Immobilization of Cellulolytic Enzymes in Mesostructured Silica Materials


Abstract
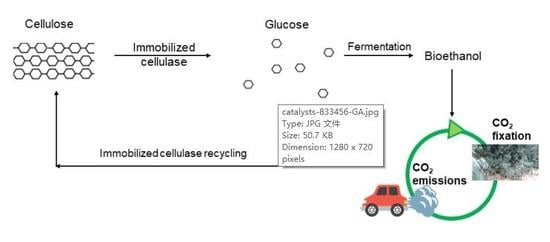
:
1. Introduction
2. Enzyme Immobilization
2.1. Adsorption
2.2. Entrapment
2.3. Cross-Linking
2.4. Covalent Bonding
3. Mesoporous and Microporous Silica Materials
3.1. Silica Nanoparticles
3.2. Functionalization
4. Enzymatic Hydrolysis of Cellulose
4.1. Lignocellulosic Biomass
4.2. Cellulolytic Enzymes
5. Immobilization of BG on Mesostructured Silica
6. Immobilization of Cellulase on Mesostructured Silica
7. Conclusions
Funding
Conflicts of Interest
References
- Chang, R.H.Y.; Jang, J.; Wu, K.C.W. Cellulase immobilized mesoporous silica nanocatalysts for efficient cellulose-to-glucose conversion. Green Chem. 2011, 13, 2844–2850. [Google Scholar] [CrossRef]
- Yin, H.; Su, Z.L.; Shao, H.; Cai, J.; Wang, X.; Yin, H. Immobilization of cellulase on modified mesoporous silica shows improved thermal stability and reusability. Afr. J. Microbiol. Res. 2013, 7, 3248–3253. [Google Scholar]
- Bhat, M. Cellulases and related enzymes in biotechnology. Biotechnol. Adv. 2000, 18, 355–383. [Google Scholar] [CrossRef]
- Bhat, M.K.; Bhat, S. Cellulose degrading enzymes and their potential industrial applications. Biotechnol. Adv. 1997, 15, 583–620. [Google Scholar] [CrossRef]
- Kitaoka, M.; Taniguchi, H.; Sasaki, T. A simple method of cellulase immobilization on a modified silica support. J. Ferment. Bioeng. 1989, 67, 182–185. [Google Scholar] [CrossRef]
- Walker, L.P.; Wilson, D.B. Enzymatic hydrolysis of cellulose: An overview. Bioresour. Technol. 1991, 36, 3–14. [Google Scholar] [CrossRef]
- Reis, C.L.B.; Sousa, E.Y.A.D.; Serpa, J.D.F.; Oliveira, R.C.; Santos, J.C.S.D. Design of immobilized enzyme biocatalysts: Drawbacks and opportunities. Quím. Nova 2019, 42, 768–783. [Google Scholar] [CrossRef]
- Khoshnevisan, K.; Poorakbar, E.; Baharifar, H.; Barkhi, M. Recent advances of cellulase immobilization onto magnetic nanoparticles: An update review. Magnetochemistry 2019, 5, 36. [Google Scholar] [CrossRef] [Green Version]
- Andriani, D.; Sunwoo, C.; Ryu, H.W.; Prasetya, B.; Park, D.H. Immobilization of cellulase from newly isolated strain Bacillus subtilis TD6 using calcium alginate as a support material. Bioprocess. Biosyst. Eng. 2012, 35, 29–33. [Google Scholar] [CrossRef]
- Anuradha, J.S.; Valli, N.C. Immobilization of Aspergillus nidulans SU04 cellulase on modified activated carbon. J. Therm. Anal. Calorim. 2012, 109, 193–202. [Google Scholar] [CrossRef]
- Magner, E. Immobilisation of enzymes on mesoporous silicate materials. Chem. Soc. Rev. 2013, 42, 6213–6222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, M.; Jung, D. Biocatalysis with enzymes immobilized on mesoporous hosts: The status quo and future trends. J. Mater. Chem. 2010, 20, 844–857. [Google Scholar] [CrossRef]
- Hartono, S.B.; Qiao, S.Z.; Liu, J.; Jack, K.; Ladewig, B.P.; Hao, Z.; Lu, G.Q.M. Functionalized mesoporous silica with very large pores for cellulase immobilization. J. Phys. Chem. C 2010, 114, 8353–8362. [Google Scholar] [CrossRef]
- Chong, A.S.M.; Zhao, X.S. Design of large-pore mesoporous materials for immobilization of penicillin G acylase biocatalyst. Catal. Today 2004, 93, 293–299. [Google Scholar] [CrossRef]
- Yiu, H.H.P.; Botting, C.H.; Botting, N.P.; Wright, P.A. Size selective protein adsorption on thiol-functionalised SBA-15 mesoporous molecular sieve. Phys. Chem. Chem. Phys. 2001, 3, 2983–2985. [Google Scholar] [CrossRef]
- Kao, H.M.; Chang, P.C.; Wu, J.D.; Chiang, A.S.; Lee, C.H. Direct synthesis, characterization and solid-state NMR spectroscopy of large-pore vinyl-functionalized cubic mesoporous silica FDU-12. Microporous Mesoporous Mater. 2006, 97, 9–20. [Google Scholar] [CrossRef]
- Koeller, K.M.; Wong, C.H. Enzymes for chemical synthesis. Nature 2001, 409, 232. [Google Scholar] [CrossRef]
- Arsalan, A.; Younus, H. Enzymes and nanoparticles: Modulation of enzymatic activity via nanoparticles. Int. J. Biol. Macromol. 2018, 118, 1833–1847. [Google Scholar] [CrossRef]
- Mateo, C.; Palomo, J.M.; Fernandez-Lorente, G.; Guisan, J.M.; Fernandez-Lafuente, R. Improvement of enzyme activity, stability and selectivity via immobilization techniques. Enzym. Microb. Technol. 2007, 40, 1451–1463. [Google Scholar] [CrossRef]
- Nelson, J.M.; Grinn, E.G. Adsorption of invertase. J. Am. Chem. Soc. 1916, 38, 1109–1115. [Google Scholar] [CrossRef] [Green Version]
- Bommarius, A.S.; Paye, M.F. Stabilizing biocatalysts. Chem. Soc. Rev. 2013, 42, 6534–6565. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A. Enzyme immobilization: The quest for optimum performance. Adv. Synth. Catal. 2007, 349, 1289–1307. [Google Scholar] [CrossRef]
- Daoud, F.B.O.; Kaddour, S.; Sadoun, T. Adsorption of cellulase Aspergillus niger on a commercial activated carbon: Kinetics and equilibrium studies. Colloids Surf. B 2010, 75, 93–99. [Google Scholar] [CrossRef]
- Kourkoutas, Y.; Bekatorou, A.; Banat, I.M.; Marchant, R.; Koutinas, A.A. Immobilization technologies and support materials suitable in alcohol beverages production: A review. Food Microbiol. 2004, 21, 377–397. [Google Scholar] [CrossRef]
- Pierre, A.C. The sol-gel encapsulation of enzymes. Biocatal. Biotransform. 2004, 22, 145–170. [Google Scholar] [CrossRef]
- Vidinha, P.; Augusto, V.; Almeida, M.; Fonseca, I.; Fidalgo, A.; Ilharco, L.; Cabral, J.M.S.; Barreiros, S. Sol–gel encapsulation: An efficient and versatile immobilization technique for cutinase in non-aqueous media. J. Biotechnol. 2006, 121, 23–33. [Google Scholar] [CrossRef]
- Zarcula, C.; Kiss, C.; Corîci, L.; Croitoru, R.; Csunderlik, C.; Peter, F. Combined sol-gel entrapment and adsorption method to obtain solid-phase lipase biocatalyst. Rev. Chim. 2009, 60, 922–927. [Google Scholar]
- Tomin, A.; Weiser, D.; Hellner, G.; Bata, Z.; Corici, L.; Peter, F.; Koczka, B.; Poppe, L. Fine-tuning the second generation sol–gel lipase immobilization with ternary alkoxysilane precursor systems. Process. Biochem. 2011, 46, 52–58. [Google Scholar] [CrossRef]
- Ungurean, M.; Paul, C.; Peter, F. Cellulase immobilized by sol–gel entrapment for efficient hydrolysis of cellulose. Bioproc. Biosys. Eng. 2013, 36, 1327–1338. [Google Scholar] [CrossRef]
- Karube, I.; Tanaka, S.; Shirai, T.; Suzuki, S. Hydrolysis of cellulose in a cellulase-bead fluidized bed reactor. Biotechnol. Bioeng. 1977, 19, 1183–1191. [Google Scholar] [CrossRef]
- Cao, L.; van Langen, L.; Sheldon, R.A. Immobilised enzymes: Carrier-bound or carrier-free? Curr. Opin. Biotech. 2003, 14, 387–394. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Sorgedrager, M.E.N.N.O.; Janssen, M.H. Use of cross-linked enzyme aggregates (CLEAs) for performing biotransformations. Chim. Oggi 2007, 25, 62. [Google Scholar]
- Li, B.; Dong, S.L.; Xie, X.L.; Xu, Z.B.; Li, L. Preparation and properties of cross-linked enzyme aggregates of cellulase. In Advanced Materials Research; Trans Tech Publications Ltd.: Bach, Switzerland, 2012; Volume 581, pp. 257–260. [Google Scholar]
- Branda, F.; Silvestri, B.; Costantini, A.; Luciani, G. Effect of exposure to growth media on size and surface charge of silica based Stöber nanoparticles: A DLS and ζ-potential study. J. Sol Gel Sci. Technol. 2015, 73, 54–61. [Google Scholar] [CrossRef]
- Thomas, J.M.; Johnson, B.F.; Raja, R.; Sankar, G.; Midgley, P.A. High-performance nanocatalysts for single-step hydrogenations. Acc. Chem. Res. 2003, 36, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.S.; Lu, M.G.; Song, C. Immobilization of aluminum chloride on MCM-41 as a new catalyst system for liquid-phase isopropylation of naphthalene. J. Mol. Catal. A Chem. 2003, 191, 67–74. [Google Scholar] [CrossRef]
- Mateo, C.; Grazu, V.; Palomo, J.M.; Lopez-Gallego, F.; Fernandez-Lafuente, R.; Guisan, J.M. Immobilization of enzymes on heterofunctional epoxy supports. Nat. Protoc. 2007, 2, 1022. [Google Scholar] [CrossRef] [PubMed]
- Popat, A.; Hartono, S.B.; Stahr, F.; Liu, J.; Qiao, S.Z.; Lu, G.Q.M. Mesoporous silica nanoparticles for bioadsorption, enzyme immobilisation, and delivery carriers. Nanoscale 2011, 3, 2801–2818. [Google Scholar] [CrossRef] [Green Version]
- McBain, J.W. The Sorption of Gases and Vapors by Solids; Routledge and Sons: London, UK, 1932; p. 169. [Google Scholar]
- Barrer, R.M.; Brook, D.W. Molecular diffusion in chabazite, mordenite, and levynite. Trans. Faraday Soc. 1953, 49, 1049–1059. [Google Scholar] [CrossRef]
- Breck, D.W.; Eversole, W.G.; Milton, R.M. New synthetic crystalline zeolites. J. Am. Chem. Soc. 1956, 78, 2338–2339. [Google Scholar] [CrossRef]
- Davis, M.E.; Lobo, R.F. Zeolite and molecular sieve synthesis. Chem. Mater. 1992, 4, 756–768. [Google Scholar] [CrossRef]
- Mitchell, P.C.H. Zeolite-encapsulated metal complexes: Biomimetic catalysts. Chem. Ind. 1991, 6, 308–311. [Google Scholar]
- Ozin, G.A. Nanochemistry: Synthesis in diminishing dimensions. Adv. Mater. 1992, 10, 612–649. [Google Scholar] [CrossRef]
- Yanagisawa, T.; Schimizu, T.; Kiroda, K.; Kato, C. The preparation of alkyltrimethylammonium-kanemite complexes and their conversion to mesoporous materials. Bull. Chem. Soc. Jpn. 1990, 63, 988–992. [Google Scholar] [CrossRef] [Green Version]
- Beck, J.S.; Calabro, D.C.; McCullen, S.B.; Pelrine, B.P.; Schmitt, K.D.; Vartuli, J.C. Method for Functionalizing Synthetic Mesoporous Crystalline Material. U.S. Patent 2,069,722, 27 May 1992. [Google Scholar]
- Kresge, C.T.; Leonowicz, M.E.; Roth, W.J.; Vartuli, J.C.; Beck, J.S. Ordered mesoporous molecular sieves synthesized by a liquid-crystal template mechanism. Nature 1992, 359, 710–712. [Google Scholar] [CrossRef]
- AlOthman, Z.A.; Apblett, A.W. Metal ion adsorption using polyamine-functionalized mesoporous materials prepared from bromopropyl-functionalized mesoporous silica. J. Hazard. Mater. 2010, 182, 581–590. [Google Scholar] [CrossRef]
- Parida, K.M.; Dash, S.S. Manganese containing MCM-41: Synthesis, characterization and catalytic activity in the oxidation of ethylbenzene. J. Mol. Catal. A 2009, 306, 54–61. [Google Scholar] [CrossRef]
- Chen, J.; Xia, N.; Zhou, T.; Tan, S.; Jiang, F. Mesoporous carbon spheres: Synthesis, characterization and supercapacitance. Int. J. Electrochem. Sci. 2009, 4, 1063–1073. [Google Scholar]
- Beck, J.S.; Vartuli, J.C.; Roth, W.J.; Leonowicz, M.E.; Kresge, C.T.; Schmitt, K.D.; Chu, C.T.W.; Olson, D.H.; Sheppard, E.W.; McCullen, S.B.; et al. A new family of mesoporous molecular sieves prepared with liquid crystal templates. J. Am. Chem. Soc. 1992, 114, 10834–10843. [Google Scholar] [CrossRef]
- Yang, X.Y.; Zhang, S.B.; Qiu, Z.M.; Tian, G.; Feng, Y.F.; Xiao, F.S. Stable ordered mesoporous silica materials templated by high-temperature stable surfactant micelle in alkaline media. J. Phys. Chem. B 2004, 108, 4696–4700. [Google Scholar] [CrossRef]
- Di Renzo, F.; Cambon, H.; Dutarte, R. A 28-year-old Synthesis of Micelle-templated mesoporous silica. Microporous Mater. 1997, 10, 283–286. [Google Scholar] [CrossRef]
- Lok, B.M.; Cannon, T.R.; Messina, C.A. The role of organic molecules in molecular sieve synthesis. Zeolites 1983, 3, 282–291. [Google Scholar] [CrossRef]
- Chen, C.Y.; Burkett, S.L.; Li, H.X.; Davis, M.E. Studies on mesoporous materials. II. Synthesis mechanism of MCM-41. Microporous Mater. 1993, 2, 27–34. [Google Scholar] [CrossRef]
- Monnier, A.; Schüth, F.; Huo, Q.; Kumar, D.; Margolese, D.; Maxwell, R.S.; Stucky, G.D.; Krishnamurty, M.; Petroff, P.; Firoouzi, A.; et al. Cooperative formation of inorganic–organic interfaces in the synthesis of silicate mesostructures. Science 1993, 261, 1299–1303. [Google Scholar] [CrossRef] [PubMed]
- Steel, A.; Carr, S.W.; Anderson, M.W. 14N NMR study of surfactant mesophases in the synthesis of mesoporous silicates. J. Chem. Soc. Chem. Commun. 1994, 13, 1571–1572. [Google Scholar] [CrossRef]
- Broekhoff, J.C.P. Mesopore determination from nitrogen sorption isotherms: Fundamentals, scope, limitations. Stud. Surf. Sci. Catal. 1979, 3, 663–684. [Google Scholar]
- Shields, J.E.; Lowell, S.; Thomas, M.A.; Thommes, M. Characterization of Porous Solids and Powders: Surface Area, Pore Size and Density; Kluwer Academic Publisher: Boston, MA, USA, 2004; pp. 43–45. [Google Scholar]
- Lawrence, M.J. Surfactant systems: Their use in drug delivery. Chem. Soc. Rev. 1994, 23, 417–424. [Google Scholar] [CrossRef]
- Myers, D. Surfactant Science and Technology; VCH: New York, NY, USA, 1992. [Google Scholar]
- Diaz, J.F.; Balkus, K.J., Jr. Enzyme immobilization in MCM-41 molecular sieve. J. Mol. Catal. B Enzym. 1996, 2, 115–126. [Google Scholar] [CrossRef]
- Washmon-Kriel, L.; Jimenez, V.L.; Balkus, K.J., Jr. Cytochrome c immobilization into mesoporous molecular sieves. J. Mol. Catal. B Enzym. 2000, 10, 453–469. [Google Scholar] [CrossRef]
- Branda, F.; Malucelli, G.; Durante, M.; Piccolo, A.; Mazzei, P.; Costantini, A.; Silvestri, B.; Pennetta, M.; Bifulco, A. Silica treatments: A fire retardant strategy for hemp fabric/epoxy composites. Polymers 2016, 8, 313. [Google Scholar] [CrossRef] [Green Version]
- Zhao, D.; Huo, Q.; Feng, J.; Chmelka, B.F.; Stucky, G.D. Nonionic triblock and star diblock copolymer and oligomeric surfactant syntheses of highly ordered, hydrothermally stable, mesoporous silica structures. J. Am. Chem. Soc. 1998, 120, 6024–6036. [Google Scholar] [CrossRef]
- Wei, Y.; Jin, D.; Ding, T.; Shih, W.H.; Liu, X.; Cheng, S.Z.D.; Fu, Q. A non-surfactant templating route to mesoporous silica materials. Adv. Mater. 1998, 10, 313–316. [Google Scholar] [CrossRef]
- Wei, Y.; Xu, J.; Dong, H.; Dong, J.; Qiu, K.; Jansen-Varnum, S.A. Preparation and physisorption characterization of D-glucose-templated mesoporous silica sol-gel materials. Chem. Mater. 1999, 11, 2023–2029. [Google Scholar] [CrossRef]
- Lü, Y.; Guo, Y.; Wang, Y.; Liu, X.; Wang, Y.; Guo, Y.; Zhang, Z.; Lu, G. Immobilized penicillin G acylase on mesoporous silica: The influence of pore size, pore volume and mesophases. Microporous Mesoporous Mater. 2008, 114, 507–510. [Google Scholar] [CrossRef]
- Serra, E.; Mayoral, Á.; Sakamoto, Y.; Blanco, R.M.; Díaz, I. Immobilization of lipase in ordered mesoporous materials: Effect of textural and structural parameters. Microporous Mesoporous Mater. 2008, 114, 201–213. [Google Scholar] [CrossRef]
- Shui, W.; Fan, J.; Yang, P.; Liu, C.; Zhai, J.; Lei, J.; Yan, Y.; Zhao, D.Y.; Chen, X. Nanopore-based proteolytic reactor for sensitive and comprehensive proteomic analyses. Anal. Chem. 2006, 78, 4811–4819. [Google Scholar]
- Fan, J.; Yu, C.; Lei, J.; Zhang, Q.; Li, T.; Tu, B.; Zhou, W.; Zhao, D.Y. Low-temperature strategy to synthesize highly ordered mesoporous silicas with very large pores. J. Am. Chem. Soc. 2005, 127, 10794–10795. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Winkel, P.; Lukens, W.W.; Yang, P.; Margolese, D.I.; Lettow, J.S.; Ying, J.Y.; Stucky, G.D. Microemulsion templating of siliceous mesostructured cellular foams with well-defined ultralarge mesopores. Chem. Mater. 2000, 12, 686–696. [Google Scholar] [CrossRef]
- Fan, J.; Yu, C.; Gao, F.; Lei, J.; Tian, B.; Wang, L.; Luo, Q.; Tu, B.; Zhou, W.; Zhao, D. Cubic mesoporous silica with large controllable entrance sizes and advanced adsorption properties. Angew. Chem. Int. Ed. 2003, 42, 3146–3150. [Google Scholar] [CrossRef]
- Kimura, T.; Suzuki, M.; Ikeda, T.; Kato, K.; Maeda, M.; Tomura, S. Silica-based mesoporous materials derived from Ti containing layered polysilicate kanemite. Micropor. Mesopor. Mat. 2006, 95, 146–153. [Google Scholar] [CrossRef]
- Baú, L.; Bártovaá, B.; Arduini, M.; Mancin, F. Surfactant-free synthesis of mesoporous and hollow silicananoparticles with an inorganic template. Chem. Commun. 2009, 7584–7586. [Google Scholar]
- Mukherjee, I.; Mylonakis, A.; Guo, Y.; Samuel, S.P.; Li, S.; Wei, R.Y.; Kojtari, A.; Wei, Y. Effect of nonsurfactant template content on the particle size and surface area of monodisperse mesoporous silica nanospheres. Microporous Mesoporous Mater. 2009, 122, 168–174. [Google Scholar] [CrossRef]
- Gao, Z.; Zharov, I. Large pore mesoporous silica nanoparticles by templating with a nonsurfactant molecule, tannic acid. Chem. Mater. 2014, 26, 2030. [Google Scholar] [CrossRef]
- Mori, T.; Rezai-Zadeh, K.; Koyama, N.; Arendash, G.W.; Yamaguchi, H.; Kakuda, N.; Horikoshi-Sakuraba, Y.; Tan, J.; Town, T. Tannic acid is a natural β-secretase inhibitor that prevents cognitive impairment and mitigates Alzheimer-like pathology in transgenic mice. J. Biol. Chem. 2012, 287, 6912–6927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagerman, A.E.; Riedl, K.M.; Jones, G.A.; Sovik, K.N.; Ritchard, N.T.; Hartzfeld, P.W.; Riechel, T.L.J. High molecular weight plant polyphenolics (Tannins) as biological antioxidants. Agric. Food Chem. 1998, 46, 1887–1892. [Google Scholar] [CrossRef]
- Moon, D.; Lee, J. Tunable synthesis of hierarchical mesoporous silica nanoparticles with radial wrinkle structure. Langmuir 2012, 28, 12341–12347. [Google Scholar] [CrossRef] [PubMed]
- Polshettiwar, V.; Thivolle-Cazat, J.; Taoufik, M.; Stoffelbach, F.; Norsic, S.; Basset, J.M. Hydro-metathesis of Olefins: A Catalytic Reaction Using a Bifunctional Single-Site Tantalum Hydride Catalyst Supported on Fibrous Silica (KCC-1) Nanospheres. Angew. Chem. Int. Ed. 2011, 50, 2747–2751. [Google Scholar] [CrossRef] [PubMed]
- Pang, J.; Zhou, G.; Liu, R.; Li, T. Esterification of oleic acid with methanol by immobilized lipase on wrinkled silica nanoparticles with highly ordered, radially oriented mesochannels. Mater. Sci. Eng. C 2016, 59, 35–42. [Google Scholar] [CrossRef]
- Pan, W.; Ye, J.; Ning, G.; Lin, Y.; Wang, J. A novel synthesis of micrometer silica hollow sphere. Mater. Res. Bull. 2009, 44, 280–283. [Google Scholar] [CrossRef]
- Zhang, H.; Li, Z.; Xu, P.; Wu, R.; Jiao, Z. A facile two step synthesis of novel chrysanthemum-like mesoporous silica nanoparticles for controlled pyrene release. Chem. Commun. 2010, 46, 6783–6785. [Google Scholar] [CrossRef]
- Al Othman, Z.A.; Apblett, A.W. Preparation of mesoporous silica with grafted chelating agents for uptake of metal ions. Chem. Eng. J. 2009, 155, 916–924. [Google Scholar] [CrossRef]
- Al Othman, Z.A.; Apblett, A.W. Synthesis and characterization of a hexagonal mesoporous silica with enhanced thermal and hydrothermal stabilities. Appl. Surf. Sci. 2010, 256, 3573–3580. [Google Scholar] [CrossRef]
- Mercier, L.; Pinnavaia, T.J. Access in mesoporous materials: Advantages of a uniform pore structure in the design of a heavy metal ion adsorbent for environmental remediation. Adv. Mater. 1997, 9, 500–503. [Google Scholar] [CrossRef]
- Feng, X.; Fryxell, G.E.; Wang, L.Q.; Kim, Y.A.; Liu, J.; Kemner, K.M. Functionalized monolayers on ordered mesoporous supports. Science 1997, 276, 923–926. [Google Scholar] [CrossRef]
- Van Rhijn, W.M.; DeVos, D.E.; Sels, B.F.; Bossaert, W.D.; Jacobs, P.A. Sulfonic acid functionalized ordered mesoporous materials as catalysts for condensation and esterification reactions. Chem. Commun. 1998, 3, 317–318. [Google Scholar] [CrossRef]
- Lin, V.S.Y.; Radu, D.R.; Han, M.K.; Deng, W.; Kuroki, S.; Shanks, B.H.; Pruski, M. Oxidative polymerization of 1,4-diethynylbenzene into highly conjugated poly(phenylene butadiynylene) within the channels of surface-functionalized mesoporous silica and alumina materials. J. Am. Chem. Soc. 2002, 124, 9040–9041. [Google Scholar] [CrossRef] [Green Version]
- Mbaraka, I.K.; Radu, D.R.; Lin, V.S.Y.; Shanks, B.H. Organosulfonic acid-functionalized mesoporous silicas for the esterification of fatty acid. J. Catal. 2003, 219, 329–336. [Google Scholar] [CrossRef]
- Anwander, R. Surface organometallic chemistry at periodic mesoporous silica. Chem. Mater. 2001, 13, 4419–4438. [Google Scholar] [CrossRef]
- Kimura, T.; Saeki, S.; Sugahara, Y.; Kuroda, K.A. Organic modification of FSM-type mesoporous silicas derived from kanemite by silylation. Langmuir 1999, 15, 2794–2798. [Google Scholar] [CrossRef]
- Lim, M.H.; Stein, A. Comparative studies of grafting and direct syntheses of inorganic-organic hybrid mesoporous materials. Chem. Mater. 1999, 11, 3285–3295. [Google Scholar] [CrossRef]
- Hudson, S.; Cooney, J.; Magner, E. Proteins in mesoporous silicates. Angew. Chem. Int. Ed. 2008, 47, 8582–8594. [Google Scholar] [CrossRef]
- Al Othman, Z.A. A Review: Fundamental Aspects of Silicate Mesoporous Materials. Materials 2012, 5, 2874–2902. [Google Scholar] [CrossRef] [Green Version]
- Dashtban, M.; Schraft, H.; Qin, W. Fungal bioconversion of lignocellulosic residues; opportunities & perspectives. Int. J. Biol. Sci. 2009, 5, 578–594. [Google Scholar] [PubMed]
- Cherubini, F. The biorefinery concept: Using biomass instead of oil for producing energy and chemicals. Energy Convers. Manag. 2010, 51, 1412–1421. [Google Scholar] [CrossRef]
- Chen, L.; Gao, K.; Zhang, C.; Lang, W. Alternative fuels for IC engines and jet engines and comparison of their gaseous and particulate matter emissions. In Advanced Biofuels; Woodhead Publishing: Sawston, UK, 2019; pp. 17–64. [Google Scholar]
- Festucci-Buselli, R.A.; Otoni, W.C.; Joshi, C.P. Structure, organization, and functions of cellulose synthase complexes in higher plants. Braz. J. Plant. Physiol. 2007, 19, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Laureano-Perez, L.; Teymouri, F.; Alizadeh, H.; Dale, B.E. Understanding factors that limit enzymatic hydrolysis of biomass: Characterization of pretreated corn stover. Appl. Biochem. Biotechnol. 2005, 121, 1081–1099. [Google Scholar] [CrossRef]
- Saha, B.C. Hemicellulose bioconversion. J. Ind. Microbiol. Biotechnol. 2003, 30, 279–291. [Google Scholar] [CrossRef]
- Avgerinos, C.G.; Wang, D.I.C. Selective delignification for fermentation enhancement. Biotechnol. Bioeng. 1983, 25, 67–83. [Google Scholar] [CrossRef]
- Holtzapple, C.M.T. Fundamental factors affecting biomass enzymatic reactivity. Appl. Biochem. Biotechnol. 2000, 84, 5–37. [Google Scholar]
- Alvira, P.; Tomas-Pejo, E.; Ballesteros, M.; Negro, M.J. Pretreatment technologies for an efficient bioethanol production process based on enzymatic hydrolysis: A review. Bioresour. Technol. 2010, 101, 4851–4861. [Google Scholar] [CrossRef]
- Davis, G.; Henrissat, B. Structure and mechanism of glycosyl hydrolases. Structure 1995, 3, 853–859. [Google Scholar] [CrossRef] [Green Version]
- Aro, N.; Pakula, T.; Pentilla, M. Transcriptional regulation of plant cell wall degradation by filamentous fungi. FEMS Microbiol. Rev. 2005, 29, 719–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.H.P.; Lynd, L.R. Toward an aggregated understanding of enzymatic hydrolysis of cellulose: Noncomplexed cellulose systems. Biotechnol. Bioeng. 2004, 88, 797–824. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Li, Z.; Fang, X.; Wang, L.; Qu, Y. Cellulolytic enzyme production and enzymatic hydrolysis for second-generation bioethanol production. Adv. Biochem. Eng. Biotechnol. 2012, 128, 1–24. [Google Scholar] [PubMed]
- Teeri, T.; Koivula, A.; Linder, M.; Wohlfahrt, G.; Divne, C.; Jones, T. Trichoderma reesei cellobiohydrolases: Why so efficient on crystalline cellulose? Biochem. Soc. Trans. 1998, 26, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, A.; Lübeck, M.; Lübeck, P.S.; Ahring, B.K. Fungal beta-glucosidases: A bottleneck in industrial use of lignocellulosic materials. Biomolecules 2013, 3, 612–631. [Google Scholar] [CrossRef] [Green Version]
- Irwin, D.C.; Spezio, M.; Walker, L.P.; Wilson, D.B. Activity studies of eight purified cellulases: Specificity, synergism, and binding domain effects. Biotech. Bioeng. 1993, 42, 1002–1013. [Google Scholar] [CrossRef]
- Wilson, D.; Irwin, D. Genetics and properties of cellulases. Adv. Biochem. Eng. Biotechnol. 1999, 65, 1–21. [Google Scholar]
- Kuhad, R.C.; Gupta, R.; Singh, A. Microbial cellulases and their industrial applications. Enzym. Res. 2011, 2011, 280696. [Google Scholar] [CrossRef] [Green Version]
- Takashima, S.; Ohno, M.; Hidaka, M.; Nakamura, A.; Masaki, H.; Uozumi, T. Correlation between cellulose binding and activity of cellulose-binding domain mutants of Humicola grisea cellobiohydrolase I. FEBS Let. 2007, 581, 5891–5896. [Google Scholar] [CrossRef] [Green Version]
- Tomme, P.; Warren, R.; Gilke, N. Cellulose hydrolysis by bacteria and fungi. Adv. Microb. Physiol. 1995, 37, 1–81. [Google Scholar]
- Divine, C.J.; Stählberg, T.T.; Teeri, T.A.; Jones, J. High-resolution crystal structures reveal how a cellulose chain is bound in the 50 A long tunnel of cellobiohydrolase I from Trichoderma reesei. Mol. Biol. 1998, 275, 309–325. [Google Scholar] [CrossRef] [PubMed]
- Bayer, E.A.; Belaich, J.P.; Shoham, Y.; Lamed, R. The cellulosomes: Multienzyme machines for degradation of plant cell wall polysaccharides. Annu. Rev. Microbiol. 2004, 58, 521–554. [Google Scholar] [CrossRef] [PubMed]
- Lynd, L.R.; Weimer, P.J.; van Zyl, W.H.; Pretorius, I.S. Microbial cellulose utilization: Fundamentals and biotechnology. Microbiol. Mol. Biol. Rev. 2002, 66, 506–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, B.; Chen, X.; Su, Y.; Wan, Y. Enzyme adsorption and recycling during hydrolysis of wheat straw lignocellulose. Bioresour. Technol. 2011, 102, 2881–2889. [Google Scholar] [CrossRef] [PubMed]
- Rosales-Calderon, O.; Trajano, H.L.; Duff, S.J. Stability of commercial glucanase and β-glucosidase preparations under hydrolysis conditions. PeerJ 2014, 2, e402. [Google Scholar] [CrossRef] [PubMed]
- Woodward, J. Immobilized cellulases for cellulose utilization. J. Biotechnol. 1989, 11, 299–312. [Google Scholar] [CrossRef]
- Sternberg, D.; Mandels, G.R. Regulation of the cellulolytic system in Trichoderma reesei by sophorose: Induction of cellulase and repression of beta-glucosidase. J. Bacteriol. 1980, 144, 1197–1199. [Google Scholar] [CrossRef] [Green Version]
- Conn, E.E. b-Glycosidases in plants: Substrate specificity. In b-Glucosidases: Biochemistry and Molecular Biology; Esen, A., Ed.; American Chemical Society: Washington, DC, USA, 1993; pp. 15–26. [Google Scholar]
- Javed, M.R.; Buthe, A.; Rashid, M.H.; Wang, P. Cost-efficient entrapment of β-glucosidase in nanoscale latex and silicone polymeric thin films for use as stable biocatalysts. Food Chem. 2016, 190, 1078–1085. [Google Scholar] [CrossRef]
- Gómez, J.M.; Romero, M.D.; Fernandéz, T.M.; García, S. Immobilization and enzymatic activity of β-glucosidase on mesoporous SBA-15 silica. J. Porous Mater. 2010, 17, 657–662. [Google Scholar] [CrossRef]
- Gómez, J.M.; Romero, M.D.; Fernandéz, T.M.; Díez, E. Immobilization of β-glucosidase in fixed bed reactor and evaluation of the enzymatic activity. Bioprocess. Biosyst. Eng. 2012, 35, 1399–1405. [Google Scholar] [CrossRef]
- Wei, Y.; Das, S.; Berke-Schlessel, D.; Ji, H.F.; McDonough, J.; Feng, L.; Zhang, X.; Zhai, W.; Cao, Y. Synthesis of a re-usable cellobiase enzyme catalyst through in situ encapsulation in nonsurfactant templated sol–gel mesoporous silica. Top. Catal. 2012, 55, 1247–1253. [Google Scholar] [CrossRef]
- Das, S.; Berke-Schlessel, D.; Ji, H.F.; McDonough, J.; Wei, Y. Enzymatic hydrolysis of biomass with recyclable use of cellobiase enzyme immobilized in sol–gel routed mesoporous silica. J. Mol. Catal. B Enzym. 2011, 70, 49–54. [Google Scholar] [CrossRef]
- Guan, L.; Di, B.; Su, M.; Qian, J. Immobilization of β-glucosidase on bifunctional periodic mesoporous organosilicas. Biotechnol. Lett. 2013, 35, 1323–1330. [Google Scholar] [CrossRef] [PubMed]
- Yiu, H.H.P.; Wright, P.A.; Botting, N.P. Enzyme immobilisation using SBA-15 mesoporous molecular sieves with functionalised surfaces. J. Mol. Catal. B Enzym. 2001, 15, 81–92. [Google Scholar] [CrossRef]
- Reshmi, R.; Sugunan, S. Improved biochemical characteristics of crosslinked b-glucosidase on nanoporous silica foams. J. Mol. Catal. B Enzym. 2013, 85, 111–118. [Google Scholar]
- Ivetić, D.Z.; Srdić, V.V.; Antov, M.G. Immobilization of b-glucosidase onto a mesoporous silica support: Physical and covalent binding of the enzyme. J. Serb. Chem. Soc. 2014, 79, 533–543. [Google Scholar] [CrossRef] [Green Version]
- Chi, H.H.; Radu, D.R.; Dezayas, G.; Penney, M.; Lai, C.Y. Stellate MSN-based dual-enzyme nano-biocatalyst for the cascade conversion of non-food feedstocks to food products. J. Thermodyn. Catal. 2017, 8, 2. [Google Scholar]
- Gascón, V.; Márquez-Álvarez, C.; Blanco, R.M. Successful encapsulation of β-glucosidase during the synthesis of siliceous mesostructured materials. J. Chem. Technol. Biotechnol. 2018, 93, 2625–2634. [Google Scholar] [CrossRef]
- Califano, V.; Sannino, F.; Costantini, A.; Avossa, J.; Cimino, S.; Aronne, A. Wrinkled silica nanoparticles: Efficient matrix for β-glucosidase immobilization. J. Phys. Chem. C 2018, 122, 8373–8379. [Google Scholar] [CrossRef]
- Califano, V.; Costantini, A.; Silvestri, B.; Venezia, V.; Cimino, S.; Sannino, F. The effect of pore morphology on the catalytic performance of β-glucosidase immobilized into mesoporous silica. Pure Appl. Chem. 2019, 91, 1583–1592. [Google Scholar] [CrossRef]
- Sannino, F.; Costantini, A.; Ruffo, F.; Aronne, A.; Venezia, V.; Califano, V. Covalent immobilization of β-glucosidase into mesoporous silica nanoparticles from anhydrous acetone enhances its catalytic performance. Nanomaterials 2020, 10, 108. [Google Scholar] [CrossRef] [Green Version]
- Venezia, V.; Sannino, F.; Costantini, A.; Silvestri, B.; Cimino, S.; Califano, V. Mesoporous silica nanoparticles for β-glucosidase immobilization by templating with a green material: Tannic acid. Microporous Mesoporous Mater. 2020, 302, 110203. [Google Scholar] [CrossRef]
- Zlateski, V.; Keller, T.C.; Pérez-Ramírez, J.; Grass, R.N. Immobilizing and de-immobilizing enzymes on mesoporous silica. RSC Adv. 2015, 5, 87706–87712. [Google Scholar] [CrossRef]
- Lei, C.; Shin, Y.; Liu, J.; Ackerman, E.J. Entrapping enzyme in a functionalized nanoporous support. J. Am. Chem. Soc. 2002, 124, 11242. [Google Scholar] [CrossRef]
- Han, H.J.; Stucky, G.D.; Butler, A. Mesoporous silicate sequestration and release of proteins. J. Am. Chem. Soc. 1999, 121, 9897–9898. [Google Scholar] [CrossRef]
- Takimoto, A.; Shiomi, T.; Ino, K.; Tsunoda, T.; Kawai, A.; Mizukami, F.; Sakaguchi, K. Encapsulation of cellulase with mesoporous silica (SBA-15). Microporous Mesoporous Mater. 2008, 116, 601–606. [Google Scholar] [CrossRef]
- Harmoko, C.; Sucipto, K.I.; Retnoningtyas, E.S.; Hartono, S.B. Vinyl functionalized cubic mesoporous silica nanoparticles as supporting materials to enhance cellulase enzyme stability. ARPN J. Eng. Appl. Sci. 2016, 11, 2981–2992. [Google Scholar]
- Dadi, A.P.; Varanasi, S.; Schall, C.A. Enhancement of cellulose saccharification kinetics using an ionic liquid pretreatment step. Biotechnol. Bioeng. 2006, 95, 904–910. [Google Scholar] [CrossRef]
- Migneault, I.; Dartiguenave, C.; Bertrand, M.J.; Waldron, K.C. Glutaraldehyde: Behavior in aqueous solution, reaction with proteins, and application to enzyme crosslinking. Biotechniques 2004, 37, 790–802. [Google Scholar] [CrossRef]
- Kannan, K.; Jasra, R.V. Improved catalytic hydrolysis of carboxy methyl cellulose using cellulase immobilized on functionalized meso cellular foam. J. Porous Mater. 2011, 18, 409–416. [Google Scholar] [CrossRef]
- Zhang, D.; Hegab, H.E.; Lvov, Y.; Snow, L.D.; Palmer, J. Immobilization of cellulase on a silica gel substrate modified using a 3-APTES self-assembled monolayer. SpringerPlus 2016, 5, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, Y.; Parashar, A.; Bressler, D.C. Highly retained enzymatic activities of two different cellulases immobilized on non-porous and porous silica particles. Biotechnol. Bioproc. Eng. 2014, 19, 621–628. [Google Scholar] [CrossRef]
- Chen, B.; Qiu, J.; Mo, H.; Yu, Y.; Ito, K.; Sakai, E.; Feng, H. Synthesis of mesoporous silica with different pore sizes for cellulase immobilization: Pure physical adsorption. N. J. Chem. 2017, 41, 9338–9345. [Google Scholar] [CrossRef]
- Jannah Sulaiman, N.; Mansor, A.F.; Rahman, R.A.; Illias, R.M.; Shaarani, S.M. Adsorption kinetics of cellulase and xylanase immobilized on magnetic mesoporous silica. Chem. Eng. Technol. 2019, 42, 1825–1833. [Google Scholar] [CrossRef]
- Lee, Y.C.; Chen, C.T.; Chiu, Y.T.; Wu, K.C.W. An effective Cellulose-to-Glucose-to-Fructose conversion sequence by using Enzyme immobilized Fe3O4-loaded mesoporous silica nanoparticles as recyclable biocatalysts. ChemCatChem 2013, 5, 2153–2157. [Google Scholar] [CrossRef]
- Lee, Y.C.; Dutta, S.; Wu, K.C.W. Integrated, cascading enzyme-/chemocatalytic cellulose conversion using catalysts based on mesoporous silica nanoparticles. ChemSusChem 2014, 7, 3241–3246. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | Average Pore Size (nm) | Immobilization Technique | Loading (mg/g) | Residual Activity (%) | Ref. |
|---|---|---|---|---|---|
| SBA-15 | 7.5 | adsorption | 430 | 95 | [126,127] |
| fructose-template | 1.5–3.5 | entrapment | - | 7.5–82.8 | [128,129] |
| bifunctional organosilicas | 7.4 | adsorption | 120 | 95.5 | [130] |
| mesoporous cellular foam | 16.1 | adsorption/CLEAs | 139 | 77.4 | [132] |
| micro-sized aggregates | 29.0 | Adsorption covalent | 20 40 | 36.6 74.8 | [133] |
| stellate macroporous nanoparticles | 12.6 | adsorption | 40 | >90 | [134] |
| mesocellular foam | 9.0 | entrapment | 175 | 56 | [135] |
| wrinkled silica nanoparticles | 12.2 | adsorption | 150 | 125 | [136,137] |
| wrinkled silica nanoparticles | 12.2 | covalent | 150 | 120 | [138] |
| tannic acid template | 3.0–8.5 | adsorption | 78–130 | - | [139] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Califano, V.; Costantini, A. Immobilization of Cellulolytic Enzymes in Mesostructured Silica Materials. Catalysts 2020, 10, 706. https://doi.org/10.3390/catal10060706
Califano V, Costantini A. Immobilization of Cellulolytic Enzymes in Mesostructured Silica Materials. Catalysts. 2020; 10(6):706. https://doi.org/10.3390/catal10060706
Chicago/Turabian StyleCalifano, Valeria, and Aniello Costantini. 2020. "Immobilization of Cellulolytic Enzymes in Mesostructured Silica Materials" Catalysts 10, no. 6: 706. https://doi.org/10.3390/catal10060706
APA StyleCalifano, V., & Costantini, A. (2020). Immobilization of Cellulolytic Enzymes in Mesostructured Silica Materials. Catalysts, 10(6), 706. https://doi.org/10.3390/catal10060706