] [CrossRef] [Green Version]
- Gissinger, J.R.; Jensen, B.D.; Wise, K.E. Modeling chemical reactions in classical molecular dynamics simulations. Polymer 2017, 128, 211–217. [Google Scholar] [CrossRef]
- Pratihar, S.; Ma, X.; Homayoon, Z.; Barnes, G.L.; Hase, W.L. Direct Chemical Dynamics Simulations. J. Am. Chem. Soc. 2017, 139, 3570–3590. [Google Scholar] [CrossRef]
- Paranjothy, M.; Sun, R.; Zhuang, Y.; Hase, W.L. Direct chemical dynamics simulations: Coupling of classical and quasiclassical trajectories with electronic structure theory. WIREs Comput. Mol. Sci. 2013, 3, 296–316. [Google Scholar] [CrossRef]
- Tsutsumi, T.; Harabuchi, Y.; Ono, Y.; Maeda, S.; Taketsugu, T. Analyses of trajectory on-the-fly based on the global reaction route map. Phys. Chem. Chem. Phys. 2018, 20, 1364–1372. [Google Scholar] [CrossRef]
- Atalay, Y.; Paquet, E.; Viktor, H.L. Computational Methods for Ab Initio Molecular Dynamics. Adv. Chem. 2018, 9839641-1–9839641-14. [Google Scholar]
- Bowman, J.M.; Czakó, G.; Fu, B. High-dimensional ab initio potential energy surfaces for reaction dynamics calculations. Phys. Chem. Chem. Phys. 2011, 13, 8094–8111. [Google Scholar] [CrossRef]
- Chmiela, S.; Sauceda, H.; Tkatchenko, A.; Müller, K. Accurate Molecular Dynamics Enabled by Efficient Physically Constrained Machine Learning Approaches. In Machine Learning Meets Quantum Physics, Lecture Notes in Physics; Schütt, K., Chmiela, S., von Lilienfeld, O., Tkatchenko, A., Tsuda, K., Müller, K., Eds.; Springer: New York, NY, USA, 2020; Volume 968. [Google Scholar]
- Jia, W.; Wang, H.; Chen, M.; Lu, D.; Liu, J.; Lin, L.; Car, R.; E, W.; Zhang, L. Pushing the limit of molecular dynamics with ab initio accuracy to 100 million atoms with machine learning. arXiv 2020, arXiv:2005.00223v1. [Google Scholar]
- Konkoli, Z.; Kraka, E.; Cremer, D. Unified Reaction Valley Approach Mechanism of the Reaction CH3 + H2→CH4 + H. J. Phys. Chem. A 1997, 101, 1742–1757. [Google Scholar] [CrossRef]
- Kraka, E. Reaction Path Hamiltonian and the Unified Reaction Valley Approach. WIREs Comput. Mol. Sci. 2011, 1, 531–556. [Google Scholar] [CrossRef]
- Kraka, E.; Cremer, D. Dieter Cremer’s Contribution to the Field of Theoretical Chemistry. Int. J. Quantum Chem. 2019, 119, e25849. [Google Scholar] [CrossRef] [Green Version]
- Kraka, E.; Cremer, D. Computational Analysis of the Mechanism of Chemical Reactions in Terms of Reaction Phases: Hidden Intermediates and Hidden Transition States. Acc. Chem. Res. 2010, 43, 591–601. [Google Scholar] [CrossRef]
- Cremer, D.; Kraka, E. From Molecular Vibrations to Bonding, Chemical Reactions, and Reaction Mechanism. Curr. Org. Chem. 2010, 14, 1524–1560. [Google Scholar] [CrossRef]
- Miller, W.H.; Handy, N.C.; Adams, J.E. Reaction path Hamiltonian for polyatomic molecules. J. Chem. Phys. 1980, 72, 99–112. [Google Scholar] [CrossRef] [Green Version]
- Kato, S.; Morokuma, K. Potential energy characteristics and energy partitioning in chemical reactions: Ab initio MO study of four-centered elimination reaction CH3CH2F + CH2→CH2 + HF. J. Chem. Phys. 1980, 73, 3900–3914. [Google Scholar] [CrossRef]
- Hofacker, L. Quantentheorie Chemischer Reaktionen. Z. Naturforsch. A 1963, 18, 60–619. [Google Scholar] [CrossRef] [Green Version]
- Marcus, R. On analytical mechanics of chemical reactions. Quantum mechanics of linear collisions. J. Chem. Phys. 1966, 45, 4493–4499. [Google Scholar] [CrossRef] [Green Version]
- Marcus, R. On analytical mechanics of chemical reactions. Classical mechanics of linear collisions. J. Chem. Phys. 1966, 45, 4500–4504. [Google Scholar] [CrossRef] [Green Version]
- Marcus, R. Analytical mechanics of chemical reactions. 3. Natural collision coordinates. J. Chem. Phys. 1968, 49, 2610–2616. [Google Scholar] [CrossRef]
- Hougen, J.; Bunker, P.; Johns, J. Vibration-rotation problem in triatomic molecules allowing for a large-amplitude bending vibration. J. Mol. Spectrosc. 1970, 34, 136–172. [Google Scholar] [CrossRef]
- Eliason, M.A.; Hirschfelder, J.O. General Collision Theory Treatment for the Rate of Bimolecular, Gas Phase Reactions. J. Chem. Phys. 1959, 30, 1426–1436. [Google Scholar] [CrossRef]
- Fukui, K. The path of chemical-reactions—The IRC approach. Acc. Chem. Res. 1981, 14, 363–368. [Google Scholar] [CrossRef]
- Wilson, E.B.; Decius, J.C.; Cross, P.C. Molecular Vibrations; McGraw-Hill: New York, NY, USA, 1955. [Google Scholar]
- Page, M.; McIver, J.W., Jr. On evaluating the Reaction path Hamiltonian. J. Chem. Phys. 1988, 88, 922–935. [Google Scholar] [CrossRef]
- Kühnel, W. Differential Geometry: Curves-Surfaces-Manifolds; American Mathematics Society, AMS: New York, NY, USA, 2005. [Google Scholar]
- Garrett, B.; Truhlar, D. Variational transition state theory. In Theory and Applications of Computational Chemistry: The First Forty Years; Dykstra, C., Frenking, G., Kim, K., Scuseria, G., Eds.; Elsevier: Amsterdam, The Netherlands, 2005; pp. 67–87. [Google Scholar]
- Gonzales, J.; Gimenez, X.; Bofill, J. A reaction path—Liouville approach to the rate constant for polyatomic chemical reactions. Phys. Chem. Chem. Phys. 2002, 4, 2921–2926. [Google Scholar] [CrossRef]
- Luckhaus, D. Large curvature tunneling on the reaction path. Phys. Chem. Chem. Phys. 2008, 10, 6215–6222. [Google Scholar] [CrossRef]
- Killelea, D.; Campbell, V.; Shuman, N.; Utz, A. Bond-selective control of a heterogeneously catalyzed reaction. Science 2008, 319, 790–793. [Google Scholar] [CrossRef]
- Kraka, E.; Dunning, T.H., Jr. Characterization of Molecular Potential Energy Surfaces: Critical Points, Reaction Paths and Reaction Valleys. In Advances in Molecular Electronic Structure Theory: The Calculation and Characterization of Molecular Potential Energy Surfaces; Dunning, T.H., Jr., Ed.; JAI Press, Inc.: Greenwich, CT, USA, 1990; pp. 129–173. [Google Scholar]
- Dunning, T.H., Jr.; Kraka, E.; Eades, R.A. Insights into the Mechanisms of Chemical Reactions—Reaction Paths for Chemical Reactions. Faraday Discuss. 1987, 84, 427–440. [Google Scholar] [CrossRef]
- Dunning, T.H., Jr.; Harding, L.B.; Kraka, E. Calculation and Characterization of Reaction Valleys for Chemical Reactions. In NATO Advanced Research Workshop on Supercomputer Algorithms for Reactivity, Dynamcis and Kinetics of Small Molecules; Dunning, T.H., Jr., Ed.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1989; pp. 57-1–57-15. [Google Scholar]
- Kraka, E. Reaction Path Hamiltonian and its Use for Investigating Reaction Mechanism. In Encyclopedia of Computational Chemistry; Schleyer, P., Allinger, N., Clark, T., Gasteiger, J., Kollman, P., Schaefer, H., III, Schreiner, P., Eds.; John Wiley & Sons: New York, NY, USA, 1998; pp. 2437–2463. [Google Scholar]
- Konkoli, Z.; Cremer, D.; Kraka, E. Diabatic Ordering of Vibrational Normal Modes in Reaction Valley Studies. J. Comput. Chem. 1997, 18, 1282–1294. [Google Scholar] [CrossRef]
- Joo, H.; Kraka, E.; Quapp, W.; Cremer, D. The Mechanism of a Barrierless Reaction: Hidden Transition State and Hidden Intermediates in the Reaction of Methylene with Ethene. Mol. Phys. 2007, 105, 2697–2717. [Google Scholar] [CrossRef]
- Cremer, D.; Wu, A.; Kraka, E. The Mechanism of the Reaction FH + H2C=CH2→H2C-CFH3. Investigation of Hidden Intermediates with the Unified Reaction Valley Approach. Phys. Chem. Chem. Phys. 2001, 3, 674–687. [Google Scholar] [CrossRef]
- Konkoli, Z.; Cremer, D. A New Way of Analyzing Vibrational Spectra. I. Derivation of Adiabatic Internal Modes. Int. J. Quantum Chem. 1998, 67, 1–9. [Google Scholar] [CrossRef]
- Konkoli, Z.; Larsson, J.A.; Cremer, D. A New Way of Analyzing Vibrational Spectra. II. Comparison of Internal Mode Frequencies. Int. J. Quantum Chem. 1998, 67, 11–27. [Google Scholar] [CrossRef]
- Konkoli, Z.; Cremer, D. A New Way of Analyzing Vibrational Spectra. III. Characterization of Normal Vibrational Modes in terms of Internal Vibrational Modes. Int. J. Quantum Chem. 1998, 67, 29–40. [Google Scholar] [CrossRef]
- Konkoli, Z.; Larsson, J.A.; Cremer, D. A New Way of Analyzing Vibrational Spectra. IV. Application and Testing of Adiabatic Modes within the Concept of the Characterization of Normal Modes. Int. J. Quantum Chem. 1998, 67, 41–55. [Google Scholar] [CrossRef]
- Cremer, D.; Larsson, J.A.; Kraka, E. New Developments in the Analysis of Vibrational Spectra on the Use of Adiabatic Internal Vibrational Modes. In Theoretical and Computational Chemistry; Parkanyi, C., Ed.; Elsevier: Amsterdam, The Netherlands, 1998; pp. 259–327. [Google Scholar]
- Kraka, E.; Zou, W.; Tao, Y. Decoding chemical information from vibrational spectroscopy data: Local vibrational mode theory. WIREs Comput. Mol. Sci. 2020, e1480. [Google Scholar] [CrossRef]
- Zou, W.; Kalescky, R.; Kraka, E.; Cremer, D. Relating Normal Vibrational Modes to Local Vibrational Modes with the Help of an Adiabatic Connection Scheme. J. Chem. Phys. 2012, 137, 084114. [Google Scholar] [CrossRef] [Green Version]
- Kraka, E.; Cremer, D. Mechanism and Dynamics of Organic Reactions: 1,2-H Shift in Methylchlorocarbene. J. Phys. Org. Chem. 2002, 15, 431–447. [Google Scholar] [CrossRef]
- Kraka, E.; Wu, A.; Cremer, D. Mechanism of the Diels-Alder Reaction Studied with the United Reaction Valley Approach: Mechanistic Differences between Symmetry-Allowed and Symmetry-Forbidden Reactions. J. Phys. Chem. A 2003, 107, 9008–9021. [Google Scholar] [CrossRef]
- Kraka, E.; Zou, W.; Freindorf, M.; Cremer, D. Energetics and Mechanism of the Hydrogenation of XHn for Group IV to Group VII Elements X. J. Chem. Theory Comput. 2012, 8, 4931–4943. [Google Scholar] [CrossRef]
- Sexton, T.; Kraka, E.; Cremer, D. Extraordinary Mechanism of the Diels-Alder Reaction: Investigation of Stereochemistry, Charge Transfer, Charge Polarization, and Biradicaloid Formation. J. Phys. Chem. A 2016, 120, 1097–1111. [Google Scholar] [CrossRef] [PubMed]
- Freindorf, M.; Sexton, T.; Kraka, E.; Cremer, D. The Mechanism of the Cycloaddition Reaction of 1,3-Dipole Molecules with Acetylene—An Investigation with the Unified Reaction Valley Approach. Theor. Chem. Acc. 2013, 133, 1423–1441. [Google Scholar] [CrossRef]
- Sexton, T.M.; Freindorf, M.; Kraka, E.; Cremer, D. A Reaction Valley Investigation of the Cycloaddition of 1,3-Dipoles with the Dipolarophiles Ethene and Acetylene: Solution of a Mechanistic Puzzle. J. Phys. Chem. A 2016, 120, 8400–8418. [Google Scholar] [CrossRef] [PubMed]
- López, C.S.; Faza, O.N.; Freindorf, M.; Kraka, E.; Cremer, D. Solving the Pericyclic-Pseudo pericyclic Puzzle in the Ring-Closure Reactions of 1,2,4,6-Heptatetraene Derivatives. J. Org. Chem. 2015, 81, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Freindorf, M.; Cremer, D.; Kraka, E. Gold(I)-Assisted Catalysis—A Comprehensive View on the [3,3]-Sigmatropic Rearrangement of Allyl Acetate. Mol. Phys. 2017, 116, 611–630. [Google Scholar] [CrossRef]
- Zou, W.; Sexton, T.; Kraka, E.; Freindorf, M.; Cremer, D. A New Method for Describing the Mechanism of a Chemical Reaction Based on the Unified Reaction Valley Approach. J. Chem. Theory Comput. 2016, 12, 650–663. [Google Scholar] [CrossRef]
- Reis, M.C.; López, C.S.; Kraka, E.; Cremer, D.; Faza, O.N. Rational Design in Catalysis: A Mechanistic Study of β-Hydride Eliminations in Gold(I) and Gold(III) Complexes Based on Features of the Reaction Valley. Inorg. Chem. 2016, 55, 8636–8645. [Google Scholar] [CrossRef]
- Nanayakkara, S.; Kraka, E. A New Way of Studying Chemical Reactions: A Hand-in-hand URVA and QTAIM Approach. Phys. Chem. Chem. Phys. 2019, 21, 15007–15018. [Google Scholar] [CrossRef]
- Cremer, D.; Pople, J.A. General Definition of Ring Puckering Coordinates. J. Am. Chem. Soc. 1975, 97, 1354–1358. [Google Scholar] [CrossRef]
- Hratchian, H.P.; Kraka, E. Improved Predictor-Corrector Integrators For Evaluating Reaction Path Curvature. J. Chem. Theory Comput. 2013, 9, 1481–1488. [Google Scholar] [CrossRef] [PubMed]
- Quapp, W.; Kraka, E.; Cremer, D. Finding the Transition State of Quasi-Barrierless Reactions by a Growing String Method for Newton Trajectories: Application to the Dissociation of Methylenecyclopropene and Cyclopropane. J. Chem. Phys. A 2007, 111, 11287–11293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraka, E.; Joo, H.; Cremer, D. A Stunning Example for a Spontaneous Reaction with a Complex Mechanism: The Vinylidene-Acetylene Cycloaddition Reaction. Mol. Phys. 2010, 108, 2667–2685. [Google Scholar] [CrossRef]
- Freindorf, M.; Tao, Y.; Sethio, D.; Cremer, D.; Kraka, E. New Mechanistic Insights into the Claisen Rearrangement of Chorismate—A Unified Reaction Valley Approach Study. Mol. Phys. 2019, 117, 1172–1192. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional Thermochemistry. III. The role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Ditchfield, D.; Hehre, W.J.; Pople, J.A. Self-Consistent Molecular Orbital Methods. 9. Extended Gaussian-type basis for molecular-orbital studies of organic molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Schwerdtfeger, P.; Dolg, M.; Schwarz, W.; Bowmaker, G.; Boyd, P. Relativistic effects in gold chemistry. I. Diatomic gold compounds. J. Chem. Phys. 1989, 91, 1762–1774. [Google Scholar] [CrossRef] [Green Version]
- Andrae, D.; Haeussermann, U.; Dolg, M.; Stoll, H.; Preuss, H. Energy-adjusted ab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Feller, D. The role of databases in support of computational chemistry calculations. J. Comput. Chem. 1996, 17, 1571–1586. [Google Scholar] [CrossRef]
- Dolg, M.; Wedig, U.; Stoll, H.; Preuss, H. Energy–adjusted ab initio pseudopotentials for the first row transition elements. J. Chem. Phys. 1987, 86, 866–872. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; Schleyer, P.V.R. Efficient diffuse function-augmented basis sets for anion calculations. III. The 3-21+G basis set for first-row elements, Li-F. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Sparta, M.; Neese, F. Chemical applications carried out by local pair natural orbital based coupled-cluster methods. Chem. Soc. Rev. 2014, 43, 5032–5041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunning, T.J. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Woon, D.; Dunning, T.J. Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J. Chem. Phys. 1993, 98, 1358–1371. [Google Scholar] [CrossRef] [Green Version]
- Woon, D.; Dunning, T.J. Gaussian basis sets for use in correlated molecular calculations. IV. Calculation of static electrical response properties. J. Chem. Phys. 1994, 100, 2975–2988. [Google Scholar] [CrossRef] [Green Version]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef]
- Chung, L.W.; Sameera, W.M.C.; Ramozzi, R.; Page, A.J.; Hatanaka, M.; Petrova, G.P.; Harris, T.V.; Li, X.; Ke, Z.; Liu, F.; et al. The ONIOM Method and Its Applications. Chem. Rev. 2015, 115, 5678–5796. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E.; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. AMBER; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Nikitin, A.M.; Milchevskiy, Y.V.; Lyubartsev, A.P. A new AMBER-compatible force field parameter set for alkanes. J. Mol. Mod. 2014, 20, 2143. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian09; Gaussian Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Kraka, E.; Zou, W.; Filatov, M.; Gräfenstein, J.; Gauss, J.; He, Y.; Wu, A.; Konkoli, Z.; He, Z.; Cremer, D. COLOGNE20. 2020. Available online: https://sites.smu.edu/dedman/catco (accessed on 1 January 2020).
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular Interactions from a Natural Bond Orbital, Donor–Acceptor Viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Kalck, P.; Berre, C.L.; Serp, P. Recent advances in the methanol carbonylation reaction into acetic acid. Coord. Chem. Rev. 2020, 402, 213078. [Google Scholar] [CrossRef]
- Credence Research, Inc. Global Acetic Acid Market; Credence Research, Inc.: San Jose, CA, USA, 2019. [Google Scholar]
- Gomes, R.J.; de Fatima Borges, M.; de Freitas Rosa, M.; Castro-Gomez, R.J.H.; Spinosa, W.A. Acetic Acid Bacteria in the Food Industry: Systematics, Characteristics and Applications. Food Technol. Biotechnol. 2018, 56, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Roth, J.F. The Production of Acetic Acid. Platinum Met. Rev. 1975, 19, 12–14. [Google Scholar]
- Foster, D. On the mechanism of a rhodium-complex-catalyzed carbonylation of methanol to acetic acid. J. Am. Chem. Soc. 1976, 98, 846–848. [Google Scholar] [CrossRef]
- Thomas, C.M.; Süfuss-Fink, G. The Production of Acetic Acid. Coord. Chem. Rev. 2003, 243, 125–142. [Google Scholar] [CrossRef]
- Pal, P.; Nayak, J. Acetic Acid Production and Purification: Critical Review Towards Process Intensification. Sep. Purif. Rev. 2017, 46, 44–61. [Google Scholar] [CrossRef]
- Dutta, D.K.; Woollins, D.; Slawin, A.M.Z.; Konwar, D.; Sharma, M.; Bhattacharyya, P.; Aucott, S.M. Rhodium(I) carbonyl complexes of mono selenium functionalized bis(diphenylphosphino)methane and bis(diphenylphosphino)amine chelating ligands and their catalytic carbonylation activity. J. Organometal. Chem. 2006, 691, 1229–1234. [Google Scholar] [CrossRef]
- Katsuki, T.; Sharpless, K.B. The First Practical Method for Asymmetric Epoxidation. J. Am. Chem. Soc. 1980, 102, 5974–5976. [Google Scholar] [CrossRef]
- Sawano, T.; Yamamoto, H. Regio-and Enantioselective Substrate-Directed Epoxidation. Eur. J. Org. Chem. 2020, 2369–2378. [Google Scholar] [CrossRef]
- Bhadra, S.; Yamamoto, H. Substrate Directed Asymmetric Reactions. Chem. Rev. 2018, 118, 3391–3446. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, K.B. Searching for New Reactivity (Nobel Lecture). Angew. Chem. Int. Ed. 2002, 41, 2024–2032. [Google Scholar] [CrossRef]
- Pedersen, S.F.; Dewan, J.C.; Eckman, R.R.; Sharpless, K.B. Unexpected Diversity in the Coordination Chemistry of Tartrate Esters with Titanium(IV). J. Am. Chem. Soc. 1987, 109, 1279–1282. [Google Scholar] [CrossRef]
- Williams, I.D.; Pedersen, S.F.; Sharpless, K.B.; Lippard, S.J. Crystal Structures of Two Titanium Tartrate Asymmetric Epoxidation Catalysts. J. Am. Chem. Soc. 1984, 106, 6430–6431. [Google Scholar] [CrossRef]
- Finn, G.M.; Sharpless, K.B. Mechanism of Asymmetric Epoxidation. 2. Catalyst Structure. J. Am. Chem. Soc. 1991, 113, 113–1262. [Google Scholar] [CrossRef]
- Marin-Luna, M.; Faza, O.N.; López, C.S. Gold-Catalyzed Homogeneous (Cyclo)Isomerization Reactions. Front. Chem. 2019, 7, 296. [Google Scholar] [CrossRef] [Green Version]
- Shahzad, S.A.; Sajid, M.A.; Khan, Z.A.; Canseco-Gonzalez, D. Gold catalysis in organic transformations: A review. Synth. Commun. 2017, 47, 735–755. [Google Scholar] [CrossRef]
- Scurrell, M.S. Thoughts on the use of gold-based catalysts in environmental protection catalysis. Gold Bull. 2017, 50, 77–84. [Google Scholar] [CrossRef]
- Zi, W.; Dean Toste, F. Recent advances in enantioselective gold catalysis. Chem. Soc. Rev. 2016, 45, 4567–4589. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Koga, H.; Okumura, M.; Haruta, M. Advances in Gold Catalysis and Understanding the Catalytic Mechanism. Chem. Rec. 2016, 16, 2278–2293. [Google Scholar] [CrossRef] [PubMed]
- Echavarren, A.M.; Hashmi, A.S.K.; Toste, F.D. Gold Catalysis—Steadily Increasing in Importance. Adv. Synth. Catal. 2016, 358, 1347. [Google Scholar] [CrossRef] [Green Version]
- Villa, A.; Dimitratos, N.; Chan-Thaw, C.E.; Hammond, C.; Veith, G.M.; Wang, D.; Manzoli, M.; Prati, L.; Hutchings, G.J. Characterisation of gold catalysts. Chem. Soc. Rev. 2016, 45, 4953–4994. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Rudolph, M.; Hashmi, A.S.K. Dual gold catalysis—An update. Chem. Commun. 2019, 55, 12127–12135. [Google Scholar] [CrossRef]
- Pflisterer, D.; Hashmi, A.S.K. Gold catalysis in total synthesis—Ecent achievements. Chem. Soc. Rev. 2016, 45, 1331–1367. [Google Scholar] [CrossRef]
- Hashmi, A.S.K. Gold-catalyzed organic reactions. Top. Organomet. Chem. 2013, 44, 143–164. [Google Scholar]
- Kennedy, C.R.; Lin, S.; Jacobsen, E.N. The Cation-π Interaction in Small-Molecule Catalysis. Angew. Chem. Int. Ed. 2016, 55, 12596–12624. [Google Scholar] [CrossRef] [Green Version]
- Vidhani, D.V.; Cran, J.W.; Krafft, M.E.; Manoharan, M.; Alabugin, I.V. Gold(I)-catalyzed claisen rearrangement of allenyl vinyl ethers: Missing transition states revealed through evolution of aromaticity, Au(I) as an oxophilic lewis acid, and lower energy barriers from a high energy complex. J. Org. Chem. 2013, 78, 2059–2073. [Google Scholar] [CrossRef]
- Sherry, B.D.; Toste, F.D. Gold ( I )-Catalyzed Propargyl Claisen Rearrangement. J. Am. Chem. Soc. 2004, 14, 15978–15979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.Y.; Horibe, T.; Jacobsen, C.B.; Toste, F.D. Stable gold(III) catalysts by oxidative addition of a carbon-carbon bond. Nature 2015, 517, 449–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comas-Vives, A.; González-Arellano, C.; Corma, A.; Iglesias, M.; Sánchez, F.; Ujaque, G. Single-site homogeneous and heterogeeized gold(III) hydrogenation catalysts: Mechanistic implications. J. Am. Chem. Soc. 2006, 128, 4756–4765. [Google Scholar] [CrossRef]
- Arduengo, A.J.; Harlow, R.L.; Kline, M. A stable crystalline carbene. J. Am. Chem. Soc. 1991, 113, 361–363. [Google Scholar] [CrossRef]
- Diez-Gonzáles, S. (Ed.) N-Heterocyclic Carbenes—From Laboratory Curiosities to Efficient Synthetic Tools; Royal Society of Chemistry: London, UK, 2017. [Google Scholar]
- Gómez-Suárez, A.; Nelson, D.J.; Nolan, S.P. Quantifying and understanding the steric properties of N-heterocyclic carbenes. Chem. Commun. 2017, 53, 2650–2660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobsen, H.; Correa, A.; Poater, A.; Costabile, C.; Cavallo, L. Understanding the M(NHC) (NHC=N-heterocyclic carbene) bond. Coord. Chem. Rev. 2009, 253, 687–703. [Google Scholar] [CrossRef]
- Mora, M.; Gimeno, M.C.; Visbal, R. Recent advances in gold-NHC complexes with biological properties. Chem. Soc. Rev. 2019, 48, 447–462. [Google Scholar] [CrossRef]
- Tang, X.T.; Yang, F.; Zhang, T.T.; Liu, Y.F.; Liu, S.Y.; Su, T.F.; Lv, D.C.; Shen, W.B. Recent Progress in N-Heterocyclic Carbene Gold-Catalyzed Reactions of Alkynes Involving Oxidation/Amination/Cycloaddition. Catalysis 2019, 10, 350. [Google Scholar] [CrossRef] [Green Version]
- Majumdar, K.C.; Alam, S.; Chattopadhyay, B. Catalysis of the Claisen rearrangement. Tetrahedron 2008, 64, 597–643. [Google Scholar] [CrossRef]
- Gourlaouen, C.; Marion, N.; Nolan, S.P. Mechanism of the [( NHC ) Au I ] -Catalyzed Rearrangement of Allylic Acetates. A DFT Study. Org. Lett. 2009, 11, 12–15. [Google Scholar] [CrossRef]
- Chen, K.; Arnold, F.H. Engineering new catalytic activities in enzymes. Nat. Catal. 2020, 3, 203–213. [Google Scholar] [CrossRef]
- Chowdhury, R.; Maranas, C.D. From directed evolution to computational enzyme engineering-A review. AIChE J. 2020, 66, e16847. [Google Scholar] [CrossRef]
- Bilal, M.; Cui, J.; Iqbal, H.M. Directed Evolution of Artificial Metalloenzymes: A Universal Means to Tune the Selectivity of Transition Metal Catalysts? Acc. Chem. Res. 2020, 52, 336–344. [Google Scholar]
- Reilley, D.J.; Hennefarth, M.R.; Alexandrova, A.N. The Case for Enzymatic Competitive Metal Affinity Methods. ACS Catal. 2020, 10, 2298–2307. [Google Scholar] [CrossRef] [Green Version]
- Bilal, M.; Cui, J.; Iqbal, H.M. The Case for Enzymatic Competitive Metal Affinity Methods. Int. J. Biol. Macromol. 2020, 130, 186–196. [Google Scholar] [CrossRef]
- Magalhaes, R.P.; Fernandes, H.S.; Sousa, S. Modeling Enzymatic Mechanisms with QM/MM Approaches: Current Status and Future Challenges. Isr. J. Chem. 2020, 60, 1–13. [Google Scholar] [CrossRef]
- Himo, F. Recent Trends in Quantum Chemical Modeling of Enzymatic Reactions. J. Am. Chem. Soc. 2017, 139, 6780–6786. [Google Scholar] [CrossRef] [Green Version]
- Quesne, M.G.; Borowski, T.; de Visser, S.P. Quantum Mechanics/Molecular Mechanics Modeling of Enzymatic Processes: Caveats and Breakthroughs. Chem. Eur. J. 2016, 22, 2562–2581. [Google Scholar] [CrossRef]
- Cui, Q. Perspective: Quantum mechanical methods in biochemistry and biophysics. J. Chem. Phys. 2016, 145, 140901–140913. [Google Scholar] [CrossRef] [Green Version]
- Warshel, A.; Bora, R.P. Perspective: Defining and quantifying the role of dynamics in enzyme catalysis. J. Chem. Phys. 2016, 144, 180901–180918. [Google Scholar] [CrossRef]
- Ramírez, C.L.; Martí, M.A.; Roitberg, A.E. Chapter Six: Steered Molecular Dynamics Methods Applied to Enzyme Mechanism and Energetics, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2016; Volume 578, pp. 123–143. [Google Scholar]
- Sokkar, P.; Boulanger, E.; Thiel, W.; Sanchez-Garcia, E. Hybrid quantum mechanics/molecular mechanics/coarse grained modeling: A triple-resolution approach for biomolecular systems. J. Chem. Theory Comput. 2015, 11, 1809–1818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lever, G. A Density-Functional Perspective on the Chorismate Mutase Enzyme. In Large-Scale Quantum-Mechanical Enzymology; Springer International Publishing: Cham, Switzerland, 2015; pp. 111–141. [Google Scholar] [CrossRef]
- Patel, S.S. Large Scale Simulation and Analysis to Understand Enzymatic Chemical Mechanisms. Ph.D. Thesis, Department of Chemistry, Massachusetts Institute of Technology, Boston, MA, USA, 2015. [Google Scholar]
- Lynch, J.H.; Dudareva, N. Aromatic Amino Acids: A Complex Network Ripe for Future Exploration. Trends Plant Sci. 2020, 1935, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Mir, R.; Jallu, S.; Singh, T.P. The shikimate pathway: Review of amino acid sequence, function and three-dimensional structures of the enzymes. Crit. Rev. Microbiol. 2015, 41, 172–189. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Dudareva, N. The Shikimate Pathway and Aromatic Amino Acid Biosynthesis in Plants. Annu. Rev. Plant Biol. 2015, 63, 73–105. [Google Scholar] [CrossRef] [PubMed]
- Claeyssens, F.; Ranaghan, K.E.; Lawan, N.; Macrae, S.J.; Manby, F.R.; Harvey, J.N.; Mulholland, A.J. Analysis of chorismate mutase catalysis by QM/MM modelling of enzyme-catalysed and uncatalysed reactions. Org. Biomol. Chem. 2011, 9, 1578–1590. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, J.; Roche, D.; Rovira, X.; Serra, J. The catalytic power of enzymes: Conformational selection or transition state stabilization? FEBS Lett. 2006, 580, 2170–2177. [Google Scholar] [CrossRef] [Green Version]
- Dong, L.; Liu, Y. Comparative studies of the catalytic mechanisms of two chorismatases: CH-fkbo and CH-Hyg5. Proteins Struct. Funct. Bioinform. 2017, 85, 1146–1158. [Google Scholar] [CrossRef]
- Gustin, D.J.; Mattei, P.; Kast, P.; Wiest, O.; Lee, L.; Cleland, W.W.; Hilvert, D. Heavy Atom Isotope Effects Reveal a Highly Polarized Transition State for Chorismate Mutase. J. Am. Chem. Soc. 1999, 121, 1756–1757. [Google Scholar] [CrossRef]
- Wiest, O.; Houk, K.N. Stabilization of the Transition State of the Chorismate-Prephenate Rearrangement: An ab Initio Study of Enzyme and Antibody Catalysis. J. Am. Chem. Soc. 1995, 117, 11628–11639. [Google Scholar] [CrossRef]
- Burschowsky, D.; Krengel, U.; Uggerud, E.; Balcells, D. Quantum chemical modeling of the reaction path of chorismate mutase based on the experimental substrate/product complex. FEBS Open Bio. 2017, 7, 789–797. [Google Scholar] [CrossRef]
- Ishida, T. Effects of point mutation on enzymatic activity: Correlation between protein electronic structure and motion in chorismate mutase reaction. J. Am. Chem. Soc. 2010, 132, 7104–7118. [Google Scholar] [CrossRef] [PubMed]
- Uyeda, C.; Jacobsen, E.N. Enantioselective Claisen Rearrangements with a Hydrogen-Bond Donor Catalyst. J. Am. Chem. Soc. 2008, 130, 9228–9229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraka, E.; Cremer, D. Weaker Bonds with Shorter Bond Lengths. Rev. Proc. Quim. 2012, 6, 39–42. [Google Scholar] [CrossRef]
- Setiawan, D.; Kraka, E.; Cremer, D. Hidden Bond Anomalies: The Peculiar Case of the Fluorinated Amine Chalcogenides. J. Phys. Chem. A 2015, 119, 9541–9556. [Google Scholar] [CrossRef] [PubMed]
- Kraka, E.; Setiawan, D.; Cremer, D. Re-Evaluation of the Bond Length-Bond Strength Rule: The Stronger Bond Is not Always the Shorter Bond. J. Comput. Chem. 2015, 37, 130–142. [Google Scholar] [CrossRef]
- Kalescky, R.; Kraka, E.; Cremer, D. Identification of the Strongest Bonds in Chemistry. J. Phys. Chem. A 2013, 117, 8981–8995. [Google Scholar] [CrossRef]
- Humason, A.; Zou, W.; Cremer, D. 11,11-Dimethyl-1,6-methano[10]annulene—An Annulene with an Ultralong CC Bond or a Fluxional Molecule? J. Phys. Chem. A. 2014, 119, 1666–1682. [Google Scholar] [CrossRef] [PubMed]
- Kalescky, R.; Zou, W.; Kraka, E.; Cremer, D. Quantitative Assessment of the Multiplicity of Carbon-Halogen Bonds: Carbenium and Halonium Ions with F, Cl, Br, and I. J. Phys. Chem. A 2014, 118, 1948–1963. [Google Scholar] [CrossRef] [PubMed]
- Kraka, E.; Cremer, D. Characterization of CF Bonds with Multiple-Bond Character: Bond Lengths, Stretching Force Constants, and Bond Dissociation Energies. ChemPhysChem 2009, 10, 686–698. [Google Scholar] [CrossRef]
- Zou, W.; Cremer, D. C2 in a Box: Determining its Intrinsic Bond Strength for the X1Σ+g Ground State. Chem. Eur. J. 2016, 22, 4087–4097. [Google Scholar] [CrossRef]
- Setiawan, D.; Sethio, D.; Cremer, D.; Kraka, E. From Strong to Weak NF Bonds: On the Design of a New Class of Fluorinating Agents. Phys. Chem. Chem. Phys. 2018, 20, 23913–23927. [Google Scholar] [CrossRef] [PubMed]
- Freindorf, M.; Kraka, E.; Cremer, D. A Comprehensive Analysis of Hydrogen Bond Interactions Based on Local Vibrational Modes. Int. J. Quantum Chem. 2012, 112, 3174–3187. [Google Scholar] [CrossRef]
- Kalescky, R.; Zou, W.; Kraka, E.; Cremer, D. Local Vibrational Modes of the Water Dimer—Comparison of Theory and Experiment. Chem. Phys. Lett. 2012, 554, 243–247. [Google Scholar] [CrossRef]
- Kalescky, R.; Kraka, E.; Cremer, D. Local Vibrational Modes of the Formic Acid Dimer—The Strength of the Double H-Bond. Mol. Phys. 2013, 111, 1497–1510. [Google Scholar] [CrossRef]
- Kalescky, R.; Zou, W.; Kraka, E.; Cremer, D. Vibrational Properties of the Isotopomers of the Water Dimer Derived from Experiment and Computations. Aust. J. Chem. 2014, 67, 426–434. [Google Scholar] [CrossRef] [Green Version]
- Tao, Y.; Zou, W.; Jia, J.; Li, W.; Cremer, D. Different Ways of Hydrogen Bonding in Water—Why Does Warm Water Freeze Faster than Cold Water? J. Chem. Theory Comput. 2017, 13, 55–76. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Zou, W.; Kraka, E. Strengthening of Hydrogen Bonding with the Push-Pull Effect. Chem. Phys. Lett. 2017, 685, 251–258. [Google Scholar] [CrossRef]
- Makoś, M.Z.; Freindorf, M.; Sethio, D.; Kraka, E. New Insights into Fe–H2 and Fe–H− Bonding of a [NiFe] Hydrogenase Mimic—A Local Vibrational Mode Study. Theor. Chem. Acc. 2019, 138, 76. [Google Scholar] [CrossRef]
- Lyu, S.; Beiranvand, N.; Freindorf, M.; Kraka, E. Interplay of Ring Puckering and Hydrogen Bonding in Deoxyribonucleosides. J. Phys. Chem. A 2019, 123, 7087–7103. [Google Scholar] [CrossRef]
- Oliveira, V.; Kraka, E.; Cremer, D. The Intrinsic Strength of the Halogen Bond: Electrostatic and Covalent Contributions Described by Coupled Cluster Theory. Phys. Chem. Chem. Phys. 2016, 18, 33031–33046. [Google Scholar] [CrossRef]
- Oliveira, V.; Kraka, E.; Cremer, D. Quantitative Assessment of Halogen Bonding Utilizing Vibrational Spectroscopy. Inorg. Chem. 2016, 56, 488–502. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, V.; Cremer, D. Transition from Metal-Ligand Bonding to Halogen Bonding Involving a Metal as Halogen Acceptor: A Study of Cu, Ag, Au, Pt, and Hg Complexes. Chem. Phys. Lett. 2017, 681, 56–63. [Google Scholar] [CrossRef]
- Yannacone, S.; Oliveira, V.; Verma, N.; Kraka, E. A Continuum from Halogen Bonds to Covalent Bonds: Where Do λ3 Iodanes Fit? Inorganics 2019, 7, 47. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, V.P.; Kraka, E.; Machado, F.B.C. Pushing 3c-4e Bonds to the Limit: A Coupled Cluster Study of Stepwise Fluorination of First-Row Atoms. Inorg. Chem. 2019, 58, 14777–14789. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, V.P.; Marcial, B.L.; Machado, F.B.C.; Kraka, E. Metal-Halogen Bonding Seen through the Eyes of Vibrational Spectroscopy. Materials 2020, 13, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Setiawan, D.; Kraka, E.; Cremer, D. Description of Pnicogen Bonding with the help of Vibrational Spectroscopy-The Missing Link Between Theory and Experiment. Chem. Phys. Lett. 2014, 614, 136–142. [Google Scholar] [CrossRef]
- Setiawan, D.; Kraka, E.; Cremer, D. Strength of the Pnicogen Bond in Complexes Involving Group VA Elements N, P, and As. J. Phys. Chem. A 2014, 119, 1642–1656. [Google Scholar] [CrossRef]
- Setiawan, D.; Cremer, D. Super-Pnicogen Bonding in the Radical Anion of the Fluorophosphine Dimer. Chem. Phys. Lett. 2016, 662, 182–187. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, V.; Cremer, D.; Kraka, E. The Many Facets of Chalcogen Bonding: Described by Vibrational Spectroscopy. J. Phys. Chem. A 2017, 121, 6845–6862. [Google Scholar] [CrossRef]
- Oliveira, V.; Kraka, E. Systematic Coupled Cluster Study of Noncovalent Interactions Involving Halogens, Chalcogens, and Pnicogens. J. Phys. Chem. A 2017, 121, 9544–9556. [Google Scholar] [CrossRef]
- Sethio, D.; Oliveira, V.; Kraka, E. Quantitative Assessment of Tetrel Bonding Utilizing Vibrational Spectroscopy. Molecules 2018, 23, 2763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraka, E.; Larsson, J.A.; Cremer, D. Generalization of the Badger Rule Based on the Use of Adiabatic Vibrational Modes. In Computational Spectroscopy; Grunenberg, J., Ed.; Wiley: New York, NY, USA, 2010; pp. 105–149. [Google Scholar]
- Burschowsky, D.; van Eerde, A.; Ökvist, M.; Kienhöfer, A.; Kast, P.; Hilvert, D.; Krengel, U. Electrostatic transition state stabilization rather than reactant destabilization provides the chemical basis for efficient chorismate mutase catalysis. Proc. Natl. Acad. Sci. USA 2014, 111, 17516. [Google Scholar] [CrossRef] [PubMed] [Green Version]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction Step | ||||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 45.0 | 2.4 | 43.5 | 2.1 | 13.2 | 12.0 | 17.1 | 2.7 |
| 2 | 20.0 | −9.3 | 19.1 | −8.5 | 0.0 | 0.9 | 3.1 | 16.0 |
| 3 | - | −8.0 | - | −7.5 | - | - | - | - |
| 4 | 29.9 | 0.5 | 25.0 | 0.1 | 0.5 | 6.5 | 12.7 | 6.2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kraka, E.; Zou, W.; Tao, Y.; Freindorf, M. Exploring the Mechanism of Catalysis with the Unified Reaction Valley Approach (URVA)—A Review. Catalysts 2020, 10, 691. https://doi.org/10.3390/catal10060691
Kraka E, Zou W, Tao Y, Freindorf M. Exploring the Mechanism of Catalysis with the Unified Reaction Valley Approach (URVA)—A Review. Catalysts. 2020; 10(6):691. https://doi.org/10.3390/catal10060691
Chicago/Turabian StyleKraka, Elfi, Wenli Zou, Yunwen Tao, and Marek Freindorf. 2020. "Exploring the Mechanism of Catalysis with the Unified Reaction Valley Approach (URVA)—A Review" Catalysts 10, no. 6: 691. https://doi.org/10.3390/catal10060691
APA StyleKraka, E., Zou, W., Tao, Y., & Freindorf, M. (2020). Exploring the Mechanism of Catalysis with the Unified Reaction Valley Approach (URVA)—A Review. Catalysts, 10(6), 691. https://doi.org/10.3390/catal10060691